Oxidative Stress and HPV Carcinogenesis

Laboratory of Virology, "Regina Elena" Italian National Cancer Institute, Via delle Messi d'Oro 156, 00158 Rome, Italy
Viruses 2013, 5(2), 708-731; https://doi.org/10.3390/v5020708
Submission received: 7 January 2013
/
Revised: 22 January 2013
/
Accepted: 5 February 2013
/
Published: 12 February 2013
Abstract
:Extensive experimental work has conclusively demonstrated that infection with certain types of human papillomaviruses, the so-called high-risk human papillomavirus (HR-HPV), represent a most powerful human carcinogen. However, neoplastic growth is a rare and inappropriate outcome in the natural history of HPV, and a number of other events have to concur in order to induce the viral infection into the (very rare) neoplastic transformation. From this perspective, a number of putative viral, host, and environmental co-factors have been proposed as potential candidates. Among them oxidative stress (OS) is an interesting candidate, yet comparatively underexplored. OS is a constant threat to aerobic organisms being generated during mitochondrial oxidative phosphorylation, as well as during inflammation, infections, ionizing irradiation, UV exposure, mechanical and chemical stresses. Epithelial tissues, the elective target for HPV infection, are heavily exposed to all named sources of OS. Two different types of cooperative mechanisms are presumed to occur between OS and HPV: I) The OS genotoxic activity and the HPV-induced genomic instability concur independently to the generation of the molecular damage necessary for the emergence of neoplastic clones. This first mode is merely a particular form of co-carcinogenesis; and II) OS specifically interacts with one or more molecular stages of neoplastic initiation and/or progression induced by the HPV infection. This manuscript was designed to summarize available data on this latter hypothesis. Experimental data and indirect evidences on promoting the activity of OS in viral infection and viral integration will be reviewed. The anti-apoptotic and pro-angiogenetic role of NO (nitric oxide) and iNOS (inducible nitric oxide synthase) will be discussed together with the OS/HPV cooperation in inducing cancer metabolism adaptation. Unexplored/underexplored aspects of the OS interplay with the HPV-driven carcinogenesis will be highlighted. The aim of this paper is to stimulate new areas of study and innovative approaches.
List of acronyms used

{kind=link}
{kind=link}
{kind=link}
| AP-1 | Activator Protein 1 |
| ATM | Ataxia telangiectasia mutated (protein) |
| ATR | ATM–Rad3-related (protein) |
| BaP | Benzo(a)pyrene |
| BER | Base excision repair complex |
| CAT | Catalase, |
| CDK1 | Ciclin dependent kinase 1 |
| ε-A | 1N6 etheno-adenine |
| ε-C | 3N4 etheno-cytosine |
| ETC | Electron transport chain |
| FAD | Flavin adenine dinucleotide |
| FapyAde | 4,6-diamino-5-formamidopyrimidine |
| FapyGua | 2,6-diamino-4-hydroxy-5-formamido- pyrimidine. |
| GSH | Glutathione |
| GPX1 | Glutathione peroxidase 1 |
| GSTP1 | Glutathione S transferase P1-1 |
| HIF-1 | Hypoxia inducible factor - 1 |
| HNE | trans-4-hydroxy-2-nonenal (HNE) |
| H2O2 | Hydrogen peroxide. |
| HPV/HR-HPV | Human papillomavirus /high-risk human papillomavirus |
| LPO | Lipid peroxydes |
| MAPK/ERK | Mitogen-activated protein kinase/Extracellular signal-activated Kinase |
| M2PK | M2 pyruvate kinase |
| NAD/NADH | Nicotinamide adenine di nucleotide/reduced |
| •NO | Nitric oxide |
| iNOS | Inducible nitric oxide synthase |
| 8-oxoAde | 8-oxo-7,8-dihydroadenine |
| 8-oxo-Gua | 8-hydroxyguanine, 8-hydroxy-2’-deoxyguanonsine |
| 1O2 | Singlet oxygen |
| O2•− | Superoxide ion |
| RNS/ROS/RONS | Reactive nitrogen/oxygen/oxygen and nitrogen species |
| SESN | Sestrins |
| SESN2 | Sestrin 2 |
| SOD | Superoxide dismutase |
| Tg | 5,6-dihydroxy-5,6-dihydrothymine (thymine glycol) |
| TGFβ-1 | Tumor growth factor β-1 |
| TopBP1 | DNA topoisomerase II beta-binding protein 1 |
| TRX | Thioredoxine reductase |
1. Introduction
Cervical cancer is one of the leading causes of cancer death among women. The identification of HR-HPV infection as the etiologic agents of cervical cancer, as well as other non-genital carcinomas, is undoubtedly among the greatest medical advances that have been made in the last twenty years [1]. According to a widely shared view, these viruses must induce an enhanced proliferative state in order to accomplish their replicative cycle in the poorly replicating host epithelial cell. This is achieved through the activity of two oncogenic proteins, E6 and E7. Their ability to target and promote degradation of p53 and pRB, respectively, is believed to be the main mechanism by which HPV oncogenes induce genomic instability and allow the cells to acquire accumulating genomic alteration, thus ultimately leading to the full neoplastic state. However, only a minor percentage of viral infections lead to invasive growth, thereby indicating that viral oncogene expression is not per se sufficient, and other events are needed for cancer to occur. These additional events could be either the consequence of long-term viral protein expression, or the results of non-viral factors. In the search for such co-factor(s), many viral, host, and environmental factors have been investigated [2]. The role of oxidative stress (OS), however, has received little attention in this regard. OS is a condition occurring whenever the generation of oxidant species (mostly reactive oxygen species (ROS) and reactive nitrogen species (RNS), collectively known as RONS) exceeds the cellular neutralizing/scavenging capabilities. ROS are constantly generated in aerobic cells by the incomplete reduction of molecular O2 to H2O during mitochondrial oxidative phosphorylation, as well as during microsomal and peroxisomal oxidations. In addition, RONS are also generated during a number of processes such as inflammation, infections, and immune reactions [3], mechanical and chemical stresses (e.g. transient metal ions)[4,5], chemical biotransformation [6], exposure to UV [7,8], and ionizing irradiation [9].
Epithelial tissues, the elective target for HPV infection, are heavily exposed to all named sources of OS. RONS, through oxidative damage, cause functional alteration of cell membrane lipids, proteins, and nucleic acids [10]. In addition, through the perturbation of the cellular redox balance, they induce the activation of several redox-sensitive transcription factors, modify the gene expression responses, and modulate the function of redox-sensitive proteins. They have therefore been implicated in various acute or chronic degenerative processes, including aging and cancer [11,12,13]. Due to their conspicuous impact on cell homeostasis, RONS levels are closely monitored by sophisticated sensing mechanisms and are strictly controlled by multiple antioxidant and scavenging systems. Moreover, they are embedded with crucial roles within multiple cell-signaling and regulation pathways [14,15,16], having either pro- or anti-proliferative potential effects. Thus, OS represents an interesting candidate as a co-factor in HPV carcinogenesis. This paper was designed to provide a brief description of RONS generation and their biological impact, to summarize available data on the impact OS has on the major stages of HPV-mediated carcinogenesis, and to promote the identification of new areas of study and innovative approaches.
2. Generation of RONS
Mitochondrial respiration is a fascinating biochemical process operating a coordinated four-electron reduction of O2 to H2O. Although highly efficient, the mitochondrial electron transport chain (ETC) is imperfect [17] and leakage of intermediate products represents the major source of OS in aerobic organisms. The following brief description of RONS generation during mitochondrial respiration offers an outline of the basic phenomena and of their chemical properties. The contribution of other sources of RONS will shortly be mentioned in order to complete the background.
Generation of the superoxide ion. The single electron (e-) reduction of molecular oxygen generates the radical superoxide ion (O2•- ) [18], according to the reaction (1).
O2 + e- → O2•-
This radical species is also produced within the cell by the action of some oxidases such as aldehyde oxidase, NADH oxidase, and xanthine oxidase, as well as under UV irradiation. Although O2•- has the potential to react with almost all biological molecules, its direct contribution to cell damage is scarce, owing to its limited rate of diffusion and its comparatively low reactivity. Nonetheless, O2•- is able to sustain other cellular reactions generating extremely powerful RONS accountable for most of its toxicity. With the reaction (2)
superoxide is rapidly dismutated (either spontaneously or enzymatically) to molecular oxygen and the much higher reactive hydrogen peroxide (H2O2). H2O2 and O2•- can further react according to the reaction (3), the “Haber–Weiss reaction,”
to generate the most reactive species hydroxyl radical (OH•). A further ROS, named singlet oxygen (1O2), is also generated as a by-product by the superoxide ion through the reactions (2) and (3) and by superoxide interaction with OH• [19]. Singlet oxygen is an excited form of molecular oxygen in which the electrons in the external shell are rearranged to confer increase oxidative activity. 1O2 attacks the double bond in lipids with formation of hydroperoxides and endoperoxides. It also attacks and oxidizes methionine, tryptophan, histidine, and cysteine.
O2•- + 2e- → ½ O2 + ½ H2O2
O2•- + H2O2 → O2 + OH- + OH•
Generation of hydrogen peroxide. In addition to being generated by the divalent reduction of O2 and protonation during electron transports (reaction 4)
and through the above reaction (2), H2O2 is the primary product of many microsomal and peroxisomal oxidases. This non-radical ROS, with an intermediate reactivity between those of O2•- and OH•, is involved in thiol groups oxidation/inactivation and in redox potential imbalance [20]. Moreover, through the reaction (5), the “Fenton reaction”
transition metal ions can catalyze the H2O2 reduction to OH- plus OH• and oxidized metal ion. The transition metal ions Me(n+1)+ can then be reduced back to Men+ by the superoxide anion with the reaction (6)
O2 + 2e- + → H2O2
H2O2 + Men+ → Me(n+1)+ + OH- + OH•
O2•- + Me(n+1)+ → O2 + Men+
This reaction, on one hand, scavenges O2•- equivalents, thus demonstrating a potentially beneficial effect, but, on the other hand, it reconstitutes the pool of reduced metal ions, thus allowing a further cycle to the Fenton reaction and further generating hydroxyl radicals.
The hydroxyl radical (OH•) is also formed under γ and x irradiation by direct lysis of water according to the reaction (7).
hν + H2O → OH• + H•
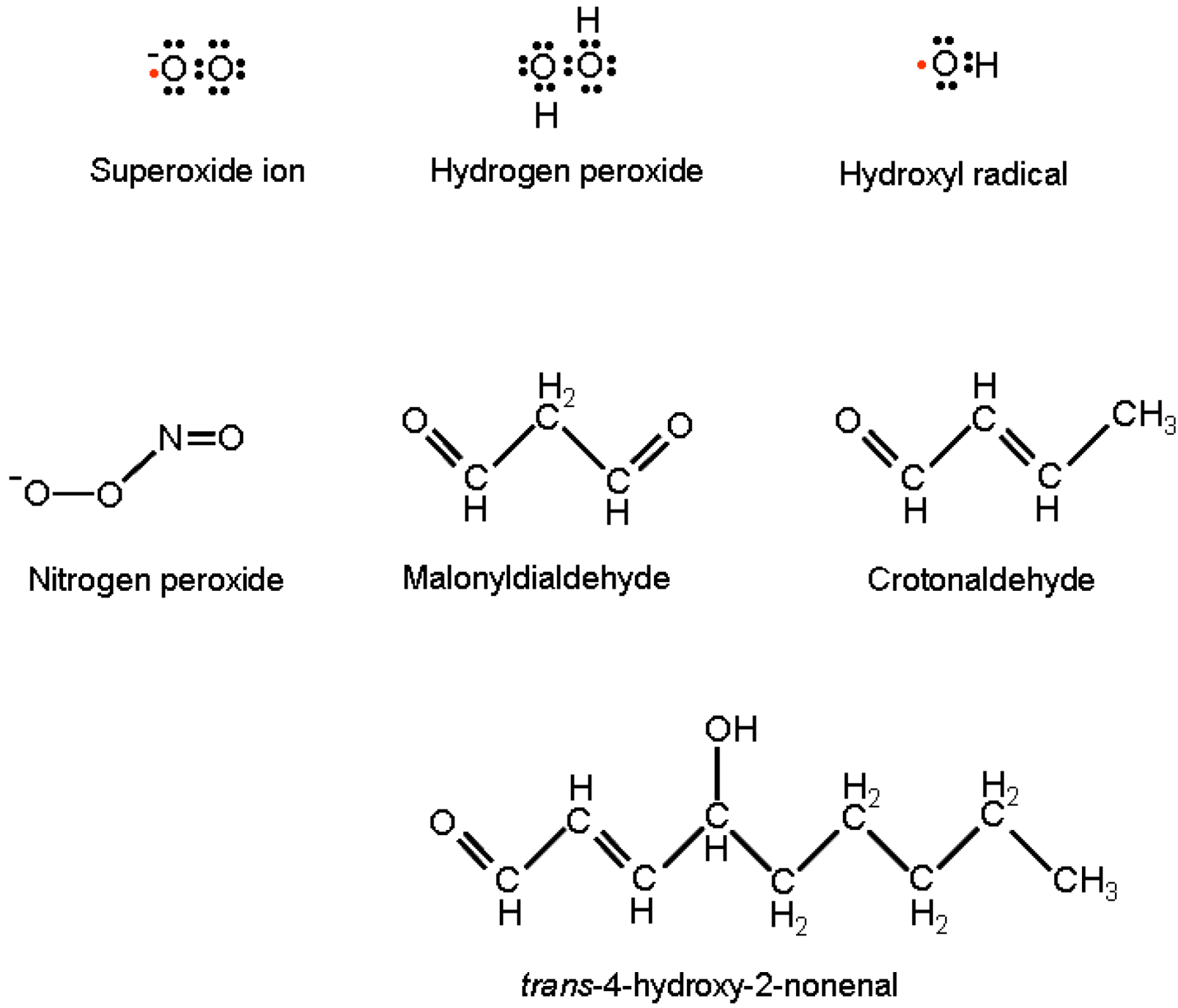
OH• is the strongest oxidizing species; it reacts with almost all biological molecules with rates close to the theoretical diffusion limit. It gives place to reactions of hydrogen extraction, hydroxyl addition, and electron transfer. OH• is the major cause of oxidative adducts on purine and pyrimidine bases, as well as on the DNA sugar moiety, resulting ultimately in nucleic acid point mutation, cross-linking and strand interruption. The hydroxyl radical further concurs to DNA modification by the activity of lipid peroxidation products (LPO) [21]. The most relevant LPOs are: malonyldialdehyde, Crotonaldehyde and trans-4-hydroxy-2-nonenal (HNE) (Figure 1).
Figure 1.
Lewis formulas of reactive oxygen and nitrogen species and structural formulas of lipid peroxidation products responsible for DNA modifications with miscoding potentials.
Figure 1.
Lewis formulas of reactive oxygen and nitrogen species and structural formulas of lipid peroxidation products responsible for DNA modifications with miscoding potentials.
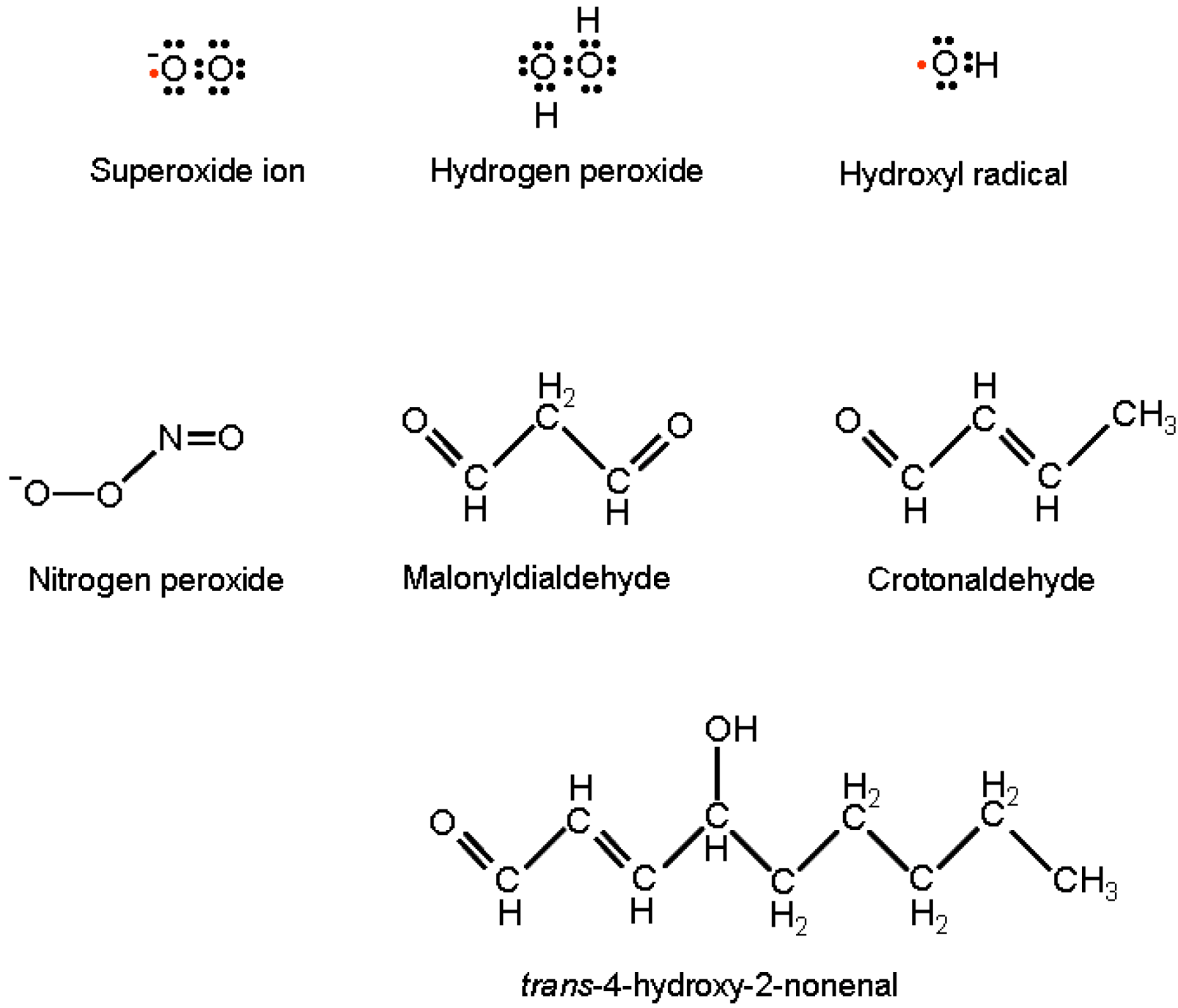
These are generated by OH• hydrogen extraction from unsaturated lipids in membranes and, following their direct purine attack, are able to produce exocyclic DNA adducts with high miscoding potentials [22].
The nitric oxide (•NO), a radical RNS, is produced endogenously from L-arginine, oxygen and NADPH by several nitric oxide synthase (NOS) enzymes in response to a number of physiological stimuli. •NO is a very highly reactive species. With its short lifetime (a few seconds) and its ability to diffuse freely across membranes, •NO represents the ideal molecular messenger. As a paracrine or autocrine signaling molecule [23], it and is involved in the regulation of a number of functions including blood pressure, endothelial and neuronal homeostasis, platelet and leukocyte adhesion, phagocytes and antimicrobial activity, immune response, apoptosis induction. According to the reaction (8)
nitric oxide, in the presence of superoxide generates the radical species peroxynitrite (OONO•) a highly reactive RNS itself able to induce lipid peroxidation and protein nitration [24].
•NO + O2•- → OONO•
2.1. Peroxisomal β-Oxidation
Another major source of RONS is the peroxisome fatty acid beta oxidation which generates H2O2 as a by-product. [25]. Despite the high concentration of catalase in peroxisomes, H2O2 leakage appears to be a physiological occurrence contributing significantly to OS as indicated by a direct correlation between the number of peroxisomes and their proliferation rate with the level of total cell OS and damage [26]. Moreover, the reported occurrence of catalase-negative peroxisomes during experimental liver regeneration [27] suggests that H2O2 leakage may be enhanced during rapid cell proliferation [28].
2.2. P450 Isoenzyme Activity
Microsomal cytochrome P-450 enzymes metabolize xenobiotic compounds, usually drugs or alkaloids of plant origin, by catalyzing their univalent oxidation or reduction. Depending on which P450 isoenzyme is involved, these reactions consist either in the direct reduction of O2 to O2•- [29] or in the single electron reduction of organic intermediates and the subsequent electron transfer to O2 with generation of O2•- and organic intermediate restoration [30].
2.3. Inflammation
Inflammation increases the risk of cancer development and, at the same time, is a constitutive trait of any cancer tissues, thus cancer itself represents a cancer-promoting agent [3]. During inflammation, the combined action of hormones, cytokines and low molecular weight messengers, the so-called “Go” signals lead to the recruitment of mast-cell and leucocytes [31]. These cells, upon a sharp increase of oxygen consumption, “the respiratory burst”, give way to a massive RONS release, including O2•-; H2O2; •NO and hypochloric acid (HClO) [32,33], at levels largely above the toxic threshold [3].
2.4. Ischemia–Reperfusion Stress
In addition to its central role in ischemic conditions such as myocardial infarction and brain stroke, ischemia-reperfusion stress is, less obviously, a relevant mechanism in peripheral areas of proliferating lesions. OS represents a major pathogenetic momentum in ischemia-reperfusion and, surprisingly enough, its generation requires both hypoxia/anoxia and reperfusion phases [34]. During ischemia–reperfusion, OS is generated by many different mechanisms, namely: leukocyte recruitment and respiratory burst; activation of xantine oxidase [35,36]; membrane NADPH oxidase activation [37]; mitochondrial impairment with increased RONS generation [38,39]; down-regulation of endogenous antioxidant mechanisms, as a consequence of mitochondrial hypoxia adaptation [34]; and, HIF-1-mediated induction of iNOS [40]. Altogether, these mechanisms have the potential to generate, directly or as secondary products, all types of RONS.
2.5. Signaling Pathways Activation
RONS are also generated during the activation of a large number of signaling pathways induced by growth factors, cytokines and tumor promoters such as: TGF alpha; TNF alpha; TGF Beta-1, etc. [39]. In turn, RONS can activate or potentiate many of these signaling pathways, often realizing a positive feedback circuit which further increases RONS generation [41].
3. Biochemical Effects of RONS
3.1. Genetic Damage
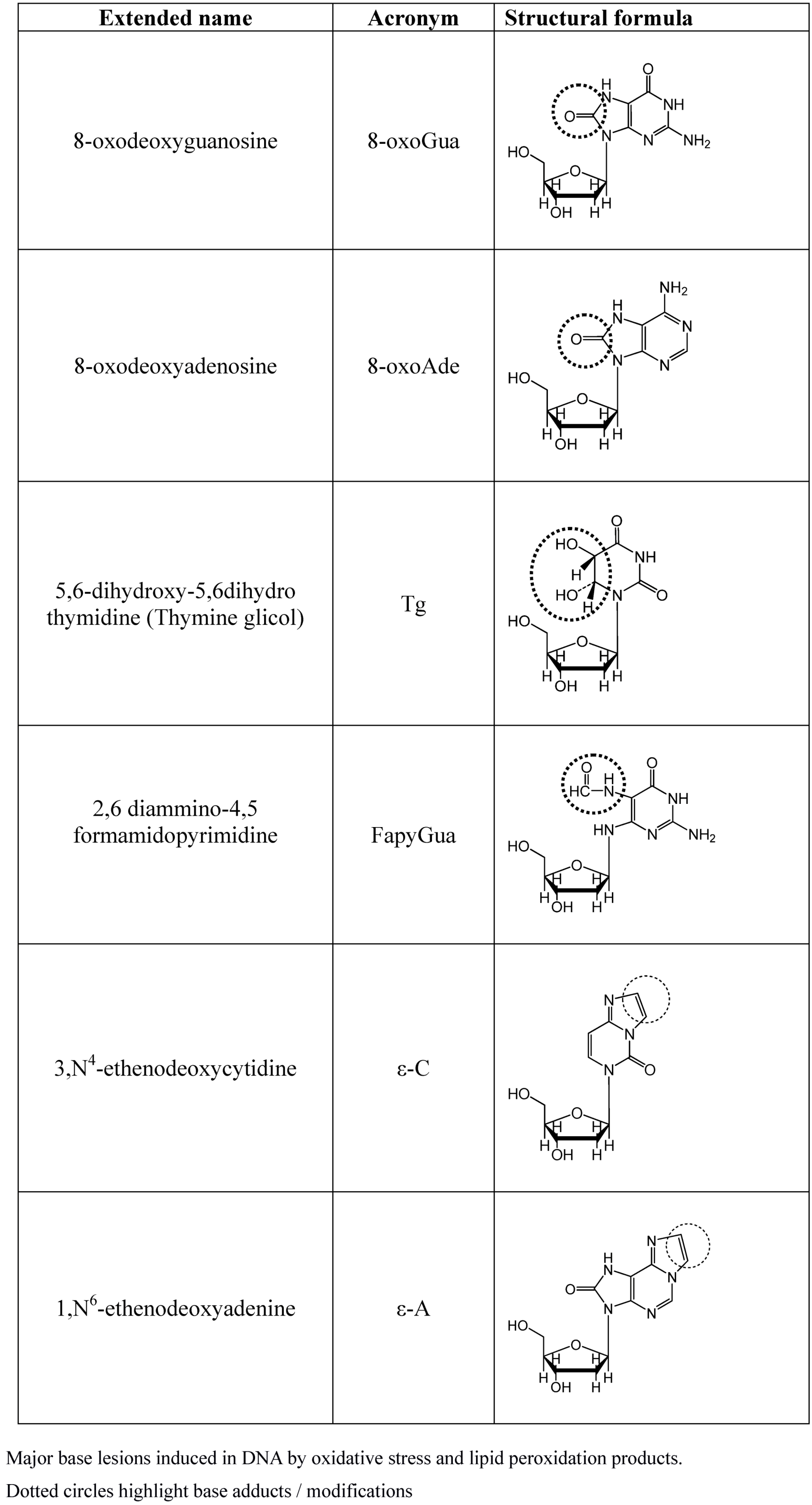
More than one hundred oxidatively induced DNA lesions have been reported so far, and the current number may reasonably be even larger [42]. However, among them the most relevant products are those reported in Figure 2, namely 8-oxo-7,8-dihydroguanine (8-oxoGua), 8-oxo-7,8-dihydroadenine (8-oxoAde), 5,6-dihydroxy-5,6-dihydrothymine (thymine glycol, Tg) and the ring-opened lesion 2,6-diamino-4-hydroxy-5-formamido-pyrimidine (FapyGua).
Figure 2.
Major base lesions induced by OS.
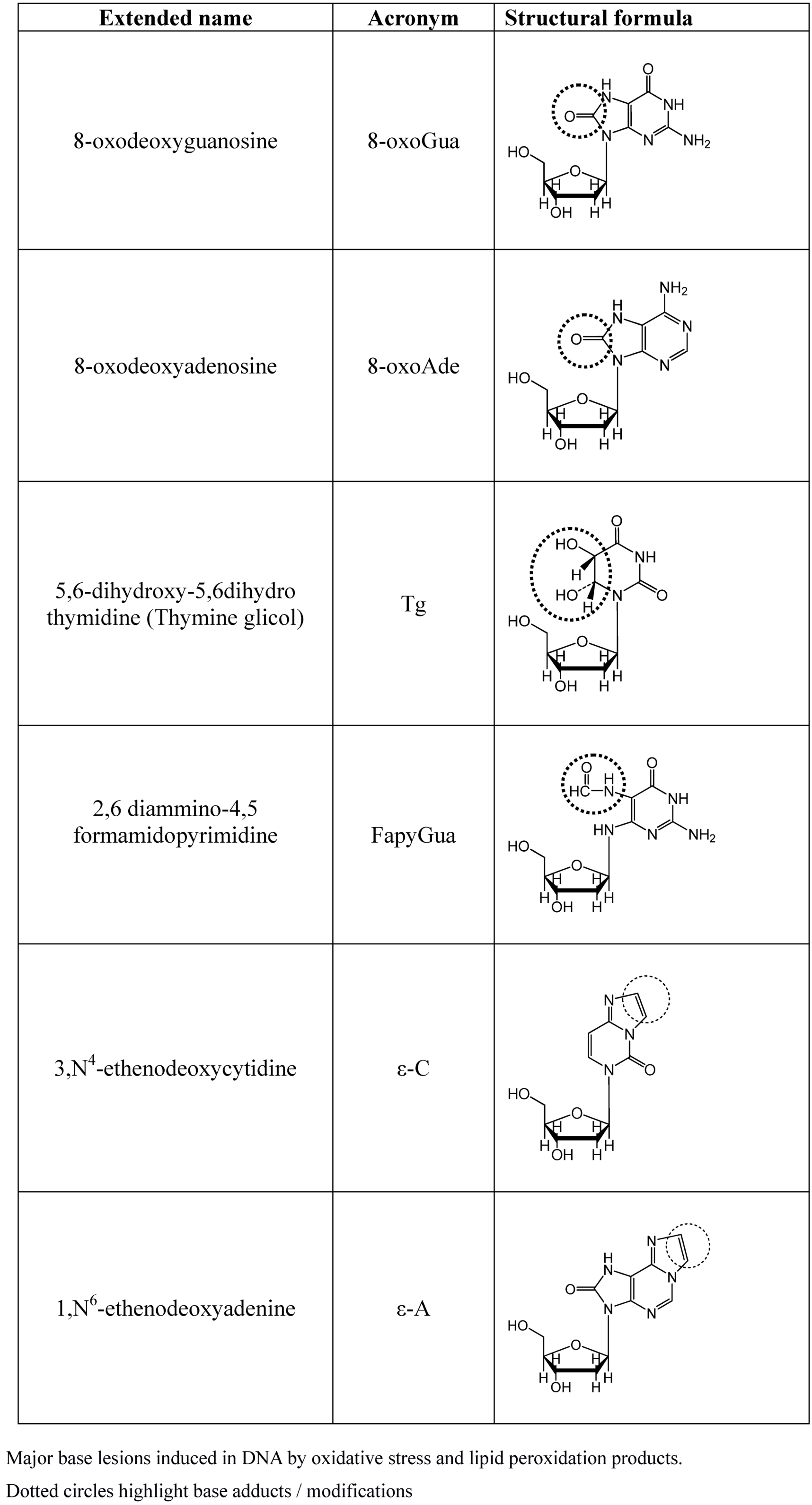
All of these have high miscoding potentials [43,44]. Modified bases on DNA may result either by a direct oxidative attack on DNA or by incorporation of modified bases by polymerases. Indeed, free mononucleotide or bases are two to three orders of magnitude more susceptible to oxidative modification than incorporated bases. Although polymerases can discriminate regular and modified bases, this ability to discriminate is incomplete, and damaged nucleotides are incorporated with still relevant rates [45,46]. The major cause of DNA base modification is the direct attack by OH• and LPO products, mostly malonyldialdehyde, crotonaldehyde and trans-4-hydroxy-2-nonenal (HNE) is another one. These aldehydes, by direct DNA interaction, generate a number of DNA etheno-adducts such as 1N6 etheno-adenine (ε-A) and 3N4 etheno-cytosine (ε-C) [47,22]. Once inserted into the DNA helix,ε-As are responsible for inducing AT→GC, AT→CG and AT→TA point mutations [48], while the ε-Cs generates the mutation CG→AT, CG→TA and CG→GC [49]. In addition, LPO lead to the formation of DNA–DNA and DNA–proteins cross-linking, thereby causing chromosome breaks and rearrangements, in addition to gross structural aberrations [50,51].
3.2. Protein Damage
It is clear that DNA is not the only molecular target of OS. Proteins are obviously prone to oxidative modification as well, and a number of oxidative modifications have been described [52]. The most relevant protein oxidative modifications are carbonyls and nitro-tyrosine adducts. These adducts have been shown to increase in a variety of conditions, including ag,ing and cancer, and are currently used to measure the levels of protein oxidation [53,54]. Protein oxidation is mostly due to the direct attack of OH• and to a smaller extent due to the attack of malonyldialdehydes and HNE, while H2O2 accounts for a very minor part of it. Oxidized proteins are not repaired and must be removed through proteolytic mechanisms. The accumulation of oxidatively modified proteins in the cell lead to altered/disrupted functions, which can obviously induce, concur, or potentiate the homeostatic derangement which is the ultimate cause and the hallmark of cancer [55,56].
3.3. Modulation of Protein Function
In addition to the structural alterations on DNA and proteins, RONS can interact with the carcinogenic process by modulating the gene transcription and the signal transduction pathways. The NF-kB; Nrf2 are two major cancer-relevant systems modulated by RONS [57,58] and, reasonably, most of the cell pathways are regulated by, and depend on, oxidant signals [41]. Generally, the free radicals •NO and O2•− act as direct signaling factors; H2O2 represents the major oxidant signal [59], and signaling occurs via oxidative activation/inactivation of phosphatases or transcription factors, activation of protein kinases; redox dependent modifications of thiol groups, etc. [59,60,61]. The modulation of multiple signaling pathways by RONS and the complementary RONS generation following activation of the signaling pathways has been extensively revised elsewhere, and their potential impact on cancer initiation, cancer promotion, and cancer progression adequately remarked upon [34,30,39,62].
In conclusion, aerobic life implies the generation of RONS. Due to their important physiological functions and sharp toxic activities, RONS represent major carcinogenic agents. It is widely accepted that HPV infection, through the expression of the E6 and E7 proteins, promote the degradation of the cellular proteins p53 and pRB, thus creating the conditions for the accumulation of cancer permitting genetic alteration. Thus, two different contributions of OS to HPV-driven carcinogenesis can be hypothesized: i) the mutagenic potentials of OS cooperate nonspecifically with the transforming activity of an HR-HPV infection. Such an additive mode can have important practical consequences, such as a shorter latency period or a higher efficiency in cancer induction; however, it does not imply any specific mechanism, rather, it represents a particular form of co-carcinogenesis; ii) OS can specifically modulate, or activate, or suppress, directly or indirectly, one or more HPV-specific molecular mechanisms orientating the viral/cell unit toward the transforming–carcinogenic process. The evidence about the latter hypothesis will now be reviewed and discussed.
4. The Interplay Between OS and HPV Carcinogenesis
4.1. OS and HPV Infection
It is commonly assumed—although not proven—that cellular conditions at the early steps of viral infection (i.e. viral adsorption, the viral entry and the initial establishment of viral expression) represent critical events for the outcome of HPV infection: spontaneously healing, productive infection versus persistent infection, and neoplastic transformation. The first data suggesting a possible involvement of OS in these early phases initially came from indirect clinical–epidemiological observations: in a study aiming to identify additional conditions for cervical cancer, the concentration of diamines and polyamines was evaluated in 187 seminal plasmas and the concentration of diamine oxidases and polyamine oxidases was evaluated in 126 cervical mucus samples. The seminal amines are oxidized by amine oxidases in the cervical mucus and the hydroxyl radical, hydrogen peroxide and LPOs are generated. The results indicated that highly variable concentrations of amines and enzymes were found in seminal plasmas and mucus, respectively [63]. It was therefore proposed that a highly divergent local burden of ROS and LPO can occur during sexual intercourse, and this variability could contribute to the fluctuating outcome of the viral infection. A recent perspective study correlating the clearance of incident HPV infection with the level of ferritin and of soluble transferrin receptor in 327 serum samples indicated that women with high levels of ferritin were less likely to clear incident HR-HPV infections than those with lower levels [64]. The authors suggested that elevated levels of iron storage may, by generating greater endogenous levels of ROS, be related to a reduced clearance rate. Along the same lines, Safaeian et al., in a very large population-based study [65], reported that the polymorphism in gene PRDX3 (a mitochondrial peroxiredoxin putatively involved in antioxidant activity and proliferation control) is associated with higher persistence and higher progression risk. Molecular data consistent with the above indirect evidences were recently reported. In a very interesting study on viral replication in organotypic “raft” viral cultures, it was shown that, following HPV16 infection, complete viral progeny assembly is dependent on a tissue spanning redox gradient. Exposition to both reducing and oxidant condition is sequentially needed for infectivity maturation and redox balance modulation in the cell microenvironment and sharply affects the infectious titer of viral progeny [66]. These results provide a molecular link connecting the OS with viral titer, a major factor potentially involved in persistent infection establishment, proposing a first mechanistic hypothesis for the connection between the oxidative status and lesion persistence/progression.
4.2. OS and Viral DNA Replication
After the viral infection, the core points of carcinogenesis are efficiency and accuracy of DNA replication. OS can strongly affect both, eventually creating the conditions for efficient viral integration. The first observation connecting OS with DNA papillomavirus replication came at the early beginning of the HPV and cancer story. In a BPV/mouse cell system, it was reported [67] that OS, either chemically or UV induced, was associated with amplification of both episomal and integrated copies of BPV. Such an amplification was linked to the DNA-damaging activity of OS, and to the occurrence of OS at the moment of viral infection, as no effect was produced by OS occurring after the infection, as well as by agents inducing cell proliferation (i.e. tumor-promoting agents) [67]. This initial observation was later extended with the report of a functional interaction between the TopBP1 protein of the base excision repair complex (BER) and the viral transcription/replication factor E2 enhancing the ability of the E2 protein to activate viral transcription and viral genome replication. [68]. Subsequently, the involvement of p53 in BER machinery sensing and repairing oxidative DNA damage was reported [69], and that in the presence of the E6 protein, the DNA repair functions are compromised and an increased 8-oxoGua accumulation takes place [70].
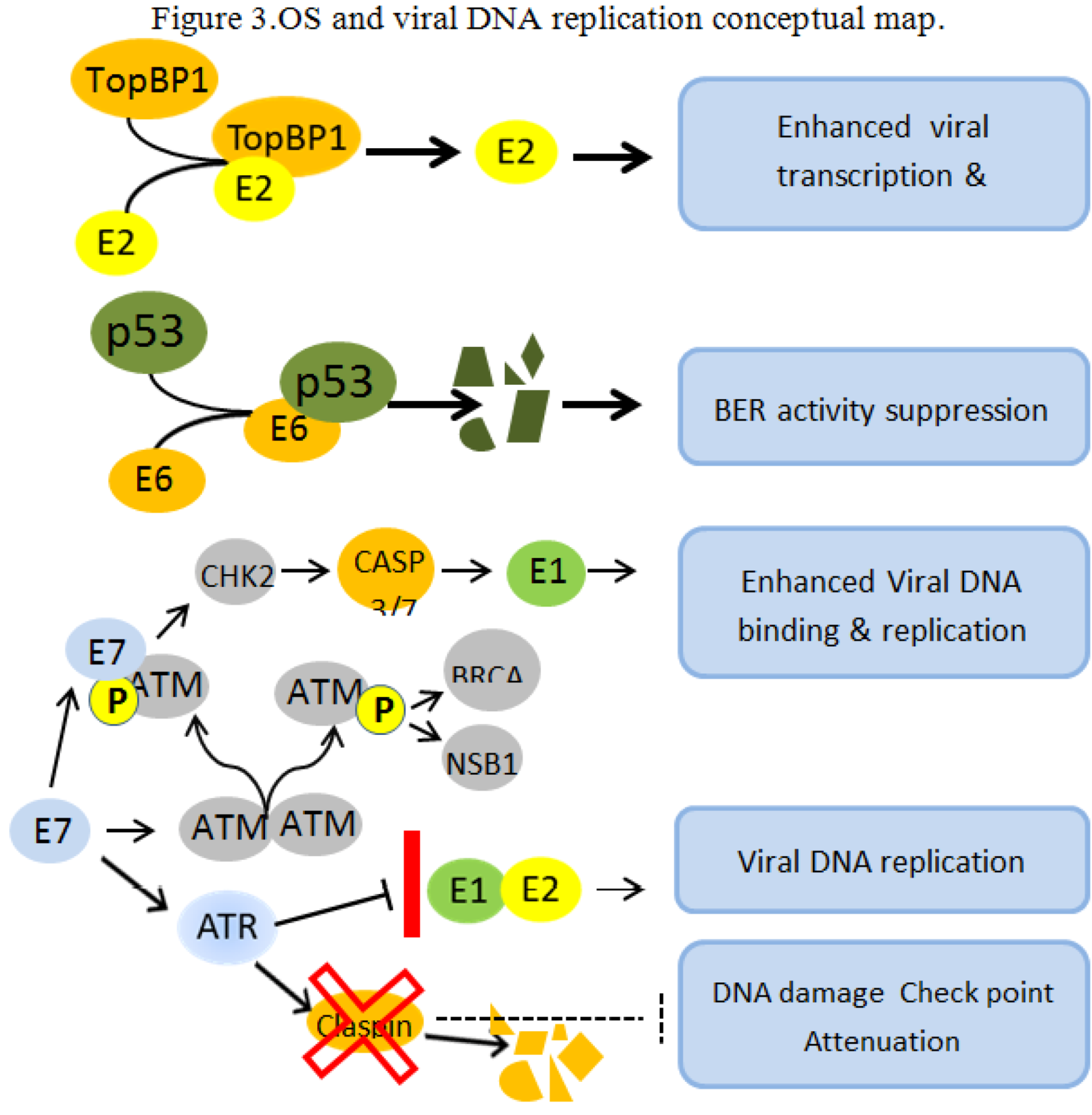
The connection between DNA damage sensing and HPV replication was further strengthened by the report of Moody and Laimins [71], showing that the activation of the ATM pathway, through the interaction with the E7 protein, specifically enhance the E1 DNA-binding and replication activity in an E2-independent manner. In addition, the E7 protein interacting with the ATR DNA damage pathway are able to alleviate the DNA damage checkpoint control by increasing the proteolytic degradation of claspin [72]. Finally, it was recently demonstrated that the E1 and E2 HPV-specific DNA replication is not arrested by ATR following DNA damage, both in vitro and in vivo, while SV40 replication is [73]. This lack of inhibition allows the HPV DNA replication to proceed in the presence of conditions inducing DNA double-strand breaks, thus strongly promoting viral integration (see Figure 3).
Taken together, the data above outline an OS-driven putative mechanism consistent with the viral DNA amplification and integration observed in cell lines derived from invasive cancers [74]. Namely, the OS activation of DNA damage pathways in the presence of HPV expression potentiates the activity of both E1 and E2 viral elements of DNA replication complex. In the presence of DNA damage, the viral replication proceeds, resulting in viral expansion and in increased production of rearranged and DS-break forms. The concomitant ATR-E7 dependent claspin degradation reinforce the release of the cell cycle checkpoint due to the p53 suppression and unrepaired cellular DNA is replicated. Thus abundant viral copies, including linearized and rearranged forms, are made available together with multiple nicks and DS breaks in the host genome, creating the mechanistic feasibility for multiple viral integrations.
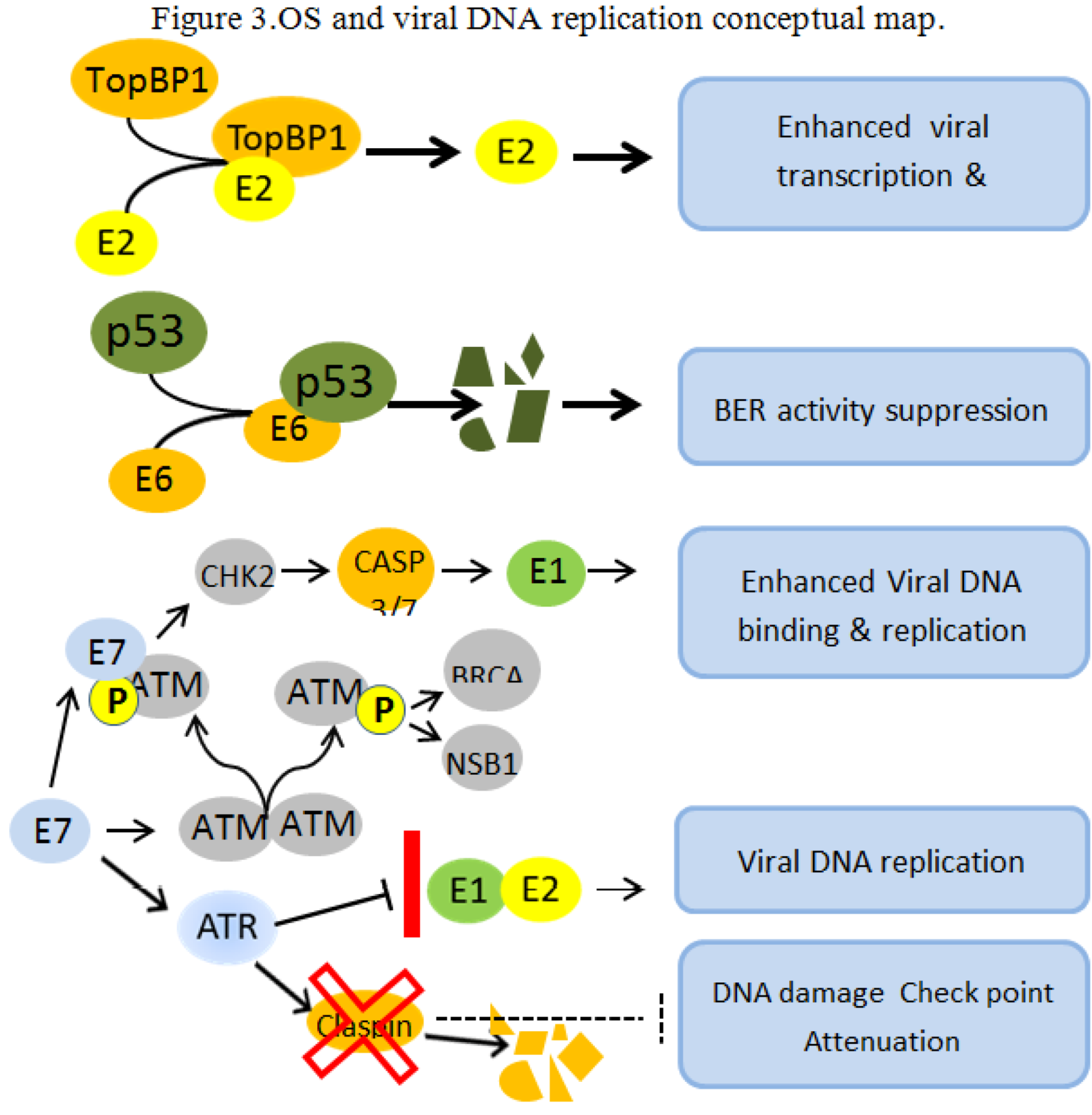
Figure 3.
OS and viral DNA replication conceptual map. OS and viral oncogenes concur to create a cellular environment conducive to viral genome amplification and integration. E2 and TopBP1 interact in vitro and in vivo and TopBP1 enhance the ability of E2 to activate transcription and replication [68]. The E6-dependent p53 degradation abolishes BER functions leading to unrepaired cellular genome [69,70]. The E7 protein promote the phosphorylation of the inactive ATM dimer causing its dissociation into phosphorilated active monomers. The E7 protein then physically interact with the activated ATM monomer activating a number of proteins including CHK2, BRCA1 and NSB1. CHK2 induce proteolytic activation of Caspase 3/7, cleaving the E1 protein, and enhancing the E1 binding to its origin and its ability to replicate in an E2-independent manner [71]. The E7 oncoprotein attenuates the DNA damage checkpoint response by accelerating the proteolytic turnover of claspin, a critical regulator of the ATR/CHK1 signaling axis, in the G2 phase of the cell cycle [72]. These conditions permit the formation of abundant regular and rearranged viral copies, while multiple nicks and DS breaks in the host genome are unrepaired, thus creating the mechanistic feasibility for multiple viral integrations.
Figure 3.
OS and viral DNA replication conceptual map. OS and viral oncogenes concur to create a cellular environment conducive to viral genome amplification and integration. E2 and TopBP1 interact in vitro and in vivo and TopBP1 enhance the ability of E2 to activate transcription and replication [68]. The E6-dependent p53 degradation abolishes BER functions leading to unrepaired cellular genome [69,70]. The E7 protein promote the phosphorylation of the inactive ATM dimer causing its dissociation into phosphorilated active monomers. The E7 protein then physically interact with the activated ATM monomer activating a number of proteins including CHK2, BRCA1 and NSB1. CHK2 induce proteolytic activation of Caspase 3/7, cleaving the E1 protein, and enhancing the E1 binding to its origin and its ability to replicate in an E2-independent manner [71]. The E7 oncoprotein attenuates the DNA damage checkpoint response by accelerating the proteolytic turnover of claspin, a critical regulator of the ATR/CHK1 signaling axis, in the G2 phase of the cell cycle [72]. These conditions permit the formation of abundant regular and rearranged viral copies, while multiple nicks and DS breaks in the host genome are unrepaired, thus creating the mechanistic feasibility for multiple viral integrations.
4.3. OS and Apoptosis Suppression in HPV-Expressing Cells
Apoptosis suppression is a fundamental mechanism for cancer development, permitting the survival of transformed clones and their selection on the basis of their fitness to the specific microenvironment. HPV-transformed cells achieve this goal expressing the E6 viral protein that promote the ubiquitination and the proteolytic degradation of the p53 cellular protein. A major contribution to the modulation of apoptosis is provided by the activation of the inducible form of nitric oxide synthase (iNOS), directing the production of •NO. As mentioned above, •NO is a powerful chemical messenger and cell regulator with a double-edged pro-survival and pro-apoptotic effect. This seemingly contradictory profile, however, can be resolved by considering the level of •NO concentration involved in each specific condition and the temporal–spatial mode of its release. At high concentration, i.e. above 10−6 M, a concentration easily attained during the macrophage/neutrophils activation, •NO induces apoptosis by nitrosating thiol groups of cellular proteins and enzymes [75]. Conversely, •NO concentrations around 2 × 10−8 M are essentially mutagenic [76] and induce the release of vascular endothelial growth factor (VEGF) [77,78]. Thus, even though initially considered a potential mechanism of neoplastic growth control, it is now evident that •NO fails to attain apoptosis-inducting concentration in the vast majority of cancers of any histological origin [75]. In the case of cervical carcinomas and HPV-positive dysplastic lesions, the reason for the inadequate level of •NO is the insufficient expression of iNOS that appears to be progressively reduced with the histological severity of lesions [79,80]. This is the result of the TGFβ-1-mediated suppression of the iNOS expression at the mRNA level [81,82]. Accordingly, the level of TGFβ-1 mRNA observed in cervical smears correlates with the progression of cervical intraepithelial neoplasia to cancer [83]. Meanwhile, the low level of •NO concentration promotes mutagenesis, that further accelerates the neoplastic progression, and promotes the VEGF mediated angiogenesis that provides optimal metabolite supply to the tumor growth. Moreover, the increased blood flow potentiates the •NO scavenging by circulating red blood cells [84], thus completely arresting tumor apoptosis. Finally, E6 proteins, in a p53-independent way, specifically up-regulate the activity of the VEGF promoter, thereby inducing a high level of VEGF mRNA expression [85].
Although the suppression of p53 functions is a pivotal event in HPV carcinogenesis, it has to be remarked that the HPV E6 expression in the context of its natural promoter does not suppress p53 activity completely, which remains measurable both in cell lines [86] and in cancer-derived cells [87]. The extent of such a residual p53 function is inversely correlated to the E6/E7 and has the physiological role to maintain partial levels of the transcriptional activation, antioxidant systems, and DNA-repairing functions still necessary for the transformed cell survival [88]. The E6 expression level controlling the previously outlined mechanism of fine p53 down-modulation can be, in turn, modulated by a number of conditions [89,90,91,92], including OS that specifically modulates both the viral RNA expression levels [93] and their splicing pattern [94]. Thus, an OS-dependent modulation of the E6-p53 axis enables the cell to activate, when appropriate, a survival response consisting in a mild growth arrest, the appropriated activation of antioxidant enzymes and DNA repair mechanism, and a limited induction of apoptosis [93,94]. Altogether, the data provided earlier depict an E6/p53 interaction much finer than commonly perceived, consisting in a delicate, but incomplete, p53 suppression adjusted by OS to match the highly dynamic OS levels faced by the dysplastic/neoplastic cell.
Other specific cooperative connections between OS and HPV carcinogenesis are sustained by the E7 protein. This oncogene increases the resistance to H2O2-induced cell death through the up-regulation of catalase activity, the activation of NF-kB, and the orchestrated modulation of the cell factors: Bcl-xL; IL-18; Fas; Bad and Cytochrome-C [95]. In addition, E7, by specifically protecting the glutathione S transferase P1-1 protein (GSTP1) from oxidation, up-regulates the GSH-mediated OS detoxification and modifies the equilibrium between the oxidized and reduced GSTP1, consequently inhibiting the JNK phosphorylation and its ability to induce apoptosis [96]. Thus, the E6 and E7 proteins, in addition to their elective oncogenic roles, seem to provide the condition to survive the increased level of OS connected with the transformed status generated by them, turning a harmful, potentially limiting condition, into a positive selection factor.
4.4. OS Cell Signaling and Metabolic Modulation of HPV Transformed Cells
A further OS-mediated regulation of viral oncogenes functions takes place at the transcription activation level. Early genes transcription is controlled by the E2 viral protein and by a wide array of cellular transcription factors [97]. Among them, a distinct role is played by the AP-1 complex. This is indeed a family of homo- or heterodimers composed of the variable association of two proteins among c-Jun; JunB; JunD; FosB; Fra-1 and Fra-2. In transformed or malignant cells with high levels of E6/E7 transcription, the AP-1 composition is mainly, but not exclusively, represented by the cJun/cFos heterodimer. However, the correct assembly of this powerful activating form is strictly dependent on the endogenous level of ROS [98] and, in conditions of low ROS levels, the variant complex cJun/Fra1 is preferentially formed. This form has a higher binding activity on the viral regulatory region and a repressive activity on transcription initiation and can therefore be associated with suppressed viral transcription and reduced oncogenic potentials. Accordingly, the cJun/Fra1 complex is preferentially expressed in non-neoplastic or reverting cell lines, while the canonical cJun/cFos form is expressed in oncogenic cell lines [99]. Conversely, the composition of the AP-1 complex seems to dictate the cell phenotype [100,101].
An elevated rate of aerobic glycolysis, the so-called “Warburg effect” [102], is a very striking metabolic feature of the cancer cell. A direct connection between the level of aerobic glycolysis in HPV neoplastic cells and the internal redox balance is provided by the physical interaction of HPV16 E7 protein with the glycolytic enzyme M2 Pyruvate Kinase (M2PK) [103]. M2PK is a regulatory enzyme occurring either in a high affinity tetrameric form or in a low affinity dimeric form. The balance of the two forms is controlled by the level of the key glycolytic intermediate fructose 1,6-biphosphate. The physical interaction with the E7 shifts the balance in favor of the dimeric form, inducing a sharp suppression of the tricarbossilic acid circle and oxidative phosphorylation, notwithstanding an elevated concentration of fructose 1,6-biphosphate. This suppression resulted in the depletion of the glycogen store and in the alteration of the redox balance between NADH and FAD [104]. The E7 protein is further able to induce an Na+/H+ exchanger-dependent alkalinization of the intracellular compartments that, presumably by modifying the activity of the many redox sensitive cytoplasmic proteins, directly modulate the cell proliferation rate, the growth rate in low serum, and the anchorage-independent growth induced by E7 [105].
4.5. Adaptation of Advanced Neoplastic Cells to OS
Data on OS in advanced cervical cancers has been steadily increasing since HPV infection was first proposed as a possible etiologic factor [106]. In a fairly large series of cervical histological samples, an increased level of oxidized protein thiol groups was found in cancer specimens compared to normal or dysplastic tissues, as well as compared to non-neoplastic areas surrounding the neoplastic lesion [107], therebysuggesting that highly oxidant conditions were acting on the cancer cells. Indications that HPV tumors experience an increased oxidative environment were then extended and confirmed by studies reporting on: an increased number of DNA adducts in histological samples forming invasive cervical cancers compared with dysplastic or normal tissue [108,109], the increased levels of thioredoxine reductase (TRX) and of apurynic apyrimidinic excision (APE)/Ref-1 in hypoxic micro-regions of cervical carcinomas [110], and by a growing number of reports correlating the activation or the increased expression of many other antioxidant enzymes with the dysplastic/neoplastic phenotype of cell lines or histological lesions [111,112,113]. However, some contradictory results were also reported [114]. Based on the reported results discussed earlier, the global oxidative status of patients has been also proposed as a potential index of efficacy in tumor treatment [115], or as a predictor of response to chemotherapy [116] or radiotherapy [117].
Indirect evidence also support the hypothesis that OS, in addition to being a hallmark of neoplastic growth, also has an active part in lesion progression. A large perspective cohort study showed that among women with a HPV-16 cervical infection, cigarette smoking, a powerful carcinogen with a wide range of different toxic activities, was associated with an increased risk of progression to CIN-III/invasive cancer [118]. Cigarette smoking, however, is also a known inducer of OS, which can be part of tobacco’s carcinogenic potential. This possible aspect is suggested by a parallel case-control study by the same group, reporting an inverse correlation of α–tocopherol plasma level with the risk of developing CIN and a protective role connected with the plasma levels of reduced ascorbic acid [119]. At the molecular level, benzo[a]pyrene (BaP), a major cigarette smoke carcinogen found in cervical mucus of women who smoke, has been shown to have a direct activity on HPV viral replication. Namely BaP, through a specific activation of the MAPK/ERK axis, activates CDK1, thus enhancing the titer of HPV16, HPV18 and HPV 31b once grown in organotypic “raft” culture [120]. These results propose a connection linking a non-mutagenic effect of BaP with the progression of HPV lesions, lending indirect support to the hypothesis that cigarette smoking may have a role as an OS inducer independent from its carcinogenic effect. Accordingly, data showed that antioxidant treatment (Vitamin C) of HeLa cells, through the modulation of the AP-1 composition, down-regulated the E6 expression and restored the p53-mediated apoptotic response to cisplatin and etoposide [121]. This suggests the possibility that antioxidant treatments could promote or restore tumor responsiveness to chemotherapeutic agents.
Complex data regarding advanced cancer and OS appear largely dispersed, somewhat recalling a patchwork of poorly related observations rather than an organic and consistent picture. Nonetheless, a direct correlation between an increasingly marked OS signature and the progression from mild dysplasia to severe dysplasia to invasive cancer is generally suggested by the majority of reports. Recent data, however, suggest a more articulated scenario indicating that, although an increasingly oxidant micro-environment is associated with tumor progression, tumor cells are characterized by a rather efficient control on RONS generation and oxidative damage attained by the appropriate up-regulation of antioxidant enzymes and detoxifying/pro-survival proteins [93,122]. This oxidative fitted modulation of cell metabolism turns the refractory, highly oxidant tumor environment into a positive selection factor for adapted cancer cells.
5. Conclusions
Theoretical consideration suggests OS and HR-HPV, two powerful initiating and promoting carcinogens, can act synergistically. Indirect clinical epidemiological evidence and initial biochemical data support the conclusion that viral infection, establishment of persistent-chronicle infection, and viral integration are indeed potentiated by OS. Both pro-survival and pro-apoptotic mechanisms have been described for OS; however, in viral dysplastic lesions, OS actually behaves as a pro-survival factor promoting the AP-1 mediated expression of E6 and E7, specifically reinforcing their anti-apoptotic mechanisms. A number of antioxidant enzymes and detoxifying pathways are consistently associated with HPV transformed cells that seem to be well equipped to fit highly oxidant environments. A number of questions, however, remain unanswered: No experimental evidence is available about the effect OS has on the regulation and functions of the E4, E5, L1 and L2 genes, as well as their proteins. Conversely, no data are available on the effect that E2, E4, E5 L1 and L2 viral proteins have on the OS generation, scavenging and repair. Apart from data on VEGF-mediated neo-angiogenesis, no conclusive data are available on the mechanisms involved in tissue invasion and remodeling, immune escape, cell shedding and metastasis homing. Only preliminary, non-systematic results are available on the role of OS in chemotherapy response/resistance and only initial attempts have been made to exploit OS signature as a potential marker for diagnosis and for clinical outcome of lesions. A large amount of work remains to be done.
Acknowledgments
I would like to point out that the literature reporting the data related to this field of research is vast. I would like to further apologize to those whose work could not be included in this first quick review. I wish to thank Mrs Tania Merlino for editing the English of this manuscript. This work was partly supported by the contribution of the Ministry of Foreign Affairs, DGSP (Direzione Generale per la Promozione del Sistema Paese).
Conflict of Interest
The author declares no conflict of interest.
References and Notes
- zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef]
- zur Hausen, H. Immortalization of human cells and their malignant conversion by high risk human papillomavirus genotypes. Semin. Cancer Biol. 1999, 9, 405–411. [Google Scholar] [CrossRef]
- Federico, A.; Morgillo, F.; Tuccillo, C.; Ciardiello, F.; Loguercio, C. Chronic inflammation and oxidative stress in human carcinogenesis. Int. J. Cancer. 2007, 121, 2381–2386. [Google Scholar] [CrossRef]
- Harrison, D.G.; Widder, J.; Grumbach, I.; Chen, W.; Weber, M.; Searles, C. Endothelial mechano-transduction, nitric oxide and vascular inflammation. J. Intern. Med. 2006, 259, 351–363. [Google Scholar] [CrossRef]
- Darley-Usmar, V.; Halliwell, B. Blood radicals: Reactive nitrogen species, reactive oxygen species, transition metal ions, and the vascular system. Pharm. Res. 1996, 13, 649–662. [Google Scholar] [CrossRef]
- Zangar, R.C.; Davydov, D.R.; Verma, S. Mechanisms that regulate production of reactive oxygen species by cytochrome P450. Toxicol. Appl. Pharmacol. 2004, 199, 316–331. [Google Scholar] [CrossRef]
- Darr, D.; Fridovich, I. Free radicals in cutaneous biology. J. Invest. Dermatol. 1994, 102, 671–675. [Google Scholar]
- Jurkiewicz, B.A.; Buettner, G.R. Ultraviolet light-induced free radical formation in skin: An electron paramagnetic resonance study. Photochem. Photobiol. 1994, 59, 1–4. [Google Scholar] [CrossRef]
- Riley, P.A. Free radicals in biology: Oxidative stress and the effects of ionizing radiation. Int. J. Radia.t Biol. 1994, 65, 27–33. [Google Scholar] [CrossRef]
- Breimer, L.H. Repair of DNA damage induced by reactive oxygen species. Free Radic. Res. Commun. 1991, 14, 159–171. [Google Scholar] [CrossRef]
- Ananthaswamy, H.N.; Pierceall, W.E. Molecular mechanisms of ultraviolet radiation carcinogenesis. Photochem. Photobiol. 1990, 52, 1119–1136. [Google Scholar] [CrossRef]
- Danno, K.; Horio, T.; Takigawa, M.; Imamura, S. Role of oxygen intermediates in UV-induced epidermal cell injury. J. Invest. Dermatol. 1984, 83, 166–168. [Google Scholar] [CrossRef]
- Miyachi, Y. Photoaging from an oxidative standpoint. J. Dermatol. Sci. 1995, 9, 79–86. [Google Scholar] [CrossRef]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol Rev. 2002, 82, 47–95. [Google Scholar]
- Hensley, K.; Robinson, K.A.; Gabbita, S.P.; Salsman, S.; Floyd, R.A. Reactive oxygen species, cell signaling, and cell injury. Free Radic. Biol. Med. 2000, 28, 1456–6142. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L1005–1028. [Google Scholar]
- Lenaz, G.; Genova, M.L. Supramolecular organisation of the mitochondrial respiratory chain: A new challenge for the mechanism and control of oxidative phosphorylation. Adv. Exp. Med. Biol. 2012, 748, 107–144. [Google Scholar] [CrossRef]
- Dröse, S.; Brandt, U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv. Exp. Med. Biol. 2012, 748, 145–169. [Google Scholar] [CrossRef]
- Di Mascio, P.; Briviba, K.; Bechara, E.J.; Medeiros, M.H.; Sies, H. Reaction of peroxynitrite and hydrogen peroxide to produce singlet molecular oxygen (1deltag). Methods Enzymol. 1996, 269, 395–400. [Google Scholar] [CrossRef]
- van Asbeck, B.S. Oxygen toxicity: Role of hydrogen peroxide and iron. Adv. Exp. Med. Biol. 1990, 264, 235–246. [Google Scholar] [CrossRef]
- Bartsch, H.; Nair, J. Oxidative stress and lipid peroxidation-derived DNA-lesions in inflammation driven carcinogenesis. Cancer Detect. Prev. 2004, 28, 385–391. [Google Scholar] [CrossRef]
- Moriya, M.; Pandya, G.A.; Johnson, F.; Grollman, A.P. Cellular response to exocyclic DNA adducts. IARC Sci. Publ. 1999, 150, 263–270. [Google Scholar]
- Martínez-Ruiz, A.; Cadenas, S.; Lamas, S. Nitric oxide signaling: classical, less classical, and nonclassical mechanisms. Free Radic. Biol. Med. 2011, 51, 17–29. [Google Scholar] [CrossRef]
- Ascenzi, P.; di Masi, A.; Sciorati, C.; Clementi, E. Peroxynitrite-An ugly biofactor? Biofactors. 2010, 36, 264–273. [Google Scholar] [CrossRef]
- Dansen, T.B.; Wirtz, K.W. The peroxisome in oxidative stress. IUBMB Life 2001, 51, 223–230. [Google Scholar] [CrossRef]
- Kasai, H.; Okada, Y.; Nishimura, S.; Rao, M.S.; Reddy, J.K. Formation of 8-hydroxy deoxyguanosine in liver DNA of rats following long-term exposure to a peroxisome proliferator. Cancer Res. 1989, 49, 2603–2605. [Google Scholar]
- Oikawa, I.; Novikoff, P.M. Catalase-negative peroxisomes: Transient appearance in rat hepatocytes during liver regeneration after partial hepatectomy. Am. J. Pathol. 1995, 146, 673–687. [Google Scholar]
- Beckman, K.B.; Ames, B.N. The free radical theory of aging matures. Physiol. Rev. 1998, 78, 547–581. [Google Scholar]
- Goeptar, A.R.; Scheerens, H.; Vermulen, N.P. Oxygen and xenobiotic reductase activities of cytochrome P450. Crit. Rev. Toxicol. 1995, 25, 25–65. [Google Scholar]
- Koop, D.R. Oxidative and reductive metabolism by cytochrome P450 2E1. FASEB J. 1992, 6, 724–730. [Google Scholar]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer. 2003, 4, 276–285. [Google Scholar] [CrossRef]
- Ohshima, H.; Bartsch, H. Chronic infections and inflammatory processes as cancer risk factors: Possible role of nitric oxide in carcinogenesis. Mutat. Res. 1994, 305, 253–264. [Google Scholar] [CrossRef]
- Bylund, J.; Brown, K.L.; Movitz, C.; Dahlgren, C.; Karlsson, A. Intracellular generation of superoxide by the phagocyte NADPH oxidase: how, where, and what for? Free Radic. Biol. Med. 2010, 49, 1834–1845. [Google Scholar] [CrossRef]
- Li, C.; Jackson, R.M. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am. J. Physiol. Cell Physiol. 2002, 282, C227–241. [Google Scholar]
- Kayyali, U.; Donaldson, C.; Huang, H.; Abdelnour, R.; Hassoun, P. Phosphorylation of xanthine dehydrogenase/oxidase in hypoxia. J. Biol. Chem. 2001, 276, 14359–14365. [Google Scholar]
- Plateel, M.; Dehouck, M.P.; Torpier, G.; Cecchelli, R.; Teissier, E. Hypoxia increases the susceptibility to oxidant stress and the permeability of the blood-brain barrier endothelial cell monolayer. J. Neurochem. 1995, 65, 2138–2145. [Google Scholar]
- Al-Mehdi, A.B; Zhao, G.; Dodia, C.; Tozawa, K; Costa, K.; Muzykantov, V.; Ross, C.; Blecha, F.; Dinauer, M.; Fisher, A.B. Endothelial NADPH oxidase as the source of oxidants in lungs exposed to ischemia or high K+. Circ Res. 1998, 83, 730–737. [Google Scholar]
- Lenaz, G. Mitochondria and reactive oxygen species. Which role in physiology and pathology? Adv. Exp. Med. Biol. 2012, 942, 93–136. [Google Scholar]
- Wu, W.S. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 2006, 25, 695–705. [Google Scholar] [CrossRef]
- Greer, S.N.; Metcalf, J.L.; Wang, Y.; Ohh, M. The updated biology of hypoxia-inducible factor. EMBO J. 2012, 31, 2448–60. [Google Scholar] [CrossRef]
- Ma, Q. Transcriptional responses to oxidative stress: Pathological and toxicological implications. Pharmacol Ther. 2010, 125, 376–393. [Google Scholar] [CrossRef]
- Cadet, J.; Loft, S.; Olinski, R.; Evans, M.D.; Bialkowski, K.; Richard Wagner, J.; Dedon, P.C.; Møller, P.; Greenberg, M.M.; Cooke, M.S. Biologically relevant oxidants and terminology, classification and nomenclature of oxidatively generated damage to nucleobases and 2-deoxyribose in nucleic acids. Free Radic. Res. 2012, 46, 367–381. [Google Scholar] [CrossRef]
- Himmelstein, M.W.; Boogaard, P.J.; Cadet, J.; Farmer, P.B.; Kim, J.H.; Martin, E.A.; Persaud, R.; Shuker, D.E. Creating context for the use of DNA adduct data in cancer risk assessment: II. Overview of methods of identification and quantitation of DNA damage. Crit. Rev. Toxicol. 2009, 39, 679–694. [Google Scholar] [CrossRef]
- Pilger, A.; Rüdiger, H.W. 8-Hydroxy-2'-deoxyguanosine as a marker of oxidative DNA damage related to occupational and environmental exposures. Int. Arch. Occup. Environ. Health 2006, 80, 1–15. [Google Scholar] [CrossRef]
- Revich, G.G.; Beattie, K.L. Utilization of 1,N6-etheno-2'-deoxyadenosine 5'-triphosphate during DNA synthesis on natural templates, catalyzed by DNA polymerase I of Escherichia coli. Carcinogenesis 1986, 7, 1569–1576. [Google Scholar] [CrossRef]
- Kamiya, H.; Kasai, H. 2-Hydroxy-dATP is incorporated opposite G by Escherichia coli DNA polymerase III resulting in high mutagenicity. Nucleic Acids Res. 2000, 28, 1640–1646. [Google Scholar] [CrossRef]
- Levine, R.L.; Yang, I.Y.; Hossain, M.; Pandya, G.A.; Grollman, A.P.; Moriya, M. Mutagenesis induced by a single 1,N6-ethenodeoxyadenosine adduct in human cells. Cancer Res. 2000, 60, 4098–4104. [Google Scholar]
- Pandya, G.A.; Moriya, M. 1,N6-ethenodeoxyadenosine, a DNA adduct highly mutagenic in mammalian cells. Biochemistry 1996, 35, 11487114–11487192. [Google Scholar]
- Moriya, M.; Zhang, W.; Johnson, F.; Grollman, A.P. Mutagenic potency of exocyclic DNA adducts: marked differences between Escherichia coli and simian kidney cells. Proc. Natl. Acad. Sci. USA 1994, 91, 11899–11903. [Google Scholar]
- Esterbauer, H.; Eckl, P.; Ortner, A. Possible mutagens derived from lipids and lipid precursors. Mutat Res. 1990, 238, 223–233. [Google Scholar] [CrossRef]
- Tudek, B.; Winczura, A.; Janik, J.; Siomek, A.; Foksinski, M.; Oliński, R. Involvement of oxidatively damaged DNA and repair in cancer development and aging. Am. J. Transl. Res. 2010, 2, 254–284. [Google Scholar]
- Stadtman, E.R.; Levine, R.L. Protein oxidation. Ann. NY Acad. Sci. 2000, 899, 191–208. [Google Scholar] [CrossRef]
- Butterfield, D.A. Oxidative stress in neurodegenerative disorders. Antioxid. Redox. Signal. 2006, 8, 1971–1973. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Keller, J.N. Antioxidants and antioxidant treatment in disease. Biochem. Biophys. Acta. 2012, 1822, 615. [Google Scholar] [CrossRef]
- England, K.; Cotter, T.G. Direct oxidative modifications of signaling proteins in mammalian cells and their effects on apoptosis. Redox Rep. 2005, 10, 237–245. [Google Scholar] [CrossRef]
- De Marco, F.; Bucaj, E.; Foppoli, C.; Fiorini, A.; Blarzino, C.; Filipi, K.; Giorgi, A.; Schininà, M.E.; di Domenico, F.; Coccia, R.; Butterfield, D.A.; Perluigi, M. Oxidative stress in HPV-Driven viral carcinogenesis: Redox proteomics analysis of HPV-16 dysplastic and neoplastic tissues. PLoS One. 2012, 7, e34366. [Google Scholar]
- Morgan, M.J.; Liu, Z.G. Reactive oxygen species in TNFalpha-induced signaling and cell death. Mol. Cells. 2010, 30, 1–12. [Google Scholar] [CrossRef]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes Cells. 2011, 16, 123–140. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Flohé, L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid Redox Signal. 2011, 15, 2335–2381. [Google Scholar] [CrossRef]
- Flohe, L.; Brigelius-Flohe, R.; Saliou, C.; Traber, M.G.; Packer, L. Redox regulation of NF-kappa B activation. Free Radic. Biol. Med. 1997, 22, 1115–1126. [Google Scholar] [CrossRef]
- Aslund, F.; Zheng, M.; Beckwith, J.; Storz, G. Regulation of the OxyR transcription factor by hydrogen peroxide and the cellular thiol-disulfide status. Proc. Natl. Acad. Sci. USA 1999, 96, 6161–6165. [Google Scholar] [CrossRef]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef]
- Fernandez, C.; Sharrard, R.M.; Talbot, M.; Reed, B.D.; Monks, N. Evaluation of the significance of polyamines and their oxidases in the aetiology of human cervical carcinoma. Br. J. Cancer 1995, 72, 1194–1199. [Google Scholar] [CrossRef]
- Siegel, E.M.; Patel, N.; Lu, B.; Lee, J.H.; Nyitray, A.G.; Huang, X.; Villa, L.L.; Franco, E.L.; Giuliano, A.R. Circulating biomarkers of iron storage and clearance of incident human papillomavirus infection. Cancer Epidemiol. Biomarkers Prev. 2012, 21, 859–865. [Google Scholar] [CrossRef]
- Safaeian, M.; Hildesheim, A.; Gonzalez, P.; Yu, K.; Porras, C.; Li, Q.; Rodriguez, A.C.; Sherman, M.E.; Schiffman, M.; Wacholder, S.; et al. Single nucleotide polymorphisms in the PRDX3 and RPS19 and risk of HPV persistence and cervical precancer/cancer. PLoS One 2012, 7, e33619. [Google Scholar]
- Conway, M.J.; Alam, S.; Ryndock, E.J.; Cruz, L.; Christensen, N.D.; Roden, R.B.; Meyers, C. Tissue-spanning redox gradient-dependent assembly of native human papillomavirus type 16 virions. J Virol. 2009, 83, 10515–10126. [Google Scholar] [CrossRef]
- Schmitt, J.; Schlehofer, J.R.; Mergener, K.; Gissmann, L.; zur Hausen, H. Amplification of bovine papillomavirus DNA by N-methyl-N'-nitro-N-nitrosoguanidine, ultraviolet irradiation, or infection with herpes simplex virus. Virology 1989, 172, 73–81. [Google Scholar]
- Boner, W.; Taylor, E.R.; Tsirimonaki, E.; Yamane, K.; Campo, M.S.; Morgan, I.M. A functional interaction between the HPV16 E2 transcription/replication factor E2 and the DNA damage response protein TopBP1. J. Biol. Chem. 2002, 277, 22297–22303. [Google Scholar]
- Achanta, G.; Huang, P. Role of p53 in sensing oxidative DNA damage in response to reactive oxygen species-generating agents. Cancer Res. 2004, 64, 6233–6239. [Google Scholar]
- Giampieri, S.; Storey, A. Repair of UV-induced thymine dimers is compromised in cells expressing the E6 protein from human papillomaviruses types 5 and 18. Br. J. Cancer. 2004, 90, 2203–2209. [Google Scholar]
- Moody, C.A.; Laimins, L.A. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009, 5, e1000605. [Google Scholar] [CrossRef]
- Spardy, N.; Covella, K.; Cha, E.; Hoskins, E.E.; Wells, S.I.; Duensing, A.; Duensing, S. Human papillomavirus 16 E7 oncoprotein attenuates DNA damage checkpoint control by increasing the proteolytic turnover of claspin. Cancer Res. 2009, 69, 7022–7029. [Google Scholar] [CrossRef]
- King, L.E.; Fisk, J.C.; Dornan, E.S.; Donaldson, M.M.; Melendy, T.; Morgan, I.M. Human papillomavirus E1 and E2 mediated DNA replication is not arrested by DNA damage signalling. Virology 2010, 406, 95–102. [Google Scholar] [CrossRef]
- Mincheva, A.; Gissmann, L.; zur Hausen, H. Chromosomal integration sites of human papillomavirus DNA in three cervical cancer cell lines mapped by in situ hybridization. Med. Microbiol. Immunol. 1987, 176, 245–256. [Google Scholar]
- Heller, A. Apoptosis-inducing high •NO concentrations are not sustained either in nascent or in developed cancers. Chem. Med. Chem. 2008, 3, 1493–1499. [Google Scholar]
- Goldstein, S.; Merényi, G. The chemistry of peroxynitrite: implications for biological activity. Methods Enzymol. 2008, 436, 49–61. [Google Scholar] [CrossRef]
- Dulak, J.; Józkowicz, A. Regulation of vascular endothelial growth factor synthesis by nitric oxide: facts and controversies. Antioxid Redox. Signal. 2003, 5, 123–132. [Google Scholar] [CrossRef]
- Morbidelli, L.; Donnini, S.; Ziche, M. Role of nitric oxide in tumor angiogenesis. Cancer Treat. Res. 2004, 117, 155–167. [Google Scholar] [CrossRef]
- Mazibrada, J.; Rittà, M.; Mondini, M.; De Andrea, M.; Azzimonti, B.; Borgogna, C.; Ciotti, M.; Orlando, A.; Surico, N.; Chiusa, L.; et al. Interaction between inflammation and angiogenesis during different stages of cervical carcinogenesis. Gynecol. Oncol. 2008, 108, 112–120. [Google Scholar] [CrossRef]
- De Marco, F.; Bucaj, E.; Foppoli, C.; Fiorini, A.; Blarzino, C.; Filipi, K.; Giorgi, A.; Schininà, M.E.; Di Domenico, F.; Coccia, R.; et al. Oxidative stress in HPV-driven viral carcinogenesis: redox proteomics analysis of HPV-16 dysplastic and neoplastic tissues. PLoS One 2012, 7, e34366. [Google Scholar]
- Vodovot, Y.; Bogdan, C.; Paik, J.; Xie, Q.W.; Nathan, C. Mechanisms of suppression of macrophage nitric oxide release by transforming growth factor beta. J. Exp. Med. 1993, 178, 605–613. [Google Scholar] [CrossRef]
- Mitani, T.; Terashima, M.; Yoshimura, H.; Nariai, Y.; Tanigawa, Y. TGF-β1 enhances degradation of IFN-γ-induced iNOS protein via proteasomes in RAW 264.7 cells. Nitric. Oxide 2005, 13, 78–87. [Google Scholar] [CrossRef]
- Baritaki, S.; Sifakis, S.; Huerta-Yepez, S.; Neonakis, I.K.; Soufla, G.; Bonavida, B.; Spandidos, D.A. Overexpression of VEGF and TGF-beta1 mRNA in Pap smears correlates with progression of cervical intraepithelial neoplasia to cancer: Implication of YY1 in cervical tumorigenesis and HPV infection. Int. J. Oncol. 2007, 31, 69–79. [Google Scholar]
- Thomas, D.D.; Liu, X.; Kantrow, S.P.; Lancaster, J.R., Jr. The biological lifetime of nitric oxide: implications for the perivascular dynamics of NO and O2. Proc. Natl. Acad. Sci. USA 2001, 8, 355–360. [Google Scholar]
- López-Ocejo, O.; Viloria-Petit, A.; Bequet-Romero, M.; Mukhopadhyay, D.; Rak, J.; Kerbel, R.S. Oncogenes and tumor angiogenesis: The HPV-16 E6 oncoprotein activates the vascular endothelial growth factor (VEGF) gene promoter in a p53 independent manner. Oncogene 2000, 19, 4611–4620. [Google Scholar]
- Butz, K.; Shahabeddin, L.; Geise, C.; Spitkovsky, D.; Ullmann, A.; Hoppe-Seyler, F. Functional p53 protein in human papillomavirus-positive cancer cells. Oncogene 1995, 10, 927–933. [Google Scholar]
- Butz, K.; Whitaker, N.; Denk, C.; Ullmann, A.; Geisen, C.; Hoppe-Seyler, F. Induction of the p53-target gene GADD45 in HPV-positive cancer cells. Oncogene 1999, 18, 2381–2386. [Google Scholar]
- Ding, B.; Chi, S.G.; Kim, S.H.; Kang, S.; Cho, J.H.; Kim, D.S.; Cho, N.H. Role of p53 in antioxidant defence of HPV-positive cervical carcinoma cells following H2O2 exposure. J. Cell. Sci. 2007, 120, 2284–2294. [Google Scholar] [CrossRef]
- De Marco, F.; Marcante, M.L. HPV-16 E6-E7 differential transcription induced in Siha cervical cancer cell line by interferons. J. Biol. Regul. Homeost. Agents 1993, 7, 15–21. [Google Scholar]
- Abdulkarim, B.; Sabri, S.; Deutsch, E.; Chagraoui, H.; Maggiorella, L.; Thierry, J.; Eschwege, F.; Vainchenker, W.; Chouaïb, S.; Bourhis, J. Antiviral agent Cidofovir restores p53 function and enhances the radio-sensitivity in HPV-associated cancers. Oncogene 2002, 21, 2334–2346. [Google Scholar] [CrossRef]
- Divya, C.S.; Pillai, M.R. Antitumor action of curcumin in human papillomavirus associated cells involves downregulation of viral oncogenes, prevention of NFkB and AP-1 translocation, and modulation of apoptosis. Mol. Carcinog. 2006, 45, 320–332. [Google Scholar] [CrossRef]
- Munagala, R.; Kausar, H.; Munjal, C.; Gupta, R.C. Withaferin A induces p53-dependent apoptosis by repression of HPV oncogenes and upregulation of tumor suppressor proteins in human cervical cancer cells. Carcinogenesis 2011, 32, 1697–1705. [Google Scholar] [CrossRef]
- De Marco, F.; Perluigi, M.; Foppoli, C.; Blarzino, C.; Cini, C.; Coccia, R.; Venuti, A. UVB irradiation down-regulates HPV-16 RNA expression: Implications for malignant progression of transformed cells. Virus Res. 2007, 130, 249–259. [Google Scholar] [CrossRef]
- Mouret, S.; Sauvaigo, S.; Peinnequin, A.; Favier, A.; Beani, J.C.; Leccia, M.T. E6* oncoprotein expression of human papillomavirus type-16 determines different ultraviolet sensitivity related to glutathione and glutathione peroxidase antioxidant defence. Exp. Dermatol. 2005, 14, 401–410. [Google Scholar] [CrossRef]
- Shim, J.H.; Kim, K.H.; Cho, Y.S.; Choi, H.S.; Song, E.Y.; Myung, P.K.; Kang, J.S.; Suh, S.K.; Park, S.N.; Yoon, D.Y. Protective effect of oxidative stress in HaCaT keratinocytes expressing E7 oncogene. Amino Acids. 2008, 34, 135–141. [Google Scholar]
- Mileo, A.M.; Abbruzzese, C.; Mattarocci, S.; Bellacchio, E.; Pisano, P.; Federico, A.; Maresca, V.; Picardo, M.; Giorgi, A.; Maras, B.; Schininà, M.E.; Paggi, M.G. Human papillomavirus-16 E7 interacts with glutathione S-transferase P1 and enhances its role in cell survival. PLoS One 2009, 13, e7254. [Google Scholar]
- Graham, S.V. Human papillomavirus: Gene expression, regulation and prospects for novel diagnostic methods and antiviral therapies. Future Microbiol. 2010, 5, 1493–1506. [Google Scholar] [CrossRef]
- Rösl, F.; Das, B.C.; Lengert, M.; Geletneky, K.; zur Hausen, H. Antioxidant-induced changes of the AP-1 transcription complex are paralleled by a selective suppression of human papillomavirus transcription. J. Virol. 1997, 71, 362–370. [Google Scholar]
- Soto, U.; Denk, C.; Finzer, P.; Hutter, K.J.; zur Hausen, H.; Rösl, F. Genetic complementation to non-tumorigenicity in cervical-carcinoma cells correlates with alterations in AP-1 composition. Int. J. Cancer. 2000, 86, 811–817. [Google Scholar] [CrossRef]
- Soto, U.; Das, B.C.; Lengert, M.; Finzer, P.; zur Hausen, H.; Rösl, F. Conversion of HPV 18 positive non-tumorigenic HeLa-fibroblast hybrids to invasive growth involves loss of TNF-alpha mediated repression of viral transcription and modification of the AP-1 transcription complex. Oncogene 1999, 18, 3187–3198. [Google Scholar] [CrossRef]
- Prusty, B.K.; Das, B.C. Constitutive activation of transcription factor AP-1 in cervical cancer and suppression of human papillomavirus (HPV) transcription and AP-1 activity in HeLa cells by curcumin. Int. J. Cancer. 2005, 113, 951–960. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar]
- Zwerschke, W.; Mazurek, S.; Massimi, P.; Banks, L.; Eigenbrodt, E.; Jansen-Dürr, P. Modulation of type M2 pyruvate kinase activity by the human papillomavirus type 16 E7 oncoprotein. Proc. Natl. Acad. Sci. USA 1999, 96, 1291–1296. [Google Scholar]
- Mazurek, S.; Zwerschke, W.; Jansen-Dürr, P.; Eigenbrodt, E. Effects of the human papilloma virus HPV-16 E7 oncoprotein on glycolysis and glutaminolysis: Role of pyruvate kinase type M2 and the glycolytic-enzyme complex. Biochem. J. 2001, 356, 247–256. [Google Scholar] [CrossRef]
- Reshkin, S.J.; Bellizzi, A.; Caldeira, S.; Albarani, V.; Malanchi, I.; Poignee, M.; Alunni-Fabbroni, M.; Casavola, V.; Tommasino, M. Na+/H+ exchanger-dependent intracellular alkalinization is an early event in malignant transformation and plays an essential role in the development of subsequent transformation-associated phenotypes. FASEB J. 2000, 14, 2185–2197. [Google Scholar] [CrossRef]
- zur Hausen, H. Human papilloma viruses and their possible role in squamous cell carcinomas. Curr. Top. Microbiol. Immunol. 1977, 78, 1–30. [Google Scholar] [CrossRef]
- Slater, T.F.; Bajardi, F.; Benedetto, C.; Bussolati, G.; Cianfano, S.; Dianzani, M.U.; Ghiringhello, B.; Nöhammer, G.; Rojanapo, W.; Schauenstein, E. Protein thiols in normal and neoplastic human uterine cervix. FEBS Lett. 1985, 187, 267–271. [Google Scholar]
- Kwasniewska, A.; Gozdzicka-Józefiak, A.; Borzecki, A.; Baranowski, W. DNA adducts in squamous cell cervical carcinomas associated with HPV infection. Eur. J. Gynaec. Oncol. 2003, 3, 359–361. [Google Scholar]
- Sgambato, A.; Zannoni, G.F.; Faraglia, B.; Camerini, A.; Tarquini, E.; Spada, D.; Cittadini, A. Decreased expression of the CDK inhibitor p27Kip1 and increased oxidative DNA damage in the multistep process of cervical carcinogenesis. Gynecol. Oncol. 2004, 92, 776–783. [Google Scholar] [CrossRef]
- Hedley, D.; Pintilie, M.; Woo, J.; Nicklee, T.; Morrison, A.; Birle, D.; Fyles, A.; Milosevic, M.; Hill, R. Up-regulation of the redox mediators thioredoxin and apurinic/apyrimidinic excision (APE)/Ref-1 in hypoxic microregions of invasive cervical carcinomas, mapped using multispectral, wide-field fluorescence image analysis. Am. J. Pathol. 2004, 164, 557–565. [Google Scholar] [CrossRef]
- Del Nonno, F.; Pisani, G.; Visca, P.; Signore, F.; Grillo, L.R.; Baiocchini, A.; Garbuglia, A.R.; Sepe, S.; Piacentini, M.; Falasca, L. Role and predictive strength of transglutaminase type 2 expression in premalignant lesions of the cervix. Mod. Pathol. 2011, 24, 855–865. [Google Scholar] [CrossRef]
- Li, L.Q.; Chen, C.L.; Cao, Z.Y.; Liao, Q.P.; Du, H.J.; Zhan, S.B.; Zhou, L.; Zeng, Y. Expression of peroxiredoxin III in cervical lesions. ZhonghuaShi Yan He Lin Chuang Bing Du Xue Za Zhi 2009, 23, 443–445. [Google Scholar]
- Perluigi, M.; Giorgi, A.; Blarzino, C.; De Marco, F.; Foppoli, C.; Di Domenico, F.; Butterfield, D.A.; Schininà, M.E.; Cini, C.; Coccia, R. Proteomics analysis of protein expression and specific protein oxidation in human papillomavirus transformed keratinocytes upon UVB irradiation. J. Cell Mol. Med. 2009, 13, 1809–1822. [Google Scholar] [CrossRef]
- Carrero, Y.; Callejas, D.; Alaña, F.; Silva, C.; Mindiola, R.; Mosquera, J. Increased vascular endothelial growth factor expression, CD3-positive cell infiltration, and oxidative stress in premalignant lesions of the cervix. Cancer 2009, 115, 3680–3688. [Google Scholar] [CrossRef]
- Maldonado, P.; Negrini, L.A.; Kaizer, R.R.; Zanin, R.F.; Araújo Mdo, C.; Battisti, V.; Morsch, V.M.; Schetinger, M.R. Oxidative status in patients submitted to conization and radiation treatments for uterine cervix neoplasia. Clin. Chim. Acta. 2006, 366, 174–178. [Google Scholar] [CrossRef]
- Sharma, A.; Rajappa, M.; Satyam, A.; Sharma, M. Oxidant/anti-oxidant dynamics in patients with advanced cervical cancer: correlation with treatment response. Mol. Cell. Biochem. 2010, 341, 65–72. [Google Scholar] [CrossRef]
- Weitmann, H.D.; Gustorff, B.; Vaupel, P.; Knocke, T.H.; Pötter, R. Oxygenation status of cervical carcinomas before and during spinal anesthesia for application of brachytherapy. Strahlenther Onkol. 2003, 179, 633–640. [Google Scholar] [CrossRef]
- Ho, G.Y.; Kadish, A.S.; Burk, R.D.; Basu, J.; Palan, P.R.; Mikhail, M.; Romney, S.L. HPV 16 and cigarette smoking as risk factors for high-grade cervical intra-epithelial neoplasia. Int. J. Cancer 1998, 78, 281–285. [Google Scholar]
- Ho, G.Y.; Palan, P.R.; Basu, J.; Romney, S.L.; Kadish, A.S.; Mikhail, M.; Wassertheil-Smoller, S.; Runowicz, C.; Burk, R.D. Viral characteristics of human papillomavirus infection and antioxidant levels as risk factors for cervical dysplasia. Int. J. Cancer. 1998, 78, 594–599. [Google Scholar] [CrossRef]
- Alam, S.; Conway, M.J.; Chen, H.S.; Meyers, C. The cigarette smoke carcinogen benzo[a]pyrene enhances human papillomavirus synthesis. J. Virol. 2008, 82, 1053–1058. [Google Scholar] [CrossRef]
- Reddy, V.G.; Khanna, N.; Singh, N. Vitamin C augments chemotherapeutic response of cervical carcinoma HeLa cells by stabilizing P53. Biochem. Biophys. Res. Commun. 2001, 282, 409–415. [Google Scholar] [CrossRef]
- López, J.; Poitevin, A.; Mendoza-Martínez, V.; Pérez-Plasencia, C.; García-Carrancá, A. Cancer-initiating cells derived from established cervical cell lines exhibit stem-cell markers and increased radioresistance. BMC Cancer 2012, 28, 12–48. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
De Marco, F. Oxidative Stress and HPV Carcinogenesis. Viruses 2013, 5, 708-731. https://doi.org/10.3390/v5020708
AMA Style
De Marco F. Oxidative Stress and HPV Carcinogenesis. Viruses. 2013; 5(2):708-731. https://doi.org/10.3390/v5020708
Chicago/Turabian StyleDe Marco, Federico. 2013. "Oxidative Stress and HPV Carcinogenesis" Viruses 5, no. 2: 708-731. https://doi.org/10.3390/v5020708