Deregulation of Epigenetic Mechanisms by the Hepatitis B Virus X Protein in Hepatocarcinogenesis

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The HBV Life Cycle
3. HBV and Hepatocellular Carcinoma (HCC)
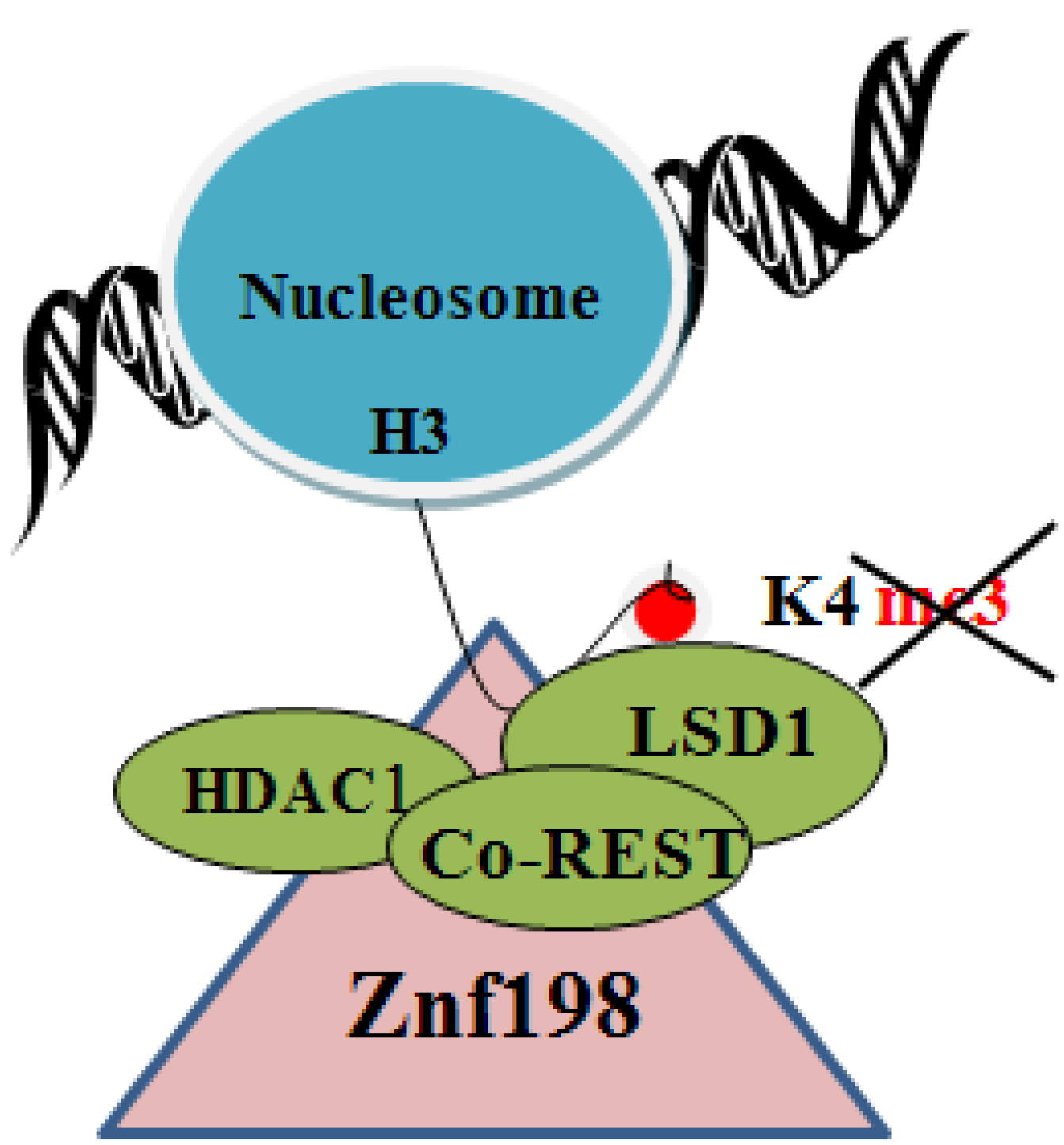
4. Chromatin Modifying Proteins Suz12 and Znf198 in the X Protein Signaling Network
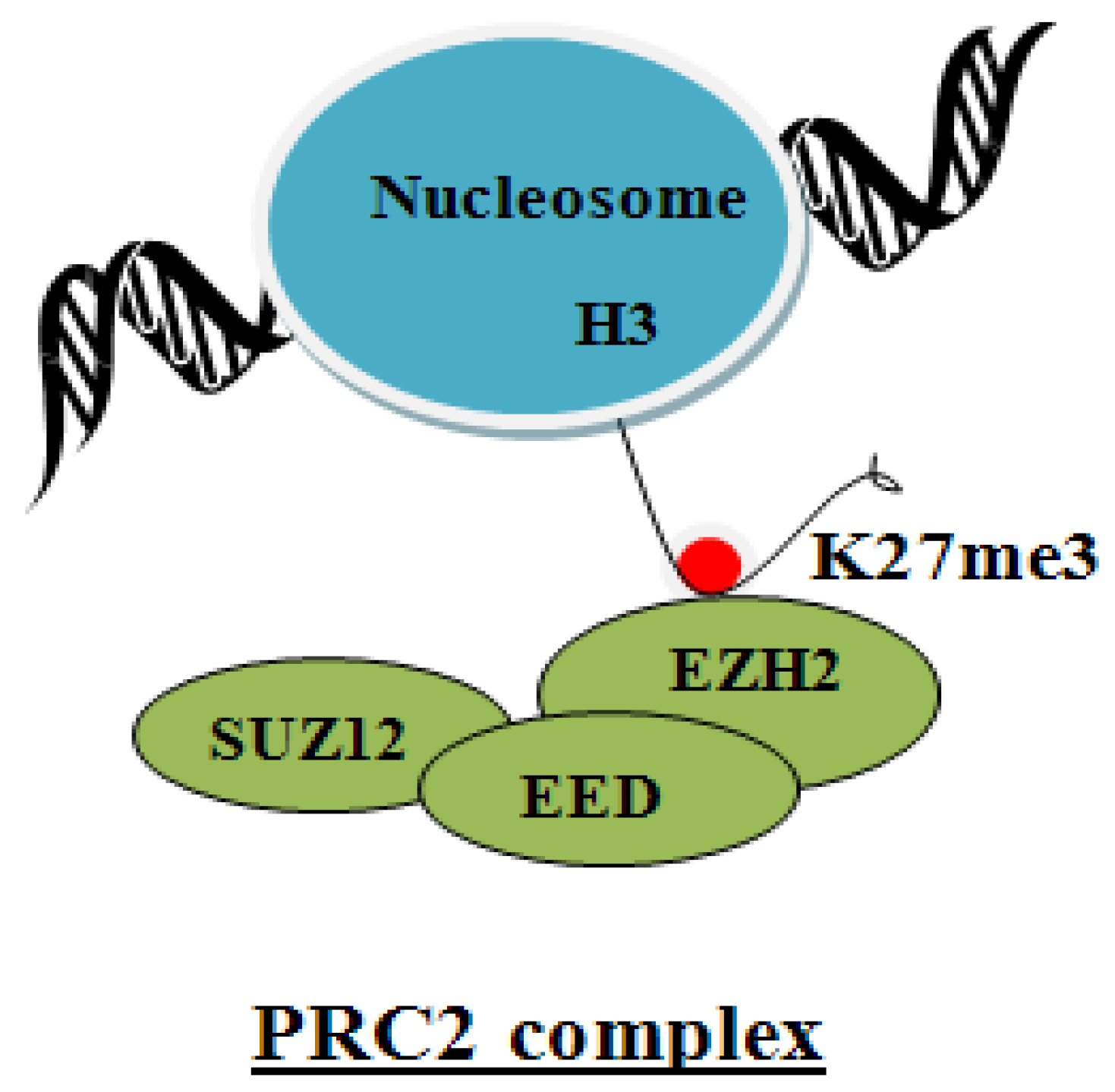
5. Suz12 Containing PRC2 Complex
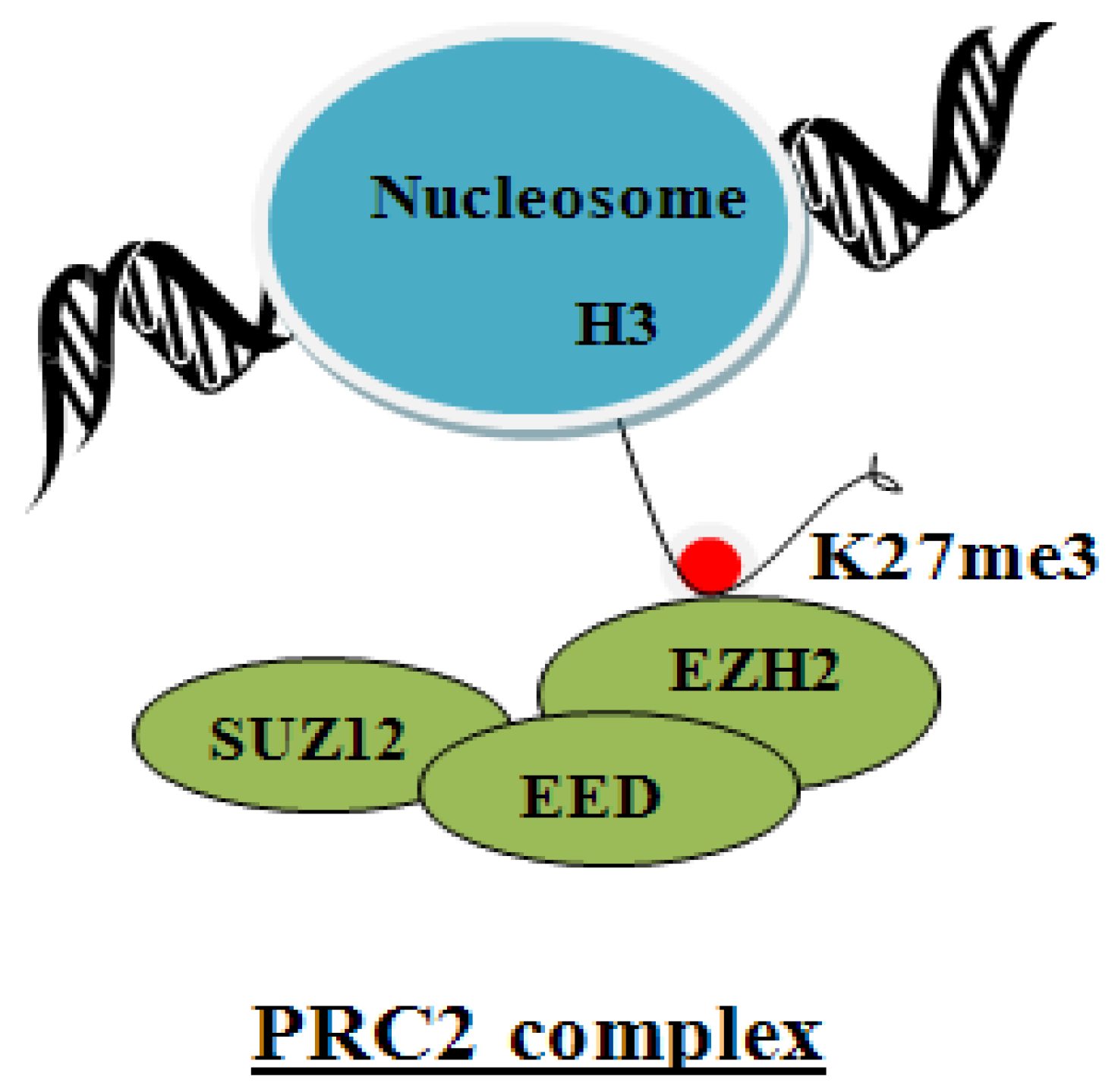
6. Regulation of the PRC2 Complex
7. Distinct Histone Modifications and DNA Methylation Landscapes in Pluripotent vs. Differentiated Cells
8. DNA Methylation and the X Protein
9. Conclusion
Acknowledgments
Conflict of Interest
References and Notes
- Beasley, R.P.; Hwang, L.Y.; Lin, C.C.; Chien, C.S. Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22,707 men in Taiwan. Lancet 1981, 2, 1129–1133. [Google Scholar]
- Bruix, J.; Boix, L.; Sala, M.; Llovet, J.M. Focus on hepatocellular carcinoma. Cancer Cell. 2004, 5, 215–219. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef]
- Parkin, D.M. International variation. Oncogene 2004, 23, 6329–6340. [Google Scholar] [CrossRef]
- Thomas, M.B.; Jaffe, D.; Choti, M.M.; Belghiti, J.; Curley, S.; Fong, Y.; Gores, G.; Kerlan, R.; Merle, P.; O'Neil, B.; et al. Hepatocellular carcinoma: Consensus recommendations of the National Cancer Institute Clinical Trials Planning Meeting. J. Clin. Onocol. 2010, 28, 3994–4005. [Google Scholar] [CrossRef]
- Hoshida, Y.; Nijman, S.M.; Kobayashi, M.; Chan, J.A.; Brunet, J.P.; Chiang, D.Y.; Villanueva, A.; Newell, P.; Ikeda, K.; Hashimoto, M.; et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009, 69, 7385–7392. [Google Scholar] [CrossRef]
- Boyault, S.; Rickman, D.S.; de Reyniès, A.; Balabaud, C.; Rebouissou, S.; Jeannot, E.; Hérault, A.; Saric, J.; Belghiti, J.; Franco, D.; et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 2007, 45, 42–52. [Google Scholar] [CrossRef] [Green Version]
- Seeger, C.; Mason, W.S. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 2000, 64, 51–68. [Google Scholar] [CrossRef]
- Zoulim, F.; Saputelli, J.; Seeger, C. Woodchuck hepatitis virus X protein is required for viral infection in vivo. J. Virol. 1994, 68, 2026–2030. [Google Scholar]
- Lucifora, J.; Arzberger, S.; Durantel, D.; Belloni, L.; Strubin, M.; Levrero, M.; Zoulim, F.; Hantz, O.; Protzer, U. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J. Hepatol. 2011, 55, 996–1003. [Google Scholar] [CrossRef]
- Rabe, B.; Vlachou, A.; Panté, N.; Helenius, A.; Kann, M. Nuclear import of hepatitis B virus capsids and release of the viral genome. Proc. Natl. Acad. Sci. USA 2003, 100, 9849–9854. [Google Scholar] [CrossRef]
- Weiser, B.; Ganem, D.; Seeger, C.; Varmus, H.E. Closed circular viral DNA and asymmetrical heterogeneous forms in livers of animals infected with ground squirrel hepatitis virus. J. Virol. 1983, 48, 1–9. [Google Scholar]
- Summers, J.; Mason, W.S. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 1982, 29, 403–415. [Google Scholar] [CrossRef]
- Beck, J.; Nassal, M. Hepatitis B virus replication. World J. Gastroenterol. 2007, 13, 48–64. [Google Scholar]
- Bock, C.T.; Schranz, P.; Schröder, C.H.; Zentgraf, H. Hepatitis B virus genome is organized into nucleosomes in the nucleus of the infected cell. Virus Genes 1994, 8, 215–229. [Google Scholar] [CrossRef]
- Pollicino, T.; Belloni, L.; Raffa, G.; Pediconi, N.; Squadrito, G.; Raimondo, G.; Levrero, M. Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound H3 and H4 histones. Gastroenterology 2006, 130, 827–837. [Google Scholar]
- Levrero, M.; Pollicino, T.; Petersen, J.; Belloni, L.; Raimondo, G.; Dandri, M. Control of cccDNA function in hepatitis B virus infection. J. Hepatol. 2009, 51, 581–592. [Google Scholar] [CrossRef]
- Belloni, L.; Pollicino, T.; de Nicola, F., Guerrieri; Raffa, G.; Fanciulli, M.; Raimondo, G.; Levrero, M. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. USA 2009, 106, 19975–19979. [Google Scholar]
- Hagen, T.M.; Huang, S.; Curnutte, J.; Fowler, P.; Martinez, V.; Wehr, C.M.; Ames, B.N.; Chisari, F.V. Extensive oxidative DNA damage in hepatocytes of transgenic mice with chronic active hepatitis destined to develop hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 1994, 91, 12808–12812. [Google Scholar] [CrossRef]
- Terradillos, O.; Billet, O.; Renard, C.A.; Levy, R.; Molina, T.; Briand, P.; Buendia, M.A. The hepatitis B virus X gene potentiates c-myc-induced liver oncogenesis in transgenic mice. Oncogene 1997, 14, 395–404. [Google Scholar]
- Madden, C.R.; Finegold, M.J.; Slagle, B.L. Hepatitis B virus X protein acts as a tumor promoter in development of diethylnitrosamine-induced preneoplastic lesions. J. Virol. 2001, 75, 3851–3858. [Google Scholar] [CrossRef]
- Yin, J.; Xie, J.; Zhang, H.; Shen, Q.; Han, L.; Lu, W.; Han, Y.; Li, C.; Ni, W.; Wang, H.; et al. Significant association of different preS mutations with hepatitis B-related cirrhosis or hepatocellular carcinoma. J. Gastroenterol. 2010, 45, 1063–1071. [Google Scholar] [CrossRef]
- Du, J.; Liang, X.; Liu, Y.; Qu, Z.; Gao, L.; Han, L.; Liu, S.; Cui, M.; Shi, Y.; Zhang, Z.; et al. Hepatitis B virus core protein inhibits TRAIL-induced apoptosis of hepatocytes by blocking DR5 expression. Cell Death Differ. 2009, 16, 219–229. [Google Scholar] [CrossRef]
- Su, Q.; Schröder, C.H.; Hofmann, W.J.; Otto, G.; Pichlmayr, R.; Bannasch, P. Expression of Hepatitis B virus X protein in HBV-infected human livers and hepatocellular carcinomas. Hepatology 1998, 27, 1109–1120. [Google Scholar] [CrossRef]
- Buendia, M.A. Genetics of hepatocellular carcinoma. Semin. Cancer Biol. 2000, 10, 185–200. [Google Scholar] [CrossRef]
- Andrisani, O.M.; Barnabas, S. The transcriptional function of the hepatitis B virus X protein and its role in hepatocarcinogenesis. Int. J. Oncol. 1999, 15, 373–379. [Google Scholar]
- Bouchard, M.J.; Schneider, R.J. The enigmatic X gene of hepatitis B virus. J. Virol. 2004, 78, 12725–12734. [Google Scholar] [CrossRef]
- Ng, S.A.; Lee, C. Hepatitis B virus X gene and hepatocarcinogenesis. J. Gastroenterol. 2011, 46, 974–990. [Google Scholar] [CrossRef]
- Rawat, S.; Clippinger, A.J.; Bouchard, M.J. Modulation of apoptotic signaling by the hepatisis B virus X protein. Viruses 2012, 4, 2945–2972. [Google Scholar] [CrossRef]
- Rakotomalala, L.; Studach, L.; Wang, W.H.; Gregori, G.; Hullinger, R.L.; Andrisani, O. Hepatitis B virus X protein increases the Cdt-1-to-geminin ratio inducing DNA re-replication and polyploidy. J. Biol. Chem. 2008, 283, 28729–28740. [Google Scholar]
- Studach, L.L.; Rakotomalala, L.; Wang, W.H.; Hullinger, R.L.; Cairo, S.; Buendia, M.A.; Andrisani, O.M. Polo-like kinase 1 inhibition suppresses hepatitis B virus X protein-induced transformation in an in vitro model of liver cancer progression. Hepatology 2009, 50, 414–423. [Google Scholar] [CrossRef]
- Studach, L.; Wang, W.H.; Weber, G.; Tang, J.; Hullinger, R.L.; Malbrue, R.; Liu, X.; Andrisani, O. Polo-like kinase 1 activated by the hepatitis B virus X protein attenuates both the DNA damage checkpoint and DNA repair resulting in partial polyploidy. J. Biol. Chem. 2010, 285, 30282–30293. [Google Scholar]
- Golsteyn, R.M.; Schultz, S.J.; Bartek, J.; Ziemiecki, A.; Ried, T.; Nigg, E.A. Cell cycle analysis and chromosomal localization of human Plk1, a putative homologue of the mitotic kinases Drosophila polo and Saccharomyces cerevisiae Cdc5. J. Cell. Sci. 1994, 107, 1509–1517. [Google Scholar]
- Mamely, I.; van Vugt, M.A.; Smits, V.A.; Semple, J.I.; Lemmens, B.; Perrakis, A.; Medema, R.H.; Freire, R. Polo-like kinase-1 controls proteasome-dependent degradation of Claspin during checkpoint recovery. Curr. Biol. 2006, 16, 1950–1955. [Google Scholar] [CrossRef]
- Kumagai, A.; Dunphy, W.G. Purification and molecular cloning of Plx1, a Cdc25-regulatory kinase from Xenopus egg extracts. Science 1996, 273, 1377–1380. [Google Scholar]
- Pelegrino, R.; Calvisi, D.F.; Ladu, S.; Ehemann, V.; Staniscia, T.; Evert, M.; Dombrowski, F.; Schirmacher, P.; Longerich, T. Oncogenic and tumor suppressive roles of Polo-like kinases in human hepatocellular carcinoma. Hepatology 2010, 51, 857–868. [Google Scholar]
- Petrelli, A.; Perra, A.; Schernhuber , K.; Cargnelutti, M.; Salvi, A.; Migliore, C.; Ghiso, E.; Benetti, A.; Barlati, S.; Ledda-Columbano, G.M.; et al. Squential analysis of multistage hepatocarcinogenesis reveals that miR-100 and PLK1 dysregulation is an early event maintained along tumor progression. Oncogene 2012, 31, 4517–4526. [Google Scholar] [CrossRef]
- Chen, X.; Cheung, S.T.; So, S.; Fan, S.T.; Barry, C.; Higgins, J.; Lai, K.M.; Ji, J.; Dudoit, S.; Ng, I.O.; et al. Gene expression patterns in human liver cancers. Mol. Biol. Cell 2002, 13, 1929–1939. [Google Scholar] [CrossRef] [Green Version]
- Bruinsma, W.; Raaijmakers, J.A.; Medema, R.H. Switching Polo-like kinase-1 on and off in time and space. Trends Biochem. Sci. 2012, 37, 534–542. [Google Scholar] [CrossRef]
- Wang, W.H.; Studach, L.L.; Andrisani, O.M. Proteins ZNF198 and SUZ12 are down-regulated in hepatitis B virus (HBV) X protein-mediated hepatocyte transformation and in HBV replication. Hepatology 2011, 53, 1137–1147. [Google Scholar] [CrossRef]
- Xiao, S.; Nalabolu, S.; Aster, J.C.; Ma, J.; Abruzzo, L.; Jaffe, E.S.; Stone, R.; Weissman, S.M.; Hudson, T.J.; Fletcher, J.A. FGFR1 is fused with novel zinc-finger gene, ZNF198, in the t(8;13) leukaemia/lymphoma syndrome. Nat. Genet. 1998, 18, 84–87. [Google Scholar]
- Li, H.; Ma, X.; Wang, J.; Koontz, J.; Nucci, M.; Sklar, J. Effects of rearrangement and allelic exclusion of JJAZ1/SUZ12 on cell proliferation and survival. Proc. Natl. Acad. Sci. USA 2007, 104, 20001–20006. [Google Scholar]
- Moinzadeh, P.; Breuhahn, K.; Stützer, H.; Schirmacher, P. Chromosome alterations in human hepatocellular carcinomas correlate with aetiology and histological grade-results of an explorative CGH meta-analysis. Br. J. Cancer 2005, 92, 935–941. [Google Scholar] [CrossRef]
- Chen, C.F.; Yeh, S.H.; Chen, D.S.; Chen, P.J.; Jou, Y.S. Molecular genetic evidence supporting a novel human hepatocellular carcinoma tumor suppressor locus at 13q12.11. Genes Chromosom. Cancer 2005, 44, 320–328. [Google Scholar] [CrossRef]
- Villa, R.; Pasini, D.; Gutierrez, A.; Morey, L.; Occhionorelli, M.; Viré, E.; Nomdedeu, J.F.; Jenuwein, T.; Pelicci, P.J.; Minucci, S.; et al. Role of the polycomb repressive complex 2 in acute promyelocytic leukemia. Cancer Cell 2007, 11, 513–525. [Google Scholar] [CrossRef]
- Kunapuli, P.; Kasyapa, C.S.; Chin, S.F.; Caldas, C.; Cowell, J.K. ZNF198, a zinc finger protein rearranged in myeloproliferative disease, localizes to the PML nuclear bodies and interacts with SUMO-1 and PML. Exp. Cell Res. 2006, 312, 3739–3751. [Google Scholar] [CrossRef]
- Bernardi, R.; Pandolfi, P.P. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 2007, 8, 1006–1016. [Google Scholar] [CrossRef]
- Torok, D.; Ching, R.W.; Bazett-Jones, D.P. PML nuclear bodies as sites of epigenetic regulation. Front. Biosci. 2009, 14, 1325–1336. [Google Scholar]
- Cho, S.; Park, J.S.; Kang, Y.K. Dual functions of histone-lysine N-methyltransferase Setdb1 protein at promyelocytic leukemia-nuclear body (PML-NB): Maintaining PML-NB structure and regulating the expression of its associated genes. J. Biol. Chem. 2011, 286, 41115–41124. [Google Scholar]
- Bernardi, R.; Papa, A.; Pandolfi, P.P. Regulation of apoptosis by PML and the PML-NBs. Oncogene 2008, 27, 6299–6312. [Google Scholar] [CrossRef]
- Dellaire, G.; Ching, R.W.; Ahmed, K.; Jalali, F.; Tse, K.C.; Bristow, R.G.; Bazett-Jones, D.P. Promyelocytic leukemia nuclear bodies behave as DNA damage sensors whose response to DNA double-strand breaks is regulated by NBS1 and the kinases ATM, Chk2, and ATR. J. Cell. Biol. 2006, 175, 55–66. [Google Scholar] [CrossRef]
- Everett, R.D.; Chelbi-Alix, M.K. PML and PML nuclear bodies: Implications in antiviral defence. Biochimie 2007, 89, 819–830. [Google Scholar] [CrossRef]
- Frappier, L. Viral disruption of promyelocytic leukemia (PML) nuclear bodies by hijacking host PML regulators. Virulence 2001, 2, 58–62. [Google Scholar] [CrossRef]
- Reineke, E.L.; Kao, H.Y. Targeting promyelocytic leukemia protein: A means to regulating PML nuclear bodies. Int. J. Biol. Sci. 2009, 5, 366–376. [Google Scholar] [CrossRef]
- Gu, H.; Roizman, B. The two functions of herpes simplex virus 1 ICP0, inhibition of silencing by the CoREST/REST/HDAC complex and degradation of PML are executed in tandem. J. Virol. 2009, 83, 181–187. [Google Scholar] [CrossRef]
- Gu, H.; Roizman, B. Engagement of the lysine-specific demethylase/HDAC1/CoREST/REST complex by herpes simplex virus 1. J. Virol. 2009, 83, 4376–4385. [Google Scholar] [CrossRef]
- Chung, Y.L.; Tsai, T.Y. Promyelocytic leukemia nuclear bodies link the DNA damage repair pathway to hepatitis B virus replication: Implications for hepatitis B virus exacerbation during chemotherapy and radiotherapy. Mol. Cancer Res. 2009, 7, 1672–1685. [Google Scholar] [CrossRef]
- Gocke, C.B.; Yu, H. ZNF198 stabilizes the LSD-1-CoREST-HDAC1 complex on chromatin through its MYM-type zinc fingers. PLoS One 2008, 3, e3255. [Google Scholar] [CrossRef]
- Cao, R.; Zhang, Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol. Cell 2004, 15, 57–67. [Google Scholar] [CrossRef]
- Squazzo, S.L.; O'Geen, H.; Komashko, V.M.; Krig, S.R.; Jin, V.X.; Jang, S.W.; Margueron, R.; Reinberg, D.; Green, R.; Farnham, P.J. Suz12 binds to silenced regions of the genome in a cell-type-specific manner. Genome Res. 2006, 16, 890–900. [Google Scholar] [CrossRef]
- Simon, J.A.; Kingston, R.E. Mechanisms of polycomb gene silencing: Knowns and unknowns. Nat. Rev. Mol. Cell Biol. 2009, 10, 697–708. [Google Scholar]
- Bracken, A.P.; Dietrich, N.; Pasini, D.; Hansen, K.H.; Helin, K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006, 20, 1123–1136. [Google Scholar] [CrossRef]
- Lee, T.I.; Jenner, R.G.; Boyer, L.A.; Guenther, M.G.; Levine, S.S.; Kumar, R.M.; Chevalier, B.; Johnstone, S.E.; Cole, M.F.; Isono, K.; et al. Control of developmentalregulators by Polycomb in human embryonic stem cells. Cell 2006, 125, 301–313. [Google Scholar] [CrossRef]
- Morey, L.; Helin, K. Polycombgroupprotein-mediated repression of transcription. Trends Biochem. Sci. 2010, 35, 323–332. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. The PolycombcomplexPRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef]
- Pasini, D.; Bracken, A.P.; Hansen, J.B.; Capillo, M.; Helin, K. The PolycombgroupproteinSuz12 is required for embryonic stem cell differentiation. Mol. Cell Biol. 2007, 27, 3769–3779. [Google Scholar] [CrossRef]
- Surface, L.E.; Thornton, S.R.; Boyer, L.A. Polycomb group proteinsset the stage for early lineage commitment. Cell Stem. Cell 2010, 7, 288–298. [Google Scholar] [CrossRef]
- Aldiri, I.; Vetter, M.L. PRC2 during vertebrate organogenesis: A complex in transition. Dev. Biol. 2012, 367, 91–99. [Google Scholar] [CrossRef]
- Studach, L.L.; Menne, S.; Cairo, S.; Buendia, M.A.; Hullinger, R.L.; Lefrançois, L.; Merle, P.; Andrisani, O.M. A subset of Suz12/PRC2 target genes is activated during hepatitis B virus replication and liver carcinogenesis associated with hepatitis B virus X protein. Hepatology 2012, 56, 1240–1251. [Google Scholar] [CrossRef]
- Andrisani, O.M.; Studach, L.; Merle, P. Gene signatures in hepatocellular carcinoma (HCC). Semin. Cancer Biol. 2001, 21, 4–9. [Google Scholar] [CrossRef]
- Yamashita, T.; Ji, J.; Budhu, A.; Forgues, M.; Yang, W.; Wang, H.Y.; Jia, H.; Ye, Q.; Qin, L.X.; Wauthier, E.; et al. EpCAM-positive hepatocellular carcinoma cells are tumor initiating cells with stem/progenitor cell features. Gastroenterology 2009, 136, 1012–1024. [Google Scholar] [CrossRef]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic non-coding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–116672. [Google Scholar] [CrossRef]
- Zhao, J.; Ohsumi, T.K.; Kung, J.T.; Ogawa, Y.; Grau, D.J.; Sarma, K.; Song, J.J.; Kingston, R.E.; Borowsky, M. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol. Cell 2010, 40, 939–953. [Google Scholar] [CrossRef]
- Toffanin, S.; Hishida, Y.; Lachenmayer, A.; Villanueva, A.; Cabellos, L.; Minguez, B.; Savic, R.; Ward, S.C.; Thung, S.; Chiang, D.Y.; et al. MicroRNA-based classification of hepatocellular carcinoma and oncogenic role of miR-517a. Gastroenterology 2011, 140, 1618–1628. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Lindahl-Allen, M.; Polytarchou, C.; Hirsch, H.A.; Tsichlis, P.N.; Struhl, K. Loss of miR-200 inhibition of Suz12 leads to polycomb-mediated repression required for the formation and maintenance of cancer stem cells. Mol. Cell 2010, 39, 761–772. [Google Scholar] [CrossRef]
- Svotelis, A.; Bianco, S.; Matore, J.; Huppé, G.; Nordell-Markovits, A.; Mes-Masson, A.M.; Gévry, N. H3K27 demethylation by JMJD3 at a poised enhancer of anti-apoptotic gene BCL2 determines ERα ligand dependency. EMBO J. 2011, 30, 3947–3961. [Google Scholar] [CrossRef]
- Pasini, D.; Cloos, P.A.; Walfridsson, J.; Olsson, L.; Bukowski, J.P.; Johansen, J.V.; Bak, M.; Tommerup, N.; Rappsilber, J.; Helin, K. JARID2 regulates binding of the Polycomb repressive complex 2 to target genes in ES cells. Nature 2010, 464, 306–310. [Google Scholar]
- Anderton, J.A.; Bose, S.; Vockerodt, M.; Vrzalikova, K.; Wei, W.; Kuo, M.; Helin, K.; Christensen, J.; Rowe, M.; Murray, P.G.; et al. The H3K27me3 demethylase, KDM6B, is induced by Epstein-Barr virus and over-expressed in Hodgkin's Lymphoma. Oncogene 2011, 30, 2037–2043. [Google Scholar] [CrossRef]
- Beausoleil, S.A.; Jedrychowski, M.; Schwartz, D.; Elias, J.E.; Villén, J.; Li, J.; Cohn, M.A.; Cantley, L.C.; Gygi, S.P. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. USA 2004, 101, 12130–12135. [Google Scholar] [CrossRef]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Mekkelsen, T.S.; Zie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef]
- Brown, D.D. The role of stable complexes that repress and activate eukaryotic genes. Philos Trans. R. Soc. Lond. B Biol. Sci. 1984, 307, 297–299. [Google Scholar] [CrossRef]
- Petruk, S.; Sedkov, Y.; Johnston, D.M.; Hodgson, J.W.; Black, K.L.; Kovermann, S.K.; Beck, S.; Canaani, E.; Brock, H.W.; Mazo, A. TrxG and PcG proteins but not methylated histones remain associated with DNA through replication. Cell 2012, 150, 922–933. [Google Scholar] [CrossRef]
- Deb-Rinker, P.; Ly, D.; Jezierski, A.; Sikorska, M.; Walker, P.R. Sequential DNA methylation of the Nanog and Oct-4 upstream regions in human NT2 cells during neuronal differentiation. J. Biol. Chem. 2005, 280, 6257–6260. [Google Scholar]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008, 454, 766–770. [Google Scholar]
- Mohn, F.; Weber, M.; Rebhan, M.; Roloff, T.C.; Richter, J.; Stadler, M.B.; Bibel, M.; Schübeler, D. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell 2008, 30, 755–766. [Google Scholar] [CrossRef]
- Hawkins, R.D.; Hon, G.C.; Lee, L.K.; Ngo, Q.; Lister, R.; Pelizzola, M.; Edsall, L.E.; Kuan, S.; Luu, Y.; Klugman, S.; et al. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell. Stem Cell. 2010, 6, 479–491. [Google Scholar] [CrossRef]
- Brunner, A.L.; Johnson, D.S.; Kim, S.W.; Valouev, A.; Reddy, T.E.; Neff, N.F.; Anton, E.; Medina, C.; Nguyen, L.; Chiao, E.; et al. Distinct DNA methylation patterns characterize differentiated human embryonic stem cells and developing human fetal liver. Genome Res. 2009, 19, 1044–1056. [Google Scholar] [CrossRef]
- Park, I.Y.; Sohn, B.H.; Yu, E.; Suh, D.J.; Chung, Y.H.; Lee, J.H.; Surzycki, S.J.; Lee, Y.I. Aberrant epigenetic modifications in hepatocarcinogenesis induced by hepatitis B virus X protein. Gastroenterology 2007, 132, 1476–1494. [Google Scholar] [CrossRef]
- Zheng, D.L.; Zhang, L.; Cheng, N.; Xu, X.; Deng, Q.; Teng, X.M.; Wang, K.S.; Zhang, X.; Huang, J.; Han, Z.G. Epigeneticmodificationinduced by hepatitis B virus X protein via interaction with de novo DNA methyltransferase DNMT3A. J. Hepatol. 2009, 50, 377–387. [Google Scholar] [CrossRef]
- Jung, K.J.; Arora, P.; Pagano, J.S.; Jang, K.L. Expression of DNA methyltransferase 1 is activated by hepatitis B virus X protein via a regulatory circuit involving the p16INK4a-cyclin D1-CDK 4/6-pRb-E2F1pathway. Cancer Res. 2007, 67, 5771–5778. [Google Scholar] [CrossRef]
- Chen, T.; Li, E. Structure and function of eukaryotic DNA methyltransferases. Curr. Top. Dev. Biol. 2004, 60, 55–89. [Google Scholar] [CrossRef]
- Vivekanandan, P.; Daniel, H.D.; Kannangai, R.; Martinez-Murillo, F.; Torbenson, M. Hepatitis B virus replication induces methylation of both host and viral DNA. J. Virol. 2010, 84, 4321–4329. [Google Scholar] [CrossRef]
- Vivekanandan, P.; Thomas, D.; Torbenson, M. Hepatitis B viralDNA is methylated in liver tissues. J. Viral Hepat. 2008, 15, 103–107. [Google Scholar]
- Vivekanandan, P.; Thomas, D.; Torbenson, M. Methylation regulates hepatitis B viral protein expression. J. Infect. Dis. 2009, 199, 1286–1291. [Google Scholar] [CrossRef]
- Akalin, A.; Garrett-Bakelman, F.E.; Kormaksson, M.; Busuttil, J.; Zhang, L.; Khrebtukova, I.; Milne, T.A.; Huang, Y.; Biswas, D.; Hess, J.L.; et al. Base-pairresolutionDNA methylationsequencingreveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet. 2012, 8, e1002781. [Google Scholar]
- Quasdorff, M.; Hösel, M.; Odenthal, M.; Zedler, U.; Bohne, F.; Gripon, P.; Dienes, H.P.; Drebber, U.; Stippel, D.; Goeser, T.; et al. A concerted action of HNF4alpha and HNF1alpha links hepatitis B virus replication to hepatocyte differentiation. Cell Microbiol. 2008, 10, 1478–1490. [Google Scholar] [CrossRef]
- Polo, J.M.; Anderssen, E.; Walsh, R.M.; Schwarz, B.A.; Nefzger, C.M.; Lim, S.M.; Borkent, M.; Apostolou, E.; Alaei, S.; Cloutier, J.; et al. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell 2012, 151, 1617–1632. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Andrisani, O.M. Deregulation of Epigenetic Mechanisms by the Hepatitis B Virus X Protein in Hepatocarcinogenesis. Viruses 2013, 5, 858-872. https://doi.org/10.3390/v5030858
Andrisani OM. Deregulation of Epigenetic Mechanisms by the Hepatitis B Virus X Protein in Hepatocarcinogenesis. Viruses. 2013; 5(3):858-872. https://doi.org/10.3390/v5030858
Chicago/Turabian StyleAndrisani, Ourania M. 2013. "Deregulation of Epigenetic Mechanisms by the Hepatitis B Virus X Protein in Hepatocarcinogenesis" Viruses 5, no. 3: 858-872. https://doi.org/10.3390/v5030858