Innate Immunity to H5N1 Influenza Viruses in Humans

Department of Microbiology and the Emerging Pathogens Institute, Mount Sinai School of Medicine, New York, New York, USA
*
Authors to whom correspondence should be addressed.
Viruses 2012, 4(12), 3363-3388; https://doi.org/10.3390/v4123363
Submission received: 3 November 2012
/
Revised: 19 November 2012
/
Accepted: 21 November 2012
/
Published: 26 November 2012
(This article belongs to the Special Issue H5N1 Influenza Virus)
Abstract
:Avian influenza virus infections in the human population are rare due to their inefficient direct human-to-human transmission. However, when humans are infected, a strong inflammatory response is usually induced, characterized by elevated levels of cytokines and chemokines in serum, believed to be important in the severe pathogenesis that develops in a high proportion of these patients. Extensive research has been performed to understand the molecular viral mechanisms involved in the H5N1 pathogenesis in humans, providing interesting insights about the virus-host interaction and the regulation of the innate immune response by these highly pathogenic viruses. In this review we summarize and discuss the most important findings in this field, focusing mainly on H5N1 virulence factors and their impact on the modulation of the innate immunity in humans.
1. H5N1 viruses and human infection
Influenza A viruses (IAV) are negative-sense and single-stranded RNA, enveloped viruses, belonging to the family Ortomixoviridae. Their genome is organized in eight RNA segments, encoding up to 13 proteins [1,2,3]. The hemagglutinin (HA) and neuraminidase (NA) are the most abundant glycoproteins in the virion. To date, 17 HA and 9 NA subtypes have been described (reviewed by [4,5]) and the combination of those proteins results in diverse subtypes of IAV.
Different subtype combinations of IAV circulated in humans during the last century. The variation in circulating subtypes is usually a consequence of reassortment (antigenic shift) that occurs in animal reservoirs, resulting in a new subtype that is able to transmit to the human population (reviewed in [6]). The circulating subtypes as of 2012 are H1N1 viruses which caused a pandemic in 2009, and H3N2 viruses. Aquatic wild birds are the natural hosts of IAV. Sixteen HA subtypes (1–16) and nine NA subtypes have been detected in different combinations in aquatic wild birds, mostly establishing short-lived subclinical enteric infections (reviewed in [7,8]). Sporadically, viruses transmit from aquatic wild birds to poultry or mammals, and new genotypes of influenza virus may become established in these new “non-natural” hosts. The most important of those influenza viruses are the H5 and H7 subtypes. Some of them evolve to become genetic variants that are able to cause severe disease in poultry and mammals.
Since 1997 there have been several outbreaks of H5N1 influenza viruses transmitted to the human population directly from poultry, showing great virulence and low rates of survival [9,10,11,12]. These viruses are known as High Pathogenic Avian Influenza Viruses (HPAIV). Common symptoms at early stages of the infection are fever, cough and dyspnea, and in the most severe cases, development of acute respiratory distress syndrome and respiratory failure [10,12]. As of August 2012, more than 600 confirmed cases of infection by H5N1 viruses have been reported to the World Health Organization (WHO), with a lethal outcome in 59% of those documented cases [13]. However, there is evidence that indicates that this lethality rate might be overestimated due to reduced sensitivity of the WHO confirmation criteria, no formal H5N1 confirmation by health providers in rural areas, and subclinical or mild infections [14]. Fortunately, avian strains of IAV are not efficient at infecting humans [15], and direct transmission from human to human has been reported only in close family clusters, with very limited spread of the virus [16].
In this review we discuss the findings that researchers in the virology and immunology fields have reported to date regarding the induction and evasion of the innate immune response to HPAIV in humans, which is believed to contribute to the severe pathogenesis that these IAV cause in the human host. Also, we focus on the virulence factors of HPAIV that might contribute to the hyper-induction of cytokines or hypercytokinemia and/or evasion of the antiviral response.
2. Hypercytokinemia Induction in Humans by H5N1: Insights from ex vivo Experimental Models
Humans infected by H5N1 frequently present high serum levels of pro-inflammatory cytokines and chemokines [11,17,18], which is believed to contribute to the disease pathogenesis. Acute respiratory distress syndrome (ARSD), which has been compared to the severe acute respiratory syndrome virus due to Coronavirus (SARS-CoV) and associated with exaggerated inflammatory responses (reviewed in [19]), has been frequently observed in patients infected by H5N1 IAV with severe degrees of disease [20]. However, the precise mechanisms of H5N1 infection leading to hypercytokinemia in humans are still unclear.
The first steps in the initiation of immune responses involve sensing of pathogen associated molecular patterns (PAMPs) from the invading microorganism by pattern recognition receptors (PRRs) in infected cells, including dendritic cells (DCs). This recognition initiates a series of signaling events that result in the secretion of inflammatory cytokines, type I IFN, chemokines and antimicrobial peptides, and also induces maturation of DCs (reviewed by [21]). PRRs are distributed in different sub-cellular localizations. Thus, some of them are in the plasma membrane, as is the case of the toll-like receptors (TLR) such as TLR2, TLR4, TLR5, TLR6 and some C-type lectins. Others are located and sense PAMPs in the endosomal compartments, such as TLR3, TLR7 and TLR9. Additionally, the cell presents cytoplasmic receptors, as RNA helicases, that are capable of sensing intracellular viral RNAs, like the retinoic acid inducible gene I (RIG-I) and the melanoma differentiation antigen 5 (MDA5), or the Nucleotide Oligomerization Domain (NOD)-like receptors (NLRs) (reviewed by [22,23]).
The respiratory upper and lower airways are continuously exposed to pathogens, and therefore viral interactions with PRRs in cells of these tissues are crucial to elicit innate immune responses in the host against those pathogens (reviewed in [24]). The main cell targets for influenza viruses are epithelial cells in the lungs, which express TLR3 and RIG-I [25,26], both of them important for the induction of type I IFN upon sensing of influenza virus replicative and genomic RNA [26,27,28,29,30]. Immune cells such as macrophages and DCs are also present in the respiratory system, and given their high expression of PRRs and efficiency at producing pro-inflammatory cytokines upon activation (that also induce antiviral responses in neighboring cells), they represent crucial elements of the innate immune response to pathogens. DCs, and macrophages to a lower extent, are professional antigen-presenting cells (APCs), and therefore they link innate and adaptive immune responses (reviewed in [31,32]). Among DCs, both myeloid and plasmacytoid DCs (pDCs), are resident in the human airway and lung tissues [33,34,35,36]. Myeloid DCs mainly sense influenza viruses through RIG-I and TLR3 [37,38], while plasmacytoid DCs express high levels of TLR7, which recognizes influenza virus ssRNA in the endosomes resulting in high and early induction of type I IFN [39,40,41]. The most abundant macrophage population in the lung is the alveolar macrophages, which express TLR2, TLR3, TLR4, TLR5, and TLR6 at high levels [24,42,43], as well as RIG-I, which is important for the recognition of influenza viruses [44].
The use of ex vivo primary human cell systems has provided interesting insights into the innate immune responses generated against IAV in general, and H5N1 in particular. Table 1 summarizes the most relevant findings reported in those types of studies. Among the most frequently used models in IAV research are the primary human tracheo-bronchial epithelial cell (HTBE) and primary bronchial epithelial cells (BECs) [45], used either non-differentiated [46] or differentiated in an air-liquid interphase. Differentiation results in pseudo-stratified and polarized cultures, containing ciliated, secretory, and basal cells that resemble human airway epithelium [47,48]. Interestingly, Matrosovich et al. [48], using this differentiated cell system, were able to show that avian and human influenza viruses infect different cell types as a consequence of their different receptor specificity (SAα2,3 and SAα2,6, discussed in the next section). Some research works on the evaluation of the cytokine responses in primary HTBE cells, as well as on the role of type-I and type II alveolar pneumocytes in response to human and avian IAV, have shown higher levels of cytokine production upon infection with H5N1 viruses than H1N1 in these cells [46,49]. Another study that compared the levels of cytokine production in HTBE upon infection with H5N1 or H1N1 showed different results depending on the differentiation state of the cells. In that study, non-polarized cells showed stronger IFN production in response to H5N1 viruses, while this response was reduced in the polarized cultures, probably due to lower infection and replication levels [50]. Two additional studies that compared the cytokine profile induced by H5N1 viruses and H3N2 human virus in bronchial epithelial cell lines (Calu-3) or using primary BECs, showed delayed type I IFN induction in H5N1 infected cells compared to H3N2 infected cells [45,51]. Therefore, although H5N1 viruses tend to induce higher level of cytokines in primary epithelial cells than H1N1, there is no clear consensus regarding the innate immune responses induced by IAV of different origin, which might be explained by the use of different strains and subtypes, high genotypic variability and the differential contribution of the viral proteins to the induction or evasion of the immune system. Also, in the real in vivo scenario, other cell types might contribute to the cytokine response induced in lung epithelial cells.

{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of cytokine induction by avian H5N1 IAV and comparison with H3N2 and H1N1 human IAV in human cell experimental models.
| Cell culture system | IAV subtype | Cytokine induction | Genes up-regulated | Refs. | |
|---|---|---|---|---|---|
| Non-immune cells | HTBE (non polarized) | H5N1 vs H1N1 | Higher in H5N1 infected cells | IFN-β, IP-10, RANTES, IL-6, MCP-1, IL-8 | [46] |
| Primary BECs | H5N1 vs H3N2 | Attenuated in H5N1 | IFN, PKR, RIG-I | [45] | |
| Calu-3 | H5N1 vs H3N2 | Attenuated in H5N1 | IFN, PKR, RIG-I | [45] | |
| Polarized HTBE | H5N1 vs H3N2 | Attenuated in H5N1 | IFN-β, ISGs | [51] | |
| Polarized Calu-3 | H5N1 vs H3N2 | Attenuated in H5N1 | IFN-β, ISGs | [51] | |
| Polarized HTBE | H5N1 vs H1N1 | Attenuated in H5N1 | IFN-β | [50] | |
| HTBE (non polarized) | H5N1 vs H1N1 | Higher in H5N1 infected cells | IFN-β | [50] | |
| Primary Type I pneumocytes | H5N1 vs H1N1 | Higher in H5N1 infected cells | IFN, IP-10, RANTES, IL-6 | [49] | |
| Primary Type II pneumocytes | H5N1 vs H1N1 | Higher in H5N1 infected cells | IFN-β, IP-10, RANTES, IL-6 | [46] | |
| HMVEC (Primary endothelial cells) | H5N1 vs H1N1 | Higher in H5N1 infected cells | IFN-β, IL-7, TNF, CCL2 | [52] | |
| HUVEC (Primary endothelial cells) | H5N1 and H1N1 | Higher in H5N1 infected cells | IFN-β, ISGs | [53] | |
| HTBE (non polarized) | H5N1 | Induction of IP-10 | IP-10 | [18] | |
| HMVEC (Primary endothelial cells) | H5N1 | Induction of IP-10 | IP-10 | [18] | |
| Immune cells | hMDMs | H5N1 vs H1N1 | Higher in H5N1 infected cells | TNF-α, MCP-1, RANTES, IP-10, IL-1β, IL-6, IFN-αβ, and TRAIL | [54] |
| hMDMs | H5N1 vs H1N1 | Higher in H5N1 infected cells | RIG-I, MDA5, TLR3, IFN-β, TNF-α, IP-10 | [55] | |
| hMDMs | H5N1 and H1N1 or H3N2 | Higher in H5N1 than H1N1/H3N2 infected cells | NF α, IFN-α/β,IL-1β, MCP-1, MIP-1α, RANTES, IL-12 | [56] | |
| hMDMs | H5N1 and H1N1 or H3N2 | Higher in H5N1/H3N1 than in H1N1 infected cells | TNF α, IL-6, MIP-1α, IP-10 | [57] | |
| hMDMs | H5N1 vs H1N1 | Higher in H1N1 infected cells | TNF-α, IFN-β, IFN-λ1,IFN-α, MCP-1 | [58] | |
| hMDMs | H5N1 vs H1N1 | Higher in H1N1 infected cells | TNF-α, IFN-β, IFN-λ1, IP-10 | [59] | |
| MDDCs, mDCs and pDCs | H5N1 | IFN-α, TNF-α | [60] | ||
| pDCs | H5N1 and H1N1 or H3N2 | Higher in H5N1 than H1N1/H3N2 infected cells | IFN-α, TNF-α | [61] |
The innate immune responses to IAV have also been studied in endothelial cells by using primary human ex vivo models as well. The study by Zeng et al. [52] showed that primary human lung micro-vascular endothelial cells (HMVEC) support viral replication of H5N1 viruses and infection results in strong up-regulation of pro-inflammatory genes. Accordingly, other studies using Human Umbilical Vein Endothelial Cells (HUVEC) showed that H5N1 viruses induced strong inflammatory responses, mostly mediated by activation of the NF-κβ (nuclear factor kappa-light-chain-enhancer) pathway [52,53,62], supporting a possible role for endothelial cells in the hyper-induction of cytokines in infected patients.
It is well established that IAV infect human monocytes and macrophages, leading to high production of cytokines [63,64,65,66,67]. These immune cell types are critical for the induction of the innate immunity in the host. An interesting report by Lee et al. [55] explored the interaction between macrophages and epithelial cells after IAV infection using a cell culture system, and showed that cytokine mediators released from H5N1 infected human monocyte derived macrophages (hMDM) induced higher levels of pro-inflammatory cascades in epithelial cells than direct viral infection, supporting the important role of these phagocytic cells in the hyper-induction of cytokines in the lung observed after H5N1 IAV infection. This has been confirmed by several studies analyzing cytokine profiling of infected hMDM, that determined that H5N1 infection results in more elevated cytokine production than H1N1 viruses in these cells [54,56,57,58,59,68,69]. Additionally, a recent report by Cheung et al. [70] analyzed and compared the proteome variation induced by H5N1 and H1N1 viruses in hMDM, showing an early and enhanced translational activity after infection with H5N1 IAV than after H1N1 IAV infection, which might be important for the increased up-regulation of pro-inflammatory cytokine expression in H5N1 infected macrophages. On the other hand, IAV is known to induce apoptosis in human macrophages, which is also a mechanism believed to contribute to pathogenesis [57,71,72,73]. However, it is not clear if infection with H5N1 results in induction of apoptosis in a different fashion than seasonal human H1N1 or H3N2 influenza viruses, since conflicting data have been obtained in independent studies, suggesting that the ability of IAV to induce apoptosis might be strain rather than subtype dependent [57].
Human myeloid DCs are also susceptible to infection by IAV [66,74], while pDCs have been shown to be resistant [60]. Monocyte-derived DCs (MDDCs) is a well-established model to study antiviral responses in myeloid DCs, although research on the pro-inflammatory responses to H5N1 IAV strains has not been performed in sufficient depth in this system. Thitithanyanont et al. [60] showed that either blood isolated myeloid DCs (mDCs) or MDDCs can be infected by H5N1 IAV, resulting on the induction of high levels of IFN-α and, importantly, leading to cell death. The same study showed that pDCs, although resistant to infection, produce high levels of IFN-α in response to H5N1. Accordingly, Sandbulte et al. [61] compared the pDC responses to H5N1 with the ones to human IAV and observed increased IFN-α and TNF-α in H5N1 infected cells, which suggests a contribution of the resident DCs to the pro-inflammatory responses in the lungs upon H5N1 infection.
Therefore, research using ex vivo cell models, most of them involving primary cell systems, have provided valuable information about the characteristics of the immune responses elicited by avian H5N1 viruses in human cells. These data indicate that endothelial cells and immune cells, like macrophages and DCs, produce elevated levels of cytokines and chemokines upon encounter with HPAIV. Some of those data have been recently confirmed with improved ex vivo tissue systems by Weinheimer et al. [75], such as human lung organ tissues, which showed elevated levels of cytokine production upon infection with avian IAV strains. Macrophages and DCs, due to their high expression levels of PRRs, are likely to significantly contribute to the induction of the rapid and severe damage observed in human infected by H5N1 viruses by recruiting other immune cells via chemokine production, as monocytes and neutrophils, and stimulating the cytokine amplification in epithelial and endothelial cells. These events result in the release of pro-inflammatory mediators that have seriously damaging effects in airway and lung tissues [76]. Although the mechanisms for lung injury development during acute distress syndrome are no yet well understood, it is known that the oxidative stress mediated by the release of reactive oxygen species (ROS), and other cytotoxic mediators as a consequence of an acute innate immune response play an important role [77,78,79] (Figure 1).
A question that is also under investigation by many groups in the IAV field is why H5N1 avian influenza viruses cause this cytokine burst in humans. In the subsequent sections of this review we will cover the current knowledge on the modulation of the innate immune response in the host by different proteins of HPAIV virus. Some of these proteins have been shown to be involved in eliciting immune responses, as the HA, while others, as the non-structural protein (NS) 1, are known to be very efficient at antagonizing the host immune response.
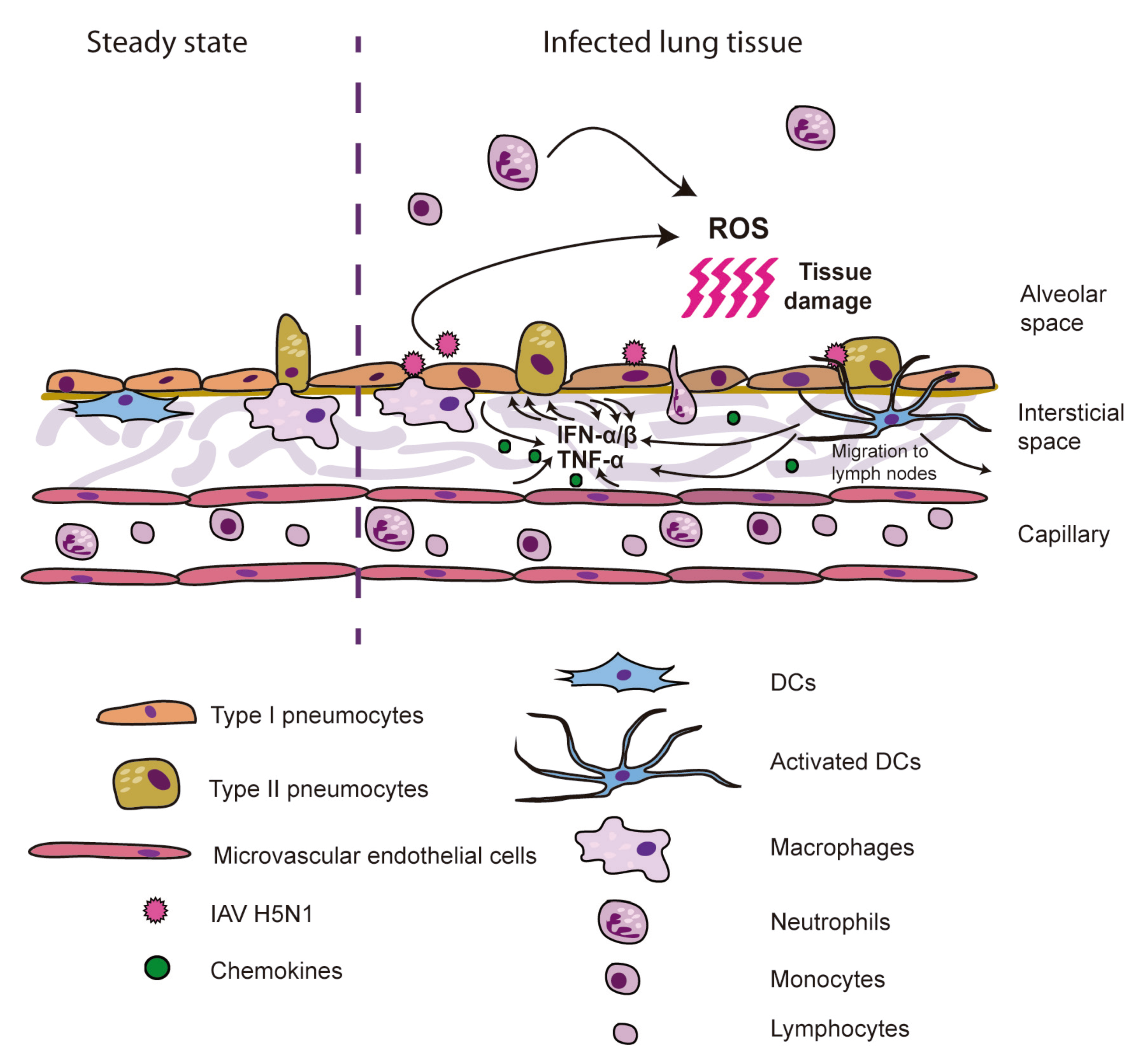
Figure 1.
Model for ARDS induction by H5N1 IAV. H5N1 viruses infect epithelial, endothelial and inmmune cells, which upon recognition of viral PAMPs produce high levels of cytokines and chemokines. Those induce increased production of cytokines by resident cells, as well as recruitment of neutrophils, monocytes and macrophages, responsible for the oxidative stress upon production of ROS, causing severe cell and tissue damage.
Figure 1.
Model for ARDS induction by H5N1 IAV. H5N1 viruses infect epithelial, endothelial and inmmune cells, which upon recognition of viral PAMPs produce high levels of cytokines and chemokines. Those induce increased production of cytokines by resident cells, as well as recruitment of neutrophils, monocytes and macrophages, responsible for the oxidative stress upon production of ROS, causing severe cell and tissue damage.

3. HA: Involvement in Entry and Cell Signaling
The HA of IAV plays an essential role in initiation of infection, since it interacts with sialic acid (N-acetylneuraminic acid) receptors (SA) on the cell surface [80], which allows for subsequent endocytosis of the viral particle. It also mediates fusion of the endosomal and viral membrane (reviewed in [81]). The main host species barrier that limits transmission of avian IAV to humans consists of the different receptor specificity of avian and human viruses [82,83,84]. SA are frequently attached through α2,3 or α2,6 (SAα2,3 or SAα2,6) linkages to the terminal galactose of the underlying sugar chains of glycoproteins in the cell membrane. It is known that HA from human isolates usually binds SAα2,6, which are abundant in the human upper respiratory tract, while the one from avian isolates usually has affinity for SAα2,3 [85]. Therefore, the low abundance of SAα2,3 in the human respiratory tract is one of the limiting factors for the lack of transmission of avian influenza viruses in humans. Recently, two independent groups have been able to identify some of the amino acid changes that allow airborne transmission of avian IAV to a mammalian host using the ferret model [86,87]. As other previous reports suggested before (reviewed by [88]), the protein that bears most of the modifications necessary for transmission in mammals is the HA, as increased affinity for SAα2,6 is necessary in order to productively infect cells in the upper respiratory tract. However, these studies also confirmed that changes in the receptor specificity are not sufficient for transmission of avian IAV to humans (or mammalian systems) and vice versa, and that modifications in the polymerase, specifically in the PB2 component [89,90], are necessary for transmission.
Although SAα2,6 are the most abundant in the respiratory system in humans, some cell types have been shown to present SAα2,3 glycoconjugates (therefore being susceptible to infection by H5N1 viruses) mainly in the lower respiratory tract, including type II pneumocytes and alveolar macrophages [82,83,84]. Additionally, human microvascular endothelial cells [52,91], human MDDCs [66] and human MDMs [68], present avian-like and human-like receptors on the surface, and therefore they are susceptible to infection by avian and human influenza viruses. Therefore, those cells bearing SAα2,3 receptors are relevant for initiation of infection by avian influenza viruses, and also for induction initial innate immune responses via PRRs, as RIG-I or TLR-dependent recognition.
Interaction of the HA with cell receptors is also believed to contribute to the induction of cytokine production (reviewed in [92]). Xu et al. [93] showed that exposure to HA from a HPAIV H5N1 (A/chicken/Guangdong/191/04) induced the activation of the Janus Kinases (JAK) 2 and 3, as well as the transcription factors STAT (signal transducer and activator of transcription) and NF-κβ of activated B cells, which correlated with induction of IP-10, IL-6, IL-8, MCP-1, MIP-1α, MIP-1β and RANTES. Another study that supports a possible role for influenza HA in cell signaling showed that recombinant H5 and H1 induce activation of human DCs [94]). Also, a study from our group showed that the avian or human-like receptor specificity might have an effect in the activation of the pro-inflammatory responses as observed after infections of primary human MDDCs, MDMs and HTBE cells with recombinant viruses bearing HA proteins with different receptor specificities [66]. Other reports have shown induction of different signaling pathways upon interaction of IAV and receptors in the cell surface. For instance, virus-receptor interaction has been reported to activate receptor tyrosine kinases (RTKs) [95], p38 mitogen-activated protein kinase (MAPK) and extracellular signal-regulated kinase (ERK) [96].
One of the most important virulence determinants of the HPAIV is the presence of a multibasic cleavage site (MBCS) as shown in avian and mammalian models [97,98]. This polybasic amino acid sequence in the cleavage site allows post-translational processing of the precursor HA0 to HA1 and HA2, which is necessary for fusion activity and virus entry, by furin and other subtilisin-like proprotein-processing endoproteases that are ubiquitinously expressed in tissues [99,100]. Cleavage of the low pathogenic avian IAV HA, on the other hand, is limited to trypsin-like proteases with a more restricted distribution, due to the presence of a single basic amino acid in the cleavage site [101]. Although avian viruses with HA with MBCS are known to have enhanced virulence, other viral factors might be necessary for this effect in mammals, since an interesting report showed that the introduction of a MBCS sequence in a human isolate H3N2 is not sufficient to increase pathogeneicity in ferrets [102]. The main role of the MBCS in pathogenesis by IAV is by allowing increased viral infection and replication in mammals [103], but it has also been associated to increased levels of cytokine production [103], probably as a result of higher detection of viral RNA by PRRs due to the increased levels of infection.
Natural Killer (NK) cells also represent important players in the innate immunity to viral infection. Upon activation and recruitment to the site of infection, NK cells produce cytokines and destroy infected cells (reviewed by [104]). There is evidence that influenza virus HA can induce activation of NK cells, by interacting with the receptors NKp46 and NKp44 [105,106,107], which is thought to be a mechanism for killing infected cells. This interaction seems to be dependent on the sialylation of the receptor, and have been demonstrated for H5 and H1 (that bind through SAα2,3 and SAα2,6, respectively). However, NKp46 interactions with H5, as opposed to the ones with a swine origin H1 2009, are insufficient to activate the NKp46-mediated killing [108], suggesting that H5 pathogeneicity might be increased by this lack of ability to activate NK cells. On the other hand, another study showed that infection with pseudotyped particles expressing influenza virus HA induced higher levels of human NK cell activation as shown by up-regulation of the activation marker CD69 [109], and in this case H5N1 and H1N1 1918 expressing pseudo-particles induced higher levels of activation that the ones expressing an H1N1 2009 HA, which suggests different outcomes depending of the viral HA. Due to the variety of sialylated glycans that HA from different IAV strains interact with [110,111], it would be possible that those interactions have diverse outcomes depending on the IAV subtype/origin, resulting in different levels of pathogenesis.
4. NS1: The Innate Immunity Suppressor
The NS1 protein from IAV is one of the strongest viral antagonists that have been described and its numerous functions have been extensively studied. However, given the complexity and great number of functional activities, inter-connection and overlapping of those functions and high strain variability, there is still much to learn about this innate immune suppressor. IAV NS1 protein has multiple different functions during cell infection, most of them related to its inhibitory effects on of type I IFN production and NF-κβ activation [112] (reviewed by [113,114]). NS1 is a dsRNA binding protein, and this feature allows it to sequester the viral RNA and prevent activation of the 2'-5' oligo (A) synthetase (OAS)/RNase L pathway and of the protein kinase RNA-activated (PKR) [115,116]. Also, the dsRNA binding domain mediates direct interaction with RIG-I, TRIM-25 and PKR, resulting on a dampened antiviral state mediated by those IFN inducible genes (ISGs) and increased viral replication [117,118,119,120]. On the other hand, the C-terminal or effector domain of NS1 presents the ability to specifically inhibit 3’ processing of cellular pre-mRNA, and therefore to reduce the protein expression of cellular proteins involved in the host cell immune response, by interacting with the 30 KDa subunit of the cleavage and polyadenylation-specificity factor (CPSF30) [121,122] and the poly(A) binding protein II [123]. Recenly, Gao et al. [124] described that the C-terminal domain of an NS1 from an H5N1 targets IKKα and IKKβ and therefore suppresses NF-κβ activation. Additionally, IAV NS1 has been described to hijack the host translational machinery to enhance viral translation [125,126], and can also interact with the p85β regulatory subunit of PI3K, resulting in subsequent Akt activation [127]. Another recent publication has shown that the C-terminal region of the NS1 of human H3N2 influenza viruses acts as a histone tail that mimics and inhibits host transcriptional elongation by interacting with the human PAF complex [128]. All these data together support the idea that the NS1 is a multifunctional innate immune inhibitor and is an important virulence factor for IAV.
Some functional differences have been described between NS1 from avian and human IAV, although due to the NS1 complexity it is difficult to determine if those differences might impact the innate immune response of HPAIV H5N1 in humans. The study from Kainov et al. [129] showed the avian IAV NS1 increased translation in an in vitro cell-free assay, in contrast to the NS1 from a human H1N1 2009 isolate. However, this same study showed that the H5N1 NS1 presented a stronger poly-adenylation inhibitory capacity (higher CPSF inhibition) than an NS1 from a human or low pathogenic avian virus. Human IAV present two highly conserved amino acids in the central region of the NS1 protein, F at 103 and M at 106, necessary for stabilization of the NS1-CPSF30 complex [130,131]. NS1 from avian H5N1 viruses isolated from human before 1997 do not present these amino acids and consequently they are unable to inhibit CPSF30 efficiently. However, it also has been shown that an H5N1 virus bearing an L103/I106 NS1 was also able to inhibit CPSF30 function to some extent since other viral proteins, specifically PA and NP, stabilize the NS1-CPSF30 complex [132].
CPSF30 inhibition has been associated with limitation of the production of type I IFN and pro-inflammatory cytokine production in cell culture [122,133] (unpublished observations from our laboratory). In contrast, in vivo studies showed that introduction of the amino acids F103/M106 in an avian H5N1 1997 isolate NS1 increases pathogeneicity, replication and cytokine production in mice [134]. So therefore, these contradictory data in cell culture vs. in vivo models seem to indicate that the NS1 antagonist function might be critical at early steps of the infection, allowing the virus to replicate more efficiently, which in subsequent stages of the infection in vivo results in increased release of type I IFN and pro-inflammatory cytokines. Interestingly, H5N1 isolated from humans after 1998 viruses present the amino acids F103/M106, correcting the defective CPSF30 binding and showing increased virulence [135].
Another feature of IAV NS1 is that the C-terminal four amino acids constitute a PDZ binding domain (PBD), as identified by Obenauer and colleagues [136] while performing a large-scale sequence analysis of avian influenza isolates. They observed that the most represented sequence in avian viruses was ESEV, while the consensus sequence for the human isolates examined was RSKV. Also, they determined that EPEV and ESEV were characteristically found in HPAIV human infections in 1997 and 2003, and KSEV in the 1918 pandemic virus. Interestingly, those motifs showed better ability to bind some known PDZ containing human proteins, and subsequent work demonstrated that these sequences are pathogenicity determinants in mice [137,138]. PDZ domains are present in a great diversity of proteins, and play important roles in multiple biological processes, such as in assembly of supra-molecular signaling complexes, protein transport and localization, and cell polarity organization (reviewed in [139]). Therefore, it is not surprising that the differential interaction of NS1 with these proteins might have multiple consequences regarding apoptosis, immune responses or viral replication. However, these processes are not well characterized yet, although some of those proteins that interact with EPEV and ESEV sequences have been already identified [140,141,142,143,144]. For instance, NS1 bearing the ESEV PBD sequence interacts with the PDZ motif of a pro-apoptotic protein named Scribble, reducing cell dead in the infected cells [144]. Also, it has been recently shown that the avian influenza NS1 protein bind and stimulate human Src tyrosine kinase through their C-terminal Src homology type 3-binding (SHB) domain. Further and ongoing research will provide better insights regarding the outcome of the HPAIV NS1 PBDs interaction with PDZ-containing proteins.
In summary, NS1 IAV is an innate immunity antagonist, which modulates the immune response in different and complex ways. The role of this protein in enhanced pro-inflammatory responses in HPAIV infected humans might be related with an early inhibition of the antiviral response that, together the interaction with PDZ-containing proteins, would lead to enhanced viral replication. In vivo, this enhanced replication would lead to an abrupt pro-inflammatory response upon recognition of viral RNA by PRRs in epithelial and endothelial cells and resident DCs and macrophages. A summary of these NS1 functions at the cellular level, as well as the role of other IAV components discussed in other sections of this review, are depicted in Figure 2.
Figure 2.
Induction and antagonist role of different components of H5N1 IAV in the innate immune response in the host-cell. ssRNA, dsRNA and HA participate in the induction of the production of IFN and pro-inflammatory cytokines, while NS1, PB1-F2, PB2 and PA have an antagonist effect.
Figure 2.
Induction and antagonist role of different components of H5N1 IAV in the innate immune response in the host-cell. ssRNA, dsRNA and HA participate in the induction of the production of IFN and pro-inflammatory cytokines, while NS1, PB1-F2, PB2 and PA have an antagonist effect.
5. PB1-F2: A Double-edged Sword
PB1-F2 is a small protein encoded in the +1 reading frame of PB1 [145] that localizes to the mitochondria and has been shown to induce apoptosis in infected cells, specifically in immune cells, by altering the mitochondrial membrane potential [145,146,147]. PB1-F2 expression increases virulence in mice [148], and it has been also reported to increase susceptibility to secondary bacterial infections in animal models [149]. Along the same lines, a serine in position 66 (instead of an arginine), has been associated with increased pathogeneicity, as shown by Conenello et al. [150] using recombinant viruses bearing a 1918 H1N1 or an H5N1 IAV PB1 segment. It is important to note that the PB1-F2 protein is not expressed by all the IAV. In many swine and human isolates, protein translation is interrupted because stop codons are introduced in the reading frame [145,151]. On the contrary, PB1-F2 is highly conserved in H5N1 avian influenza viruses [151] and the residue 66S is present in some of the HPAIV [150]. Therefore, the expression of PB1-F2 and the presence of the amino acid 66S are considered virulence markers, which is critical for surveillance purposes. Interestingly, the pandemic H1N1 that jumped into the human population in 2009 did not expressed PB1-F2, correlating with a low virulent profile. This is an excellent example of how the research on influenza pathogenesis can provide important and useful information regarding the threats that we might face in a pandemic situation. Related to this, an important fact to take into account when considering PB1-F2 as a virulence factor, is that its pathogenic potential is strain-specific. Thus, in the case of the H1N1 2009 pandemic virus, Hai et al. [152] showed that the modification of the H1N1 2009 pandemic virus in order to allow the expression of PB1-F2 had a very slight impact in pathogenesis in mice.
Zamarin et al. [147] unraveled the mechanism by which this small protein induces apoptosis. They showed that PB1-F2 interacts with the adenine nucleotide translocator 3 (ANT3) and the membrane voltage-dependent anion channel 1 (VDAC1) of the mitochondrial permeability transition pore complex (PTPC). They also found that PB1-F2 also renders the cells more sensitive to pro-apoptotic stimuli, like TNF-α. On the other hand, other group showed that PB1-F2 can also oligomerize to form pores in the mitochondrial membrane [153].
In addition, another virulence mechanism for PB1-F2 has been recently identified and characterized, namely the ability of PB1-F2 to inhibit type I IFN production both in cell culture and in vivo [150,154]. The C-terminal domain of PB1-F2 interacts with the mitochondrial antiviral signaling (MAVS) protein, decreasing the mitochondrial membrane potential and resulting in the inhibition of RIG-I-mediated of IFN-α/β production [155]. Data from our laboratory contributed to a report that confirmed the effect of the 66S residue on the inhibition of type I IFN in human primary DCs, indicating a potential important contribution to the pathogenesis of PB1-F2 66S in the human host [154]. Due to the small size of PB1-F2 and wondering how such a small protein may have such a strong impact on two important cell events such as apoptosis induction and IFN production, it has been suggested that those two functions might be linked at the level the MAVS protein [156].
Therefore, the PB1-F2 protein, expressed by most of the HPAIV H5N1, is believed to have an important impact on the innate immune response in humans by two different processes. One the one hand, it promotes apoptosis of immune cells and, on the other hand, it inhibits IFN production at early stages after infection. So, as it may be the case of the NS1 protein, this might result in early inhibition of the antiviral state followed by and increased replication and subsequent hypercytokinemia in infected humans.
6. Polymerase
Another element from IAV known to be involved in the deregulation of the antiviral immune response is the polymerase complex. One of the polymerase components, the PB2 bearing a lysine in position 627, which is predominant in human IAV and in some HPAIV strains, is considered a virulence determinant in mammals for H5N1 IAV, since viruses bearing this residue are more virulent and pathogenic in mice than those with a glutamic acid, encoded in PB2 from low pathogenic avian IAV strains [157]. Interestingly, 627K has been also demonstrated to be associated with strong production of TNF-α, IFN-β and IP-10 in human primary macrophages and type 1–like pneumocytes infected with HPAIV H5N1, and therefore is thought to contribute to the elevation of cytokines in serum in H5N1 infected patients [158], although the mechanisms for this phenotype are yet unknown.
Nevertheless, components of the polymerase complex are also involved in the evasion of the innate immunity. Each component of the viral polymerase complex, PA, PB1 and PB2, has been shown to interact with MAVS. However, PB2 has the strongest MAVS-induced IFN-α/β production inhibitory ability [159]. This function seems to be common to H1N1 and H5N1 viruses isolated from humans, but H5N1 isolated from birds does not have mitochondrial localization, and therefore do not associate with MAVS, due to a polymorphism in the amino acid position 9, indicating that the PB2 mitochondrial localization might be necessary for human host adaptation of H5N1 [160].
IAV polymerase also contributes to the host protein shut-off by two independent mechanisms. On the one hand, it indirectly participates in the evasion of the antiviral response by supporting the NS1 protein in the inhibition of CPSF30, as discussed above (Section 4). Kuo and Krug [132] showed that PA is able to stabilize the NS1-CPSF30 complex of an H5N1 NS1 lacking the residues F103/M106, which allows the inhibition of the post-transcriptional processing of cellular pre-mRNA. On the other hand, it is known that, in order to hijack the host transcriptional system, IAV polymerase associates with the host RNA polymerase II (Pol II). It has been described that this interaction results in the degradation and inhibition of Pol II, which seems to contribute to the inhibition of the cellular gene expression and therefore, of the antiviral response [161] (reviewed in [162]).
Hence, the influenza polymerase proteins have been associated both with hyper-induction of the inflammatory response and with evasion of the innate immunity, and therefore contribute to the idea that the “cytokine storm” induced in humans upon infection with HPAIV H5N1 might be a consequence of a compendium of altered innate immune functions at different times and at different levels.
7. Concluding Remarks
HPAIV H5N1 viruses induce severe disease in humans, probably as a consequence of the induction of an exacerbated innate immune response. Research using ex vivo cell models indicates that H5N1 IAV induce a high concentration of cytokines and chemokines in primary endothelial cells, as well as in immune cell types such as macrophages and DCs, indicating that the strong cytokine induction might be mediated by those cells. H5N1 components have been involved in both induction and inhibition of the innate immune response, and for that reason the contribution of these different elements at different stages of the infection in vivo could lead to failure of the innate immune response to control virus spread and a subsequent tissue damaging pro-inflammatory burst. In Figure 3 we show that while the desired outcome of infection is viral clearance, in the case of H5N1 IAV infections the balance is tilted due to higher virus replication and cytokine production, which result in more severe disease.
The acute pathology and severe disease related to H5N1 infection are thought to be a consequence of the hypercytokinemia observed in patients, suggesting that treatment with immuno-modulator drugs might help to diminish tissue damage and mitigate symptoms. The use of steroids has been proposed, however there is no empirical evidence that indicates benefits of this type of anti-inflammatory therapy, and dosage levels should be administered carefully, since high doses might be detrimental instead of beneficial (reviewed in [163,164]). The treatment of choice for influenza H5N1 viruses up to date is oseltamivir, which has been shown to be effective when administered early after infection, particularly before respiratory failure [164]. Nevertheless, there is a great need for new and optimized antiviral treatments or drug combinations that work more efficiently, especially in those patients diagnosed in late stages of the disease.
Figure 3.
Induction of hypercytokinemia by H5N1 influenza viruses results from an imbalanced innate immune response. H5N1 influenza viruses are able to dysregulate the innate immune response, either by inducing high production of cytokines or by inhibiting the antiviral response. The evasion of the innate immunity might have as a consequence increased levels of viral replication, which contributes to increased PAMPs detection and cytokine induction. A combination of these events in vivo might explain how H5N1 innate immunity antagonists contribute to hypercytokinemia development in humans.
Figure 3.
Induction of hypercytokinemia by H5N1 influenza viruses results from an imbalanced innate immune response. H5N1 influenza viruses are able to dysregulate the innate immune response, either by inducing high production of cytokines or by inhibiting the antiviral response. The evasion of the innate immunity might have as a consequence increased levels of viral replication, which contributes to increased PAMPs detection and cytokine induction. A combination of these events in vivo might explain how H5N1 innate immunity antagonists contribute to hypercytokinemia development in humans.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
Research on influenza viruses in the AF-S lab is supported by an NIH/ NIAID contract, Center for Research on Influenza Pathogenesis (CRIP, HHSN266200700010C), one of the Centers of Excellence in influenza Research and Surveillance (CEIRS).
References
- Palese, P.; Shaw, M.L. Orthomyxoviridae: The viruses and their replication. In Fields virology, 5th; Howley, D.M.K.P.M., Ed.; Lippincott Williams & Wilkins: Philadelphia, PA, 2007; pp. 1647–1689. [Google Scholar]
- Wise, H.M.; Foeglein, A.; Sun, J.; Dalton, R.M.; Patel, S.; Howard, W.; Anderson, E.C.; Barclay, W.S.; Digard, P. A complicated message: Identification of a novel pb1-related protein translated from influenza a virus segment 2 mrna. J. Virol. 2009, 83, 8021–8031. [Google Scholar] [CrossRef]
- Jagger, B.W.; Wise, H.M.; Kash, J.C.; Walters, K.A.; Wills, N.M.; Xiao, Y.L.; Dunfee, R.L.; Schwartzman, L.M.; Ozinsky, A.; Bell, G.L.; et al. An overlapping protein-coding region in influenza a virus segment 3 modulates the host response. Science 2012, 337, 199–204. [Google Scholar]
- Mubareka, S.; Palese, P. Influenza vaccines for the future. Rino Rappuoli, G.D.G., Ed.; 2011; pp. 3–21. [Google Scholar]
- Tong, S.; Li, Y.; Rivailler, P.; Conrardy, C.; Castillo, D.A.; Chen, L.M.; Recuenco, S.; Ellison, J.A.; Davis, C.T.; York, I.A.; et al. A distinct lineage of influenza a virus from bats. Proc. Nat. Acad. Sci. USA 2012, 109, 4269–4274. [Google Scholar]
- Medina, R.A.; Garcia-Sastre, A. Influenza a viruses: New research developments. Nature Rev. Microbiol. 2011, 9, 590–603. [Google Scholar] [CrossRef]
- Krauss, S.; Webster, R.G. Avian influenza virus surveillance and wild birds: Past and present. Avian. Dis. 2010, 54, 394–398. [Google Scholar] [CrossRef]
- Webster, R.G.; Hulse, D.J. Microbial adaptation and change: Avian influenza. Rev. Sci. Tech. 2004, 23, 453–465. [Google Scholar]
- de Jong, J.C.; Claas, E.C.; Osterhaus, A.D.; Webster, R.G.; Lim, W.L. A pandemic warning? Nature 1997, 389, 554. [Google Scholar]
- Claas, E.C.; de Jong, J.C.; van Beek, R.; Rimmelzwaan, G.F.; Osterhaus, A.D. Human influenza virus a/hongkong/156/97 (h5n1) infection. Vaccine 1998, 16, 977–978. [Google Scholar] [CrossRef]
- To, K.F.; Chan, P.K.; Chan, K.F.; Lee, W.K.; Lam, W.Y.; Wong, K.F.; Tang, N.L.; Tsang, D.N.; Sung, R.Y.; Buckley, T.A.; et al. Pathology of fatal human infection associated with avian influenza a h5n1 virus. J. Med. Virol. 2001, 63, 242–246. [Google Scholar] [CrossRef]
- Liem, N.T.; Tung, C.V.; Hien, N.D.; Hien, T.T.; Chau, N.Q.; Long, H.T.; Hien, N.T.; Mai le, Q.; Taylor, W.R.; Wertheim, H.; et al. Clinical features of human influenza a (h5n1) infection in vietnam: 2004-2006. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 2009, 48, 1639–1646. [Google Scholar] [CrossRef]
- WHO. Cumulative number of confirmed human cases of avian influenza a/(h5n1) reported to who. 2012. Available online: Http://www.Who.Int/influenza/human_animal_interface/h5n1_cumulative_table_archives/en/index.Html.
- Wang, T.T.; Parides, M.K.; Palese, P. Seroevidence for h5n1 influenza infections in humans: Meta-analysis. Science 2012, 335, 1463. [Google Scholar] [CrossRef]
- Beare, A.S.; Webster, R.G. Replication of avian influenza viruses in humans. Arch. Virol. 1991, 119, 37–42. [Google Scholar]
- Yang, Y.; Halloran, M.E.; Sugimoto, J.D.; Longini, I.M., Jr. Detecting human-to-human transmission of avian influenza a (h5n1). Emerg. Inf. Dis. 2007, 13, 1348–1353. [Google Scholar]
- de Jong, M.D.; Simmons, C.P.; Thanh, T.T.; Hien, V.M.; Smith, G.J.; Chau, T.N.; Hoang, D.M.; Chau, N.V.; Khanh, T.H.; Dong, V.C.; et al. Fatal outcome of human influenza a (h5n1) is associated with high viral load and hypercytokinemia. Nat. Med. 2006, 12, 1203–1207. [Google Scholar] [CrossRef]
- Thitithanyanont, A.; Engering, A.; Uiprasertkul, M.; Ekchariyawat, P.; Wiboon-Ut, S.; Kraivong, R.; Limsalakpetch, A.; Kum-Arb, U.; Yongvanitchit, K.; Sa-Ard-Iam, N.; et al. Antiviral immune responses in h5n1-infected human lung tissue and possible mechanisms underlying the hyperproduction of interferon-inducible protein ip-10. Biochem. Biophys. Res. Commun. 2010, 398, 752–758. [Google Scholar]
- Ng, W.F.; To, K.F.; Lam, W.W.; Ng, T.K.; Lee, K.C. The comparative pathology of severe acute respiratory syndrome and avian influenza a subtype h5n1--a review. Hum. Pathol. 2006, 37, 381–390. [Google Scholar] [CrossRef]
- Yuen, K.Y.; Chan, P.K.; Peiris, M.; Tsang, D.N.; Que, T.L.; Shortridge, K.F.; Cheung, P.T.; To, W.K.; Ho, E.T.; Sung, R.; et al. Clinical features and rapid viral diagnosis of human disease associated with avian influenza a h5n1 virus. Lancet 1998, 351, 467–471. [Google Scholar]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nature Immun. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef]
- Kanneganti, T.D. Central roles of nlrs and inflammasomes in viral infection. Nat. Rev. Immunol. 2010, 10, 688–698. [Google Scholar]
- Kovach, M.A.; Standiford, T.J. Toll like receptors in diseases of the lung. Int. Immunopharmacol. 2011, 11, 1399–1406. [Google Scholar] [CrossRef]
- Liu, P.; Jamaluddin, M.; Li, K.; Garofalo, R.P.; Casola, A.; Brasier, A.R. Retinoic acid-inducible gene i mediates early antiviral response and toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells. J. Virol. 2007, 81, 1401–1411. [Google Scholar]
- Matsukura, S.; Kokubu, F.; Kurokawa, M.; Kawaguchi, M.; Ieki, K.; Kuga, H.; Odaka, M.; Suzuki, S.; Watanabe, S.; Takeuchi, H.; et al. Synthetic double-stranded rna induces multiple genes related to inflammation through toll-like receptor 3 depending on nf-kappab and/or irf-3 in airway epithelial cells. Clin. Exp. Allergy 2006, 36, 1049–1062. [Google Scholar]
- Matsukura, S.; Kokubu, F.; Kurokawa, M.; Kawaguchi, M.; Ieki, K.; Kuga, H.; Odaka, M.; Suzuki, S.; Watanabe, S.; Homma, T.; et al. Role of rig-i, mda-5, and pkr on the expression of inflammatory chemokines induced by synthetic dsrna in airway epithelial cells. Int. Arch. Allergy Immunol. 2007, 143 Suppl 1, 80–83. [Google Scholar] [CrossRef]
- Le Goffic, R.; Pothlichet, J.; Vitour, D.; Fujita, T.; Meurs, E.; Chignard, M.; Si-Tahar, M. Cutting edge: Influenza a virus activates tlr3-dependent inflammatory and rig-i-dependent antiviral responses in human lung epithelial cells. J. Immun. 2007, 178, 3368–3372. [Google Scholar]
- Guillot, L.; Le Goffic, R.; Bloch, S.; Escriou, N.; Akira, S.; Chignard, M.; Si-Tahar, M. Involvement of toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded rna and influenza a virus. J. Biol. Chem. 2005, 280, 5571–5580. [Google Scholar]
- Baum, A.; Sachidanandam, R.; Garcia-Sastre, A. Preference of rig-i for short viral rna molecules in infected cells revealed by next-generation sequencing. Proc. Nat. Acad. Sci. USA 2010, 107, 16303–16308. [Google Scholar]
- Pulendran, B.; Palucka, K.; Banchereau, J. Sensing pathogens and tuning immune responses. Science 2001, 293, 253–256. [Google Scholar]
- McGill, J.; Heusel, J.W.; Legge, K.L. Innate immune control and regulation of influenza virus infections. J. Leukoc. Biol. 2009, 86, 803–812. [Google Scholar]
- Demedts, I.K.; Brusselle, G.G.; Vermaelen, K.Y.; Pauwels, R.A. Identification and characterization of human pulmonary dendritic cells. Am. J. Respir. Cell Mol. Biol. 2005, 32, 177–184. [Google Scholar] [CrossRef]
- van Haarst, J.M.; de Wit, H.J.; Drexhage, H.A.; Hoogsteden, H.C. Distribution and immunophenotype of mononuclear phagocytes and dendritic cells in the human lung. Am. J. Respir. Cell Mol. Biol. 1994, 10, 487–492. [Google Scholar]
- Sertl, K.; Takemura, T.; Tschachler, E.; Ferrans, V.J.; Kaliner, M.A.; Shevach, E.M. Dendritic cells with antigen-presenting capability reside in airway epithelium, lung parenchyma, and visceral pleura. J. Exp. Med. 1986, 163, 436–451. [Google Scholar] [CrossRef]
- Cochand, L.; Isler, P.; Songeon, F.; Nicod, L.P. Human lung dendritic cells have an immature phenotype with efficient mannose receptors. Am. J. Respir. Cell Mol. Biol. 1999, 21, 547–554. [Google Scholar]
- Perrot, I.; Deauvieau, F.; Massacrier, C.; Hughes, N.; Garrone, P.; Durand, I.; Demaria, O.; Viaud, N.; Gauthier, L.; Blery, M.; et al. Tlr3 and rig-like receptor on myeloid dendritic cells and rig-like receptor on human nk cells are both mandatory for production of ifn-gamma in response to double-stranded rna. J. Immun. 2010, 185, 2080–2088. [Google Scholar] [CrossRef]
- Matsumoto, M.; Funami, K.; Tanabe, M.; Oshiumi, H.; Shingai, M.; Seto, Y.; Yamamoto, A.; Seya, T. Subcellular localization of toll-like receptor 3 in human dendritic cells. J. Immun. 2003, 171, 3154–3162. [Google Scholar]
- Jarrossay, D.; Napolitani, G.; Colonna, M.; Sallusto, F.; Lanzavecchia, A. Specialization and complementarity in microbial molecule recognition by human myeloid and plasmacytoid dendritic cells. Eur. J. Immunol. 2001, 31, 3388–3393. [Google Scholar] [CrossRef]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis e Sousa, C. Innate antiviral responses by means of tlr7-mediated recognition of single-stranded rna. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef]
- Lund, J.M.; Alexopoulou, L.; Sato, A.; Karow, M.; Adams, N.C.; Gale, N.W.; Iwasaki, A.; Flavell, R.A. Recognition of single-stranded rna viruses by toll-like receptor 7. Proc. Nat. Acad. Sci. USA 2004, 101, 5598–5603. [Google Scholar]
- Maris, N.A.; Dessing, M.C.; de Vos, A.F.; Bresser, P.; van der Zee, J.S.; Jansen, H.M.; Spek, C.A.; van der Poll, T. Toll-like receptor mrna levels in alveolar macrophages after inhalation of endotoxin. Eur. Respir. J. 2006, 28, 622–626. [Google Scholar] [CrossRef]
- Striz, I.; Wang, Y.M.; Svarcova, I.; Trnka, L.; Sorg, C.; Costabel, U. The phenotype of alveolar macrophages and its correlation with immune cells in bronchoalveolar lavage. Eur. Respir. J. 1993, 6, 1287–1294. [Google Scholar]
- Ohman, T.; Rintahaka, J.; Kalkkinen, N.; Matikainen, S.; Nyman, T.A. Actin and rig-i/mavs signaling components translocate to mitochondria upon influenza a virus infection of human primary macrophages. J. Immun. 2009, 182, 5682–5692. [Google Scholar] [CrossRef]
- Hsu, A.C.; Parsons, K.; Barr, I.; Lowther, S.; Middleton, D.; Hansbro, P.M.; Wark, P.A. Critical role of constitutive type i interferon response in bronchial epithelial cell to influenza infection. PLoS One 2012, 7, e32947. [Google Scholar]
- Chan, M.C.; Cheung, C.Y.; Chui, W.H.; Tsao, S.W.; Nicholls, J.M.; Chan, Y.O.; Chan, R.W.; Long, H.T.; Poon, L.L.; Guan, Y.; et al. Proinflammatory cytokine responses induced by influenza a (h5n1) viruses in primary human alveolar and bronchial epithelial cells. Respir. Res. 2005, 6, 135. [Google Scholar]
- Gray, T.E.; Guzman, K.; Davis, C.W.; Abdullah, L.H.; Nettesheim, P. Mucociliary differentiation of serially passaged normal human tracheobronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 1996, 14, 104–112. [Google Scholar]
- Matrosovich, M.N.; Matrosovich, T.Y.; Gray, T.; Roberts, N.A.; Klenk, H.D. Human and avian influenza viruses target different cell types in cultures of human airway epithelium. Proc. Nat. Acad. Sci. USA 2004, 101, 4620–4624. [Google Scholar]
- Chan, M.C.; Chan, R.W.; Yu, W.C.; Ho, C.C.; Chui, W.H.; Lo, C.K.; Yuen, K.M.; Guan, Y.I.; Nicholls, J.M.; Peiris, J.S. Influenza h5n1 virus infection of polarized human alveolar epithelial cells and lung microvascular endothelial cells. Resp. Res. 2009, 10, 102. [Google Scholar]
- Chan, R.W.; Yuen, K.M.; Yu, W.C.; Ho, C.C.; Nicholls, J.M.; Peiris, J.S.; Chan, M.C. Influenza h5n1 and h1n1 virus replication and innate immune responses in bronchial epithelial cells are influenced by the state of differentiation. PLoS One 2010, 5, e8713. [Google Scholar]
- Zeng, H.; Goldsmith, C.; Thawatsupha, P.; Chittaganpitch, M.; Waicharoen, S.; Zaki, S.; Tumpey, T.M.; Katz, J.M. Highly pathogenic avian influenza h5n1 viruses elicit an attenuated type i interferon response in polarized human bronchial epithelial cells. J. Virol. 2007, 81, 12439–12449. [Google Scholar] [CrossRef]
- Zeng, H.; Pappas, C.; Belser, J.A.; Houser, K.V.; Zhong, W.; Wadford, D.A.; Stevens, T.; Balczon, R.; Katz, J.M.; Tumpey, T.M. Human pulmonary microvascular endothelial cells support productive replication of highly pathogenic avian influenza viruses: Possible involvement in the pathogenesis of human h5n1 virus infection. J. Virol. 2012, 86, 667–678. [Google Scholar]
- Schmolke, M.; Viemann, D.; Roth, J.; Ludwig, S. Essential impact of nf-kappab signaling on the h5n1 influenza a virus-induced transcriptome. J. Immun. 2009, 183, 5180–5189. [Google Scholar] [CrossRef]
- Hui, K.P.; Lee, S.M.; Cheung, C.Y.; Ng, I.H.; Poon, L.L.; Guan, Y.; Ip, N.Y.; Lau, A.S.; Peiris, J.S. Induction of proinflammatory cytokines in primary human macrophages by influenza a virus (h5n1) is selectively regulated by ifn regulatory factor 3 and p38 mapk. J. Immun. 2009, 182, 1088–1098. [Google Scholar]
- Lee, S.M.; Cheung, C.Y.; Nicholls, J.M.; Hui, K.P.; Leung, C.Y.; Uiprasertkul, M.; Tipoe, G.L.; Lau, Y.L.; Poon, L.L.; Ip, N.Y.; et al. Hyperinduction of cyclooxygenase-2-mediated proinflammatory cascade: A mechanism for the pathogenesis of avian influenza h5n1 infection. J. Infect. Dis. 2008, 198, 525–535. [Google Scholar]
- Cheung, C.Y.; Poon, L.L.; Lau, A.S.; Luk, W.; Lau, Y.L.; Shortridge, K.F.; Gordon, S.; Guan, Y.; Peiris, J.S. Induction of proinflammatory cytokines in human macrophages by influenza a (h5n1) viruses: A mechanism for the unusual severity of human disease? Lancet 2002, 360, 1831–1837. [Google Scholar]
- Geiler, J.; Michaelis, M.; Sithisarn, P.; Cinatl, J., Jr. Comparison of pro-inflammatory cytokine expression and cellular signal transduction in human macrophages infected with different influenza a viruses. Med. Microbiol. Immunol. 2011, 200, 53–60. [Google Scholar] [CrossRef]
- Lee, D.C.; Law, A.H.; Hui, K.; Tam, A.H.; Peiris, J.S.; Lau, A.S. Interferon dysregulation and virus-induced cell death in avian influenza h5n1 virus infections. Hong Kong Med. J. 2012, 18 Suppl 2, 12–16. [Google Scholar]
- Hui, K.P.; Lee, S.M.; Cheung, C.Y.; Mao, H.; Lai, A.K.; Chan, R.W.; Chan, M.C.; Tu, W.; Guan, Y.; Lau, Y.L.; et al. H5n1 influenza virus-induced mediators upregulate rig-i in uninfected cells by paracrine effects contributing to amplified cytokine cascades. J. Infect. Dis. 2011, 204, 1866–1878. [Google Scholar] [CrossRef]
- Thitithanyanont, A.; Engering, A.; Ekchariyawat, P.; Wiboon-ut, S.; Limsalakpetch, A.; Yongvanitchit, K.; Kum-Arb, U.; Kanchongkittiphon, W.; Utaisincharoen, P.; Sirisinha, S.; et al. High susceptibility of human dendritic cells to avian influenza h5n1 virus infection and protection by ifn-alpha and tlr ligands. J. Immun. 2007, 179, 5220–5227. [Google Scholar]
- Sandbulte, M.R.; Boon, A.C.; Webby, R.J.; Riberdy, J.M. Analysis of cytokine secretion from human plasmacytoid dendritic cells infected with h5n1 or low-pathogenicity influenza viruses. Virology 2008, 381, 22–28. [Google Scholar]
- Viemann, D.; Schmolke, M.; Lueken, A.; Boergeling, Y.; Friesenhagen, J.; Wittkowski, H.; Ludwig, S.; Roth, J. H5n1 virus activates signaling pathways in human endothelial cells resulting in a specific imbalanced inflammatory response. J. Immun. 2011, 186, 164–173. [Google Scholar] [CrossRef]
- Peschke, T.; Bender, A.; Nain, M.; Gemsa, D. Role of macrophage cytokines in influenza a virus infections. Immunobiol. 1993, 189, 340–355. [Google Scholar] [CrossRef]
- Hofmann, P.; Sprenger, H.; Kaufmann, A.; Bender, A.; Hasse, C.; Nain, M.; Gemsa, D. Susceptibility of mononuclear phagocytes to influenza a virus infection and possible role in the antiviral response. J. Leukoc. Biol. 1997, 61, 408–414. [Google Scholar]
- Lehmann, C.; Sprenger, H.; Nain, M.; Bacher, M.; Gemsa, D. Infection of macrophages by influenza a virus: Characteristics of tumour necrosis factor-alpha (tnf alpha) gene expression. Res. Virol. 1996, 147, 123–130. [Google Scholar]
- Ramos, I.; Bernal-Rubio, D.; Durham, N.; Belicha-Villanueva, A.; Lowen, A.C.; Steel, J.; Fernandez-Sesma, A. Effects of receptor binding specificity of avian influenza virus on the human innate immune response. J. Virol. 2011, 85, 4421–4431. [Google Scholar] [CrossRef]
- Wang, J.; Nikrad, M.P.; Travanty, E.A.; Zhou, B.; Phang, T.; Gao, B.; Alford, T.; Ito, Y.; Nahreini, P.; Hartshorn, K.; et al. Innate immune response of human alveolar macrophages during influenza a infection. PLoS One 2012, 7, e29879. [Google Scholar]
- Sakabe, S.; Iwatsuki-Horimoto, K.; Takano, R.; Nidom, C.A.; Le, M.; Nagamura-Inoue, T.; Horimoto, T.; Yamashita, N.; Kawaoka, Y. Cytokine production by primary human macrophages infected with highly pathogenic h5n1 or pandemic h1n1 2009 influenza viruses. J. Virol. 2011, 92, 1428–1434. [Google Scholar] [CrossRef]
- Lee, S.M.; Gardy, J.L.; Cheung, C.Y.; Cheung, T.K.; Hui, K.P.; Ip, N.Y.; Guan, Y.; Hancock, R.E.; Peiris, J.S. Systems-level comparison of host-responses elicited by avian h5n1 and seasonal h1n1 influenza viruses in primary human macrophages. PLoS One 2009, 4, e8072. [Google Scholar]
- Cheung, C.Y.; Chan, E.Y.; Krasnoselsky, A.; Purdy, D.; Navare, A.T.; Bryan, J.T.; Leung, C.K.; Hui, K.P.; Peiris, J.S.; Katze, M.G. H5N1 virus causes significant perturbations in host proteome very early in influenza virus-infected primary human monocyte-derived macrophages. J. Infect. Dis. 2012, 206, 640–645. [Google Scholar]
- Zhou, J.; Law, H.K.; Cheung, C.Y.; Ng, I.H.; Peiris, J.S.; Lau, Y.L. Differential expression of chemokines and their receptors in adult and neonatal macrophages infected with human or avian influenza viruses. J. Infect. Dis. 2006, 194, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Ekchariyawat, P.; Thitithanyanont, A.; Sirisinha, S.; Utaisincharoen, P. Apoptosis induced by avian h5n1 virus in human monocyte-derived macrophages involves trail-inducing caspase-10 activation. Innate Immun. 2012, 18, 390–397. [Google Scholar] [CrossRef]
- Mok, C.K.; Lee, D.C.; Cheung, C.Y.; Peiris, M.; Lau, A.S. Differential onset of apoptosis in influenza a virus h5n1- and h1n1-infected human blood macrophages. J. Gen. Virol. 2007, 88, 1275–1280. [Google Scholar] [CrossRef]
- Haye, K.; Burmakina, S.; Moran, T.; Garcia-Sastre, A.; Fernandez-Sesma, A. The ns1 protein of a human influenza virus inhibits type i interferon production and the induction of antiviral responses in primary human dendritic and respiratory epithelial cells. J. Virol. 2009, 83, 6849–6862. [Google Scholar]
- Weinheimer, V.K.; Becher, A.; Tonnies, M.; Holland, G.; Knepper, J.; Bauer, T.T.; Schneider, P.; Neudecker, J.; Ruckert, J.C.; Szymanski, K.; et al. Influenza a viruses target type ii pneumocytes in the human lung. J. Infect. Dis. 2012, 206, 1685–1694. [Google Scholar] [CrossRef]
- Chabot, F.; Mitchell, J.A.; Gutteridge, J.M.; Evans, T.W. Reactive oxygen species in acute lung injury. Eur. Respir. J. 1998, 11, 745–757. [Google Scholar]
- Imai, Y.; Kuba, K.; Neely, G.G.; Yaghubian-Malhami, R.; Perkmann, T.; van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.H.; Wang, H.; et al. Identification of oxidative stress and toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef]
- Smits, S.L.; van den Brand, J.M.; de Lang, A.; Leijten, L.M.; van Ijcken, W.F.; van Amerongen, G.; Osterhaus, A.D.; Andeweg, A.C.; Haagmans, B.L. Distinct severe acute respiratory syndrome coronavirus-induced acute lung injury pathways in two different nonhuman primate species. J. Virol. 2011, 85, 4234–4245. [Google Scholar]
- Chow, C.W.; Herrera Abreu, M.T.; Suzuki, T.; Downey, G.P. Oxidative stress and acute lung injury. Am. J. Respir. Cell Mol. Biol. 2003, 29, 427–431. [Google Scholar]
- Weis, W.; Brown, J.H.; Cusack, S.; Paulson, J.C.; Skehel, J.J.; Wiley, D.C. Structure of the influenza virus haemagglutinin complexed with its receptor, sialic acid. Nature 1988, 333, 426–431. [Google Scholar] [CrossRef]
- Wiley, D.C.; Skehel, J.J. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Ann. Rev. Biochem. 1987, 56, 365–394. [Google Scholar]
- Shinya, K.; Ebina, M.; Yamada, S.; Ono, M.; Kasai, N.; Kawaoka, Y. Avian flu: Influenza virus receptors in the human airway. Nature 2006, 440, 435–436. [Google Scholar]
- van Riel, D.; Munster, V.J.; de Wit, E.; Rimmelzwaan, G.F.; Fouchier, R.A.; Osterhaus, A.D.; Kuiken, T. H5n1 virus attachment to lower respiratory tract. Science 2006, 312, 399. [Google Scholar] [CrossRef]
- Nicholls, J.M.; Chan, M.C.; Chan, W.Y.; Wong, H.K.; Cheung, C.Y.; Kwong, D.L.; Wong, M.P.; Chui, W.H.; Poon, L.L.; Tsao, S.W.; et al. Tropism of avian influenza a (h5n1) in the upper and lower respiratory tract. Nat. Med. 2007, 13, 147–149. [Google Scholar]
- Rogers, G.N.; Paulson, J.C. Receptor determinants of human and animal influenza virus isolates: Differences in receptor specificity of the h3 hemagglutinin based on species of origin. Virology 1983, 127, 361–373. [Google Scholar] [CrossRef]
- Herfst, S.; Schrauwen, E.J.; Linster, M.; Chutinimitkul, S.; de Wit, E.; Munster, V.J.; Sorrell, E.M.; Bestebroer, T.M.; Burke, D.F.; Smith, D.J.; et al. Airborne transmission of influenza a/h5n1 virus between ferrets. Science 2012, 336, 1534–1541. [Google Scholar]
- Imai, M.; Watanabe, T.; Hatta, M.; Das, S.C.; Ozawa, M.; Shinya, K.; Zhong, G.; Hanson, A.; Katsura, H.; Watanabe, S.; et al. Experimental adaptation of an influenza h5 ha confers respiratory droplet transmission to a reassortant h5 ha/h1n1 virus in ferrets. Nature 2012, 486, 420–428. [Google Scholar]
- Imai, M.; Kawaoka, Y. The role of receptor binding specificity in interspecies transmission of influenza viruses. Curr. Opin. Virol. 2012, 2, 160–167. [Google Scholar]
- Steel, J.; Lowen, A.C.; Mubareka, S.; Palese, P. Transmission of influenza virus in a mammalian host is increased by pb2 amino acids 627k or 627e/701n. PLoS Pathog. 2009, 5, e1000252. [Google Scholar] [CrossRef]
- Van Hoeven, N.; Pappas, C.; Belser, J.A.; Maines, T.R.; Zeng, H.; Garcia-Sastre, A.; Sasisekharan, R.; Katz, J.M.; Tumpey, T.M. Human ha and polymerase subunit pb2 proteins confer transmission of an avian influenza virus through the air. Proc. Nat. Acad. Sci. USA 2009, 106, 3366–3371. [Google Scholar]
- Ocana-Macchi, M.; Bel, M.; Guzylack-Piriou, L.; Ruggli, N.; Liniger, M.; McCullough, K.C.; Sakoda, Y.; Isoda, N.; Matrosovich, M.; Summerfield, A. Hemagglutinin-dependent tropism of h5n1 avian influenza virus for human endothelial cells. J. Virol. 2009, 83, 12947–12955. [Google Scholar]
- Ramos, I.; Fernandez-Sesma, A. Cell receptors for influenza a viruses and the innate immune response. Front. Microbiol. 2012, 3, 117. [Google Scholar]
- Xu, W.; Chen, M.; Ge, N.; Xu, J. Hemagglutinin from the h5n1 virus activates janus kinase 3 to dysregulate innate immunity. PLoS One 2012, 7, e31721. [Google Scholar]
- Liu, W.C.; Lin, S.C.; Yu, Y.L.; Chu, C.L.; Wu, S.C. Dendritic cell activation by recombinant hemagglutinin proteins of h1n1 and h5n1 influenza a viruses. J. Vvirol. 2010, 84, 12011–12017. [Google Scholar] [CrossRef]
- Eierhoff, T.; Hrincius, E.R.; Rescher, U.; Ludwig, S.; Ehrhardt, C. The epidermal growth factor receptor (egfr) promotes uptake of influenza a viruses (iav) into host cells. PLoS Pathog. 2010, 6, e1001099. [Google Scholar] [CrossRef]
- Marchant, D.; Singhera, G.K.; Utokaparch, S.; Hackett, T.L.; Boyd, J.H.; Luo, Z.; Si, X.; Dorscheid, D.R.; McManus, B.M.; Hegele, R.G. Toll-like receptor 4-mediated activation of p38 mitogen-activated protein kinase is a determinant of respiratory virus entry and tropism. J. Virol. 2010, 84, 11359–11373. [Google Scholar]
- Suguitan, A.L., Jr.; Matsuoka, Y.; Lau, Y.F.; Santos, C.P.; Vogel, L.; Cheng, L.I.; Orandle, M.; Subbarao, K. The multibasic cleavage site of the hemagglutinin of highly pathogenic a/vietnam/1203/2004 (h5n1) avian influenza virus acts as a virulence factor in a host-specific manner in mammals. J. Virol. 2012, 86, 2706–2714. [Google Scholar]
- Horimoto, T.; Nakayama, K.; Smeekens, S.P.; Kawaoka, Y. Proprotein-processing endoproteases pc6 and furin both activate hemagglutinin of virulent avian influenza viruses. J. Virol. 1994, 68, 6074–6078. [Google Scholar]
- Horimoto, T.; Kawaoka, Y. Reverse genetics provides direct evidence for a correlation of hemagglutinin cleavability and virulence of an avian influenza a virus. J. Virol. 1994, 68, 3120–3128. [Google Scholar]
- Stieneke-Grober, A.; Vey, M.; Angliker, H.; Shaw, E.; Thomas, G.; Roberts, C.; Klenk, H.D.; Garten, W. Influenza virus hemagglutinin with multibasic cleavage site is activated by furin, a subtilisin-like endoprotease. EMBO J. 1992, 11, 2407–2414. [Google Scholar]
- Barbey-Morel, C.L.; Oeltmann, T.N.; Edwards, K.M.; Wright, P.F. Role of respiratory tract proteases in infectivity of influenza a virus. J. Infect. Dis. 1987, 155, 667–672. [Google Scholar]
- Schrauwen, E.J.; Bestebroer, T.M.; Munster, V.J.; de Wit, E.; Herfst, S.; Rimmelzwaan, G.F.; Osterhaus, A.D.; Fouchier, R.A. Insertion of a multibasic cleavage site in the haemagglutinin of human influenza h3n2 virus does not increase pathogenicity in ferrets. J. Gen. Virol. 2011, 92, 1410–1415. [Google Scholar] [CrossRef]
- Schrauwen, E.J.; Herfst, S.; Leijten, L.M.; van Run, P.; Bestebroer, T.M.; Linster, M.; Bodewes, R.; Kreijtz, J.H.; Rimmelzwaan, G.F.; Osterhaus, A.D.; et al. The multibasic cleavage site in h5n1 virus is critical for systemic spread along the olfactory and hematogenous routes in ferrets. J. Virol. 2012, 86, 3975–3984. [Google Scholar]
- Brandstadter, J.D.; Yang, Y. Natural killer cell responses to viral infection. J. Innate Immun. 2011, 3, 274–279. [Google Scholar]
- Arnon, T.I.; Lev, M.; Katz, G.; Chernobrov, Y.; Porgador, A.; Mandelboim, O. Recognition of viral hemagglutinins by nkp44 but not by nkp30. Eur. J. Immunol. 2001, 31, 2680–2689. [Google Scholar] [CrossRef]
- Mandelboim, O.; Lieberman, N.; Lev, M.; Paul, L.; Arnon, T.I.; Bushkin, Y.; Davis, D.M.; Strominger, J.L.; Yewdell, J.W.; Porgador, A. Recognition of haemagglutinins on virus-infected cells by nkp46 activates lysis by human nk cells. Nature 2001, 409, 1055–1060. [Google Scholar]
- Ho, J.W.; Hershkovitz, O.; Peiris, M.; Zilka, A.; Bar-Ilan, A.; Nal, B.; Chu, K.; Kudelko, M.; Kam, Y.W.; Achdout, H.; et al. H5-type influenza virus hemagglutinin is functionally recognized by the natural killer-activating receptor nkp44. J. Virol. 2008, 82, 2028–2032. [Google Scholar] [CrossRef]
- Achdout, H.; Meningher, T.; Hirsh, S.; Glasner, A.; Bar-On, Y.; Gur, C.; Porgador, A.; Mendelson, M.; Mandelboim, M.; Mandelboim, O. Killing of avian and swine influenza virus by natural killer cells. J. Virol. 2010, 84, 3993–4001. [Google Scholar]
- Du, N.; Zhou, J.; Lin, X.; Zhang, Y.; Yang, X.; Wang, Y.; Shu, Y. Differential activation of nk cells by influenza a pseudotype h5n1 and 1918 and 2009 pandemic h1n1 viruses. J. Virol. 2010, 84, 7822–7831. [Google Scholar]
- Stevens, J.; Blixt, O.; Paulson, J.C.; Wilson, I.A. Glycan microarray technologies: Tools to survey host specificity of influenza viruses. Nat. Rev. Microbiol. 2006, 4, 857–864. [Google Scholar] [CrossRef]
- Stevens, J.; Blixt, O.; Tumpey, T.M.; Taubenberger, J.K.; Paulson, J.C.; Wilson, I.A. Structure and receptor specificity of the hemagglutinin from an h5n1 influenza virus. Science 2006, 312, 404–410. [Google Scholar] [CrossRef]
- Wang, X.; Li, M.; Zheng, H.; Muster, T.; Palese, P.; Beg, A.A.; Garcia-Sastre, A. Influenza a virus ns1 protein prevents activation of nf-kappab and induction of alpha/beta interferon. J. Virol. 2000, 74, 11566–11573. [Google Scholar]
- Garcia-Sastre, A. Induction and evasion of type i interferon responses by influenza viruses. Virus Res. 2011, 162, 12–18. [Google Scholar]
- Hale, B.G.; Randall, R.E.; Ortin, J.; Jackson, D. The multifunctional ns1 protein of influenza a viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef]
- Lu, Y.; Wambach, M.; Katze, M.G.; Krug, R.M. Binding of the influenza virus ns1 protein to double-stranded rna inhibits the activation of the protein kinase that phosphorylates the elf-2 translation initiation factor. Virology 1995, 214, 222–228. [Google Scholar] [CrossRef]
- Min, J.Y.; Krug, R.M. The primary function of rna binding by the influenza a virus ns1 protein in infected cells: Inhibiting the 2'-5' oligo (a) synthetase/rnase l pathway. Proc. Nat. Acad. Sci. USA 2006, 103, 7100–7105. [Google Scholar]
- Bergmann, M.; Garcia-Sastre, A.; Carnero, E.; Pehamberger, H.; Wolff, K.; Palese, P.; Muster, T. Influenza virus ns1 protein counteracts pkr-mediated inhibition of replication. J. Virol. 2000, 74, 6203–6206. [Google Scholar] [CrossRef]
- Tan, S.L.; Katze, M.G. Biochemical and genetic evidence for complex formation between the influenza a virus ns1 protein and the interferon-induced pkr protein kinase. J. Interferon. Cytokine Res. 1998, 18, 757–766. [Google Scholar] [CrossRef]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.S.; Huang, I.C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; Garcia-Sastre, A. Influenza a virus ns1 targets the ubiquitin ligase trim25 to evade recognition by the host viral rna sensor rig-i. Cell Host Microbe 2009, 5, 439–449. [Google Scholar]
- Mibayashi, M.; Martinez-Sobrido, L.; Loo, Y.M.; Cardenas, W.B.; Gale, M., Jr.; Garcia-Sastre, A. Inhibition of retinoic acid-inducible gene i-mediated induction of beta interferon by the ns1 protein of influenza a virus. J. Virol. 2007, 81, 514–524. [Google Scholar] [CrossRef]
- Nemeroff, M.E.; Barabino, S.M.; Li, Y.; Keller, W.; Krug, R.M. Influenza virus ns1 protein interacts with the cellular 30 kda subunit of cpsf and inhibits 3'end formation of cellular pre-mrnas. Mol. Cell 1998, 1, 991–1000. [Google Scholar]
- Noah, D.L.; Twu, K.Y.; Krug, R.M. Cellular antiviral responses against influenza a virus are countered at the posttranscriptional level by the viral ns1a protein via its binding to a cellular protein required for the 3' end processing of cellular pre-mrnas. Virology 2003, 307, 386–395. [Google Scholar] [CrossRef]
- Chen, Z.; Li, Y.; Krug, R.M. Influenza a virus ns1 protein targets poly(a)-binding protein ii of the cellular 3'-end processing machinery. EMBO J. 1999, 18, 2273–2283. [Google Scholar]
- Gao, S.; Song, L.; Li, J.; Zhang, Z.; Peng, H.; Jiang, W.; Wang, Q.; Kang, T.; Chen, S.; Huang, W. Influenza a virus-encoded ns1 virulence factor protein inhibits innate immune response by targeting ikk. Cell Microbiol. 2012, 14, 1849–1866. [Google Scholar] [CrossRef]
- Burgui, I.; Aragon, T.; Ortin, J.; Nieto, A. Pabp1 and eif4gi associate with influenza virus ns1 protein in viral mrna translation initiation complexes. J. Gen. Virol. 2003, 84, 3263–3274. [Google Scholar]
- Aragon, T.; de la Luna, S.; Novoa, I.; Carrasco, L.; Ortin, J.; Nieto, A. Eukaryotic translation initiation factor 4gi is a cellular target for ns1 protein, a translational activator of influenza virus. Mol. Cell. Biol. 2000, 20, 6259–6268. [Google Scholar] [CrossRef]
- Hale, B.G.; Jackson, D.; Chen, Y.H.; Lamb, R.A.; Randall, R.E. Influenza a virus ns1 protein binds p85beta and activates phosphatidylinositol-3-kinase signaling. Proc. Nat. Acad. Sci. USA 2006, 103, 14194–14199. [Google Scholar]
- Marazzi, I.; Ho, J.S.; Kim, J.; Manicassamy, B.; Dewell, S.; Albrecht, R.A.; Seibert, C.W.; Schaefer, U.; Jeffrey, K.L.; Prinjha, R.K.; et al. Suppression of the antiviral response by an influenza histone mimic. Nature 2012, 483, 428–433. [Google Scholar] [CrossRef]
- Kainov, D.E.; Muller, K.H.; Theisen, L.L.; Anastasina, M.; Kaloinen, M.; Muller, C.P. Differential effects of ns1 proteins of human pandemic h1n1/2009, avian highly pathogenic h5n1, and low pathogenic h5n2 influenza a viruses on cellular pre-mrna polyadenylation and mrna translation. J. Biol. Chem. 2011, 286, 7239–7247. [Google Scholar]
- Kochs, G.; Garcia-Sastre, A.; Martinez-Sobrido, L. Multiple anti-interferon actions of the influenza a virus ns1 protein. J. Virol. 2007.
- Das, K.; Ma, L.C.; Xiao, R.; Radvansky, B.; Aramini, J.; Zhao, L.; Marklund, J.; Kuo, R.L.; Twu, K.Y.; Arnold, E.; et al. Structural basis for suppression of a host antiviral response by influenza a virus. Proc. Nat. Acad. Sci. USA 2008, 105, 13093–13098. [Google Scholar]
- Kuo, R.L.; Krug, R.M. Influenza a virus polymerase is an integral component of the cpsf30-ns1a protein complex in infected cells. J. Virol. 2009, 83, 1611–1616. [Google Scholar] [CrossRef]
- Twu, K.Y.; Noah, D.L.; Rao, P.; Kuo, R.L.; Krug, R.M. The cpsf30 binding site on the ns1a protein of influenza a virus is a potential antiviral target. J. Virol. 2006, 80, 3957–3965. [Google Scholar]
- Spesock, A.; Malur, M.; Hossain, M.J.; Chen, L.M.; Njaa, B.L.; Davis, C.T.; Lipatov, A.S.; York, I.A.; Krug, R.M.; Donis, R.O. The virulence of 1997 h5n1 influenza viruses in the mouse model is increased by correcting a defect in their ns1 proteins. J. Virol. 2011, 85, 7048–7058. [Google Scholar]
- Twu, K.Y.; Kuo, R.L.; Marklund, J.; Krug, R.M. The h5n1 influenza virus ns genes selected after 1998 enhance virus replication in mammalian cells. J. Virol. 2007, 81, 8112–8121. [Google Scholar]
- Obenauer, J.C.; Denson, J.; Mehta, P.K.; Su, X.; Mukatira, S.; Finkelstein, D.B.; Xu, X.; Wang, J.; Ma, J.; Fan, Y.; et al. Large-scale sequence analysis of avian influenza isolates. Science 2006, 311, 1576–1580. [Google Scholar] [CrossRef]
- Zielecki, F.; Semmler, I.; Kalthoff, D.; Voss, D.; Mauel, S.; Gruber, A.D.; Beer, M.; Wolff, T. Virulence determinants of avian h5n1 influenza a virus in mammalian and avian hosts: Role of the c-terminal esev motif in the viral ns1 protein. J. Virol. 2010, 84, 10708–10718. [Google Scholar] [CrossRef]
- Jackson, D.; Hossain, M.J.; Hickman, D.; Perez, D.R.; Lamb, R.A. A new influenza virus virulence determinant: The ns1 protein four c-terminal residues modulate pathogenicity. Proc. Natl. Acad. Sci. USA 2008, 105, 4381–4386. [Google Scholar]
- Sheng, M.; Sala, C. Pdz domains and the organization of supramolecular complexes. Annu Rev Neurosci. 2001, 24, 1–29. [Google Scholar]
- Zhang, H.; Li, W.; Wang, G.; Su, Y.; Zhang, C.; Chen, X.; Xu, Y.; Li, K. The distinct binding properties between avian/human influenza a virus ns1 and postsynaptic density protein-95 (psd-95), and inhibition of nitric oxide production. J. Virol. 2011, 8, 298. [Google Scholar] [CrossRef]
- Bavagnoli, L.; Dundon, W.G.; Garbelli, A.; Zecchin, B.; Milani, A.; Parakkal, G.; Baldanti, F.; Paolucci, S.; Volmer, R.; Tu, Y.; et al. The pdz-ligand and src-homology type 3 domains of epidemic avian influenza virus ns1 protein modulate human src kinase activity during viral infection. PLoS One 2011, 6, e27789. [Google Scholar]
- Golebiewski, L.; Liu, H.; Javier, R.T.; Rice, A.P. The avian influenza virus ns1 esev pdz binding motif associates with dlg1 and scribble to disrupt cellular tight junctions. J. Virol. 2011, 85, 10639–10648. [Google Scholar]
- Yu, J.; Li, X.; Wang, Y.; Li, B.; Li, H.; Li, Y.; Zhou, W.; Zhang, C.; Rao, Z.; Bartlam, M.; et al. Pdlim2 selectively interacts with the pdz binding motif of highly pathogenic avian h5n1 influenza a virus ns1. PLoS One 2011, 6, e19511. [Google Scholar]
- Liu, H.; Golebiewski, L.; Dow, E.C.; Krug, R.M.; Javier, R.T.; Rice, A.P. The esev pdz-binding motif of the avian influenza a virus ns1 protein protects infected cells from apoptosis by directly targeting scribble. J. Virol. 2010, 84, 11164–11174. [Google Scholar]
- Chen, W.; Calvo, P.A.; Malide, D.; Gibbs, J.; Schubert, U.; Bacik, I.; Basta, S.; O'Neill, R.; Schickli, J.; Palese, P.; et al. A novel influenza a virus mitochondrial protein that induces cell death. Nat. Med. 2001, 7, 1306–1312. [Google Scholar] [CrossRef]
- Gibbs, J.S.; Malide, D.; Hornung, F.; Bennink, J.R.; Yewdell, J.W. The influenza a virus pb1-f2 protein targets the inner mitochondrial membrane via a predicted basic amphipathic helix that disrupts mitochondrial function. J. Virol. 2003, 77, 7214–7224. [Google Scholar] [CrossRef]
- Zamarin, D.; Garcia-Sastre, A.; Xiao, X.; Wang, R.; Palese, P. Influenza virus pb1-f2 protein induces cell death through mitochondrial ant3 and vdac1. PLoS Pathog 2005, 1, e4. [Google Scholar]
- Zamarin, D.; Ortigoza, M.B.; Palese, P. Influenza a virus pb1-f2 protein contributes to viral pathogenesis in mice. J. Virol. 2006, 80, 7976–7983. [Google Scholar] [CrossRef]
- McAuley, J.L.; Hornung, F.; Boyd, K.L.; Smith, A.M.; McKeon, R.; Bennink, J.; Yewdell, J.W.; McCullers, J.A. Expression of the 1918 influenza a virus pb1-f2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell. Host. Microbe. 2007, 2, 240–249. [Google Scholar] [CrossRef]
- Conenello, G.M.; Zamarin, D.; Perrone, L.A.; Tumpey, T.; Palese, P. A single mutation in the pb1-f2 of h5n1 (hk/97) and 1918 influenza a viruses contributes to increased virulence. PLoS Pathog. 2007, 3, 1414–1421. [Google Scholar]
- Zell, R.; Krumbholz, A.; Eitner, A.; Krieg, R.; Halbhuber, K.J.; Wutzler, P. Prevalence of pb1-f2 of influenza a viruses. J. Gen. Virol. 2007, 88, 536–546. [Google Scholar]
- Hai, R.; Schmolke, M.; Varga, Z.T.; Manicassamy, B.; Wang, T.T.; Belser, J.A.; Pearce, M.B.; Garcia-Sastre, A.; Tumpey, T.M.; Palese, P. Pb1-f2 expression by the 2009 pandemic h1n1 influenza virus has minimal impact on virulence in animal models. J. Virol. 2010, 84, 4442–4450. [Google Scholar]
- Bruns, K.; Studtrucker, N.; Sharma, A.; Fossen, T.; Mitzner, D.; Eissmann, A.; Tessmer, U.; Roder, R.; Henklein, P.; Wray, V.; et al. Structural characterization and oligomerization of pb1-f2, a proapoptotic influenza a virus protein. J. Biol. Chem. 2007, 282, 353–363. [Google Scholar]
- Varga, Z.T.; Ramos, I.; Hai, R.; Schmolke, M.; Garcia-Sastre, A.; Fernandez-Sesma, A.; Palese, P. The influenza virus protein pb1-f2 inhibits the induction of type i interferon at the level of the mavs adaptor protein. PLoS Pathog. 2011, 7, e1002067. [Google Scholar] [CrossRef]
- Varga, Z.T.; Grant, A.; Manicassamy, B.; Palese, P. Influenza virus protein pb1-f2 inhibits the induction of type i interferon by binding to mavs and decreasing mitochondrial membrane potential. J. Virol. 2012, 86, 8359–8366. [Google Scholar]
- Varga, Z.T.; Palese, P. The influenza a virus protein pb1-f2: Killing two birds with one stone? Virulence 2011, 2, 542–546. [Google Scholar] [CrossRef]
- Hatta, M.; Gao, P.; Halfmann, P.; Kawaoka, Y. Molecular basis for high virulence of hong kong h5n1 influenza a viruses. Science 2001, 293, 1840–1842. [Google Scholar]
- Mok, K.P.; Wong, C.H.; Cheung, C.Y.; Chan, M.C.; Lee, S.M.; Nicholls, J.M.; Guan, Y.; Peiris, J.S. Viral genetic determinants of h5n1 influenza viruses that contribute to cytokine dysregulation. J. Infect. Dis. 2009, 200, 1104–1112. [Google Scholar] [CrossRef] [Green Version]
- Iwai, A.; Shiozaki, T.; Kawai, T.; Akira, S.; Kawaoka, Y.; Takada, A.; Kida, H.; Miyazaki, T. Influenza a virus polymerase inhibits type i interferon induction by binding to interferon beta promoter stimulator 1. J. Biol. Chem. 2010, 285, 32064–32074. [Google Scholar]
- Graef, K.M.; Vreede, F.T.; Lau, Y.F.; McCall, A.W.; Carr, S.M.; Subbarao, K.; Fodor, E. The pb2 subunit of the influenza virus rna polymerase affects virulence by interacting with the mitochondrial antiviral signaling protein and inhibiting expression of beta interferon. J. Virol. 2010, 84, 8433–8445. [Google Scholar] [CrossRef]
- Vreede, F.T.; Chan, A.Y.; Sharps, J.; Fodor, E. Mechanisms and functional implications of the degradation of host rna polymerase ii in influenza virus infected cells. Virology 2010, 396, 125–134. [Google Scholar]
- Vreede, F.T.; Fodor, E. The role of the influenza virus rna polymerase in host shut-off. Virulence 2010, 1, 436–439. [Google Scholar] [CrossRef]
- Carter, M.J. A rationale for using steroids in the treatment of severe cases of h5n1 avian influenza. J. Med. Microbiol. 2007, 56, 875–883. [Google Scholar]
- Chan, P.K.; Lee, N.; Zaman, M.; Adisasmito, W.; Coker, R.; Hanshaoworakul, W.; Gasimov, V.; Oner, A.F.; Dogan, N.; Tsang, O.; et al. Determinants of antiviral effectiveness in h5n1 avian influenza. J. Iinfect. Dis. 2012, 206, 1359–1366. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Ramos, I.; Fernandez-Sesma, A. Innate Immunity to H5N1 Influenza Viruses in Humans. Viruses 2012, 4, 3363-3388. https://doi.org/10.3390/v4123363
AMA Style
Ramos I, Fernandez-Sesma A. Innate Immunity to H5N1 Influenza Viruses in Humans. Viruses. 2012; 4(12):3363-3388. https://doi.org/10.3390/v4123363
Chicago/Turabian StyleRamos, Irene, and Ana Fernandez-Sesma. 2012. "Innate Immunity to H5N1 Influenza Viruses in Humans" Viruses 4, no. 12: 3363-3388. https://doi.org/10.3390/v4123363