Modulation of Apoptotic Signaling by the Hepatitis B Virus X Protein

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
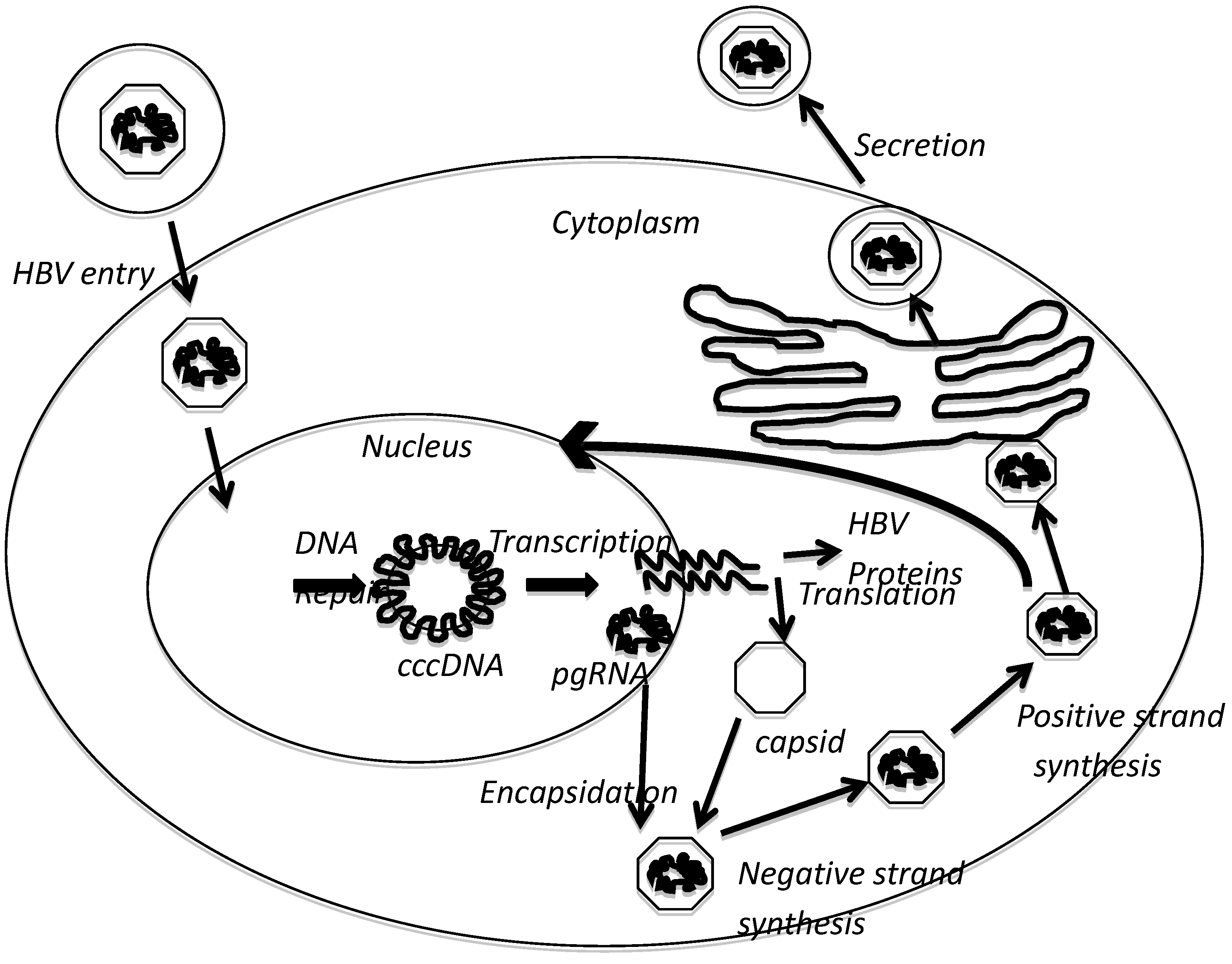
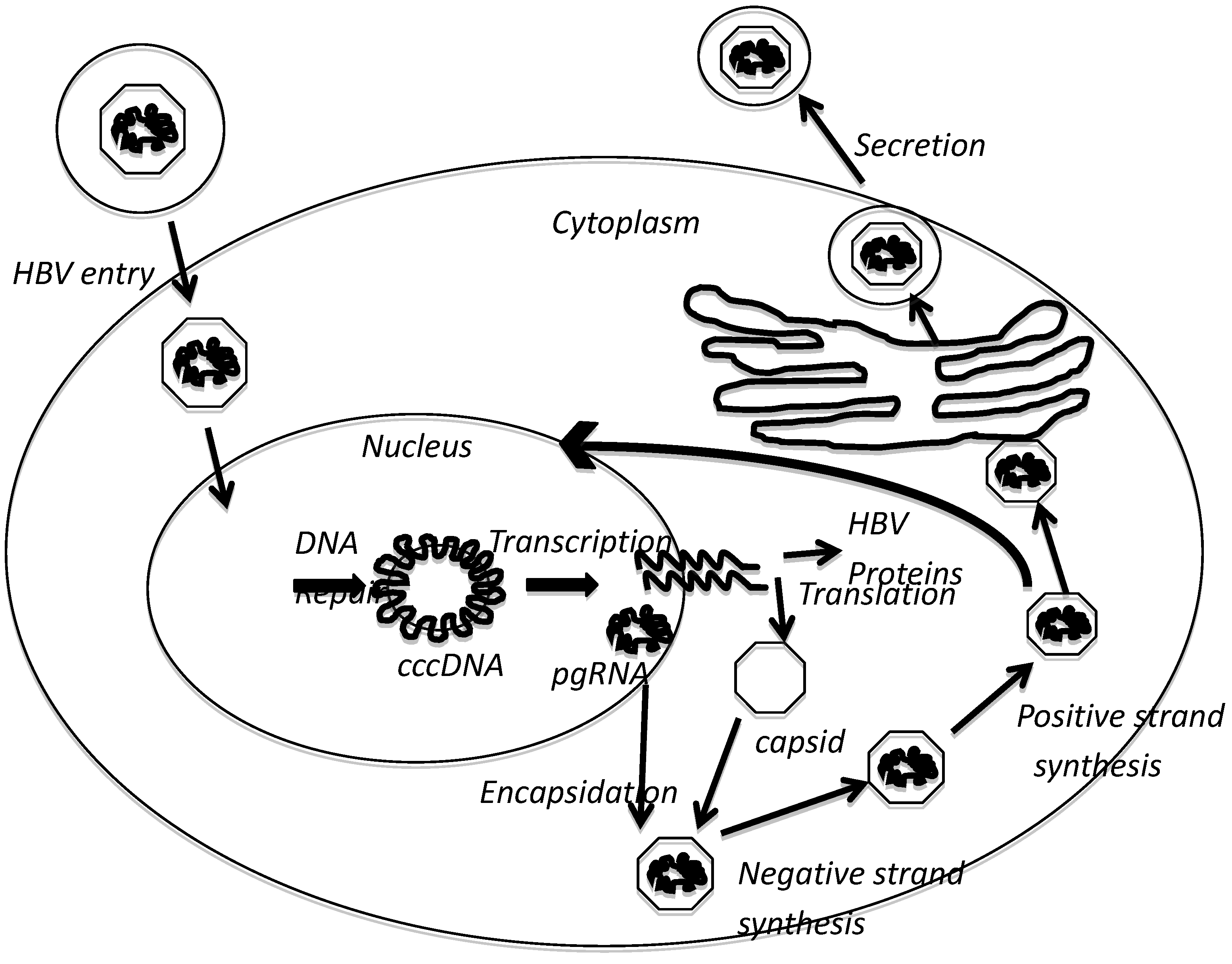
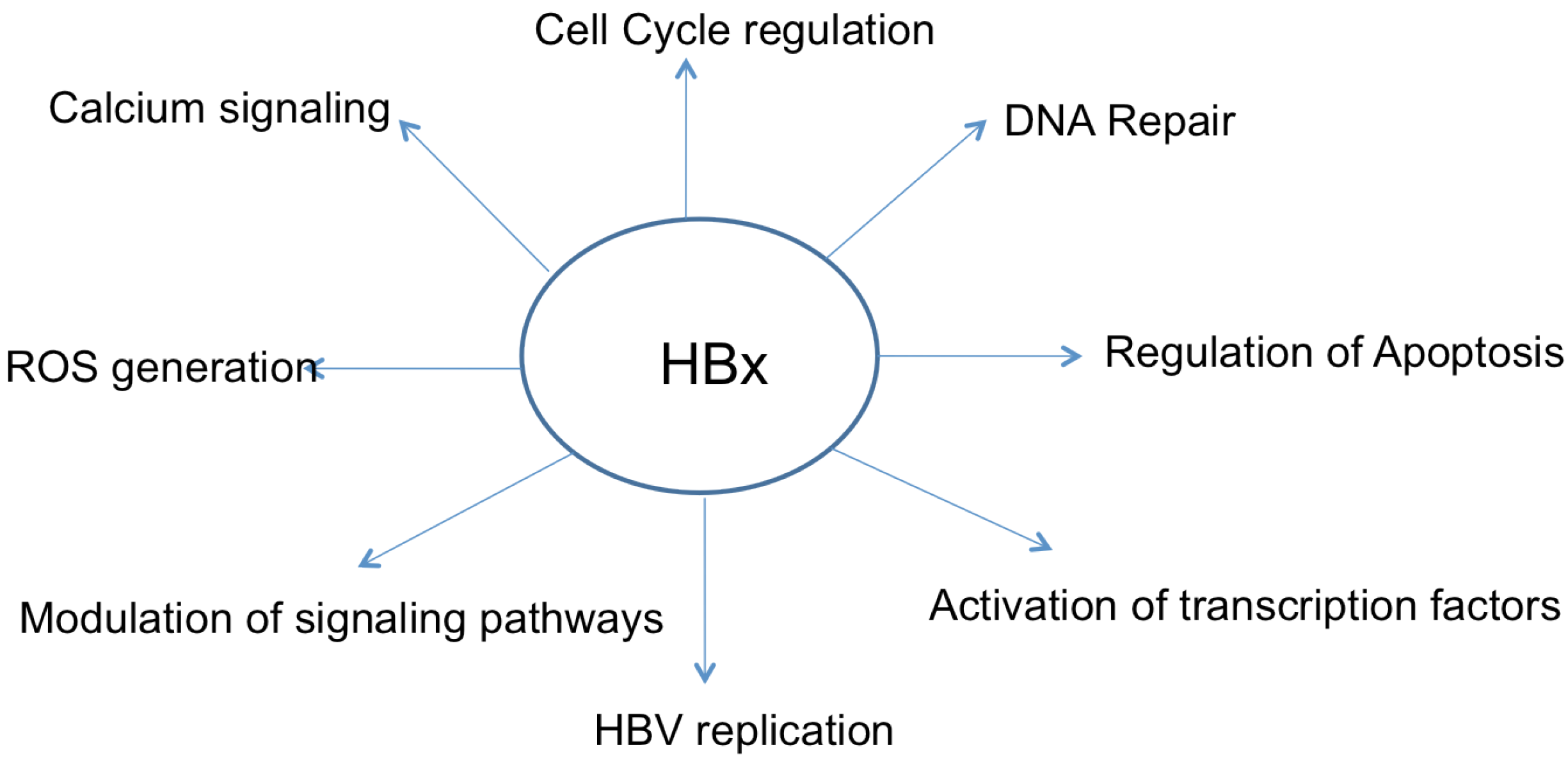
2. HBx Overview
3. HBx and HCC
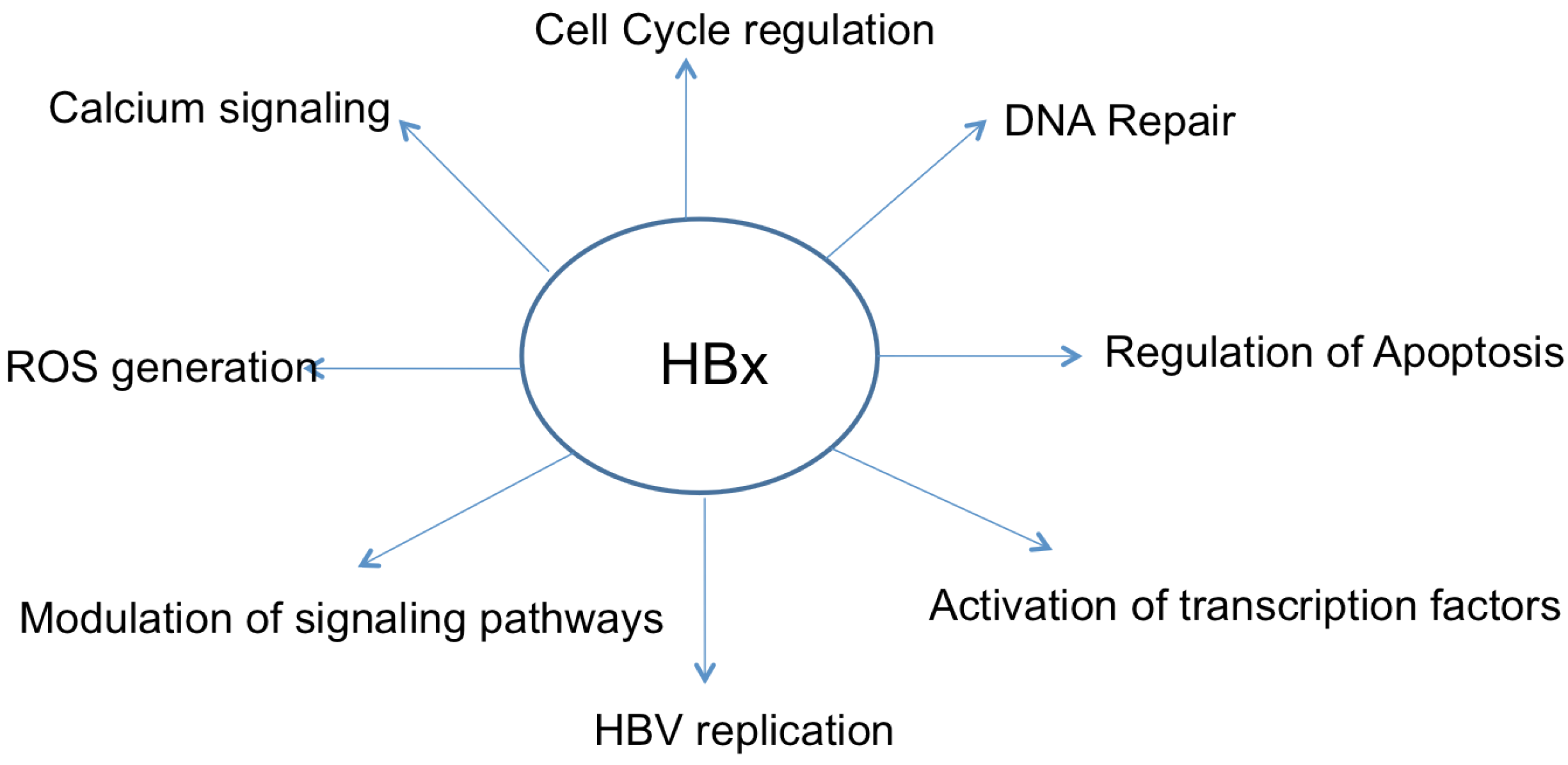
4. Apoptosis
5. HBx and Apoptosis
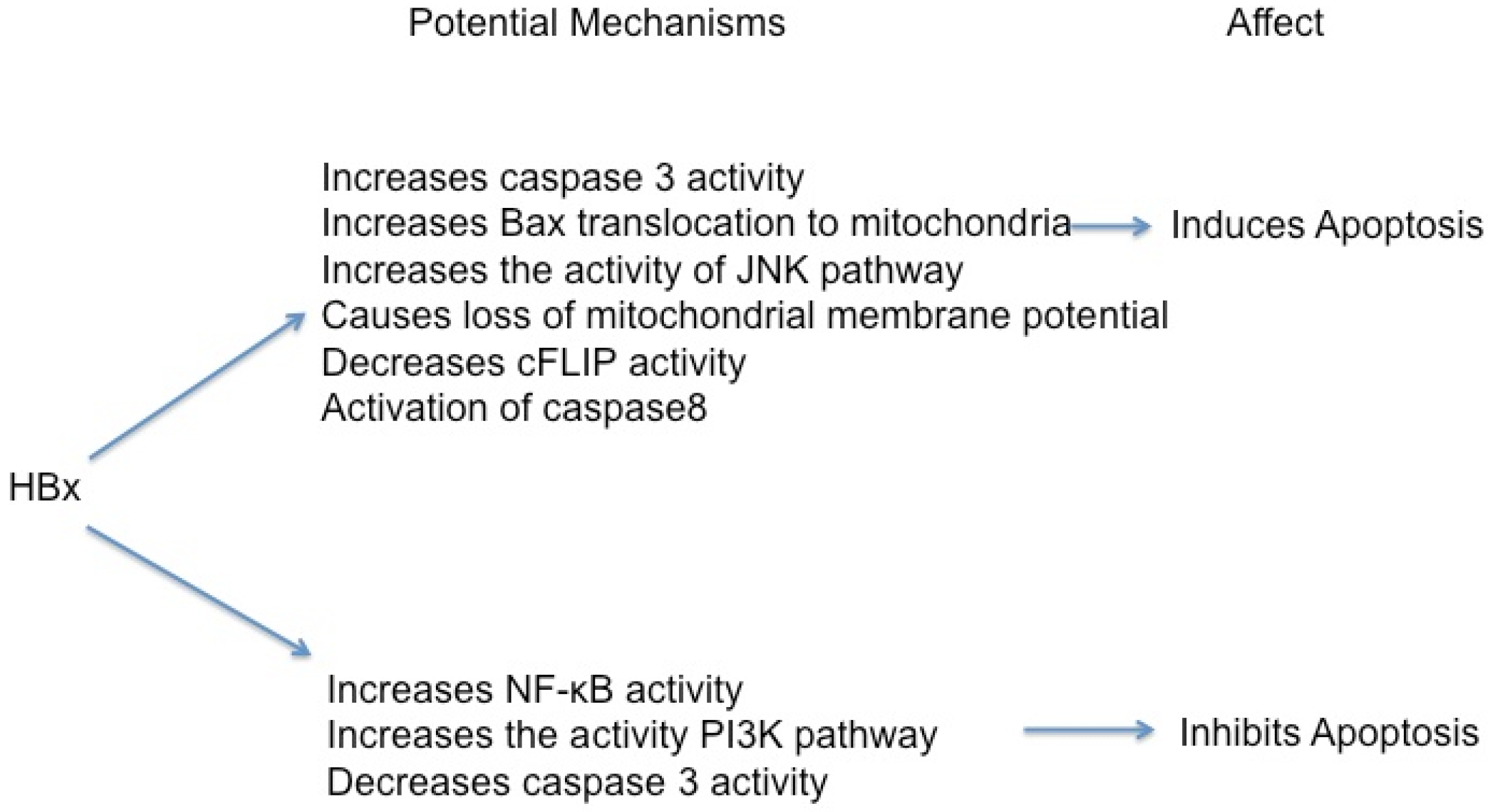
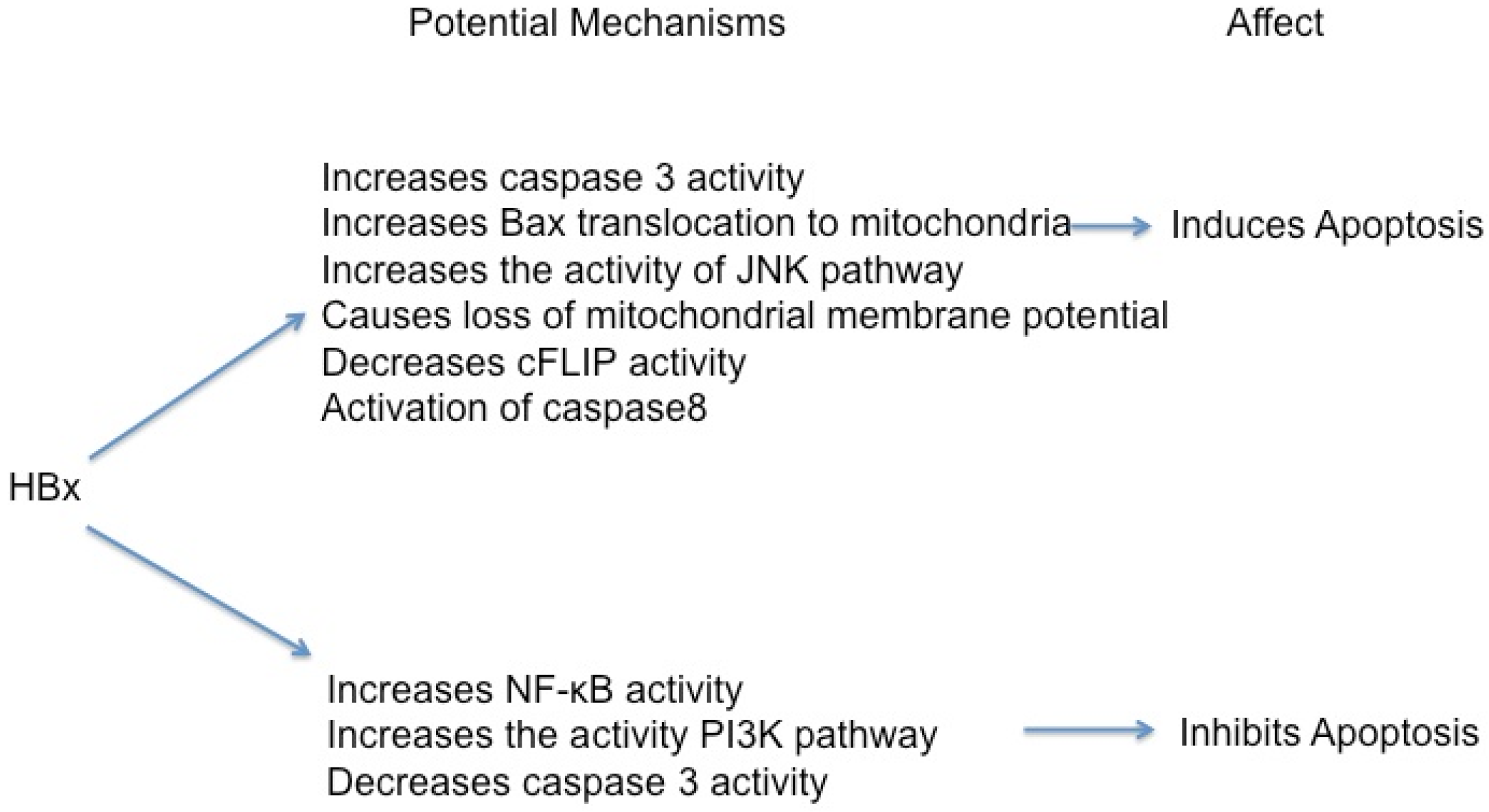
5.1. HBx Can Activate Apoptosis
5.2. HBx Can Inhibit Apoptosis
5.3. HBx, NF-κB, and Apoptosis
5.4. HBx, p53, and Apoptosis
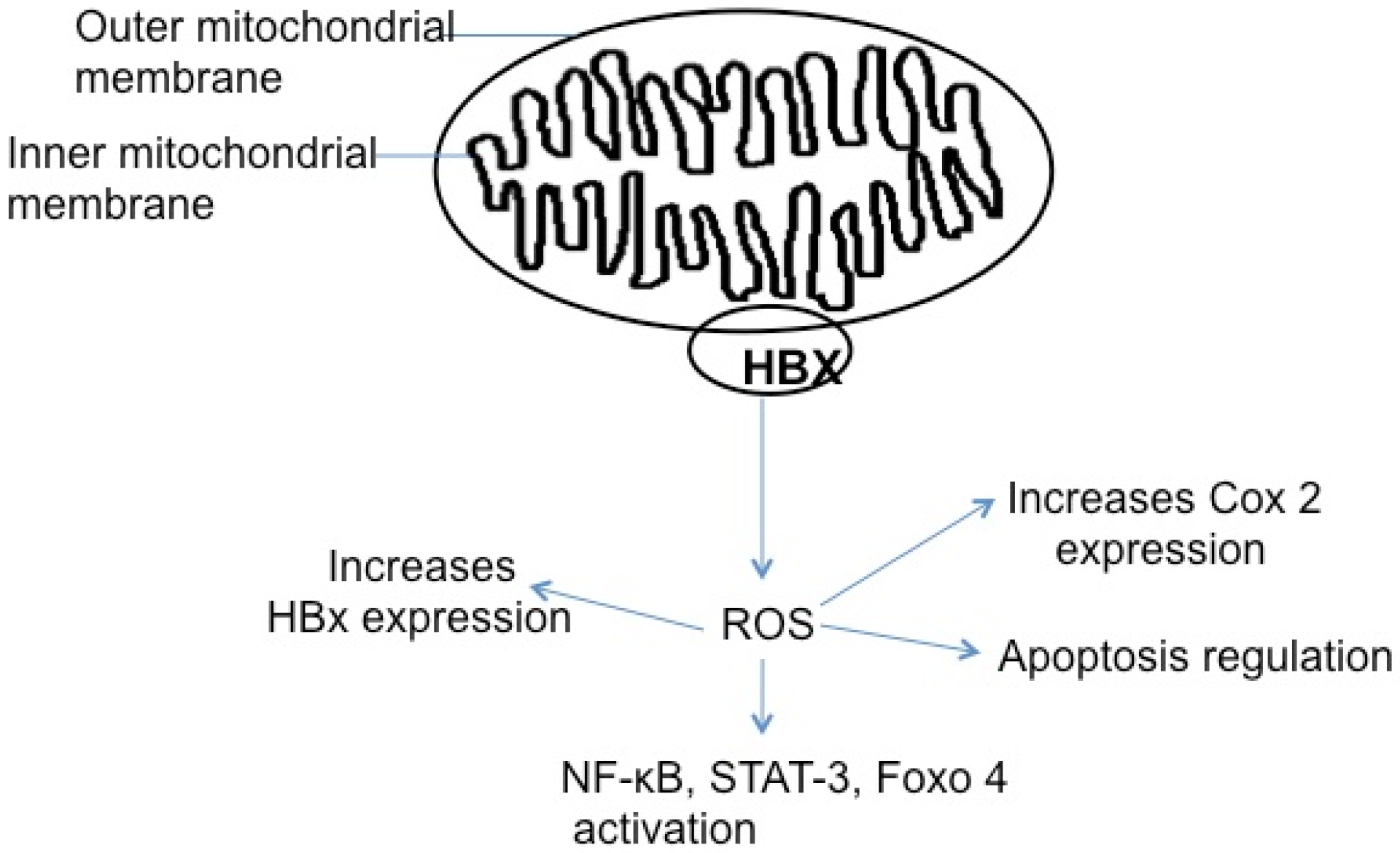
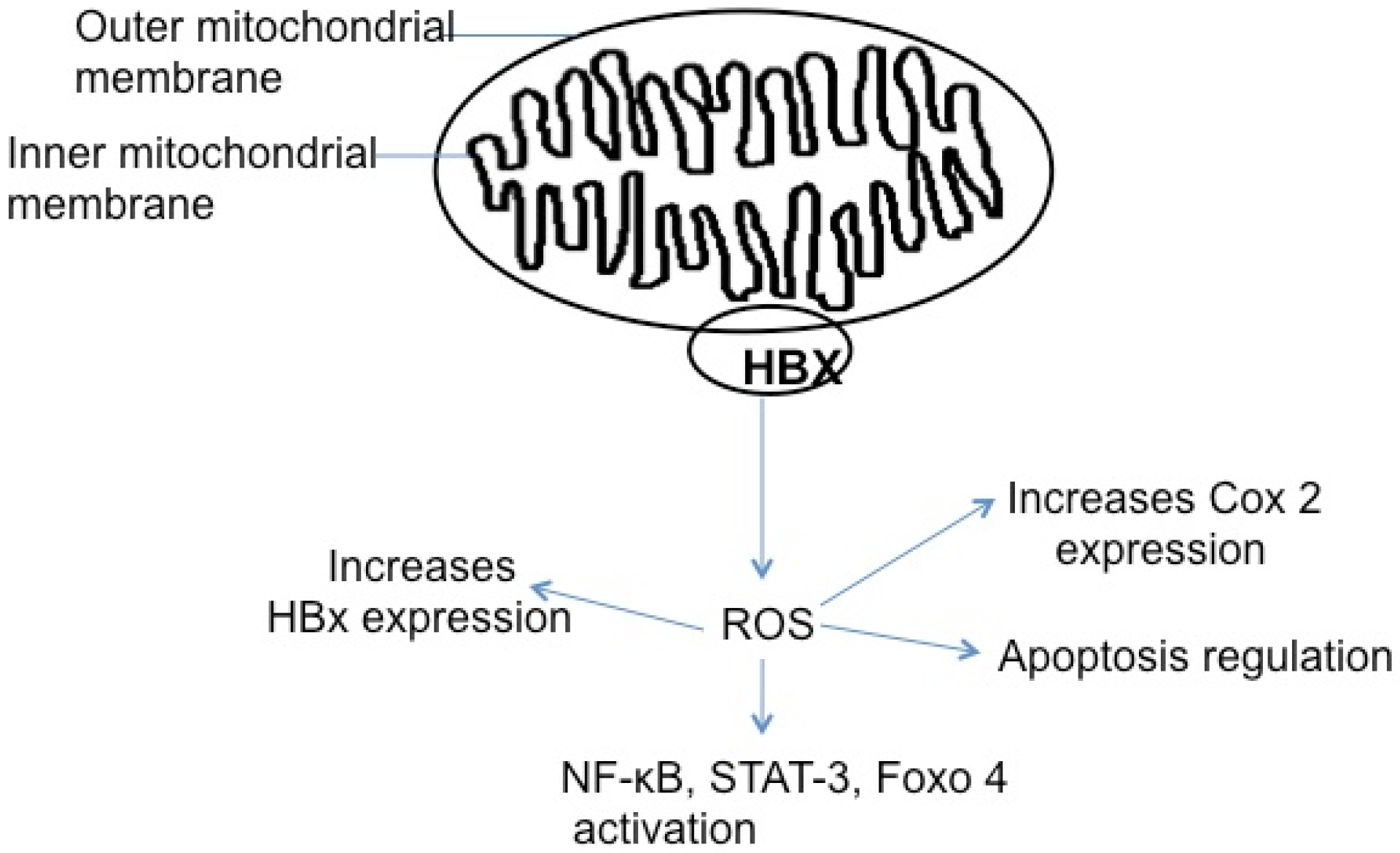
5.5. HBx, Reactive Oxygen Species, and Apoptosis
5.6. Effect of HBx on Apoptosis in HBx-Transgenic Mice
6. Conclusions and Perspective
References and Notes
- Nguyen, V.T.; Law, M.G.; Dore, G.J. Hepatitis B-related hepatocellular carcinoma: Epidemiological characteristics and disease burden. J. Viral Hepat. 2009, 16, 453–463. [Google Scholar] [CrossRef]
- Barrero, P.R.; Mistchenko, A.S. Genetic analysis of dengue virus type 3 isolated in Buenos Aires, Argentina. Virus Res. 2008, 135, 83–88. [Google Scholar] [CrossRef]
- Seeger, C.; Mason, W.S. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 2000, 64, 51–68. [Google Scholar] [CrossRef]
- Bertoletti, A.; Gehring, A.J. The immune response during hepatitis B virus infection. J. Gen. Virol. 2006, 87, 1439–1449. [Google Scholar] [CrossRef]
- De Meyer, S.; Gong, Z.J.; Suwandhi, W.; van Pelt, J.; Soumillion, A.; Yap, S.H. Organ and species specificity of hepatitis B virus (HBV) infection: A review of literature with a special reference to preferential attachment of HBV to human hepatocytes. J. Viral Hepat. 1997, 4, 145–153. [Google Scholar]
- Ganem, D.; Schnider, R.J. The Molecular Biology of the Hepatitis B Viruses, 4th ed; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2001; Volume 2. [Google Scholar]
- Huang, H.C.; Chen, C.C.; Chang, W.C.; Tao, M.H.; Huang, C. Entry of hepatitis B virus into immortalized human primary hepatocytes by clathrin-dependent endocytosis. J. Virol. 2012, 86, 9443–9453. [Google Scholar]
- Ganem, D.; Prince, A.M. Hepatitis B virus infection—Natural history and clinical consequences. New Engl. J. Med. 2004, 350, 1118–1129. [Google Scholar] [CrossRef]
- Watanabe, T.; Sorensen, E.M.; Naito, A.; Schott, M.; Kim, S.; Ahlquist, P. Involvement of host cellular multivesicular body functions in hepatitis B virus budding. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 10205–10210. [Google Scholar]
- Feitelson, M.A.; Lee, J. Hepatitis B virus integration, fragile sites, and hepatocarcinogenesis. Canc. Lett. 2007, 252, 157–170. [Google Scholar] [CrossRef]
- Bonilla Guerrero, R.; Roberts, L.R. The role of hepatitis B virus integrations in the pathogenesis of human hepatocellular carcinoma. J. Hepatol. 2005, 42, 760–777. [Google Scholar] [CrossRef]
- Berasain, C.; Castillo, J.; Perugorria, M.J.; Latasa, M.U.; Prieto, J.; Avila, M.A. Inflammation and liver cancer: New molecular links. Ann. New York Acad. Sci. 2009, 1155, 206–221. [Google Scholar]
- Bertoletti, A.; Gehring, A. Immune response and tolerance during chronic hepatitis B virus infection. Hepatol. Res. 2007, 37, S331–S338. [Google Scholar] [CrossRef]
- Bouchard, M.J.; Navas-Martin, S. Hepatitis B and C virus hepatocarcinogenesis: Lessons learned and future challenges. Canc. Lett. 2011, 305, 123–143. [Google Scholar] [CrossRef]
- Brechot, C.; Hadchouel, M.; Scotto, J.; Fonck, M.; Potet, F.; Vyas, G.N.; Tiollais, P. State of hepatitis B virus DNA in hepatocytes of patients with hepatitis B surface antigen-positive and -negative liver diseases. Proc. Natl. Acad. Sci. U. S. A. 1981, 78, 3906–3910. [Google Scholar] [CrossRef]
- Shafritz, D.A.; Kew, M.C. Identification of integrated hepatitis B virus DNA sequences in human hepatocellular carcinomas. Hepatology 1981, 1, 1–8. [Google Scholar] [CrossRef]
- Shafritz, D.A.; Shouval, D.; Sherman, H.I.; Hadziyannis, S.J.; Kew, M.C. Integration of hepatitis B virus DNA into the genome of liver cells in chronic liver disease and hepatocellular carcinoma. Studies in percutaneous liver biopsies and post-mortem tissue specimens. New Engl. J. Med. 1981, 305, 1067–1073. [Google Scholar] [CrossRef]
- Yaginuma, K.; Kobayashi, H.; Kobayashi, M.; Morishima, T.; Matsuyama, K.; Koike, K. Multiple integration site of hepatitis B virus DNA in hepatocellular carcinoma and chronic active hepatitis tissues from children. J. Virol. 1987, 61, 1808–1813. [Google Scholar]
- Paterlini-Brechot, P.; Saigo, K.; Murakami, Y.; Chami, M.; Gozuacik, D.; Mugnier, C.; Lagorce, D.; Brechot, C. Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene 2003, 22, 3911–3916. [Google Scholar]
- Murakami, Y.; Saigo, K.; Takashima, H.; Minami, M.; Okanoue, T.; Brechot, C.; Paterlini-Brechot, P. Large scaled analysis of hepatitis B virus (HBV) DNA integration in HBV related hepatocellular carcinomas. Gut 2005, 54, 1162–1168. [Google Scholar] [CrossRef]
- Wang, J.; Chenivesse, X.; Henglein, B.; Brechot, C. Hepatitis B virus integration in a cyclin A gene in a hepatocellular carcinoma. Nature 1990, 343, 555–557. [Google Scholar] [CrossRef]
- Tokino, T.; Matsubara, K. Chromosomal sites for hepatitis B virus integration in human hepatocellular carcinoma. J. Virol. 1991, 65, 6761–6764. [Google Scholar]
- Unsal, H.; Yakicier, C.; Marcais, C.; Kew, M.; Volkmann, M.; Zentgraf, H.; Isselbacher, K.J.; Ozturk, M. Genetic heterogeneity of hepatocellular carcinoma. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 822–826. [Google Scholar] [CrossRef]
- Paterlini, P.; Poussin, K.; Kew, M.; Franco, D.; Brechot, C. Selective accumulation of the X transcript of hepatitis B virus in patients negative for hepatitis B surface antigen with hepatocellular carcinoma. Hepatology 1995, 21, 313–321. [Google Scholar]
- Bill, C.A.; Summers, J. Genomic DNA double-strand breaks are targets for hepadnaviral DNA integration. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 11135–11140. [Google Scholar] [CrossRef]
- Guidotti, L.G.; Chisari, F.V. Immunobiology and pathogenesis of viral hepatitis. Annu. Rev. Pathol. 2006, 1, 23–61. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- He, G.; Karin, M. NF-kappaB and STAT3—Key players in liver inflammation and cancer. Cell Res. 2011, 21, 159–168. [Google Scholar] [CrossRef]
- Na, B.; Huang, Z.; Wang, Q.; Qi, Z.; Tian, Y.; Lu, C.C.; Yu, J.; Hanes, M.A.; Kakar, S.; Huang, E.J.; et al. Transgenic expression of entire hepatitis B virus in mice induces hepatocarcinogenesis independent of chronic liver injury. PLoS One 2011, 6, e26240. [Google Scholar]
- Kim, C.M.; Koike, K.; Saito, I.; Miyamura, T.; Jay, G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 1991, 351, 317–320. [Google Scholar] [CrossRef]
- Chung, T.W.; Lee, Y.C.; Kim, C.H. Hepatitis B viral HBx induces matrix metalloproteinase-9 gene expression through activation of ERK and PI-3K/AKT pathways: Involvement of invasive potential. FASEB J. 2004, 18, 1123–1125. [Google Scholar]
- Moon, E.J.; Jeong, C.H.; Jeong, J.W.; Kim, K.R.; Yu, D.Y.; Murakami, S.; Kim, C.W.; Kim, K.W. Hepatitis B virus X protein induces angiogenesis by stabilizing hypoxia-inducible factor-1alpha. FASEB J. 2004, 18, 382–384. [Google Scholar]
- Bouchard, M.J.; Schneider, R.J. The enigmatic X gene of hepatitis B virus. J. Virol. 2004, 78, 12725–12734. [Google Scholar] [CrossRef]
- Benhenda, S.; Cougot, D.; Buendia, M.A.; Neuveut, C. Hepatitis B virus X protein molecular functions and its role in virus life cycle and pathogenesis. Adv. Canc. Res. 2009, 103, 75–109. [Google Scholar] [CrossRef]
- Seeger, C.; Zoulim, F.; Mason, W. Hepadnaviruses. In Field's Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; Volume 2, pp. 2977–3029. [Google Scholar]
- Takada, S.; Shirakata, Y.; Kaneniwa, N.; Koike, K. Association of hepatitis B virus X protein with mitochondria causes mitochondrial aggregation at the nuclear periphery, leading to cell death. Oncogene 1999, 18, 6965–6973. [Google Scholar]
- Koike, K.; Moriya, K.; Yotsuyanagi, H.; Shintani, Y.; Fujie, H.; Tsutsumi, T.; Kimura, S. Compensatory apoptosis in preneoplastic liver of a transgenic mouse model for viral hepatocarcinogenesis. Canc. Lett. 1998, 134, 181–186. [Google Scholar] [CrossRef]
- Clippinger, A.J.; Gearhart, T.L.; Bouchard, M.J. Hepatitis B virus X protein modulates apoptosis in primary rat hepatocytes by regulating both NF-kappaB and the mitochondrial permeability transition pore. J. Virol. 2009, 83, 4718–4731. [Google Scholar] [CrossRef]
- Diao, J.; Khine, A.A.; Sarangi, F.; Hsu, E.; Iorio, C.; Tibbles, L.A.; Woodgett, J.R.; Penninger, J.; Richardson, C.D. X protein of hepatitis B virus inhibits Fas-mediated apoptosis and is associated with up-regulation of the SAPK/JNK pathway. J. Biol. Chem. 2001, 276, 8328–8340. [Google Scholar]
- Madden, C.R.; Finegold, M.J.; Slagle, B.L. Expression of hepatitis B virus X protein does not alter the accumulation of spontaneous mutations in transgenic mice. J. Virol. 2000, 74, 5266–5272. [Google Scholar] [CrossRef]
- Klein, A.; Guhl, E.; Tzeng, Y.J.; Fuhrhop, J.; Levrero, M.; Graessmann, M.; Graessmann, A. HBX causes cyclin D1 overexpression and development of breast cancer in transgenic animals that are heterozygous for p53. Oncogene 2003, 22, 2910–2919. [Google Scholar] [CrossRef]
- Guicciardi, M.E.; Gores, G.J. Apoptosis: A mechanism of acute and chronic liver injury. Gut 2005, 54, 1024–1033. [Google Scholar] [CrossRef]
- Clippinger, A.J.; Bouchard, M.J. Hepatitis B virus HBx protein localizes to mitochondria in primary rat hepatocytes and modulates mitochondrial membrane potential. J. Virol. 2008, 82, 6798–6811. [Google Scholar] [CrossRef]
- Rahmani, Z.; Huh, K.-W.; Lasher, R.; Siddiqui, A. Hepatitis B virus X protein colocalizes to mitochondria with human voltage-dependent anion channel, HVDAC3, and alters its transmembrane potential. J. Virol. 2000, 74, 2840–2846. [Google Scholar] [CrossRef]
- Kim, S.; Kim, H.-Y.; Lee, S.; Kim, S.; Sohn, S.; Kim, K.; Cho, H. Hepatitis B Virus X Protein induces perinuclear mitochondrial clustering in microtubule- and dynein-dependent manners. J. Virol. 2007, 81, 1714–1726. [Google Scholar] [CrossRef]
- Huh, K.; Siddiqui, A. Characterization of the mitochondrial association of hepatitis B virus X protein, HBx. Mitochondrion 2002, 1, 349–359. [Google Scholar] [CrossRef]
- Henkler, F.; Hoare, J.; Waseem, N.; Goldin, R.; McGarvey, M.; Koshy, R.; King, I. Intracellular localization of the hepatitis B virus HBx protein. J. Gen. Virol. 2001, 82, 871–882. [Google Scholar]
- Tanaka, Y.; Kanai, F.; Kawakami, T.; Tateishi, K.; Ijichi, H.; Kawabe, T.; Aradawa, Y.; Kawakami, T.; Nishimura, T.; Shirakata, Y.; et al. Interaction of the hepatitis B virus X protein (HBx) with heat shock protein 60 enhances HBx-mediated apoptosis. Biochem. Biophys. Res. Comm. 2004, 318, 461–469. [Google Scholar] [CrossRef]
- Shirakata, Y.; Koike, K. Hepatitis B virus X protein induces cell death by causing loss of mitochondrial membrane potential. J. Biol. Chem. 2003, 278, 22071–22078. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Israelson, A.; Brdiczka, D.; Sheu, S.S. The voltage-dependent anion channel (VDAC): Function in intracellular signalling, cell life and cell death. Curr. Pharmaceut. Des. 2006, 12, 2249–2270. [Google Scholar] [CrossRef]
- Blum, H.; Zhang, Z.-S.; Galun, E.; von Weizsacker, F.; Garner, B.; Liang, T.; Wands, J. Hepatitis B virus X protein is not central to the viral life cycle in vitro. J. Virol. 1992, 66, 1223–1227. [Google Scholar]
- Melegari, M.; Scaglioni, P.-P.; Wands, J. Cloning and characterization of a novel hepatitis B virus x binding protein that inhibits viral replication. J. Virol. 1998, 72, 1737–1743. [Google Scholar]
- Leupin, O.; Bontron, S.; Schaeffer, C.; Strubin, M. Hepatitis B virus X protein stimulates viral genome replication via a DDB1-dependent pathway distinct from that leading to cell death. J. Virol. 2005, 79, 4238–4245. [Google Scholar] [CrossRef]
- Tang, H.; Delgermaa, L.; Huang, F.; Oishi, N.; Liu, L.; He, F.; Zhao, L.; Murakami, S. The transcriptional transactivation function of HBx protein is important for its augmentation role in hepatitis B virus replication. J. Virol. 2005, 79, 5548–5556. [Google Scholar] [CrossRef]
- Bouchard, M.J.; Wang, L.H.; Schneider, R.J. Calcium signaling by HBx protein in hepatitis B virus DNA replication. Science 2001, 294, 2376–2378. [Google Scholar] [CrossRef]
- Lim, W.; Kwon, S.H.; Cho, H.; Kim, S.; Lee, S.; Ryu, W.S.; Cho, H. HBx targeting to mitochondria and ROS generation are necessary but insufficient for HBV-induced cyclooxygenase-2 expression. J. Mol. Med. (Berl) 2010, 88, 359–369. [Google Scholar] [CrossRef]
- Xu, Z.; Yen, T.; Wu, L.; Madden, C.; Tan, W.; Slagle, B.; Ou, J.-H. Enhancement of hepatitis B virus replication by its X protein in transgenic mice. J. Virol. 2002, 76, 2579–2584. [Google Scholar] [CrossRef]
- Keasler, V.V.; Hodgson, A.J.; Madden, C.R.; Slagle, B.L. Enhancement of hepatitis B virus replication by the regulatory X protein in vitro and in vivo. J. Virol. 2007, 81, 2656–2662. [Google Scholar] [CrossRef]
- Tsuge, M.; Hiraga, N.; Akiyama, R.; Tanaka, S.; Matsushita, M.; Mitsui, F.; Abe, H.; Kitamura, S.; Hatakeyama, T.; Kimura, T.; et al. HBx protein is indispensable for development of viraemia in human hepatocyte chimeric mice. J. Gen. Virol. 2010, 91, 1854–1864. [Google Scholar] [CrossRef]
- Lucifora, J.; Arzberger, S.; Durantel, D.; Belloni, L.; Strubin, M.; Levrero, M.; Zoulim, F.; Hantz, O.; Protzer, U. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J. Hepatol. 2011, 55, 996–1003. [Google Scholar] [CrossRef]
- Yoo, Y.G.; Oh, S.H.; Park, E.S.; Cho, H.; Lee, N.; Park, H.; Kim, D.K.; Yu, D.Y.; Seong, J.K.; Lee, M.O. Hepatitis B virus X protein enhances transcriptional activity of hypoxia-inducible factor-1alpha through activation of mitogen-activated protein kinase pathway. J. Biol. Chem. 2003, 278, 39076–39084. [Google Scholar]
- Wang, H.D.; Trivedi, A.; Johnson, D.L. Regulation of RNA polymerase I-dependent promoters by the hepatitis B virus X protein via activated Ras and TATA-binding protein. Mol. Cell. Biol. 1998, 18, 7086–7094. [Google Scholar]
- Becker, S.A.; Lee, T.H.; Butel, J.S.; Slagle, B.L. Hepatitis B virus X protein interferes with cellular DNA repair. J. Virol. 1998, 72, 266–272. [Google Scholar]
- Martin-Lluesma, S.; Schaeffer, C.; Robert, E.I.; van Breugel, P.C.; Leupin, O.; Hantz, O.; Strubin, M. Hepatitis B virus X protein affects S phase progression leading to chromosome segregation defects by binding to damaged DNA binding protein 1. Hepatology 2008, 48, 1467–1476. [Google Scholar] [CrossRef]
- Gearhart, T.L.; Bouchard, M.J. The hepatitis B virus X protein modulates hepatocyte proliferation pathways to stimulate viral replication. J. Virol. 2010, 84, 2675–2686. [Google Scholar] [CrossRef]
- Jung, J.K.; Kwun, H.J.; Lee, J.O.; Arora, P.; Jang, K.L. Hepatitis B virus X protein differentially affects the ubiquitin-mediated proteasomal degradation of beta-catenin depending on the status of cellular p53. J. Gen. Virol. 2007, 88, 2144–2154. [Google Scholar] [CrossRef]
- Bouchard, M.J.; Puro, R.J.; Wang, L.; Schneider, R.J. Activation and inhibition of cellular calcium and tyrosine kinase signaling pathways identify targets of the HBx protein involved in hepatitis B virus replication. J. Virol. 2003, 77, 7713–7719. [Google Scholar] [CrossRef]
- Gearhart, T.L.; Bouchard, M.J. Replication of the hepatitis B virus requires a calcium-dependent HBx-induced G1 phase arrest of hepatocytes. Virology 2010, 407, 14–25. [Google Scholar] [CrossRef]
- Gearhart, T.L.; Bouchard, M.J. The hepatitis B virus HBx protein modulates cell cycle regulatory proteins in cultured primary human hepatocytes. Virus Res. 2011, 155, 363–367. [Google Scholar] [CrossRef]
- McClain, S.L.; Clippinger, A.J.; Lizzano, R.; Bouchard, M.J. Hepatitis B virus replication is associated with an HBx-dependent mitochondrion-regulated increase in cytosolic calcium levels. J. Virol. 2007, 81, 12061–12065. [Google Scholar] [CrossRef]
- Koike, K.; Moriya, K.; Iino, S.; Yotsuyanagi, H.; Endo, Y.; Miyamura, T.; Kurokawa, K. High-level expression of hepatitis B virus HBx gene and hepatocarcinogenesis in transgenic mice. Hepatology 1994, 19, 810–819. [Google Scholar] [CrossRef]
- Dragani, T.A.; Manenti, G.; Farza, H.; Della Porta, G.; Tiollais, P.; Pourcel, C. Transgenic mice containing hepatitis B virus sequences are more susceptible to carcinogen-induced hepatocarcinogenesis. Carcinogenesis 1990, 11, 953–956. [Google Scholar] [CrossRef]
- Slagle, B.L.; Lee, T.H.; Medina, D.; Finegold, M.J.; Butel, J.S. Increased sensitivity to the hepatocarcinogen diethylnitrosamine in transgenic mice carrying the hepatitis B virus X gene. Mol. Carcinog. 1996, 15, 261–269. [Google Scholar] [CrossRef]
- Zheng, Y.; Chen, W.L.; Louie, S.G.; Yen, T.S.; Ou, J.H. Hepatitis B virus promotes hepatocarcinogenesis in transgenic mice. Hepatology 2007, 45, 16–21. [Google Scholar] [CrossRef]
- Kojima, H.; Kaita, K.D.; Xu, Z.; Ou, J.H.; Gong, Y.; Zhang, M.; Minuk, G.Y. The absence of up-regulation of telomerase activity during regeneration after partial hepatectomy in hepatitis B virus X gene transgenic mice. J. Hepatol. 2003, 39, 262–268. [Google Scholar] [CrossRef]
- Terradillos, O.; Billet, O.; Renard, C.A.; Levy, R.; Molina, T.; Briand, P.; Buendia, M.A. The hepatitis B virus X gene potentiates c-myc-induced liver oncogenesis in transgenic mice. Oncogene 1997, 14, 395–404. [Google Scholar]
- Madden, C.; Finegold, M.; Slagle, B. Hepatitis B virus X protein acts as a tumor promoter in development of diethylnitrosamine-induced preneoplastic lesions. J. Virol. 2001, 75, 3851–3858. [Google Scholar] [CrossRef]
- Madden, C.R.; Slagle, B.L. Stimulation of cellular proliferation by hepatitis B virus X protein. Dis. Markers 2001, 17, 153–157. [Google Scholar]
- Yu, D.Y.; Moon, H.B.; Son, J.K.; Jeong, S.; Yu, S.L.; Yoon, H.; Han, Y.M.; Lee, C.S.; Park, J.S.; Lee, C.H.; et al. Incidence of hepatocellular carcinoma in transgenic mice expressing the hepatitis B virus X-protein. J. Hepatol. 1999, 31, 123–132. [Google Scholar] [CrossRef]
- Nguyen, D.H.; Ludgate, L.; Hu, J. Hepatitis B virus-cell interactions and pathogenesis. J. Cell. Physiol. 2008, 216, 289–294. [Google Scholar] [CrossRef]
- Mason, W.S.; Litwin, S.; Xu, C.; Jilbert, A.R. Hepatocyte turnover in transient and chronic hepadnavirus infections. J. Viral Hepat. 2007, 14, 22–28. [Google Scholar]
- Whalley, S.A.; Murray, J.M.; Brown, D.; Webster, G.J.; Emery, V.C.; Dusheiko, G.M.; Perelson, A.S. Kinetics of acute hepatitis B virus infection in humans. J. Exp. Med. 2001, 193, 847–854. [Google Scholar] [CrossRef]
- Fabregat, I.; Roncero, C.; Fernandez, M. Survival and apoptosis: A dysregulated balance in liver cancer. Liver Int. 2007, 27, 155–162. [Google Scholar] [CrossRef]
- Schattenberg, J.M.; Schuchmann, M.; Galle, P.R. Cell death and hepatocarcinogenesis: Dysregulation of apoptosis signaling pathways. J. Gastroenterol. Hepatol. 2011, 26, 213–219. [Google Scholar]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef]
- Riedl, S.J.; Shi, Y. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell Biol. 2004, 5, 897–907. [Google Scholar] [CrossRef]
- Luo, X.; Budihardjo, I.; Zou, H.; Slaughter, C.; Wang, X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998, 94, 481–490. [Google Scholar] [CrossRef]
- Li, H.; Zhu, H.; Xu, C.J.; Yuan, J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef]
- Yin, X.M.; Wang, K.; Gross, A.; Zhao, Y.; Zinkel, S.; Klocke, B.; Roth, K.A.; Korsmeyer, S.J. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 1999, 400, 886–891. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Moldoveanu, T.; Llambi, F.; Parsons, M.J.; Green, D.R. The BCL-2 family reunion. Mol. Cell 2010, 37, 299–310. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Kim, K.H.; Seong, B.L. Pro-apoptotic function of HBV X protein is mediated by interaction with c-FLIP and enhancement of death-inducing signal. EMBO J. 2003, 22, 2104–2116. [Google Scholar] [CrossRef]
- Liang, X.; Liu, Y.; Zhang, Q.; Gao, L.; Han, L.; Ma, C.; Zhang, L.; Chen, Y.H.; Sun, W. Hepatitis B virus sensitizes hepatocytes to TRAIL-induced apoptosis through Bax. J. Immunol. 2007, 178, 503–510. [Google Scholar]
- Miao, J.; Chen, G.G.; Chun, S.Y.; Lai, P.P. Hepatitis B virus X protein induces apoptosis in hepatoma cells through inhibiting Bcl-xL expression. Canc. Lett. 2006, 236, 115–124. [Google Scholar] [CrossRef]
- Shintani, Y.; Yotsuyanagi, H.; Moriya, K.; Fujie, H.; Tsutsumi, T.; Kanegae, Y.; Kimura, S.; Saito, I.; Koike, K. Induction of apoptosis after switch-on of the hepatitis B virus X gene mediated by the Cre/loxP recombination system. J. Gen. Virol. 1999, 80, 3257–3265. [Google Scholar]
- Su, F.; Schneider, R. Hepatitis B virus HBx protein sensitizes cells to apoptotic killing by tumor necrosis factor α. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 8744–8749. [Google Scholar] [CrossRef]
- Terradillos, O.; de La Coste, A.; Pollicino, T.; Neuveut, C.; Sitterlin, D.; Lecoeur, H.; Gougeon, M.L.; Kahn, A.; Buendia, M.A. The hepatitis B virus X protein abrogates Bcl-2-mediated protection against Fas apoptosis in the liver. Oncogene 2002, 21, 377–386. [Google Scholar]
- Wang, W.H.; Gregori, G.; Hullinger, R.L.; Andrisani, O.M. Sustained activation of p38 mitogen-activated protein kinase and c-Jun N-terminal kinase pathways by hepatitis B virus X protein mediates apoptosis via induction of Fas/FasL and tumor necrosis factor (TNF) receptor 1/TNF-alpha expression. Mol. Cell. Biol. 2004, 24, 10352–10365. [Google Scholar] [CrossRef]
- Chirillo, P.; Pagano, S.; Natoli, G.; Puri, P.; Burgio, V.; Balsano, C.; Levrero, M. The hepatitis B virus X gene induces p53-mediated programmed cell death. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 8162–8167. [Google Scholar]
- Terradillos, O.; Pollicino, T.; Lecoeur, H.; Tripodi, M.; Gougeon, M.L.; Tiollais, P.; Buendia, M.A. p53-independent apoptotic effects of the hepatitis B virus HBx protein in vivo and in vitro. Oncogene 1998, 17, 2115–2123. [Google Scholar]
- Su, F.; Theodosis, C.; Schneider, R. Role of NF-κB and myc proteins in apoptosis induced by hepatitis B virus HBx protein. J. Virol. 2001, 75, 215–225. [Google Scholar] [CrossRef]
- Kim, H.; Kim, S.; Kim, J.; Lee, H.; Choi, M.; Kim, J.; Ahn, J. Hepatitis B virus X protein induces apoptosis by enhancing translocation of Bax to mitochondria. IUBMB 2008, 60, 473–480. [Google Scholar] [CrossRef]
- Chami, M.; Ferrari, D.; Nicotera, P.; Paterlini-Brechot, P.; Rizzuto, R. Caspase-dependent alterations of Ca2+ signaling in the induction of apoptosis by hepatitis B virus X protein. J. Biol. Chem. 2003, 278, 31745–31755. [Google Scholar]
- Kim, H.; Lee, H.; Yun, Y. X-gene product of hepatitis B virus induces apoptosis in liver cells. J. Biol. Chem. 1998, 273, 381–385. [Google Scholar] [CrossRef]
- Bergametti, F.; Prigent, S.; Luber, B.; Benoit, A.; Tiollais, P.; Sarasin, A.; Transy, C. The proapoptotic effect of hepatitis B virus HBx protein correlates with transactivation activity in stably transfected cell lines. Oncogene 1999, 18, 2860–2871. [Google Scholar] [CrossRef]
- Lee, Y.I.; Hwang, J.M.; Im, J.H.; Lee, Y.I.; Kim, N.S.; Kim, D.G.; Yu, D.Y.; Moon, H.B.; Park, S.K. Human hepatitis B virus-X protein alters mitochondrial function and physiology in human liver cells. J. Biol. Chem. 2004, 279, 15460–15471. [Google Scholar]
- Lee, Y.I.; Kang-Park, S.; Do, S.-I.; Lee, Y.I. The hepatitis B virus-X protein activates a phosphatidylinositol 3-kinase-dependent survival signaling cascade. J. Biol. Chem. 2001, 276, 16969–16977. [Google Scholar] [CrossRef]
- Shih, W.-L.; Kuo, M.-L.; Chuang, S.-E.; Cheng, A.-L.; Doong, S.-L. Hepatitis B virus X protein inhibits transforming growth factor-β-induced apoptosis through the activation of phosphatidylinositol 3-kinase pathway. J. Biol. Chem. 2000, 275, 25858–25864. [Google Scholar]
- Pan, J.; Duan, L.X.; Sun, B.S.; Feitelson, M.A. Hepatitis B virus X protein protects against anti-Fas-mediated apoptosis in human liver cells by inducing NF-kappa B. J. Gen. Virol. 2001, 82, 171–182. [Google Scholar]
- Marusawa, H.; Matsuzawa, S.; Welsh, K.; Zou, H.; Armstrong, R.; Tamm, I.; Reed, J.C. HBXIP functions as a cofactor of survivin in apoptosis suppression. EMBO J. 2003, 22, 2729–2740. [Google Scholar] [CrossRef]
- Gottlob, K.; Fulco, M.; Levrero, M.; Graessmann, A. The hepatitis B virus HBx protein inhibits caspase 3 activity. J. Biol. Chem. 1998, 273, 33347–33353. [Google Scholar] [CrossRef]
- Elmore, L.W.; Hancock, A.R.; Chang, S.F.; Wang, X.W.; Chang, S.; Callahan, C.P.; Geller, D.A.; Will, H.; Harris, C.C. Hepatitis B virus X protein and p53 tumor suppressor interactions in the modulation of apoptosis. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 14707–14712. [Google Scholar]
- Yun, C.; Um, H.-R.; Jin, Y.; Wang, J.-H.; Lee, M.-O.; Park, S.; Lee, J.-H.; Hyeseong, C. NF-κB activation by hepatitis B virus X (HBx) protein shifts the cellular fate toward survival. Canc. Lett. 2002, 184, 97–104. [Google Scholar] [CrossRef]
- Wu, J.C.; Merlino, G.; Fausto, N. Establishment and characterization of differentiated, nontransformed hepatocyte cell lines derived from mice transgenic for transforming growth factor alpha. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 674–678. [Google Scholar] [CrossRef]
- ATCC. Available online: http://www.atcc.org/ATCCAdvancedCatalogSearch/ProductDetails/tabid/452/Default.aspx?ATCCNum=CCL-13&Template=cellBiology (accessed on 28 October 2012).
- Twist, E.M.; Clark, H.F.; Aden, D.P.; Knowles, B.B.; Plotkin, S.A. Integration pattern of hepatitis B virus DNA sequences in human hepatoma cell lines. J. Virol. 1981, 37, 239–243. [Google Scholar]
- Meyer, J.L.; Fournier, J.G.; Bouteille, M. Expression of integrated hepatitis B virus DNA in PLC/PRF/5, Hep 3B, and L6EC3 cell lines detected by in situ hybridisation. Med. Biol. 1986, 64, 367–371. [Google Scholar]
- Wang, X.W.; Gibson, M.K.; Vermeulen, W.; Yeh, H.; Forrester, K.; Sturzbecher, H.W.; Hoeijmakers, J.H.; Harris, C.C. Abrogation of p53-induced apoptosis by the hepatitis B virus X gene. Canc. Res. 1995, 55, 6012–6016. [Google Scholar]
- Kang-Park, S.; Im, J.H.; Lee, J.H.; Lee, Y.I. PTEN modulates hepatitis B virus-X protein induced survival signaling in Chang liver cells. Virus Res. 2006, 122, 53–60. [Google Scholar] [CrossRef]
- Arzberger, S.; Hosel, M.; Protzer, U. Apoptosis of hepatitis B virus-infected hepatocytes prevents release of infectious virus. J. Virol. 2010, 84, 11994–12001. [Google Scholar] [CrossRef]
- Guidotti, L.G.; Matzke, B.; Schaller, H.; Chisari, F.V. High-level hepatitis B virus replication in transgenic mice. J. Virol. 1995, 69, 6158–6169. [Google Scholar]
- Tang, X.; Liu, D.; Shishodia, S.; Ozburn, N.; Behrens, C.; Lee, J.J.; Hong, W.K.; Aggarwal, B.B.; Wistuba, II. Nuclear factor-kappaB (NF-kappaB) is frequently expressed in lung cancer and preneoplastic lesions. Cancer 2006, 107, 2637–2646. [Google Scholar] [CrossRef]
- Garg, A.K.; Hortobagyi, G.N.; Aggarwal, B.B.; Sahin, A.A.; Buchholz, T.A. Nuclear factor-kappa B as a predictor of treatment response in breast cancer. Curr. Opin. Oncol. 2003, 15, 405–411. [Google Scholar] [CrossRef]
- Xiong, H.Q.; Abbruzzese, J.L.; Lin, E.; Wang, L.; Zheng, L.; Xie, K. NF-kappaB activity blockade impairs the angiogenic potential of human pancreatic cancer cells. Int. J. Canc. 2004, 108, 181–188. [Google Scholar] [CrossRef]
- Maeda, S.; Kamata, H.; Luo, J.L.; Leffert, H.; Karin, M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef]
- Sakurai, T.; Maeda, S.; Chang, L.; Karin, M. Loss of hepatic NF-kappa B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 10544–10551. [Google Scholar] [CrossRef]
- Li, W.; Tan, D.; Zenali, M.J.; Brown, R.E. Constitutive activation of nuclear factor-kappa B (NF-kB) signaling pathway in fibrolamellar hepatocellular carcinoma. Int. J. Clin. Exp. Pathol. 2010, 3, 238–243. [Google Scholar]
- Chan, C.F.; Yau, T.O.; Jin, D.Y.; Wong, C.M.; Fan, S.T.; Ng, I.O. Evaluation of nuclear factor-kappaB, urokinase-type plasminogen activator, and HBx and their clinicopathological significance in hepatocellular carcinoma. Clin. Canc. Res. 2004, 10, 4140–4149. [Google Scholar] [CrossRef]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef]
- Chirillo, P.; Falco, M.; Puri, P.L.; Artini, M.; Balsano, C.; Levrero, M.; Natoli, G. Hepatitis B virus pX activates NF-kappa B-dependent transcription through a Raf-independent pathway. J. Virol. 1996, 70, 641–646. [Google Scholar]
- Su, F.; Schneider, R. Hepatitis B virus HBx protein activates transcription factor NF-κB by acting on multiple cytoplasmic inhibitors of rel-related proteins. J. Virol. 1996, 70, 4558–4566. [Google Scholar]
- Waris, G.; Huh, K.-W.; Siddiqui, A. Mitochondrially associated hepatitis B virus X protein constitutively activates transcription factors STAT-3 and NF-κB via oxidative stress. MCB 2001, 21, 7721–7730. [Google Scholar]
- Andrisani, O.; Barnabas, S. The transcriptional function of the hepatitis B virus X protein and its role in hepatocarcinogenesis (Review). Int. J. Oncol. 1999, 15, 1–8. [Google Scholar]
- Lucito, R.; Schneider, R. Hepatitis B virus X protein activates transcription factor NF-κB without a requirement for protein kinase C. J. Virol. 1992, 66, 983–991. [Google Scholar]
- Mahe, Y.; Mukaida, N.; Kuno, K.; Akiyama, M.; Ikeda, N.; Matsushima, K.; Murakami, S. Hepatitis B virus X protein transactivates human interleukin-8 gene through acting on nuclear factor kB and CCAAT/enhancer-binding protein-like cis-elements. J. Biol. Chem. 1991, 266, 13759–13763. [Google Scholar]
- Bouchard, M.; Wang, L.; Schneider, R. Activation of Focal Adhesion Kinase by Hepatitis B Virus HBx Protein: Multiple functions in viral replication. J. Virol. 2006, 80, 4406–4414. [Google Scholar] [CrossRef]
- Purcell, N.; Yu, C.; He, D.; Xiang, J.; Paran, N.; DiDonato, J.; Yamaoka, S.; Shaul, Y.; Lin, A. Activation of NF-κB by hepatitis B virus X protein through an IkB kinase-independent mechansism. Am. J. Physiol. Gastointest. Liver Physiol. 2001, 280, G669–G677. [Google Scholar]
- Cong, Y.-S.; Yao, Y.-L.; Yang, W.-M.; Kuzhandaivelu, N.; Seto, E. The hepatitis B virus X-associated protein, XAP3, is a protein kinase C-binding protein. J. Biol. Chem. 1997, 272, 16482–16489. [Google Scholar] [CrossRef]
- Kekulé, A.; Lauer, U.; Weiss, L.; Luber, B.; Hofschneider, P. Hepatitis B virus transactivator HBx uses a tumour promoter signalling pathway. Nature 1993, 361, 742–745. [Google Scholar] [CrossRef]
- DiDonato, J.; Mercurio, F.; Rosette, C.; Wu-Li, J.; Suyang, H.; Ghosh, S.; Karin, M. Mapping of the inducible IkappaB phosphorylation sites that signal its ubiquitination and degradation. Mol. Cell. Biol. 1996, 16, 1295–1304. [Google Scholar]
- Liu, Q.; Chen, J.; Liu, L.; Zhang, J.; Wang, D.; Ma, L.; He, Y.; Liu, Y.; Liu, Z.; Wu, J. The X protein of hepatitis B virus inhibits apoptosis in hepatoma cells through enhancing the methionine adenosyltransferase 2A gene expression and reducing S-adenosylmethionine production. J. Biol. Chem. 2011, 286, 17168–17180. [Google Scholar]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef]
- Bressac, B.; Kew, M.; Wands, J.; Ozturk, M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature 1991, 350, 429–431. [Google Scholar]
- Yoon, Y.J.; Chang, H.Y.; Ahn, S.H.; Kim, J.K.; Park, Y.K.; Kang, D.R.; Park, J.Y.; Myoung, S.M.; Kim do, Y.; Chon, C.Y.; et al. MDM2 and p53 polymorphisms are associated with the development of hepatocellular carcinoma in patients with chronic hepatitis B virus infection. Carcinogenesis 2008, 29, 1192–1196. [Google Scholar] [CrossRef]
- Feitelson, M.A.; Zhu, M.; Duan, L.X.; London, W.T. Hepatitis B x antigen and p53 are associated in vitro and in liver tissues from patients with primary hepatocellular carcinoma. Oncogene 1993, 8, 1109–1117. [Google Scholar]
- Wang, X.W.; Forrester, K.; Yeh, H.; Feitelson, M.A.; Gu, J.R.; Harris, C.C. Hepatitis B virus X protein inhibits p53 sequence-specific DNA binding, transcriptional activity, and association with transcription factor ERCC3. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 2230–2234. [Google Scholar] [CrossRef]
- Lee, S.G.; Rho, H.M. Transcriptional repression of the human p53 gene by hepatitis B viral X protein. Oncogene 2000, 19, 468–471. [Google Scholar] [CrossRef]
- Wang, W.-H.; Hullinger, R.L.; Andrisani, O.M. Hepatitis B virus X protein via the p38MAPK pathway induces E2F1 release and ATR kinase activation mediating p53 apoptosis. J. Biol. Chem. 2008, 283, 25455–25467. [Google Scholar] [CrossRef]
- Wang, W.H.; Studach, L.L.; Andrisani, O.M. Proteins ZNF198 and SUZ12 are down-regulated in hepatitis B virus (HBV) X protein-mediated hepatocyte transformation and in HBV replication. Hepatology 2011, 53, 1137–1147. [Google Scholar] [CrossRef]
- Studach, L.L.; Menne, S.; Cairo, S.; Buendia, M.A.; Hullinger, R.L.; Lefrancois, L.; Merle, P.; Andrisani, O.M. Subset of Suz12/PRC2 target genes is activated during hepatitis B virus replication and liver carcinogenesis associated with HBV X protein. Hepatology 2012, 56, 1240–1251. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Mikkelsen, R.B.; Wardman, P. Biological chemistry of reactive oxygen and nitrogen and radiation-induced signal transduction mechanisms. Oncogene 2003, 22, 5734–5754. [Google Scholar] [CrossRef]
- Jiang, F.; Zhang, Y.; Dusting, G.J. NADPH oxidase-mediated redox signaling: Roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol. Rev. 2011, 63, 218–242. [Google Scholar] [CrossRef]
- Gwinn, M.R.; Vallyathan, V. Respiratory burst: Role in signal transduction in alveolar macrophages. J. Toxicol. Environ. Health B Crit. Rev. 2006, 9, 27–39. [Google Scholar] [CrossRef]
- Wang, J.H.; Yun, C.; Kim, S.; Lee, J.H.; Yoon, G.; Lee, M.O.; Cho, H. Reactive oxygen species modulates the intracellular level of HBx viral oncoprotein. Biochem. Biophys. Res. Comm. 2003, 310, 32–39. [Google Scholar] [CrossRef]
- Ha, H.L.; Yu, D.Y. HBx-induced reactive oxygen species activates hepatocellular carcinogenesis via dysregulation of PTEN/Akt pathway. World J. Gastroenterol. 2010, 16, 4932–4937. [Google Scholar] [CrossRef]
- Gu, J.M.; Lim, S.O.; Oh, S.J.; Yoon, S.M.; Seong, J.K.; Jung, G. HBx modulates iron regulatory protein 1-mediated iron metabolism via reactive oxygen species. Virus Res. 2008, 133, 167–177. [Google Scholar] [CrossRef]
- Mohammed, N.A.; Abd El-Aleem, S.A.; El-Hafiz, H.A.; McMahon, R.F. Distribution of constitutive (COX-1) and inducible (COX-2) cyclooxygenase in postviral human liver cirrhosis: A possible role for COX-2 in the pathogenesis of liver cirrhosis. J. Clin. Pathol. 2004, 57, 350–354. [Google Scholar] [CrossRef]
- Hu, L.; Chen, L.; Yang, G.; Li, L.; Sun, H.; Chang, Y.; Tu, Q.; Wu, M.; Wang, H. HBx sensitizes cells to oxidative stress-induced apoptosis by accelerating the loss of Mcl-1 protein via caspase-3 cascade. Mol. Canc. 2011, 10, 43. [Google Scholar] [CrossRef]
- Srisuttee, R.; Koh, S.S.; Park, E.H.; Cho, I.R.; Min, H.J.; Jhun, B.H.; Yu, D.Y.; Park, S.; Park do, Y.; Lee, M.O.; et al. Up-regulation of Foxo4 mediated by hepatitis B virus X protein confers resistance to oxidative stress-induced cell death. Int. J. Mol. Med. 2011, 28, 255–260. [Google Scholar]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog. 2006, 5, 14. [Google Scholar] [CrossRef]
- Brenner, D. Signal transduction during liver regeneration. J. Gastroenterol. Hepatol. 1998, 13, 93–95. [Google Scholar]
- FitzGerald, M.; Webber, E.; Donovan, J.; Fausto, N. Rapid DNA binding by nuclear factor kappa B in hepatocytes at the start of liver regeneration. Cell Growth Differ. 1995, 6, 417–427. [Google Scholar]
- Locaputo, S.; Carrick, T.; Bezerra, J. Zonal regulation of gene expression during liver regeneration of urokinase transgenic mice. Hepatology 1999, 29, 1106–1113. [Google Scholar] [CrossRef]
- Lindros, K.O.; Oinonen, T.; Issakainen, J.; Nagy, P.; Thorgeirsso, S.S. Zonal distribution of transcripts of four hepatic transcription factors in the mature rat liver. Cell Biol. Toxicol. 1997, 13, 257–262. [Google Scholar] [CrossRef]
- Fausto, N.; Webber, E.M. Liver regeneration. In The Liver: Biology and Pathobiology, 3rd; Arias, I.M., Boyer, J.L., Fausto, N., Jakoby, W.B., Schchter, D.A., Shafritz, D.A., Eds.; Raven: New York, NY, USA, 1994; pp. 1059–1084. [Google Scholar]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, K.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-kB functions as a tumor promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef]
- Sodunke, T.R.; Bouchard, M.J.; Noh, H.M. Microfluidic platform for hepatitis B viral replication study. Biomed. Microdevices 2008, 10, 393–402. [Google Scholar] [CrossRef]
- Tsang, V.L.; Bhatia, S.N. Fabrication of three-dimensional0 tissues. Adv. Biochem. Eng. Biot. 2007, 103, 189–205. [Google Scholar]
- Ploss, A.; Khetani, S.R.; Jones, C.T.; Syder, A.J.; Trehan, K.; Gaysinskaya, V.A.; Mu, K.; Ritola, K.; Rice, C.M.; Bhatia, S.N. Persistent hepatitis C virus infection in microscale primary human hepatocyte cultures. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 3141–3145. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rawat, S.; Clippinger, A.J.; Bouchard, M.J. Modulation of Apoptotic Signaling by the Hepatitis B Virus X Protein. Viruses 2012, 4, 2945-2972. https://doi.org/10.3390/v4112945
Rawat S, Clippinger AJ, Bouchard MJ. Modulation of Apoptotic Signaling by the Hepatitis B Virus X Protein. Viruses. 2012; 4(11):2945-2972. https://doi.org/10.3390/v4112945
Chicago/Turabian StyleRawat, Siddhartha, Amy J. Clippinger, and Michael J. Bouchard. 2012. "Modulation of Apoptotic Signaling by the Hepatitis B Virus X Protein" Viruses 4, no. 11: 2945-2972. https://doi.org/10.3390/v4112945