Modulation of Apoptotic Pathways by Human Papillomaviruses (HPV): Mechanisms and Implications for Therapy


{kind=link}
Abstract
:1. Introduction
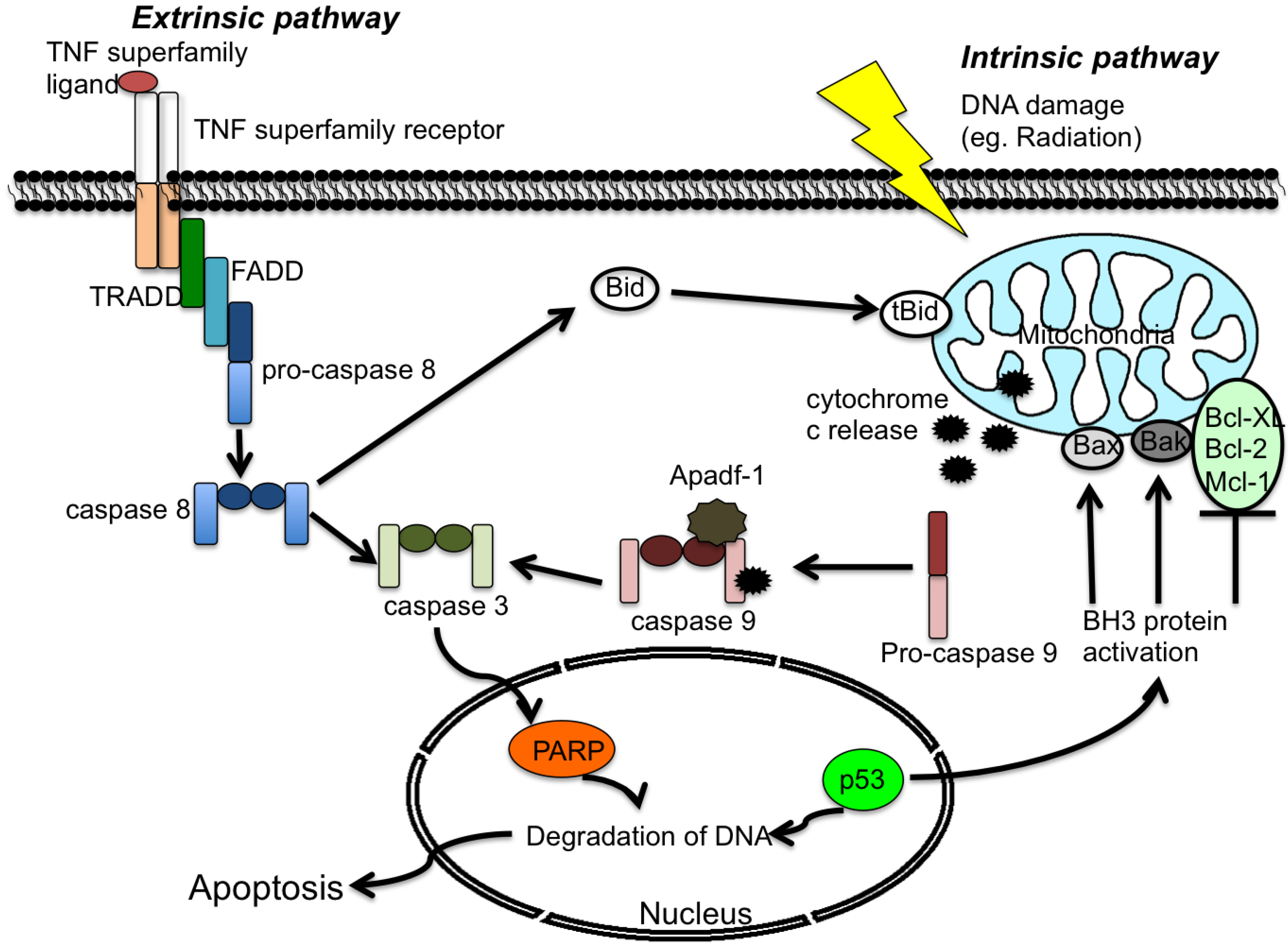
2. Apoptosis
2.1 Extrinsic Pathway
2.2 Intrinsic Pathway
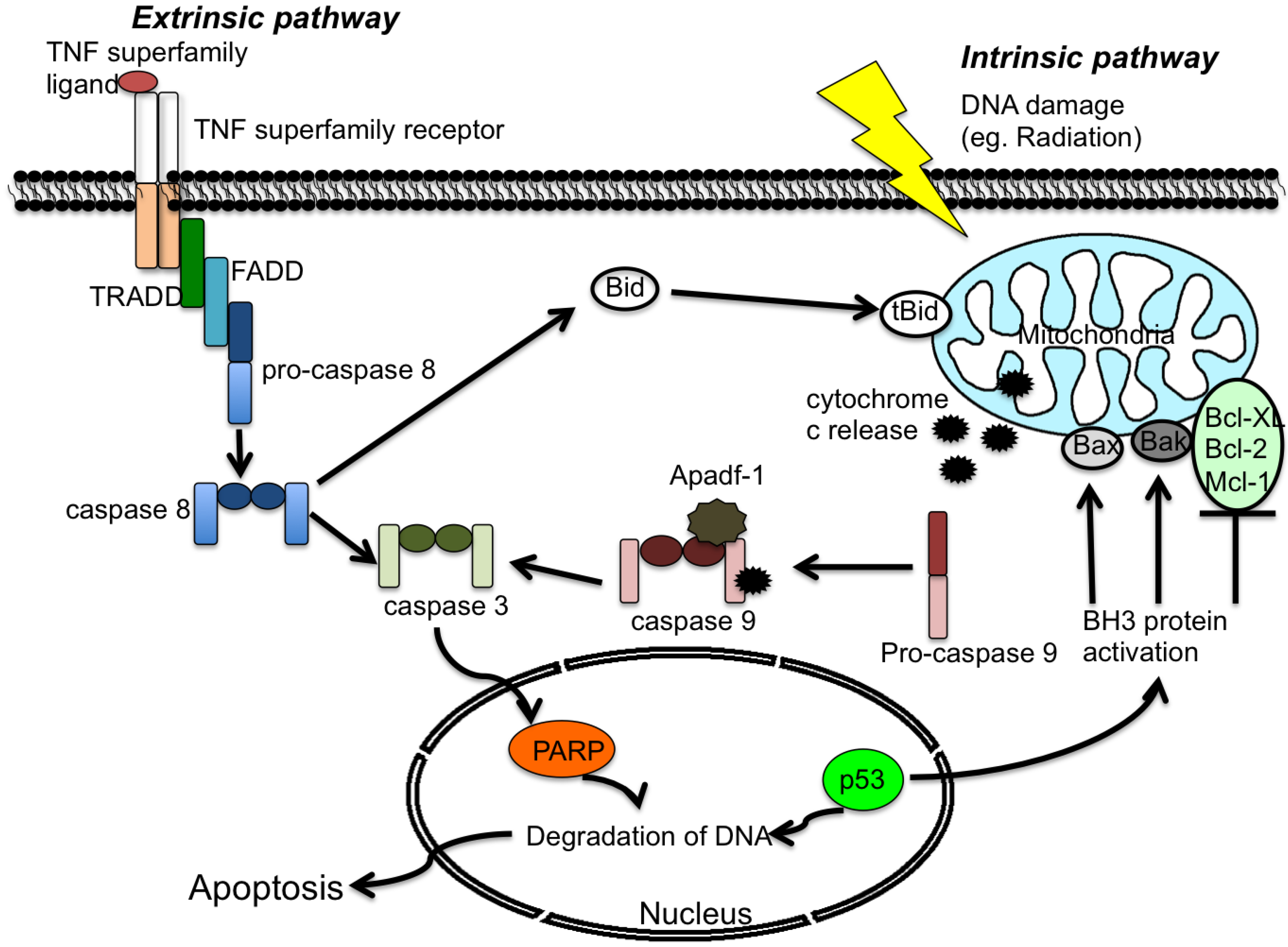
3. Viruses and Apoptosis
4. The HPV Life Cycle
5. HPV Proteins that Modulate Apoptosis
5.1. HPV E2
5.2. HPV E5
5.3. HPV E6
5.4. HPV E6*
5.5. HPV E7
6. Re-Engagement of Apoptotic Pathways as a Therapeutic Approach to HPV-Induced Cancers
Conflict of Interest
References
- Stanley, M.A. Genital human papillomavirus infections: current and prospective therapies. J. Gen. Virol 2012, 93, 681–691. [Google Scholar] [CrossRef]
- Goldstein, M.A.; Goodman, A.; del Carmen, M.G.; Wilbur, D.C. Case records of the Massachusetts General Hospital. Case 10–2009. A 23-year-old woman with an abnormal Papanicolaou smear. N. Engl. J. Med. 2009, 360, 1337–1344. [Google Scholar] [CrossRef]
- Tungteakkhun, S.S.; Filippova, M.; Neidigh, J.W.; Fodor, N.; Duerksen-Hughes, P.J. The interaction between human papillomavirus type 16 and FADD is mediated by a novel E6 binding domain. J. Virol. 2008, 82, 9600–9614. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef]
- Grm, H.S.; Massimi, P.; Gammoh, N.; Banks, L. Crosstalk between the human papillomavirus E2 transcriptional activator and the E6 oncoprotein. Oncogene 2005, 24, 5149–5164. [Google Scholar] [CrossRef]
- Rudin, C.M.; Thompson, C.B. Apoptosis and disease: regulation and clinical relevance of programmed cell death. Annu. Rev. Med. 1997, 48, 267–281. [Google Scholar] [CrossRef]
- Mundt, B.; Kuhnel, F.; Zender, L.; Paul, Y.; Tillmann, H.; Trautwein, C.; Manns, M.P.; Kubicka, S. Involvement of TRAIL and its receptors in viral hepatitis. Faseb. J. 2003, 17, 94–96. [Google Scholar]
- White, S.; Rosen, A. Apoptosis in systemic lupus erythematosus. Curr Opin Rheumatol 2003, 15, 557–562. [Google Scholar] [CrossRef]
- Chen, W.; Sulcove, J.; Frank, I.; Jaffer, S.; Ozdener, H.; Kolson, D.L. Development of a human neuronal cell model for human immunodeficiency virus (HIV)-infected macrophage-induced neurotoxicity: apoptosis induced by HIV type 1 primary isolates and evidence for involvement of the Bcl-2/Bcl-xL-sensitive intrinsic apoptosis pathway. J. Virol. 2002, 76, 9407–9419. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef]
- Taylor, J.M.; Barry, M. Near death experiences: poxvirus regulation of apoptotic death. Virology 2006, 344, 139–150. [Google Scholar] [CrossRef]
- Gaur, U.; Aggarwal, B.B. Regulation of proliferation, survival and apoptosis by members of the TNF superfamily. Biochem. Pharmacol. 2003, 66, 1403–1408. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Holland, P.; Eckhardt, S.G. Ligand-based targeting of apoptosis in cancer: The potential of recombinant human apoptosis ligand 2/Tumor necrosis factor-related apoptosis-inducing ligand (rhApo2L/TRAIL). J. Clin. Oncol. 2008, 26, 3621–3630. [Google Scholar] [CrossRef]
- Scaffidi, C.; Fulda, S.; Srinivasan, A.; Friesen, C.; Li, F.; Tomaselli, K.J.; Debatin, K.M.; Krammer, P.H.; Peter, M.E. Two CD95 (APO-1/Fas) signaling pathways. Embo J. 1998, 17, 1675–1687. [Google Scholar] [CrossRef]
- Schmitz, I.; Walczak, H.; Krammer, P.H.; Peter, M.E. Differences between CD95 type I and II cells detected with the CD95 ligand. Cell. Death Differ. 1999, 6, 821–822. [Google Scholar]
- Wang, J.; Chun, H.J.; Wong, W.; Spencer, D.M.; Lenardo, M.J. Caspase-10 is an initiator caspase in death receptor signaling. Proc. Natl. Acad. Sci. USA 2001, 98, 13884–13888. [Google Scholar]
- Tinel, A.; Tschopp, J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science 2004, 304, 843–846. [Google Scholar] [CrossRef]
- Henshall, D.C.; Skradski, S.L.; Bonislawski, D.P.; Lan, J.Q.; Simon, R.P. Caspase-2 activation is redundant during seizure-induced neuronal death. J. Neurochem. 2001, 77, 886–895. [Google Scholar] [CrossRef]
- Sohn, D.; Budach, W.; Janicke, R.U. Caspase-2 is required for DNA damage-induced expression of the CDK inhibitor p21(WAF1/CIP1). Cell. Death Differ. 2011, 18, 1664–1674. [Google Scholar] [CrossRef]
- Rich, T.; Allen, R.L.; Wyllie, A.H. Defying death after DNA damage. Nature 2000, 407, 777–783. [Google Scholar] [CrossRef]
- Vermeulen, K.; Van Bockstaele, D.R.; Berneman, Z.N. Apoptosis: Mechanisms and relevance in cancer. Ann. Hematol. 2005, 84, 627–639. [Google Scholar] [CrossRef]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997, 91, 479–489. [Google Scholar] [CrossRef]
- Zou, H.; Li, Y.; Liu, X.; Wang, X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol Chem 1999, 274, 11549–11556. [Google Scholar] [CrossRef]
- Hill, M.M.; Adrain, C.; Duriez, P.J.; Creagh, E.M.; Martin, S.J. Analysis of the composition, assembly kinetics and activity of native Apaf-1 apoptosomes. Embo J. 2004, 23, 2134–2145. [Google Scholar] [CrossRef]
- Kothakota, S.; Azuma, T.; Reinhard, C.; Klippel, A.; Tang, J.; Chu, K.; McGarry, T.J.; Kirschner, M.W.; Koths, K.; Kwiatkowski, D.J.; et al. Caspase-3-generated fragment of gelsolin: Effector of morphological change in apoptosis. Science 1997, 278, 294–298. [Google Scholar] [CrossRef]
- Kato, K.; Yamanouchi, D.; Esbona, K.; Kamiya, K.; Zhang, F.; Kent, K.C.; Liu, B. Caspase-mediated protein kinase C-delta cleavage is necessary for apoptosis of vascular smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H2253–2261. [Google Scholar] [CrossRef]
- Zilli, D.; Voelkel-Johnson, C.; Skinner, T.; Laster, S.M. The adenovirus E3 region 14.7 kDa protein, heat and sodium arsenite inhibit the TNF-induced release of arachidonic acid. Biochem. Biophys. Res. Commun. 1992, 188, 177–183. [Google Scholar] [CrossRef]
- Rahman, M.M.; Lucas, A.R.; McFadden, G. Viral TNF inhibitors as potential therapeutics. Adv. Exp. Med. Biol. 2009, 666, 64–77. [Google Scholar] [CrossRef]
- Bertin, J.; Armstrong, R.C.; Ottilie, S.; Martin, D.A.; Wang, Y.; Banks, S.; Wang, G.H.; Senkevich, T.G.; Alnemri, E.S.; Moss, B.; Lenardo; et al. Death effector domain-containing herpesvirus and poxvirus proteins inhibit both Fas- and TNFR1-induced apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 1172–1176. [Google Scholar]
- Zhang, Y.; Zhao, H.; He, X.; Zheng, S.; Wang, T.; Yan, D.; Sun, J.; Lu, X.; Wen, J.; Lau, W.Y. Effects of Epstein-Barr virus infection on the development of multiple myeloma after liver transplantation. Sci. China Life Sci. 2012, 55, 735–743. [Google Scholar] [CrossRef]
- Cai, Q.; Guo, Y.; Xiao, B.; Banerjee, S.; Saha, A.; Lu, J.; Glisovic, T.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C stabilizes Gemin3 to block p53-mediated apoptosis. PLoS Pathog. 2011, 7, e1002418. [Google Scholar] [CrossRef]
- Stubenrauch, F.; Lim, H.B.; Laimins, L.A. Differential requirements for conserved E2 binding sites in the life cycle of oncogenic human papillomavirus type 31. J. Virol 1998, 72, 1071–1077. [Google Scholar]
- Thomas, J.T.; Hubert, W.G.; Ruesch, M.N.; Laimins, L.A. Human papillomavirus type 31 oncoproteins E6 and E7 are required for the maintenance of episomes during the viral life cycle in normal human keratinocytes. Proc. Natl. Acad. Sci. USA 1999, 96, 8449–8454. [Google Scholar] [CrossRef]
- Park, R.B.; Androphy, E.J. Genetic analysis of high-risk e6 in episomal maintenance of human papillomavirus genomes in primary human keratinocytes. J. Virol. 2002, 76, 11359–11364. [Google Scholar] [CrossRef]
- De Geest, K.; Turyk, M.E.; Hosken, M.I.; Hudson, J.B.; Laimins, L.A.; Wilbanks, G.D. Growth and differentiation of human papillomavirus type 31b positive human cervical cell lines. Gynecol Oncol 1993, 49, 303–310. [Google Scholar] [CrossRef]
- Stanley, M. The immunology of genital human papilloma virus infection. Eur. J. Dermatol. 1998, 8, 8–12, discussion 20-12. [Google Scholar]
- Doorbar, J. The papillomavirus life cycle. J. Clin. Virol. 2005, 32, S7–S15. [Google Scholar] [CrossRef]
- Munger, K.; Basile, J.R.; Duensing, S.; Eichten, A.; Gonzalez, S.L.; Grace, M.; Zacny, V.L. Biological activities and molecular targets of the human papillomavirus E7 oncoprotein. Oncogene 2001, 20, 7888–7898. [Google Scholar] [CrossRef]
- McBride, A.A.; Oliveira, J.G.; McPhillips, M.G. Partitioning viral genomes in mitosis: Same idea, different targets. Cell. Cycle 2006, 5, 1499–1502. [Google Scholar] [CrossRef]
- Peh, W.L.; Brandsma, J.L.; Christensen, N.D.; Cladel, N.M.; Wu, X.; Doorbar, J. The viral E4 protein is required for the completion of the cottontail rabbit papillomavirus productive cycle in vivo. J. Virol. 2004, 78, 2142–2151. [Google Scholar] [CrossRef]
- Genther, S.M.; Sterling, S.; Duensing, S.; Munger, K.; Sattler, C.; Lambert, P.F. Quantitative role of the human papillomavirus type 16 E5 gene during the productive stage of the viral life cycle. J. Virol 2003, 77, 2832–2842. [Google Scholar] [CrossRef]
- Fehrmann, F.; Laimins, L.A. Human papillomaviruses: Targeting differentiating epithelial cells for malignant transformation. Oncogene 2003, 22, 5201–5207. [Google Scholar] [CrossRef]
- Hummel, M.; Hudson, J.B.; Laimins, L.A. Differentiation-induced and constitutive transcription of human papillomavirus type 31b in cell lines containing viral episomes. J. Virol 1992, 66, 6070–6080. [Google Scholar]
- Ozbun, M.A.; Meyers, C. Characterization of late gene transcripts expressed during vegetative replication of human papillomavirus type 31b. J. Virol. 1997, 71, 5161–5172. [Google Scholar]
- Davy, C.; McIntosh, P.; Jackson, D.J.; Sorathia, R.; Miell, M.; Wang, Q.; Khan, J.; Soneji, Y.; Doorbar, J. A novel interaction between the human papillomavirus type 16 E2 and E1--E4 proteins leads to stabilization of E2. Virology 2009, 394, 266–275. [Google Scholar] [CrossRef]
- McIntosh, P.B.; Laskey, P.; Sullivan, K.; Davy, C.; Wang, Q.; Jackson, D.J.; Griffin, H.M.; Doorbar, J. E1--E4-mediated keratin phosphorylation and ubiquitylation: A mechanism for keratin depletion in HPV16-infected epithelium. J. Cell Sci. 2010, 123, 2810–2822. [Google Scholar] [CrossRef]
- Venuti, A.; Paolini, F.; Nasir, L.; Corteggio, A.; Roperto, S.; Campo, M.S.; Borzacchiello, G. Papillomavirus E5: The smallest oncoprotein with many functions. Mol. Cancer 2011, 10, 140. [Google Scholar] [CrossRef]
- Gandarillas, A.; Goldsmith, L.A.; Gschmeissner, S.; Leigh, I.M.; Watt, F.M. Evidence that apoptosis and terminal differentiation of epidermal keratinocytes are distinct processes. Exp. Dermatol. 1999, 8, 71–79. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Giri, I.; Yaniv, M. Structural and mutational analysis of E2 trans-activating proteins of papillomaviruses reveals three distinct functional domains. Embo. J. 1988, 7, 2823–2829. [Google Scholar]
- Desaintes, C.; Demeret, C.; Goyat, S.; Yaniv, M.; Thierry, F. Expression of the papillomavirus E2 protein in HeLa cells leads to apoptosis. Embo. J. 1997, 16, 504–514. [Google Scholar] [CrossRef]
- Demeret, C.; Garcia-Carranca, A.; Thierry, F. Transcription-independent triggering of the extrinsic pathway of apoptosis by human papillomavirus 18 E2 protein. Oncogene 2003, 22, 168–175. [Google Scholar] [CrossRef]
- Thierry, F.; Demeret, C. Direct activation of caspase 8 by the proapoptotic E2 protein of HPV18 independent of adaptor proteins. Cell Death Differ. 2008, 15, 1356–1363. [Google Scholar] [CrossRef]
- Massimi, P.; Pim, D.; Bertoli, C.; Bouvard, V.; Banks, L. Interaction between the HPV-16 E2 transcriptional activator and p53. Oncogene 1999, 18, 7748–7754. [Google Scholar]
- Webster, K.; Parish, J.; Pandya, M.; Stern, P.L.; Clarke, A.R.; Gaston, K. The human papillomavirus (HPV) 16 E2 protein induces apoptosis in the absence of other HPV proteins and via a p53-dependent pathway. J. Biol. Chem. 2000, 275, 87–94. [Google Scholar]
- Green, K.L.; Brown, C.; Roeder, G.E.; Southgate, T.D.; Gaston, K. A cancer cell-specific inducer of apoptosis. Hum. Gene Ther. 2007, 18, 547–561. [Google Scholar] [CrossRef]
- Straight, S.W.; Hinkle, P.M.; Jewers, R.J.; McCance, D.J. The E5 oncoprotein of human papillomavirus type 16 transforms fibroblasts and effects the downregulation of the epidermal growth factor receptor in keratinocytes. J. Virol. 1993, 67, 4521–4532. [Google Scholar]
- Genther Williams, S.M.; Disbrow, G.L.; Schlegel, R.; Lee, D.; Threadgill, D.W.; Lambert, P.F. Requirement of epidermal growth factor receptor for hyperplasia induced by E5, a high-risk human papillomavirus oncogene. Cancer Res. 2005, 65, 6534–6542. [Google Scholar]
- Zhang, B.; Srirangam, A.; Potter, D.A.; Roman, A. HPV16 E5 protein disrupts the c-Cbl-EGFR interaction and EGFR ubiquitination in human foreskin keratinocytes. Oncogene 2005, 24, 2585–2588. [Google Scholar] [CrossRef]
- Suprynowicz, F.A.; Krawczyk, E.; Hebert, J.D.; Sudarshan, S.R.; Simic, V.; Kamonjoh, C.M.; Schlegel, R. The human papillomavirus type 16 E5 oncoprotein inhibits epidermal growth factor trafficking independently of endosome acidification. J. Virol. 2010, 84, 10619–10629. [Google Scholar] [CrossRef]
- Dannenberg, A.J.; Lippman, S.M.; Mann, J.R.; Subbaramaiah, K.; DuBois, R.N. Cyclooxygenase-2 and epidermal growth factor receptor: Pharmacologic targets for chemoprevention. J. Clin Oncol 2005, 23, 254–266. [Google Scholar]
- Kabsch, K.; Alonso, A. The human papillomavirus type 16 E5 protein impairs TRAIL- and FasL-mediated apoptosis in HaCaT cells by different mechanisms. J. Virol 2002, 76, 12162–12172. [Google Scholar] [CrossRef]
- Zhang, B.; Spandau, D.F.; Roman, A. E5 protein of human papillomavirus type 16 protects human foreskin keratinocytes from UV B-irradiation-induced apoptosis. J. Virol 2002, 76, 220–231. [Google Scholar] [CrossRef]
- Oh, J.M.; Kim, S.H.; Cho, E.A.; Song, Y.S.; Kim, W.H.; Juhnn, Y.S. Human papillomavirus type 16 E5 protein inhibits hydrogen-peroxide-induced apoptosis by stimulating ubiquitin-proteasome-mediated degradation of Bax in human cervical cancer cells. Carcinogenesis 2010, 31, 402–410. [Google Scholar] [CrossRef]
- Kim, M.K.; Kim, H.S.; Kim, S.H.; Oh, J.M.; Han, J.Y.; Lim, J.M.; Juhnn, Y.S.; Song, Y.S. Human papillomavirus type 16 E5 oncoprotein as a new target for cervical cancer treatment. Biochem. Pharmacol. 2010, 80, 1930–1935. [Google Scholar] [CrossRef]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. Embo. J. 1991, 10, 4129–4135. [Google Scholar]
- Crook, T.; Tidy, J.A.; Vousden, K.H. Degradation of p53 can be targeted by HPV E6 sequences distinct from those required for p53 binding and trans-activation. Cell. 1991, 67, 547–556. [Google Scholar] [CrossRef]
- Lechner, M.S.; Laimins, L.A. Inhibition of p53 DNA binding by human papillomavirus E6 proteins. J. Virol. 1994, 68, 4262–4273. [Google Scholar]
- Massimi, P.; Shai, A.; Lambert, P.; Banks, L. HPV E6 degradation of p53 and PDZ containing substrates in an E6AP null background. Oncogene 2008, 27, 1800–1804. [Google Scholar] [CrossRef]
- Shai, A.; Pitot, H.C.; Lambert, P.F. E6-associated protein is required for human papillomavirus type 16 E6 to cause cervical cancer in mice. Cancer Res. 2010, 70, 5064–5073. [Google Scholar] [CrossRef]
- Thomas, M.; Banks, L. Inhibition of Bak-induced apoptosis by HPV-18 E6. Oncogene 1998, 17, 2943–2954. [Google Scholar]
- Gross-Mesilaty, S.; Reinstein, E.; Bercovich, B.; Tobias, K.E.; Schwartz, A.L.; Kahana, C.; Ciechanover, A. Basal and human papillomavirus E6 oncoprotein-induced degradation of Myc proteins by the ubiquitin pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 8058–8063. [Google Scholar]
- Filippova, M.; Brown-Bryan, T.A.; Casiano, C.A.; Duerksen-Hughes, P.J. The human papillomavirus 16 E6 protein can either protect or further sensitize cells to TNF: Effect of dose. Cell. Death Differ. 2005, 12, 1622–1635. [Google Scholar] [CrossRef]
- Filippova, M.; Parkhurst, L.; Duerksen-Hughes, P.J. The human papillomavirus 16 E6 protein binds to Fas-associated death domain and protects cells from Fas-triggered apoptosis. J. Biol. Chem. 2004, 279, 25729–25744. [Google Scholar] [CrossRef]
- Filippova, M.; Filippov, V.A.; Kagoda, M.; Garnett, T.; Fodor, N.; Duerksen-Hughes, P.J. Complexes of human papillomavirus type 16 E6 proteins form pseudo-death-inducing signaling complex structures during tumor necrosis factor-mediated apoptosis. J. Virol 2009, 83, 210–227. [Google Scholar]
- Garnett, T.O.; Filippova, M.; Duerksen-Hughes, P.J. Accelerated degradation of FADD and procaspase 8 in cells expressing human papilloma virus 16 E6 impairs TRAIL-mediated apoptosis. Cell. Death Differ. 2006, 13, 1915–1926. [Google Scholar] [CrossRef]
- Du, J.; Chen, G.G.; Vlantis, A.C.; Chan, P.K.; Tsang, R.K.; van Hasselt, C.A. Resistance to apoptosis of HPV 16-infected laryngeal cancer cells is associated with decreased Bak and increased Bcl-2 expression. Cancer Lett. 2004, 205, 81–88. [Google Scholar] [CrossRef]
- Tungteakkhun, S.S.; Filippova, M.; Fodor, N.; Duerksen-Hughes, P.J. The full-length isoform of human papillomavirus 16 E6 and its splice variant E6* bind to different sites on the procaspase 8 death effector domain. J. Virol. 2010, 84, 1453–1463. [Google Scholar]
- Filippova, M.; Song, H.; Connolly, J.L.; Dermody, T.S.; Duerksen-Hughes, P.J. The human papillomavirus 16 E6 protein binds to tumor necrosis factor (TNF) R1 and protects cells from TNF-induced apoptosis. J. Biol. Chem. 2002, 277, 21730–21739. [Google Scholar]
- Borbely, A.A.; Murvai, M.; Konya, J.; Beck, Z.; Gergely, L.; Li, F.; Veress, G. Effects of human papillomavirus type 16 oncoproteins on survivin gene expression. J. Gen. Virol. 2006, 87, 287–294. [Google Scholar] [CrossRef]
- Jabbar, S.F.; Park, S.; Schweizer, J.; Berard-Bergery, M.; Pitot, H.C.; Lee, D.; Lambert, P.F. Cervical cancers require the continuous expression of the human papillomavirus type 16 E7 oncoprotein even in the presence of the viral E6 oncoprotein. Cancer Res. 2012, 72, 4008–4016. [Google Scholar] [CrossRef]
- Smotkin, D.; Prokoph, H.; Wettstein, F.O. Oncogenic and nononcogenic human genital papillomaviruses generate the E7 mRNA by different mechanisms. J. Virol 1989, 63, 1441–1447. [Google Scholar]
- Stacey, S.N.; Jordan, D.; Snijders, P.J.; Mackett, M.; Walboomers, J.M.; Arrand, J.R. Translation of the human papillomavirus type 16 E7 oncoprotein from bicistronic mRNA is independent of splicing events within the E6 open reading frame. J. Virol. 1995, 69, 7023–7031. [Google Scholar]
- Pim, D.; Tomaic, V.; Banks, L. The human papillomavirus (HPV) E6* proteins from high-risk, mucosal HPVs can direct degradation of cellular proteins in the absence of full-length E6 protein. J. Virol. 2009, 83, 9863–9874. [Google Scholar] [CrossRef]
- Mesplede, T.; Gagnon, D.; Bergeron-Labrecque, F.; Azar, I.; Senechal, H.; Coutlee, F.; Archambault, J. p53 degradation activity, expression, and subcellular localization of E6 proteins from 29 human papillomavirus genotypes. J. Virol 2012, 86, 94–107. [Google Scholar] [CrossRef]
- Filippova, M.; Johnson, M.M.; Bautista, M.; Filippov, V.; Fodor, N.; Tungteakkhun, S.S.; Williams, K.; Duerksen-Hughes, P.J. The large and small isoforms of human papillomavirus type 16 E6 bind to and differentially affect procaspase 8 stability and activity. J. Virol 2007, 81, 4116–4129. [Google Scholar] [CrossRef]
- Münger, K.; Basile, J.; Duensing, S.; Eichten, A.; Gonzalez, S.; Grace, M.; Zacny, L. Biological activities and molecular targets of the human papillomavirus E7 oncoprotein. Oncogene 2001, 20, 7888–7898. [Google Scholar] [CrossRef]
- Berezutskaya, E.; Yu, B.; Morozov, A.; Raychaudhuri, P.; Bagchi, S. Differential regulation of the pocket domains of the retinoblastoma family proteins by the HPV16 E7 oncoprotein. Cell Growth Differ. 1997, 8, 1277–1286. [Google Scholar]
- Howes, K.A.; Ransom, N.; Papermaster, D.S.; Lasudry, J.G.; Albert, D.M.; Windle, J.J. Apoptosis or retinoblastoma: Alternative fates of photoreceptors expressing the HPV-16 E7 gene in the presence or absence of p53. Genes Dev. 1994, 8, 1300–1310. [Google Scholar] [CrossRef]
- Pan, H.; Griep, A.E. Temporally distinct patterns of p53-dependent and p53-independent apoptosis during mouse lens development. Genes Dev. 1995, 9, 2157–2169. [Google Scholar] [CrossRef]
- Pan, H.; Griep, A.E. Altered cell cycle regulation in the lens of HPV-16 E6 or E7 transgenic mice: implications for tumor suppressor gene function in development. Genes Dev. 1994, 8, 1285–1299. [Google Scholar] [CrossRef]
- Alunni-Fabbroni, M.; Littlewood, T.; Deleu, L.; Caldeira, S.; Giarre, M.; Dell' Orco, M.; Tommasino, M. Induction of S phase and apoptosis by the human papillomavirus type 16 E7 protein are separable events in immortalized rodent fibroblasts. Oncogene 2000, 19, 2277–2285. [Google Scholar] [CrossRef]
- Kaznelson, D.W.; Bruun, S.; Monrad, A.; Gjerlov, S.; Birk, J.; Ropke, C.; Norrild, B. Simultaneous human papilloma virus type 16 E7 and cdk inhibitor p21 expression induces apoptosis and cathepsin B activation. Virology 2004, 320, 301–312. [Google Scholar] [CrossRef]
- Stoppler, H.; Stoppler, M.C.; Johnson, E.; Simbulan-Rosenthal, C.M.; Smulson, M.E.; Iyer, S.; Rosenthal, D.S.; Schlegel, R. The E7 protein of human papillomavirus type 16 sensitizes primary human keratinocytes to apoptosis. Oncogene 1998, 17, 1207–1214. [Google Scholar]
- Thomas, M.; Glaunsinger, B.; Pim, D.; Javier, R.; Banks, L. HPV E6 and MAGUK protein interactions: determination of the molecular basis for specific protein recognition and degradation. Oncogene 2001, 20, 5431–5439. [Google Scholar] [CrossRef]
- Magal, S.S.; Jackman, A.; Pei, X.F.; Schlegel, R.; Sherman, L. Induction of apoptosis in human keratinocytes containing mutated p53 alleles and its inhibition by both the E6 and E7 oncoproteins. Int. J. Cancer 1998, 75, 96–104. [Google Scholar] [CrossRef]
- Severino, A.; Abbruzzese, C.; Manente, L.; Valderas, A.A.; Mattarocci, S.; Federico, A.; Starace, G.; Chersi, A.; Mileo, A.M.; Paggi, M.G. Human papillomavirus-16 E7 interacts with Siva-1 and modulates apoptosis in HaCaT human immortalized keratinocytes. J. Cell. Physiol. 2007, 212, 118–125. [Google Scholar] [CrossRef]
- Aguilar-Lemarroy, A.; Gariglio, P.; Whitaker, N.J.; Eichhorst, S.T.; zur Hausen, H.; Krammer, P.H.; Rosl, F. Restoration of p53 expression sensitizes human papillomavirus type 16 immortalized human keratinocytes to CD95-mediated apoptosis. Oncogene 2002, 21, 165–175. [Google Scholar] [CrossRef]
- Kim, C.Y.; Tsai, M.H.; Osmanian, C.; Graeber, T.G.; Lee, J.E.; Giffard, R.G.; DiPaolo, J.A.; Peehl, D.M.; Giaccia, A.J. Selection of human cervical epithelial cells that possess reduced apoptotic potential to low-oxygen conditions. Cancer Res. 1997, 57, 4200–4204. [Google Scholar]
- Brink, A.A.; Zielinski, G.D.; Steenbergen, R.D.; Snijders, P.J.; Meijer, C.J. Clinical relevance of human papillomavirus testing in cytopathology. Cytopathology 2005, 16, 7–12. [Google Scholar] [CrossRef]
- Longworth, M.S.; Laimins, L.A. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol. Mol. Biol. Rev. 2004, 68, 362–372. [Google Scholar]
- Parkin, D.M. The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer 2006, 118, 3030–3044. [Google Scholar] [CrossRef]
- Widdice, L.E. Human papillomavirus disease in adolescents: Management and prevention. Adolesc Med. State Art Rev. 2012, 23, 192–206, xii-xiii. [Google Scholar]
- Paavonen, J.; Jenkins, D.; Bosch, F.X.; Naud, P.; Salmeron, J.; Wheeler, C.M.; Chow, S.N.; Apter, D.L.; Kitchener, H.C.; Castellsague, X.; et al. Efficacy of a prophylactic adjuvanted bivalent L1 virus-like-particle vaccine against infection with human papillomavirus types 16 and 18 in young women: An interim analysis of a phase III double-blind, randomised controlled trial. Lancet 2007, 369, 2161–2170. [Google Scholar]
- Oehler-Janne, C.; Huguet, F.; Provencher, S.; Seifert, B.; Negretti, L.; Riener, M.O.; Bonet, M.; Allal, A.S.; Ciernik, I.F. HIV-specific differences in outcome of squamous cell carcinoma of the anal canal: a multicentric cohort study of HIV-positive patients receiving highly active antiretroviral therapy. J. Clin. Oncol. 2008, 26, 2550–2557. [Google Scholar]
- Hagensee, M.E.; Cameron, J.E.; Leigh, J.E.; Clark, R.A. Human papillomavirus infection and disease in HIV-infected individuals. Am. J. Med. Sci 2004, 328, 57–63. [Google Scholar] [CrossRef]
- Beutner, K.R.; Ferenczy, A. Therapeutic approaches to genital warts. Am. J. Med. 1997, 102, 28–37. [Google Scholar] [CrossRef]
- Beutner, K.R.; Tyring, S.K.; Trofatter, K.F., Jr.; Douglas, J.M., Jr.; Spruance, S.; Owens, M.L.; Fox, T.L.; Hougham, A.J.; Schmitt, K.A. Imiquimod, a patient-applied immune-response modifier for treatment of external genital warts. Antimicrob Agents Chemother 1998, 42, 789–794. [Google Scholar]
- Scheinfeld, N.; Lehman, D.S. An evidence-based review of medical and surgical treatments of genital warts. Dermatol. Online J. 2006, 12, 5. [Google Scholar]
- D'Abramo, C.M.; Archambault, J. Small molecule inhibitors of human papillomavirus protein-protein interactions. Open Virol. J. 2011, 5, 80–95. [Google Scholar] [CrossRef]
- Phelps, W.C.; Barnes, J.A.; Lobe, D.C. Molecular targets for human papillomaviruses: Prospects for antiviral therapy. Antivir. Chem. Chemother. 1998, 9, 359–377. [Google Scholar]
- Faucher, A.M.; White, P.W.; Brochu, C.; Grand-Maitre, C.; Rancourt, J.; Fazal, G. Discovery of small-molecule inhibitors of the ATPase activity of human papillomavirus E1 helicase. J. Med. Chem. 2004, 47, 18–21. [Google Scholar] [CrossRef]
- Hebner, C.; Beglin, M.; Laimins, L.A. Human papillomavirus E6 proteins mediate resistance to interferon-induced growth arrest through inhibition of p53 acetylation. J. Virol. 2007, 81, 12740–12747. [Google Scholar] [CrossRef]
- Scheffner, M.; Whitaker, N.J. Human papillomavirus-induced carcinogenesis and the ubiquitin-proteasome system. Semin Cancer Biol. 2003, 13, 59–67. [Google Scholar] [CrossRef]
- Thomas, M.C.; Chiang, C.M. E6 oncoprotein represses p53-dependent gene activation via inhibition of protein acetylation independently of inducing p53 degradation. Mol. Cell. 2005, 17, 251–264. [Google Scholar] [CrossRef]
- Baleja, J.D.; Cherry, J.J.; Liu, Z.; Gao, H.; Nicklaus, M.C.; Voigt, J.H.; Chen, J.J.; Androphy, E.J. Identification of inhibitors to papillomavirus type 16 E6 protein based on three-dimensional structures of interacting proteins. Antiviral Res. 2006, 72, 49–59. [Google Scholar] [CrossRef]
- Bellail, A.C.; Qi, L.; Mulligan, P.; Chhabra, V.; Hao, C. TRAIL agonists on clinical trials for cancer therapy: the promises and the challenges. Rev. Recent Clin. Trials 2009, 4, 34–41. [Google Scholar] [CrossRef]
- El-Zawahry, A.; McKillop, J.; Voelkel-Johnson, C. Doxorubicin increases the effectiveness of Apo2L/TRAIL for tumor growth inhibition of prostate cancer xenografts. BMC Cancer 2005, 5, 2. [Google Scholar] [CrossRef]
- Mom, C.H.; Verweij, J.; Oldenhuis, C.N.; Gietema, J.A.; Fox, N.L.; Miceli, R.; Eskens, F.A.; Loos, W.J.; de Vries, E.G.; Sleijfer, S. Mapatumumab, a fully human agonistic monoclonal antibody that targets TRAIL-R1, in combination with gemcitabine and cisplatin: a phase I stud. Clin. Cancer Res. 2009, 15, 5584–5590. [Google Scholar] [CrossRef]
- Naka, T.; Sugamura, K.; Hylander, B.L.; Widmer, M.B.; Rustum, Y.M.; Repasky, E.A. Effects of tumor necrosis factor-related apoptosis-inducing ligand alone and in combination with chemotherapeutic agents on patients' colon tumors grown in SCID mice. Cancer Res. 2002, 62, 5800–5806. [Google Scholar]
- Yi, C.; Maksimoska, J.; Marmorstein, R.; Kissil, J.L. Development of small-molecule inhibitors of the group I p21-activated kinases, emerging therapeutic targets in cancer. Biochem. Pharmacol. 2010, 80, 683–689. [Google Scholar] [CrossRef]
- Yuan, C.H.; Filippova, M.; Tungteakkhun, S.S.; Duerksen-Hughes, P.J.; Krstenansky, J.L. Small molecule inhibitors of the HPV16-E6 interaction with caspase 8. Bioorg. Med. Chem. Lett. 2012, 22, 2125–2129. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yuan, C.-H.; Filippova, M.; Duerksen-Hughes, P. Modulation of Apoptotic Pathways by Human Papillomaviruses (HPV): Mechanisms and Implications for Therapy. Viruses 2012, 4, 3831-3850. https://doi.org/10.3390/v4123831
Yuan C-H, Filippova M, Duerksen-Hughes P. Modulation of Apoptotic Pathways by Human Papillomaviruses (HPV): Mechanisms and Implications for Therapy. Viruses. 2012; 4(12):3831-3850. https://doi.org/10.3390/v4123831
Chicago/Turabian StyleYuan, Chung-Hsiang, Maria Filippova, and Penelope Duerksen-Hughes. 2012. "Modulation of Apoptotic Pathways by Human Papillomaviruses (HPV): Mechanisms and Implications for Therapy" Viruses 4, no. 12: 3831-3850. https://doi.org/10.3390/v4123831