Nanotechnology and the Treatment of HIV Infection



Abstract
:
1. Introduction
2. HIV Pathogenesis and Treatment: Challenges and Opportunities
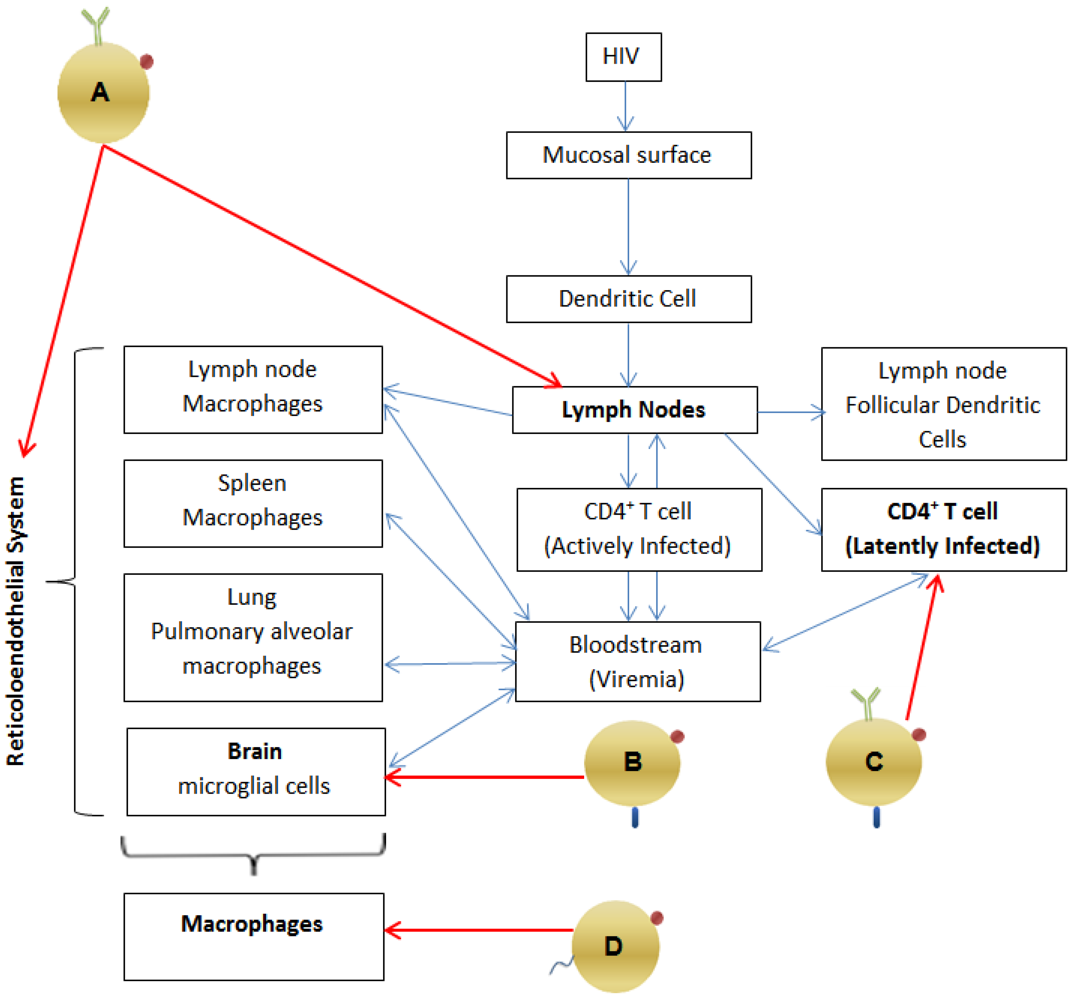
2.1. Pathogenesis: Viral Reservoirs (See Figure 1)
- Cells of the monocyte-macrophage lineage [12,13] (which constitute the reticulo-endothelial system) and which includes the microglial cells of the brain, pulmonary alveolar macrophages of the lung [12] and macrophages within the spleen and lymph nodes [10,12], Antiretrovirals have poor activity in chronically infected macrophages, requiring much higher intracellular concentrations to achieve inhibition [14]. Macrophages are relatively long-lived cells, and HIV has minimal cytopathic effects on them [12,14,15]. Therefore, macrophages are a persistent reservoir of HIV, even in the presence of HAART.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
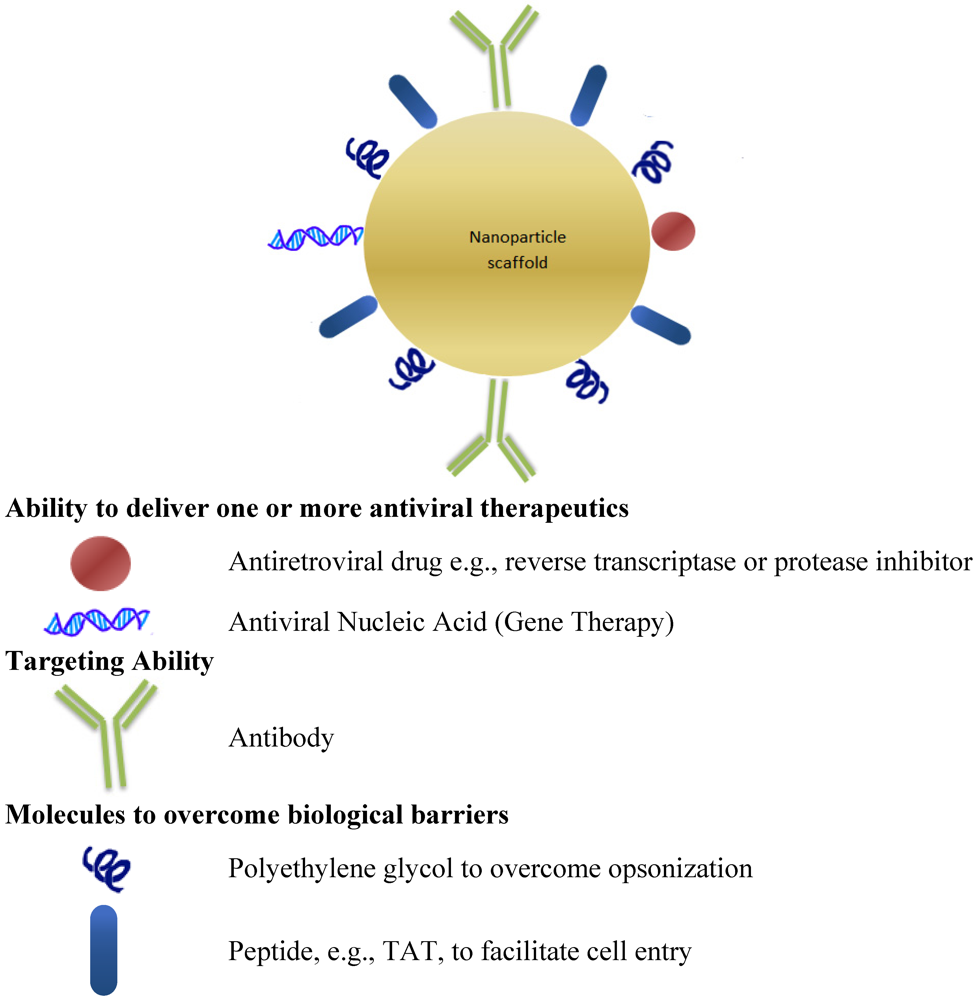{kind=link}
| Physical Property | Biological Implications | Potential Benefit for HIV Therapy |
|---|---|---|
| Particle size | Particle size affects bioavailability and circulation time [2]. Particles <5–10 nm are removed by renal clearance [21] while those >200 nm are sequestered by the spleen[21]. Particles up to 70 nm can penetrate capillaries [2] while nanocomplexes between 35–120 nm localize in lymph nodes [22,23]. | Nanocomplexes containing indinavir were designed so that they were in the size range that allowed localization within lymphatic tissues. Following subcutaneous injection in macaques, the particles drain into the lymphatic system, and because of their size, remain trapped there, rather than entering the bloodstream—this strategy avoids undesirable, excessive peaks in plasma concentration [22,23,24]. |
| Size determines mechanism of internalization (phagocytosis versus endocytosis versus pinocytosis) and therefore subcellular localization [21]; depending on their size, particles may be opsonized by plasma proteins, phagocytosed by macrophages and removed by the RES* [21]. | Liposomes are phagocytosed by macrophages and deliver drugs such as AZT+ [25] and ddI# [26,27], which are carried in their aqueous core, to murine RES*. | |
| Large surface area to volume ratio [4] | Dissolution of poorly soluble drugs is greatly dependent on the surface area of the particle. Nanosized particles therefore display enhanced solubility compared to larger particles [2,28,29,30,31]. | Engineering drugs in the nanorange, in the form of nanocrystals or nanosuspensions [28,29,30,31,32], for example, allow for clinical development of lead compounds that would not otherwise be considered viable due to poor solubility; Improved solubility enhances bioavailability and dosing of poorly water-soluble antiretroviral drugs, such as rilpivirine [33,34]. |
| Surface charge of particle [2,21] | The cell membrane is negatively charged and repels like-charged molecules. Positively charged nanoparticles may shield such molecules, allowing them to enter the cell [35]. | Allows cellular entry of antiretroviral agents, which are negatively charged, such as phosphorylated nucleotide analogues [35] and nucleic acids (See Table 3) [36,37,38,39,40,41,42]. |
| Formation of stable structures which are able to encapsulate drugs [2] | Encapsulation improves solubility and protects against degradation (within the gastrointestinal tract, for example) [43,44]. | Polymeric micelles encapsulate efavirenz and improve its solubility [45,46,47,48,49]. |
| In in vitro experiments, a polymeric nanocapsule was used to deliver AZT in its triphosphorylated form directly into the cytoplasm [50,51]. | ||
| Biofunctionalized nanoparticles, [2,5,52] whereby particles may be functionalized by attachment of bioactive moieties | Nanomedicines are easily tagged by coating them with moieties that bind to biomarkers, thus directing them to cells, tissues or even organelles that exhibit the biomarker [4,21,52]. | In animal experiments, liposomes coated with galactose or lectin (“immunoliposomes”) target cells of the RES* that bear receptors for these moieties, and may thus be employed to deliver antiretroviral drugs specifically to these sites [53,54,55,56] (thus decreasing side-effects caused by distribution of drugs to non-specific sites [5]). |
| Conjugation to polyethylene glycol (PEGylation) improves solubility and reduces interaction with opsonizing proteins, thus modulating phagocytosis and bioavailability [4]. | Sterically stabilized (PEGylated) liposomes and solid lipid nanoparticles, containing ddI# [57] and AZT+ [58,59] respectively, result in extended half-lives of these drugs in rodents. | |
| Multifunctionality (combining several beneficial features in a stable construct) [4,7,52] | Currently available antiretrovirals have no effect on latent virus. Nanomedicines may be designed to simultaneously stimulate the replication of latent virus and deliver an antiviral to the activated cell [60]. | Lipid nanoparticles loaded with bryostatin-2 (which activates primary CD4+ T cells) and nelfinavir may be capable of simultaneously activating latent virus and inhibiting viral spread [60]. |
| The “stealth” properties of polyethylene glycol, which allow drugs to remain longer in the systemic circulation, may be combined with peptides that promote cellular uptake [61]. | An HIV TAT**-based peptide (known to have cell penetrating properties), polyethylene glycol and the cell-uptake enhancer, biotin, were conjugated in various combinations and assessed as carriers of saquinavir. The multifunctional bioconjugates had significantly different in vitro cellular uptake and anti-HIV potency compared to the prodrug alone [62]. | |
| Biomimetic properties [7] | Nanomedicines may “mimic” the properties of biological entities, such as antibodies, receptors, nucleic acids or proteins, by binding to functional sites, such as the active site of an enzyme, thus exerting antiviral effects [7]. | Several nanomedicines may have intrinsic antiviral properties (Table 4). |
| Synthetic, nanoparticle-based multivalent displays mimic the ubiquitous biological property of multivalency that enhances affinity between naturally occurring molecules (between receptors and ligands, for example) [63]. | SDC-1721, a derivative of a known CCR5**** antagonist, does not by itself inhibit viral replication. However, when conjugated to gold nanoparticles, at a ratio of 12 SDC-1721 molecules per gold nanoparticle, activity with an IC50 of 10 nM was demonstrated in PBMCs *** infected with the CCR5-tropic HIV-1 [63]. Further results are eagerly awaited. |
- Targeting anatomical reservoirs
- Targeting cellular reservoirs
- ○ by optimizing the intracellular concentration of antiretroviral drugs into macrophages (Section 7);
- ○ by activating latent HIV (Section 10).
3. The “Nano” Landscape: From Nanomaterials to Nanopharmaceuticals
4. Nanopharmaceuticals and Biological Barriers
- Enzymatic degradation and poor stability: [87] Nanopharmaceuticals which are stable in experimental or in vitro systems may not necessarily be stable when administered to patients.
- Immunological barriers—opsonization and uptake by the reticuloendothelial system [86]. Opsonization leads to aggregation of nanoparticles and activation of defense mechanisms, including phagocytosis, which removes nanoparticles from the circulation and results in their accumulation in the reticuloendothelial system [89,90].
- Cellular barriers: Inability to traverse the cell membrane [87].
- Extracellular barriers: e.g., inability to penetrate mucin and extracellular matrix [87].
- Intracellular barriers: Entrapment within endosomes, ejection of nanopharaceuticals from the target cell by efflux pumps [88].

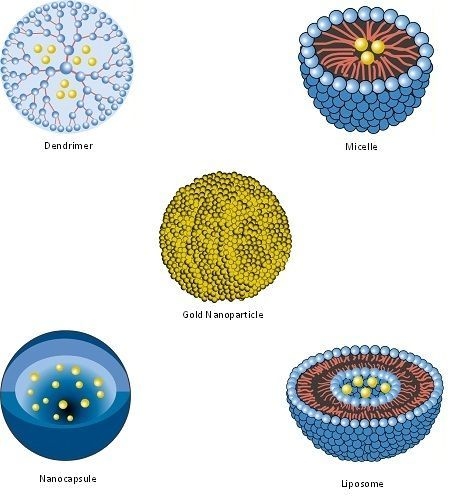
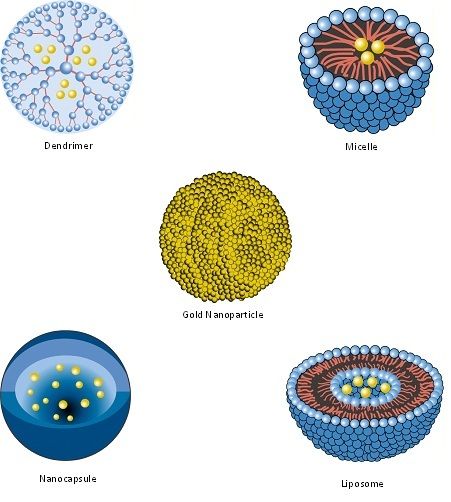
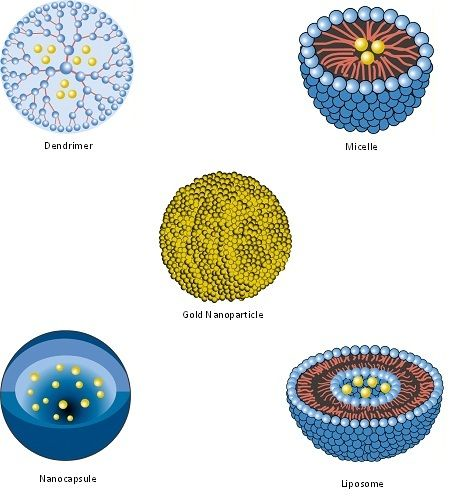
| Nanopharmaceutical | Definition |
|---|---|
| Bucky Ball (Buckminsterfullerene) | A series of carbon atoms arranged in a closed cage structure that resembles a nanosized soccer ball [109]. |
| Dendrimer | Synthetic, nanosized structure made up of multiple branched monomeric units radiating from a central core [110,111]. |
| Liposome | Vesicular nanosized structures made up of one or more phospholipid bilayer membranes surrounding an aqueous core [112]. |
| Micelle | Nanosized structure consisting of a shell and a core (made up of a water-soluble and hydrophobic polymer, respectively) [113,114]. |
| Nanoassembly | Generally, any assembly of hydrophobic and hydrophilic groups that form nanosized aggregates [115]; this article refers to such assemblies brought about by conjugation to squalene* [35,116,117]. |
| Nanoemulsion | Dispersion of immiscible droplets with sizes in the ‘nano’ range [118] |
| Nanocapsule | A nanosized structure consisting of a shell surrounding a space within which drugs are placed [119]. |
| Nanocarrier | Any nanosized entity used for the controlled and targeted delivery of pharmaceutical agents [2,120]. This is a functional definition; many of the pharmaceuticals listed in this table (including nanoparticles, micelles, dendrimers, liposomes and solid lipid nanoparticles) are defined structurally (in terms of their composition) but may function as, and can be additionally defined as nanocarriers. Further details are provided in Section 7. |
| Nanocrystal | Drug crystals with a size in the nanometer range [30]. |
| Nanoparticle | Structure with all three dimensions <100 nm [121]. Nanoparticles commonly consist of metals or polymers. |
| Nanopharmaceutical | Any nanomaterial with therapeutic potential [122]. |
| Quantum dots and rods | Semiconductor nanocrystals [123] having the shape of dots or rods [124]. |
| Solid Lipid Nanoparticle | Particle with a solid lipid matrix and a diameter in the nanometer range [125]. |
5. Nanocrystals
6. Nanoassemblies
7. Nanocarriers
8. Targeting the Reticulo-Endothelial System (RES)
9. Targeting the Brain
10. Targeting Latent HIV
11. Bio-Functionalized Nanoparticles

12. Gene Therapy: Nanocarriers of RNA and DNA
| Nanocarrier | Nucleic acid | Target | Effect |
| Poly(L-lysine), a cationic peptide | antisense oligonucleotide | primer binding site and U5 region of the viral genome | In cell culture, the antisense oligonucleotide covalently linked to poly(L-lysine) inhibited HIV-1 reverse transcriptase mediated elongation of cDNA, in a sequence and dose-dependent manner [168]. |
| Protamine, a cationic peptide | antisense oligonucleotides | tat*** mRNA | Protamine bound to the antisense oligonucleotide led to specific inhibition of tat-mediated HIV-1 transactivation in lymphocytes [169]. |
| Quantum rod | siRNA# | poly A/TAR (transactivator of the HIV-1 LTR**) site | The quantum rod-siRNA nanoplex suppressed HIV-1 viral replication in a THP-1 cell line [170]. |
| Amino-terminated carbosilane dendrimers | siRNA# | p24 region of gag or the nef sequence | Dendrimers formed dendriplexes with siRNA#, which were delivered to human astrocytes, where they reduced the replication of HIV-1 [171]. |
| Amino-terminated carbosilane dendrimers | siRNA# | p24, gag and nef | Dendriplexes were able to transfect the lymphocytic cell line SupT1 and hard-to-transfect HIV-infected peripheral blood mononuclear cells (PBMCs), where they reduced HIV replication [172]. |
| pH-sensitive liposomes | Antisense oligodeoxynucleotide; ribozyme | Rev responsive element and 5'-LTR**, respectively | Inhibited virus replication in monocyte-derived macrophages [173]. However, delivery of functional ribozymes by liposomes is relatively inefficient [174]. |
| LFA-1*- targeted and stabilized immunoliposome nanoparticles | siRNA# | CCR5 | siRNA# administered to humanized mice using immunoliposome nanoparticles resulted in selective uptake of siRNA# by T-cells and macrophages and reduction in HIV plasma viral load [175]. |
| Nanopharmaceuticals | Proposed Mechanism of Action | Activity |
|---|---|---|
| Silver nanoparticles [176] | The nanoparticles interact preferentially, and in a size-dependent manner [177], with gp120 knobs and prevents CD4-dependent virion binding, fusion, and infectivity [178]. | TI** = 8.9 [178] |
| Phenyldicarboxylic acid and naphthyldisulfonic acid polyanionic dendrimers | Interacts with gp120 and interferes with virus-cell binding. | EC50* of 0.1 and 0.3 µg/mL, respectively [179] |
| One-tailed, long-chain, water-soluble, dendritic tricarboxylato amphiphiles | They most likely act by blocking viral fusion [77]. | TI** = 4 and EC50* = 110–740 µM [180] |
| Phosphorus-containing dendrimers bearing Galβ1cer*** analogues | Block HIV entry. | IC50 values of 1.1 and 0.12 µM, respectively[181] |
| Polylysine-sulfated cellobiose## glycodendrimer | Electrostatic interaction between negatively charged sulfated oligosaccharide and positively charged gp120 on the surface of HIV [182]. | EC50* = 3.2 µg/mL [182] |
| Mannose hyperbranched dendritic polymers | Inhibits binding of HIV gp120 to DC-SIGN### [183]. | IC50 = 50 µM [183] |
| Polyamidoamine (PAMAM) dendrimers | Binds to TAR# RNA and prevents its interaction with Tat**** protein [184]. | not known |
| Multivalent glycosphingolipid-derived carbohydrate head groups covalently attached to a dendrimer core | Inhibits interaction between HIV gp120 and glycosphingolipids, which are alternate receptors for HIV-1 on the surface of immune cells [185]. | IC50 between 0.1 and 7.4 μg/mL [185] |
| Sialic Acid-Polyamidoamine (PAMAM) Glycodendrimers | Probably bind to and down-regulate CD4 antigen on surface of T-cells [186] | IC50 between 1.6 and 5.1 μM [187] |
| Water soluble dendrimic fullerene | Dendrimers seem to bind to HIV-1 protease. | EC50* = 0.22 µM [188] |
| “Bucky Ball” (C60 fullerene) structures | Computational docking models and kinetic analysis suggest binding of Bucky Ball derivatives to the active site of HIV-1 protease [189]. | not known |
14. Limitations
15. Perspectives
Acknowledgments
Conflict of Interest
References and Notes
- Picraux, T. Nanotechnology. In Encyclopaedia Britannica Deluxe Edition; Encyclopaedia Britannica: Chicago, IL, USA, 2010. [Google Scholar]
- Goldberg, M.; Langer, R.; Xinqiao, J. Nanostructured materials for applications in drug delivery and tissue engineering. J. Biomat. Sci. Polym. E 2007, 18, 241–268. [Google Scholar]
- Tegart, G. Nanotechnology: The technology for the 21st Century. In Proceedings of the Second International Conference on Technology Foresight, APEC Center for Technology Foresight, Bangkok, Thailand, 27–28 February 2001.
- McNeil, S.E. Unique benefits of nanotechnology to drug delivery and diagnostics. Methods Mol. Biol. 2011, 697, 3–8. [Google Scholar]
- Ochekpe, N.A.; Olorunfemi, P.O.; Ngwuluka, N.C. Nanotechnology and drug delivery part 1: Background and applications. Trop. J. Pharm. Res. 2009, 8, 265–274. [Google Scholar]
- Williams, D. The relationship between biomaterials and nanotechnology. Biomaterials 2008, 29, 1737–1738. [Google Scholar]
- Sanvicens, N.; Marco, M.P. Multifunctional nanoparticles—Properties and prospects for their use in human medicine. Trends Biotechnol. 2008, 26, 425–433. [Google Scholar]
- United States National Nanotechnology Initiative. Nanotechnology 101: What is it and how it works. Available online: http://www.nano.gov/nanotech-101/what (accessed on 5 December 2011).
- Fields, B.N.; Knipe, D.M.; Howley, P.M. Fields' Virology, 5th ed; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007. [Google Scholar]
- Braunwald, E.; Hauser, S.L.; Fauci, A.S.; Longo, D.L.; Kasper, D.L.; Jameson, J.L. Harrison's Principles of Internal Medicine, 15th ed; McGraw-Hill: New York, NY, USA, 2001. [Google Scholar]
- Panel on Antiretroviral Guidelines for Adults and Adolescents. United States Department of Health and Human Services Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Available online: http://www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf (accessed on 9 March 2012).
- Clarke, J.R.; White, N.C.; Weber, J.N. HIV compartmentalization: Pathogenesis and clinical implications. AIDS Rev. 2000, 2, 15–22. [Google Scholar]
- Stevenson, M. HIV-1 pathogenesis. Nat. Med. 2003, 9, 853–860. [Google Scholar]
- Sonza, S.; Crowe, S.M. Reservoirs for HIV infection and their persistence in the face of undetectable viral load. AIDS Patient Care STDS 2001, 15, 511–518. [Google Scholar]
- Crowe, S.M. Macrophages and residual HIV infection. Curr. Opin. HIV AIDS 2006, 1, 129. [Google Scholar]
- Pomerantz, R. Reservoirs, sanctuaries, and residual disease: The hiding spots of HIV-1. HIV Clin. Trials 2003, 4, 137–143. [Google Scholar] [CrossRef]
- Palmer, S.; Josefsson, L.; Coffin, J.M. HIV reservoirs and the possibility of a cure for HIV infection. J. Intern. Med. 2011, 270, 550–560. [Google Scholar]
- Dahl, V.; Josefsson, L.; Palmer, S. HIV reservoirs, latency, and reactivation: Prospects for eradication. Antivir. Res. 2010, 85, 286–294. [Google Scholar] [CrossRef]
- Farokhzad, O.C. Nanotechnology for drug delivery: The perfect partnership. Expert Opin. Drug Del. 2008, 5, 927–929. [Google Scholar]
- Farokhzad, O.C.; Langer, R. Impact of nanotechnology on drug delivery. ACS Nano 2009, 3, 16–20. [Google Scholar]
- Petros, R.A.; DeSimone, J.M. Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. Drug Discov. 2010, 9, 615–627. [Google Scholar]
- Kinman, L.; Brodie, S.J.; Tsai, C.C.; Bui, T.; Larsen, K.; Schmidt, A.; Anderson, D.; Morton, W.R.; Hu, S.L.; Ho, R.J. Lipid-drug association enhanced HIV-1 protease inhibitor indinavir localization in lymphoid tissues and viral load reduction: A proof of concept study in HIV-2287-infected macaques. J. Acquir. Immune Defic. Syndr. 2003, 34, 387–397. [Google Scholar]
- Kinman, L.; Bui, T.; Larsen, K.; Tsai, C.C.; Anderson, D.; Morton, W.R.; Hu, S.L.; Ho, R.J. Optimization of lipid-indinavir complexes for localization in lymphoid tissues of HIV-infected macaques. J. Acquir. Immune Defic. Syndr. 2006, 42, 155–161. [Google Scholar]
- Choi, S.U.; Bui, T.; Ho, R.J. pH-dependent interactions of indinavir and lipids in nanoparticles and their ability to entrap a solute. J. Pharm. Sci. 2008, 97, 931–943. [Google Scholar]
- Phillips, N.C.; Tsoukas, C. Liposomal encapsulation of azidothymidine results in decreased hematopoietic toxicity and enhanced activity against murine acquired immunodeficiency syndrome. Blood 1992, 79, 1137–1143. [Google Scholar]
- Harvie, P.; Desormeaux, A.; Gagne, N.; Tremblay, M.; Poulin, L.; Beauchamp, D.; Bergeron, M.G. Lymphoid tissues targeting of liposome-encapsulated 2',3'-dideoxyinosine. AIDS 1995, 9, 701–707. [Google Scholar]
- Désormeaux, A.; Harvie, P.; Perron, S.; Makabi-Panzu, B.; Beauchamp, D.; Tremblay, M.; Poulin, L.; Bergeron, M.G. Antiviral efficacy, intracellular uptake and pharmacokinetics of free and liposome-encapsulated 2', 3'-dideoxyinosine. AIDS 1994, 8, 1545. [Google Scholar] [CrossRef]
- Sharma, P.; Garg, S. Pure drug and polymer based nanotechnologies for the improved solubility, stability, bioavailability and targeting of anti-HIV drugs. Adv. Drug Deliv. Rev. 2010, 62, 491–502. [Google Scholar] [CrossRef]
- Merisko-Liversidge, E.; Liversidge, G.G.; Cooper, E.R. Nanosizing: A formulation approach for poorly-water-soluble compounds. Eur. J. Pharm. Sci. 2003, 18, 113–120. [Google Scholar]
- Junghanns, J.U.A.H.; Müller, R.H. Nanocrystal technology, drug delivery and clinical applications. Int. J. Nanomed. 2008, 3, 295–309. [Google Scholar]
- Müller, R.H.; Gohla, S.; Keck, C.M. State of the art of nanocrystals - Special features, production, nanotoxicology aspects and intracellular delivery. Eur. J. Pharm. Biopharm. 2011, 78, 1–9. [Google Scholar] [CrossRef]
- Rabinow, B.E. Nanosuspensions in drug delivery. Nat. Rev. Drug Discov. 2004, 3, 785–796. [Google Scholar]
- Baert, L.; van 't Klooster, G.; Dries, W.; Francois, M.; Wouters, A.; Basstanie, E.; Iterbeke, K.; Stappers, F.; Stevens, P.; Schueller, L.; et al. Development of a long-acting injectable formulation with nanoparticles of rilpivirine (TMC278) for HIV treatment. Eur. J. Pharm. Biopharm. 2009, 72, 502–508. [Google Scholar] [CrossRef]
- van 't Klooster, G.; Hoeben, E.; Borghys, H.; Looszova, A.; Bouche, M.P.; van Velsen, F.; Baert, L. Pharmacokinetics and disposition of rilpivirine (TMC278) nanosuspension as a long-acting injectable antiretroviral formulation. Antimicrob. Agents Chemother. 2010, 54, 2042–2050. [Google Scholar]
- Caron, J.; Reddy, L.H.; Lepetre-Mouelhi, S.; Wack, S.; Clayette, P.; Rogez-Kreuz, C.; Yousfi, R.; Couvreur, P.; Desmaele, D. Squalenoyl nucleoside monophosphate nanoassemblies: New prodrug strategy for the delivery of nucleotide analogues. Bioorg. Med. Chem. Lett. 2010, 20, 2761–2764. [Google Scholar]
- Fattal, E.; Barratt, G. Nanotechnologies and controlled release systems for the delivery of antisense oligonucleotides and small interfering RNA. Brit. J. Pharmacol. 2009, 157, 179–194. [Google Scholar]
- Howard, K.A. Delivery of RNA interference therapeutics using polycation-based nanoparticles. Adv. Drug Deliv. Rev. 2009, 61, 710–720. [Google Scholar]
- Singha, K.; Namgung, R.; Kim, W.J. Polymers in small-interfering RNA delivery. Nucleic Acid Ther. 2011, 21, 133–147. [Google Scholar]
- Gao, K.; Huang, L. Nonviral methods for siRNA delivery. Mol. Pharm. 2009, 6, 651–658. [Google Scholar]
- Zhang, S.; Zhao, B.; Jiang, H.; Wang, B.; Ma, B. Cationic lipids and polymers mediated vectors for delivery of siRNA. J. Control. Release 2007, 123, 1–10. [Google Scholar]
- de Martimprey, H.; Vauthier, C.; Malvy, C.; Couvreur, P. Polymer nanocarriers for the delivery of small fragments of nucleic acids: Oligonucleotides and siRNA. Eur. J. Pharm. Biopharm. 2009, 71, 490–504. [Google Scholar]
- Luo, D.; Saltzman, W.M. Synthetic DNA delivery systems. Nat. Biotech. 2000, 18, 33–37. [Google Scholar]
- Kayser, O.; Lemke, A.; Hernandez-Trejo, N. The impact of nanobiotechnology on the development of new drug delivery systems. Curr. Pharm. Biotechno. 2005, 6, 3–5. [Google Scholar]
- Gupta, U.; Jain, N.K. Non-polymeric nano-carriers in HIV/AIDS drug delivery and targeting. Adv. Drug Deliv. Rev. 2010, 62, 478–490. [Google Scholar]
- Chiappetta, D.A.; Facorro, G.; de Celis, E.R.; Sosnik, A. Synergistic encapsulation of the anti-HIV agent efavirenz within mixed poloxamine/poloxamer polymeric micelles. Nanomedicine 2011, 7, 624–637. [Google Scholar]
- Chiappetta, D.A.; Hocht, C.; Sosnik, A. A highly concentrated and taste-improved aqueous formulation of efavirenz for a more appropriate pediatric management of the anti-HIV therapy. Curr. HIV Res. 2010, 8, 223–231. [Google Scholar]
- Chiappetta, D.A.; Hocht, C.; Taira, C.; Sosnik, A. Efavirenz-loaded polymeric micelles for pediatric anti-HIV pharmacotherapy with significantly higher oral bioavailability [corrected]. Nanomedicine (Lond.) 2010, 5, 11–23. [Google Scholar] [CrossRef]
- Chiappetta, D.A.; Hocht, C.; Taira, C.; Sosnik, A. Oral pharmacokinetics of the anti-HIV efavirenz encapsulated within polymeric micelles. Biomaterials 2011, 32, 2379–2387. [Google Scholar]
- Chiappetta, D.A.; Sosnik, A. Poly (ethylene oxide)-poly (propylene oxide) block copolymer micelles as drug delivery agents: Improved hydrosolubility, stability and bioavailability of drugs. Eur. J. Pharm. Biopharm. 2007, 66, 303–317. [Google Scholar]
- Hillaireau, H.; Le Doan, T.; Appel, M.; Couvreur, P. Hybrid polymer nanocapsules enhance in vitro delivery of azidothymidine-triphosphate to macrophages. J. Control. Release 2006, 116, 346–352. [Google Scholar]
- Hillaireau, H.; Le Doan, T.; Besnard, M.; Chacun, H.; Janin, J.; Couvreur, P. Encapsulation of antiviral nucleotide analogues azidothymidine-triphosphate and cidofovir in poly(iso-butylcyanoacrylate) nanocapsules. Int. J. Pharm. 2006, 324, 37–42. [Google Scholar]
- Muthu, M.S.; Singh, S. Targeted nanomedicines: Effective treatment modalities for cancer, AIDS and brain disorders. Nanomedicine (Lond.) 2009, 4, 105–118. [Google Scholar] [CrossRef]
- Garg, M.; Asthana, A.; Agashe, H.B.; Agrawal, G.P.; Jain, N.K. Stavudine loaded mannosylated liposomes: In vitro anti HIV I activity, tissue distribution and pharmacokinetics. J. Pharm. Pharmacol. 2006, 58, 605–616. [Google Scholar]
- Garg, M.; Dutta, T.; Jain, N.K. Reduced hepatic toxicity, enhanced cellular uptake and altered pharmacokinetics of stavudine loaded galactosylated liposomes. Eur. J. Pharm. Biopharm. 2007, 67, 76–85. [Google Scholar]
- Garg, M.; Garg, B.R.; Jain, S.; Mishra, P.; Sharma, R.K.; Mishra, A.K.; Dutta, T.; Jain, N.K. Radiolabeling, pharmacoscintigraphic evaluation and antiretroviral efficacy of stavudine loaded 99mTc labeled galactosylated liposomes. Eur. J. Pharm. Sci. 2008, 33, 271–281. [Google Scholar]
- Wu, H.B.; Deng, Y.H.; Wang, S.N.; Zhou, X.Y.; Wang, N.; Shi, L. The distribution of azidothymidine palmitate galactosylated liposomes in mice. Yao Xue Xue Bao 2007, 42, 538–544. [Google Scholar]
- Harvie, P.; Desormeaux, A.; Bergeron, M.C.; Tremblay, M.; Beauchamp, D.; Poulin, L.; Bergeron, M.G. Comparative pharmacokinetics, distributions in tissue, and interactions with blood proteins of conventional and sterically stabilized liposomes containing 2',3'-dideoxyinosine. Antimicrob. Agents Ch. 1996, 40, 225–229. [Google Scholar]
- Heiati, H.; Tawashi, R.; Phillips, N.C. Solid lipid nanoparticles as drug carriers: II. Plasma stability and biodistribution of solid lipid nanoparticles containing the lipophilic prodrug 3'-azido-3'-deoxythymidine palmitate in mice. Int. J. Pharm. 1998, 174, 71–80. [Google Scholar] [CrossRef]
- Heiati, H.; Tawashi, R.; Shivers, R.R.; Phillips, N.C. Solid lipid nanoparticles as drug carriers. I. Incorporation and retention of the lipophilic prodrug 3'-azido-3'-deoxythymidine palmitate. Int. J. Pharm. 1997, 146, 123–131. [Google Scholar] [CrossRef]
- Kovochich, M.; Marsden, M.D.; Zack, J.A. Activation of latent HIV using drug-loaded nanoparticles. PLoS One 2011, 6, e18270. [Google Scholar]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar]
- Wan, L.; Zhang, X.; Gunaseelan, S.; Pooyan, S.; Debrah, O.; Leibowitz, M.J.; Rabson, A.B.; Stein, S.; Sinko, P.J. Novel multi-component nanopharmaceuticals derived from poly(ethylene) glycol, retro-inverso-Tat nonapeptide and saquinavir demonstrate combined anti-HIV effects. AIDS Res. Ther. 2006, 3, 12. [Google Scholar]
- Bowman, M.C.; Ballard, T.E.; Ackerson, C.J.; Feldheim, D.L.; Margolis, D.M.; Melander, C. Inhibition of HIV fusion with multivalent gold nanoparticles. J. Am. Chem. Soc. 2008, 130, 6896–6897. [Google Scholar]
- Esté, J.A.; Cihlar, T. Current status and challenges of antiretroviral research and therapy. Antivir. Res. 2010, 85, 25–33. [Google Scholar]
- Marsden, M.D.; Zack, J.A. Eradication of HIV: Current challenges and new directions. J. Antimicrob. Chemother. 2009, 63, 7–10. [Google Scholar]
- Ma, X.; Wang, D.; Wu, Y.; Ho, R.J.; Jia, L.; Guo, P.; Hu, L.; Xing, G.; Zeng, Y.; Liang, X.J. AIDS treatment with novel anti-HIV compounds improved by nanotechnology. AAPS J. 2010, 12, 272–278. [Google Scholar]
- Amiji, M.; Vyas, T.; Shah, L. Role of nanotechnology in HIV/AIDS treatment: Potential to overcome the viral reservoir challenge. Discov. Med. 2006, 6, 157–162. [Google Scholar]
- NICNAS NICNAS Information Sheet Nanomaterials. Available online: http://www.nicnas.gov.au/publications/information_sheets/general_information_sheets/nis_nanomaterials_pdf.pdf (accessed on 1 December 2011).
- Lines, M. Nanomaterials for practical functional uses. J. Alloy Compd. 2008, 449, 242–245. [Google Scholar]
- Aitken, R.; Chaudhry, M.; Boxall, A.; Hull, M. Manufacture and use of nanomaterials: Current status in the UK and global trends. Occup. Med. 2006, 56, 300–306. [Google Scholar]
- Niemeyer, C.; Mirkin, C. Nanobiotechnology: Concepts, Applications and Perspectives; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Medepalli, K.K. Advanced Nanomaterials for Biomedical Applications; ProQuest: Cambridge, UK, 2008. [Google Scholar]
- Schulz, M.J.; Shanov, V.N. Nanomedicine Design of Particles, Sensors, Motors, Implants, Robots, and Devices; Artech House Publishers: Boston, MA, USA, 2009. [Google Scholar]
- Bawarski, W.E.; Chidlowsky, E.; Bharali, D.J.; Mousa, S.A. Emerging nanopharmaceuticals. Nanomedicine 2008, 4, 273–282. [Google Scholar]
- Gunaseelan, S.; Gunaseelan, K.; Deshmukh, M.; Zhang, X.; Sinko, P.J. Surface modifications of nanocarriers for effective intracellular delivery of anti-HIV drugs. Adv. Drug Deliv. Rev. 2010, 62, 518–531. [Google Scholar]
- Nowacek, A.; Gendelman, H.E. NanoART, neuroAIDS and CNS drug delivery. Nanomedicine 2009, 4, 557–574. [Google Scholar]
- Mallipeddi, R.; Rohan, L.C. Progress in antiretroviral drug delivery using nanotechnology. Int. J. Nanomedicine 2010, 5, 533–547. [Google Scholar]
- Ojewole, E.; Mackraj, I.; Naidoo, P.; Govender, T. Exploring the use of novel drug delivery systems for antiretroviral drugs. Eur. J. Pharm. Biopharm. 2008, 70, 697–710. [Google Scholar]
- Sosnik, A.; Chiappetta, D.A.; Carcaboso, Á.M. Drug delivery systems in HIV pharmacotherapy: What has been done and the challenges standing ahead. J. Contr. Release 2009, 138, 2–15. [Google Scholar] [CrossRef]
- Wong, H.L.; Chattopadhyay, N.; Wu, X.Y.; Bendayan, R. Nanotechnology applications for improved delivery of antiretroviral drugs to the brain. Adv. Drug Deliver. Rev. 2010, 62, 503–517. [Google Scholar]
- Govender, T.; Ojewole, E.; Naidoo, P.; Mackraj, I. Polymeric nanoparticles for enhancing antiretroviral drug therapy. Drug. Deliv. 2008, 15, 493–501. [Google Scholar]
- Kim, P.S.; Read, S.W. Nanotechnology and HIV: Potential applications for treatment and prevention. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2010, 2, 693–702. [Google Scholar]
- Mamo, T.; Moseman, E.A.; Kolishetti, N.; Salvador-Morales, C.; Shi, J.; Kuritzkes, D.R.; Langer, R.; von Andrian, U.; Farokhzad, O.C. Emerging nanotechnology approaches for HIV/AIDS treatment and prevention. Nanomedicine 2010, 5, 269–285. [Google Scholar]
- das Neves, J.; Amiji, M.M.; Bahia, M.F.; Sarmento, B. Nanotechnology-based systems for the treatment and prevention of HIV/AIDS. Adv. Drug Deliv. Rev. 2010, 62, 458–477. [Google Scholar]
- Fischer, H.C.; Chan, W.C.W. Nanotoxicity: The growing need for in vivo study. Curr. Opin. Biotech. 2007, 18, 565–571. [Google Scholar] [CrossRef]
- Ferrari, M. Cancer nanotechnology: Opportunities and challenges. Nat. Rev. Cancer 2005, 5, 161–171. [Google Scholar]
- Duncan, R.; Gaspar, R. Nanomedicine(s) under the microscope. Mol. Pharm. 2011, 8, 2101–2141. [Google Scholar]
- Sanhai, W.R.; Sakamoto, J.H.; Canady, R.; Ferrari, M. Seven challenges for nanomedicine. Nat. Nanotechnol. 2008, 3, 242–244. [Google Scholar]
- Karmali, P.P.; Simberg, D. Interactions of nanoparticles with plasma proteins: Implication on clearance and toxicity of drug delivery systems. Expert Opin. Drug Del. 2011, 1–15. [Google Scholar]
- Aggarwal, P.; Hall, J.B.; McLeland, C.B.; Dobrovolskaia, M.A.; McNeil, S.E. Nanoparticle interaction with plasma proteins as it relates to particle biodistribution, biocompatibility and therapeutic efficacy. Adv. Drug Deliv. Rev. 2009, 61, 428–437. [Google Scholar]
- Nie, S. Understanding and overcoming major barriers in cancer nanomedicine. Nanomedicine (Lond.) 2010, 5, 523. [Google Scholar] [CrossRef]
- Chrastina, A.; Massey, K.A.; Schnitzer, J.E. Overcoming in vivo barriers to targeted nanodelivery. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2011, 3, 421–437. [Google Scholar] [CrossRef]
- He, C.; Hu, Y.; Yin, L.; Tang, C.; Yin, C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials 2010, 31, 3657–3666. [Google Scholar]
- Spitzenberger, T.J.; Heilman, D.; Diekmann, C.; Batrakova, E.V.; Kabanov, A.V.; Gendelman, H.E.; Elmquist, W.F.; Persidsky, Y. Novel delivery system enhances efficacy of antiretroviral therapy in animal model for HIV-1 encephalitis. J. Cereb. Blood Flow Metab. 2007, 27, 1033–1042. [Google Scholar]
- Pereira de Oliveira, M.; Garcion, E.; Venisse, N.; Benoit, J.P.; Couet, W.; Olivier, J.C. Tissue distribution of indinavir administered as solid lipid nanocapsule formulation in mdr1a (+/+) and mdr1a (-/-) CF-1 mice. Pharm. Res. 2005, 22, 1898–1905. [Google Scholar]
- Koo, O.M.; Rubinstein, I.; Onyuksel, H. Role of nanotechnology in targeted drug delivery and imaging: A concise review. Nanomedicine 2005, 1, 193–212. [Google Scholar]
- Vyas, T.K.; Shah, L.; Amiji, M.M. Nanoparticulate drug carriers for delivery of HIV/AIDS therapy to viral reservoir sites. Expert Opin. Drug Deliv. 2006, 3, 613–628. [Google Scholar]
- Shegokar, R.; Singh, K.K. Surface modified nevirapine nanosuspensions for viral reservoir targeting: In vitro and in vivo evaluation. Int. J. Pharm. 2011, 421, 341–352. [Google Scholar] [CrossRef]
- Kaur, A.; Jain, S.; Tiwary, A.K. Mannan-coated gelatin nanoparticles for sustained and targeted delivery of didanosine: In vitro and in vivo evaluation. Acta Pharm. 2008, 58, 61–74. [Google Scholar] [CrossRef]
- Gagné, J.F.; Désormeaux, A.; Perron, S.; Tremblay, M.J.; Bergeron, M.G. Targeted delivery of indinavir to HIV-1 primary reservoirs with immunoliposomes. Biochim. Biophys. Acta 2002, 1558, 198–210. [Google Scholar] [CrossRef]
- Garg, M.; Dutta, T.; Jain, N.K. Stability study of stavudine-loaded O-palmitoyl-anchored carbohydrate-coated liposomes. AAPS PharmSciTech 2007, 8, 86–93. [Google Scholar]
- Dutta, T.; Jain, N.K. Targeting potential and anti-HIV activity of lamivudine loaded mannosylated poly (propyleneimine) dendrimer. BBA Gen. Subjects 2007, 1770, 681–686. [Google Scholar] [CrossRef]
- Flasher, D.; Konopka, K.; Chamow, S.M.; Dazin, P.; Ashkenazi, A.; Pretzer, E.; Duzgunes, N. Liposome targeting to human immunodeficiency virus type 1-infected cells via recombinant soluble CD4 and CD4 immunoadhesin (CD4-IgG). Biochim. Biophys. Acta 1994, 1194, 185–196. [Google Scholar] [CrossRef]
- Pollock, S.; Dwek, R.A.; Burton, D.R.; Zitzmann, N. N-Butyldeoxynojirimycin is a broadly effective anti-HIV therapy significantly enhanced by targeted liposome delivery. AIDS 2008, 22, 1961–1969. [Google Scholar]
- Clayton, R.; Ohagen, A.; Nicol, F.; Del Vecchio, A.M.; Jonckers, T.H.; Goethals, O.; Van Loock, M.; Michiels, L.; Grigsby, J.; Xu, Z.; et al. Sustained and specific in vitro inhibition of HIV-1 replication by a protease inhibitor encapsulated in gp120-targeted liposomes. Antivir. Res. 2009, 84, 142–149. [Google Scholar]
- Dutta, T.; Agashe, H.B.; Garg, M.; Balasubramanium, P.; Kabra, M.; Jain, N.K. Poly (propyleneimine) dendrimer based nanocontainers for targeting of efavirenz to human monocytes/macrophages in vitro. J. Drug Target. 2007, 15, 89–98. [Google Scholar] [CrossRef]
- Dutta, T.; Garg, M.; Jain, N.K. Targeting of efavirenz loaded tuftsin conjugated poly (propyleneimine) dendrimers to HIV infected macrophages in vitro. Eur. J. Pharm. Sci. 2008, 34, 181–189. [Google Scholar] [CrossRef]
- Harvie, P.; Désormeaux, A.; Gagné, N.; Tremblay, M.; Poulin, L.; Beauchamp, D.; Bergeron, M.G. Lymphoid tissues targeting of liposome-encapsulated 2',3'-dideoxyinosine. AIDS 1995, 9, 701–707. [Google Scholar]
- Kroto, H.W.; Walton, D.R.M. Fullerene. In Encyclopaedia Britannica Deluxe Edition; Encyclopaedia Britannica: Chicago, IL, USA, 2010. [Google Scholar]
- Pedziwiatr-Werbicka, E.; Ferenc, M.; Zaborski, M.; Gabara, B.; Klajnert, B.; Bryszewska, M. Characterization of complexes formed by polypropylene imine dendrimers and anti-HIV oligonucleotides. Colloid. Surface. B 2011, 83, 360–366. [Google Scholar]
- Lee, C.C.; MacKay, J.A.; Fréchet, J.M.J.; Szoka, F.C. Designing dendrimers for biological applications. Nat. Biotechnol. 2005, 23, 1517–1526. [Google Scholar]
- Chrai, S.S.; Murari, R.; Ahmad, I. Liposomes: A review. Pharmaceut. Tech. 2002, 26, 28–34. [Google Scholar]
- Kwon, G.S.; Forrest, M.L. Amphiphilic block copolymer micelles for nanoscale drug delivery. Drug Develop. Res. 2006, 67, 15–22. [Google Scholar]
- Croy, S.; Kwon, G. Polymeric micelles for drug delivery. Curr. Pharm. Design 2006, 12, 4669–4684. [Google Scholar]
- Zhang, J.; Li, S.; Li, X. Polymeric nano-assemblies as emerging delivery carriers for therapeutic applications: A review of recent patents. Recent Pat. Nanotechnol. 2009, 3, 225–231. [Google Scholar]
- Bekkara-Aounallah, F.; Gref, R.; Othman, M.; Reddy, L.H.; Pili, B.; Allain, V.; Bourgaux, C.; Hillaireau, H.; Lepêtre-Mouelhi, S.; Desmaële, D.; et al. Novel PEGylated Nanoassemblies Made of Self-Assembled Squalenoyl Nucleoside Analogues. Adv. Funct. Mater. 2008, 18, 3715–3725. [Google Scholar]
- Couvreur, P.; Stella, B.; Reddy, L.H.; Hillaireau, H.; Dubernet, C.; Desmaele, D.; Lepetre-Mouelhi, S.; Rocco, F.; Dereuddre-Bosquet, N.; Clayette, P.; et al. Squalenoyl nanomedicines as potential therapeutics. Nano Lett. 2006, 6, 2544–2548. [Google Scholar]
- Mason, T.G.; Wilking, J.; Meleson, K.; Chang, C.; Graves, S. Nanoemulsions: Formation, structure, and physical propertie. J. Phys. Condens. Mat. 2006, 18, R635–R666. [Google Scholar]
- AZoNano. Nanocapsules and Dendrimers—Properties and Future Applications. Available online: http://www.azonano.com/article.aspx?ArticleID=1649 (accessed on 2 December 2011).
- Torchilin, V.P. Nanoparticulates as Drug Carriers; Imperial College Press; Distributed by World Scientific Pub.: London, UK/Hackensack, NJ, USA, 2006; p. xxix, 724. [Google Scholar]
- British Standards Institution Vocabulary: Nanoparticles. Available online: http://www.bsigroup.com/sectorsandservices/Forms/PAS-712011-Download/ (accessed on 6 December 2011).
- Bawa, R. Nanopharmaceuticals: Nanopharmaceuticals. European Journal of Nanomedicine 2010, 3, 34–40. [Google Scholar]
- Fu, A.; Gu, W.; Boussert, B.; Koski, K.; Gerion, D.; Manna, L.; Le Gros, M.; Larabell, C.A.; Alivisatos, A.P. Semiconductor quantum rods as single molecule fluorescent biological labels. Nano Lett. 2006, 7, 179–182. [Google Scholar]
- Buhro, W.E.; Colvin, V.L. Shape matters. Nat. Mater. 2003, 2, 138–139. [Google Scholar]
- Wissing, S.; Kayser, O.; Muller, R. Solid lipid nanoparticles for parenteral drug delivery. Adv. Drug Deliv. Rev. 2004, 56, 1257–1272. [Google Scholar]
- Pozniak, A.L.; Morales-Ramirez, J.; Katabira, E.; Steyn, D.; Lupo, S.H.; Santoscoy, M.; Grinsztejn, B.; Ruxrungtham, K.; Rimsky, L.T.; Vanveggel, S.; et al. Efficacy and safety of TMC278 in antiretroviral-naive HIV-1 patients: Week 96 results of a phase IIb randomized trial. AIDS 2010, 24, 55–65. [Google Scholar]
- Schrijvers, R.; Desimmie, B.A.; Debyser, Z. Rilpivirine: A step forward in tailored HIV treatment. Lancet 2011, 378, 201–203. [Google Scholar]
- Kiser, J.J. Pharmacologic characteristics of investigational and recently approved agents for the treatment of HIV. Curr. Opin. HIV AIDS 2008, 3, 330–341. [Google Scholar]
- Kilby, J.M.; Lalezari, J.P.; Eron, J.J.; Carlson, M.; Cohen, C.; Arduino, R.C.; Goodgame, J.C.; Gallant, J.E.; Volberding, P.; Murphy, R.L. The safety, plasma pharmacokinetics, and antiviral activity of subcutaneous enfuvirtide (T-20), a peptide inhibitor of gp41-mediated virus fusion, in HIV-infected adults. AIDS Res. Hum. Retrovir. 2002, 18, 685–693. [Google Scholar] [CrossRef]
- Desmaële, D.; Gref, R.; Couvreur, P. Squalenoylation: A generic platform for nanoparticular drug delivery. J. Contr. Release 2012, in press. [Google Scholar] [CrossRef]
- Shah, L.K.; Amiji, M.M. Intracellular delivery of saquinavir in biodegradable polymeric nanoparticles for HIV/AIDS. Pharm. Res. 2006, 23, 2638–2645. [Google Scholar]
- Buchanan, C.M.; Buchanan, N.L.; Edgar, K.J.; Little, J.L.; Ramsey, M.G.; Ruble, K.M.; Wacher, V.J.; Wempe, M.F. Pharmacokinetics of saquinavir after intravenous and oral dosing of saquinavir: Hydroxybutenyl-beta-cyclodextrin formulations. Biomacromolecules 2008, 9, 305–313. [Google Scholar]
- Sathigari, S.; Chadha, G.; Lee, Y.H.; Wright, N.; Parsons, D.L.; Rangari, V.K.; Fasina, O.; Babu, R.J. Physicochemical characterization of efavirenz-cyclodextrin inclusion complexes. AAPS PharmSciTech 2009, 10, 81–87. [Google Scholar]
- Wagner, C.R.; Iyer, V.V.; McIntee, E.J. Pronucleotides: Toward the in vivo delivery of antiviral and anticancer nucleotides. Med. Res. Rev. 2000, 20, 417–451. [Google Scholar]
- Kreuter, J.R.; Täuber, U.; Illi, V. Distribution and elimination of poly (methyl 2 14C methacrylate) nanoparticle radioactivity after injection in rats and mice. J. Pharm. Sci. 1979, 68, 1443–1447. [Google Scholar]
- Ahsan, F.; Rivas, I.P.; Khan, M.A.; Torres Suárez, A.I. Targeting to macrophages: Role of physicochemical properties of particulate carriers—liposomes and microspheres—on the phagocytosis by macrophages. J. Contr. Release 2002, 79, 29–40. [Google Scholar]
- Feinberg, M.B. Changing the natural history of HIV disease. Lancet 1996, 348, 239–246. [Google Scholar]
- Bestman-Smith, J.; Gourde, P.; Desormeaux, A.; Tremblay, M.J.; Bergeron, M.G. Sterically stabilized liposomes bearing anti-HLA-DR antibodies for targeting the primary cellular reservoirs of HIV-1. Biochim. Biophys. Acta 2000, 1468, 161–174. [Google Scholar] [CrossRef]
- Sarin, H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J. Angiogenes. Res. 2010, 2, 1–19. [Google Scholar]
- Destache, C.J.; Belgum, T.; Christensen, K.; Shibata, A.; Sharma, A.; Dash, A. Combination antiretroviral drugs in PLGA nanoparticle for HIV-1. BMC Infect. Dis. 2009, 9, 198. [Google Scholar]
- Lobenberg, R.; Araujo, L.; von Briesen, H.; Rodgers, E.; Kreuter, J. Body distribution of azidothymidine bound to hexyl-cyanoacrylate nanoparticles after i.v. injection to rats. J. Contr. Release 1998, 50, 21–30. [Google Scholar] [CrossRef]
- Lobenberg, R.; Kreuter, J. Macrophage targeting of azidothymidine: A promising strategy for AIDS therapy. AIDS Res. Hum. Retroviruses 1996, 12, 1709–1715. [Google Scholar]
- Löbenberg, R.; Araujo, L.; Kreuter, J. Body distribution of azidothymidine bound to nanoparticles after oral administration. Eur. J. Pharm. Biopharm. 1997, 44, 127–132. [Google Scholar]
- Jain, S.; Tiwary, A.K.; Jain, N.K. PEGylated elastic liposomal formulation for lymphatic targeting of zidovudine. Curr. Drug. Deliv. 2008, 5, 275–281. [Google Scholar]
- Mainardes, R.M.; Gremião, M.P.D.; Brunetti, I.L.; da Fonseca, L.M.; Khalil, N.M. Zidovudine loaded PLA and PLA–PEG blend nanoparticles: Influence of polymer type on phagocytic uptake by polymorphonuclear cells. J. Pharm. Sci. 2009, 98, 257–267. [Google Scholar]
- Batrakova, E.V.; Li, S.; Vinogradov, S.V.; Alakhov, V.Y.; Miller, D.W.; Kabanov, A.V. Mechanism of pluronic effect on P-glycoprotein efflux system in blood-brain barrier: Contributions of energy depletion and membrane fluidization. J. Pharmacol. Exp. Ther. 2001, 299, 483–493. [Google Scholar]
- Chattopadhyay, N.; Zastre, J.; Wong, H.L.; Wu, X.Y.; Bendayan, R. Solid lipid nanoparticles enhance the delivery of the HIV protease inhibitor, atazanavir, by a human brain endothelial cell line. Pharm. Res. 2008, 25, 2262–2271. [Google Scholar] [CrossRef]
- Vyas, T.K.; Shahiwala, A.; Amiji, M.M. Improved oral bioavailability and brain transport of Saquinavir upon administration in novel nanoemulsion formulations. Int. J. Pharm. 2008, 347, 93–101. [Google Scholar]
- Kuo, Y.C.; Chen, H.H. Effect of nanoparticulate polybutylcyanoacrylate and methylmethacrylate-sulfopropylmethacrylate on the permeability of zidovudine and lamivudine across the in vitro blood-brain barrier. Int. J. Pharm. 2006, 327, 160–169. [Google Scholar] [CrossRef]
- Kuo, Y.C.; Su, F.L. Transport of stavudine, delavirdine, and saquinavir across the blood-brain barrier by polybutylcyanoacrylate, methylmethacrylate-sulfopropylmethacrylate, and solid lipid nanoparticles. Int. J. Pharm. 2007, 340, 143–152. [Google Scholar] [CrossRef]
- Kuo, Y.C.; Kuo, C.Y. Electromagnetic interference in the permeability of saquinavir across the blood-brain barrier using nanoparticulate carriers. Int. J. Pharm. 2008, 351, 271–281. [Google Scholar]
- Rao, K.S.; Reddy, M.K.; Horning, J.L.; Labhasetwar, V. TAT-conjugated nanoparticles for the CNS delivery of anti-HIV drugs. Biomaterials 2008, 29, 4429–4438. [Google Scholar]
- McNeil, S.E. Nanotechnology for the biologist. J. Leukoc. Biol. 2005, 78, 585–594. [Google Scholar]
- Jabr-Milane, L.; Van Vlerken, L.; Devalapally, H.; Shenoy, D.; Komareddy, S.; Bhavsar, M.; Amiji, M. Multi-functional nanocarriers for targeted delivery of drugs and genes. J. Contr. Release 2008, 130, 121–128. [Google Scholar]
- Wan, L.; Zhang, X.; Pooyan, S.; Palombo, M.S.; Leibowitz, M.J.; Stein, S.; Sinko, P.J. Optimizing size and copy number for PEG-fMLF (N-formyl-methionyl-leucyl-phenylalanine) nanocarrier uptake by macrophages. Bioconjug. Chem. 2008, 19, 28–38. [Google Scholar] [CrossRef]
- Dorman, N.; Lever, A.M. RNA-based gene therapy for HIV infection. HIV Med. 2001, 2, 114–122. [Google Scholar]
- Reyes-Darias, J.A.; Sanchez-Luque, F.J.; Berzal-Herranz, A. Inhibition of HIV-1 replication by RNA-based strategies. Curr. HIV Res. 2008, 6, 500–514. [Google Scholar]
- McManus, M.T.; Sharp, P.A. Gene silencing in mammals by small interfering RNAs. Nat. Rev. Genet. 2002, 3, 737–747. [Google Scholar]
- Akhtar, S.; Hughes, M.D.; Khan, A.; Bibby, M.; Hussain, M.; Nawaz, Q.; Double, J.; Sayyed, P. The Delivery of Antisense Therapeutics. Adv. Drug Deliv. Rev. 2000, 44, 3–21. [Google Scholar]
- Akhtar, S.; Benter, I. Nonviral delivery of synthetic siRNAs in vivo. J. Clin. Invest. 2007, 117, 3623–3632. [Google Scholar] [CrossRef]
- Brown, M.D.; Schatzlein, A.G.; Uchegbu, I.F. Gene delivery with synthetic (non viral) carriers. Int. J. Pharm. 2001, 229, 1–21. [Google Scholar]
- Fattal, E.; Bochot, A. State of the art and perspectives for the delivery of antisense oligonucleotides and siRNA by polymeric nanocarriers. Int. J. Pharm. 2008, 364, 237–248. [Google Scholar]
- Hughes, M.D.; Hussain, M.; Nawaz, Q.; Sayyed, P.; Akhtar, S. The cellular delivery of antisense oligonucleotides and ribozymes. Drug Discov. Today 2001, 6, 303–315. [Google Scholar]
- Jaaskelainen, I.; Urtti, A. Cell membranes as barriers for the use of antisense therapeutic agents. Mini-Rev. Med. Chem. 2002, 2, 307–318. [Google Scholar] [CrossRef]
- Juliano, R.; Bauman, J.; Kang, H.; Ming, X. Biological barriers to therapy with antisense and siRNA oligonucleotides. Mol. Pharm. 2009, 6, 686–695. [Google Scholar]
- Whitehead, K.; Langer, R.; Anderson, D. Knocking down barriers: Advances in siRNA delivery. Nat. Rev. Drug Discov. 2009, 8, 129–138. [Google Scholar]
- Ozpolat, B.; Sood, A.K.; Lopez-Berestein, G. Nanomedicine based approaches for the delivery of siRNA in cancer. J. Intern. Med. 2010, 267, 44–53. [Google Scholar]
- Bordier, B.; Perala-Heape, M.; Degols, G.; Lebleu, B.; Litvak, S.; Sarih-Cottin, L.; Hélène, C. Sequence-specific inhibition of human immunodeficiency virus (HIV) reverse transcription by antisense oligonucleotides: Comparative study in cell-free assays and in HIV-infected cells. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 9383–9387. [Google Scholar]
- Dinauer, N.; Lochmann, D.; Demirhan, I.; Bouazzaoui, A.; Zimmer, A.; Chandra, A.; Kreuter, J.; von Briesen, H. Intracellular tracking of protamine/antisense oligonucleotide nanoparticles and their inhibitory effect on HIV-1 transactivation. J. Contr. Release 2004, 96, 497–507. [Google Scholar]
- Mahajan, S.D.; Aalinkeel, R.; Reynolds, J.L.; Nair, B.; Sykes, D.E.; Law, W.C.; Ding, H.; Bergey, E.J.; Prasad, P.N.; Schwartz, S.A. Nanotherapeutics using an HIV-1 Poly A and transactivator of the HIV-1 LTR-(TAR-) specific siRNA. Patholog. Res. Int. 2011, 2011, 719139. [Google Scholar]
- Jimenez, J.; Clemente, M.; Weber, N.; Sanchez, J.; Ortega, P.; de la Mata, F.; Gomez, R.; Garcia, D.; Lopez-Fernandez, L.; Munoz-Fernandez, M. Carbosilane dendrimers to transfect human astrocytes with small interfering RNA targeting human immunodeficiency virus. BioDrugs 2010, 24, 331–343. [Google Scholar]
- Weber, N.; Ortega, P.; Clemente, M.I.; Shcharbin, D.; Bryszewska, M.; de la Mata, F.J.; Gomez, R.; Munoz-Fernandez, M.A. Characterization of carbosilane dendrimers as effective carriers of siRNA to HIV-infected lymphocytes. J. Contr. Release 2008, 132, 55–64. [Google Scholar]
- Duzgunes, N.; Simoes, S.; Slepushkin, V.; Pretzer, E.; Rossi, J.J.; De Clercq, E.; Antao, V.P.; Collins, M.L.; de Lima, M.C. Enhanced inhibition of HIV-1 replication in macrophages by antisense oligonucleotides, ribozymes and acyclic nucleoside phosphonate analogs delivered in pH-sensitive liposomes. Nucleos. Nucleot. Nucleic Acids 2001, 20, 515–523. [Google Scholar]
- Konopka, K.; Rossi, J.J.; Swiderski, P.; Slepushkin, V.A.; Duzgunes, N. Delivery of an anti-HIV-1 ribozyme into HIV-infected cells via cationic liposomes. Biochim. Biophys. Acta 1998, 1372, 55–68. [Google Scholar] [CrossRef]
- Kim, S.S.; Peer, D.; Kumar, P.; Subramanya, S.; Wu, H.; Asthana, D.; Habiro, K.; Yang, Y.G.; Manjunath, N.; Shimaoka, M.; et al. RNAi-mediated CCR5 silencing by LFA-1-targeted nanoparticles prevents HIV infection in BLT mice. Mol. Ther. 2010, 18, 370–376. [Google Scholar] [CrossRef]
- Sun, R.W.; Chen, R.; Chung, N.P.; Ho, C.M.; Lin, C.L.; Che, C.M. Silver nanoparticles fabricated in Hepes buffer exhibit cytoprotective activities toward HIV-1 infected cells. Chem. Commun. (Camb.) 2005, 5059–5061. [Google Scholar]
- Elechiguerra, J.L.; Burt, J.L.; Morones, J.R.; Camacho-Bragado, A.; Gao, X.; Lara, H.H.; Yacaman, M.J. Interaction of silver nanoparticles with HIV-1. J. Nanobiotechnology 2005, 3, 6. [Google Scholar]
- Lara, H.H.; Ayala-Nunez, N.V.; Ixtepan-Turrent, L.; Rodriguez-Padilla, C. Mode of antiviral action of silver nanoparticles against HIV-1. J. Nanobiotechnology 2010, 8, 1. [Google Scholar]
- Witvrouw, M.; Fikkert, V.; Pluymers, W.; Matthews, B.; Mardel, K.; Schols, D.; Raff, J.; Debyser, Z.; De Clercq, E.; Holan, G. Polyanionic (i.e., polysulfonate) dendrimers can inhibit the replication of human immunodeficiency virus by interfering with both virus adsorption and later steps (reverse transcriptase/integrase) in the virus replicative cycle. Mol. Pharmacol. 2000, 58, 1100–1108. [Google Scholar]
- Macri, R.V.; Karlovska, J.; Doncel, G.F.; Du, X.; Maisuria, B.B.; Williams, A.A.; Sugandhi, E.W.; Falkinham, J.O., 3rd; Esker, A.R.; Gandour, R.D. Comparing anti-HIV, antibacterial, antifungal, micellar, and cytotoxic properties of tricarboxylato dendritic amphiphiles. Bioorg. Med. Chem. 2009, 17, 3162–3168. [Google Scholar]
- Blanzat, M.; Turrin, C.O.; Aubertin, A.M.; Couturier-Vidal, C.; Caminade, A.M.; Majoral, J.P.; Rico-Lattes, I.; Lattes, A. Dendritic catanionic assemblies: In vitro anti-HIV activity of phosphorus-containing dendrimers bearing galbeta1cer analogues. Chembiochem 2005, 6, 2207–2213. [Google Scholar] [CrossRef]
- Han, S.; Yoshida, D.; Kanamoto, T.; Nakashima, H.; Uryu, T.; Yoshida, T. Sulfated oligosaccharide cluster with polylysine core scaffold as a new anti-HIV dendrimer. Carbohyd. Polym. 2010, 80, 1111–1115. [Google Scholar]
- Tabarani, G.; Reina, J.J.; Ebel, C.; Vives, C.; Lortat-Jacob, H.; Rojo, J.; Fieschi, F. Mannose hyperbranched dendritic polymers interact with clustered organization of DC-SIGN and inhibit gp120 binding. FEBS Lett. 2006, 580, 2402–2408. [Google Scholar]
- Wang, W.; Guo, Z.; Chen, Y.; Liu, T.; Jiang, L. Influence of generation 2-5 of PAMAM dendrimer on the inhibition of Tat peptide/ TAR RNA binding in HIV-1 transcription. Chem. Biol. Drug Des. 2006, 68, 314–318. [Google Scholar]
- Rosa Borges, A.; Wieczorek, L.; Johnson, B.; Benesi, A.J.; Brown, B.K.; Kensinger, R.D.; Krebs, F.C.; Wigdahl, B.; Blumenthal, R.; Puri, A.; et al. Multivalent dendrimeric compounds containing carbohydrates expressed on immune cells inhibit infection by primary isolates of HIV-1. Virology 2010, 408, 80–88. [Google Scholar] [CrossRef]
- Yang, D.W.; Ohta, Y.; Yamaguchi, S.; Tsukada, Y.; Haraguchi, Y.; Hoshino, H.; Amagai, H.; Kobayashi, I. Sulfated colominic acid: An antiviral agent that inhibits the human immunodeficiency virus type 1 in vitro. Antivir. Res. 1996, 31, 95–104. [Google Scholar]
- Clayton, R.; Hardman, J.; LaBranche, C.C.; McReynolds, K.D. Evaluation of the synthesis of sialic acid-PAMAM glycodendrimers without the use of sugar protecting groups, and the anti-HIV-1 properties of these compounds. Bioconjug. Chem. 2011, 22, 2186–2197. [Google Scholar]
- Schinazi, R.F.; Brettreich, M.; Hirsch, A. Water-soluble dendrimeric fullerene as anti-HIV therapeutic. US Patent App 20,030/036,562, 2001. [Google Scholar]
- Friedman, S.H.; Decamp, D.L.; Sijbesma, R.P.; Srdanov, G.; Wudl, F.; Kenyon, G.L. Inhibition of the HIV-1 protease by fullerene derivatives—Model-building studies and experimental-verification. J. Am. Chem. Soc. 1993, 115, 6506–6509. [Google Scholar]
- Weislow, O.S.; Kiser, R.; Fine, D.L.; Bader, J.; Shoemaker, R.H.; Boyd, M.R. New soluble-formazan assay for HIV-1 cytopathic effects: Application to high-flux screening of synthetic and natural products for AIDS-antiviral activity. J. Natl. Cancer I. 1989, 81, 577–586. [Google Scholar]
- European Medicines Agency Scientific Discussion for the Approval of Kaletra. Available online: http://www.ema.europa.eu (accessed on 31 October 2011).
- Li, M.; Al-Jamal, K.T.; Kostarelos, K.; Reineke, J. Physiologically based pharmacokinetic modeling of nanoparticles. ACS Nano 2010, 4, 6303–6317. [Google Scholar]
- Ochekpe, N.A.; Olorunfemi, P.O.; Ngwuluka, N.C. Nanotechnology and drug delivery part 2: Nanostructures for drug delivery. Trop. J. Pharm. Res. 2009, 8, 275–287. [Google Scholar]
- European Medicines Agency 1st International Workshop on Nanomedicines 2010 Summary Report. Available online: http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2010/09/news_detail_001108.jsp&murl=menus/news_and_events/news_and_events.jsp&mid=WC0b01ac058004d5c1 (accessed on 12 March 2012).
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Parboosing, R.; Maguire, G.E.M.; Govender, P.; Kruger, H.G. Nanotechnology and the Treatment of HIV Infection. Viruses 2012, 4, 488-520. https://doi.org/10.3390/v4040488
Parboosing R, Maguire GEM, Govender P, Kruger HG. Nanotechnology and the Treatment of HIV Infection. Viruses. 2012; 4(4):488-520. https://doi.org/10.3390/v4040488
Chicago/Turabian StyleParboosing, Raveen, Glenn E. M. Maguire, Patrick Govender, and Hendrik G. Kruger. 2012. "Nanotechnology and the Treatment of HIV Infection" Viruses 4, no. 4: 488-520. https://doi.org/10.3390/v4040488