Correlation of Naturally Occurring HIV-1 Resistance to DEB025 with Capsid Amino Acid Polymorphisms

Abstract
:1. Introduction

2. Results and Discussion
2.1. HIV-1 CA Amino Acid Polymorphisms Associated with Resistance to DEB025

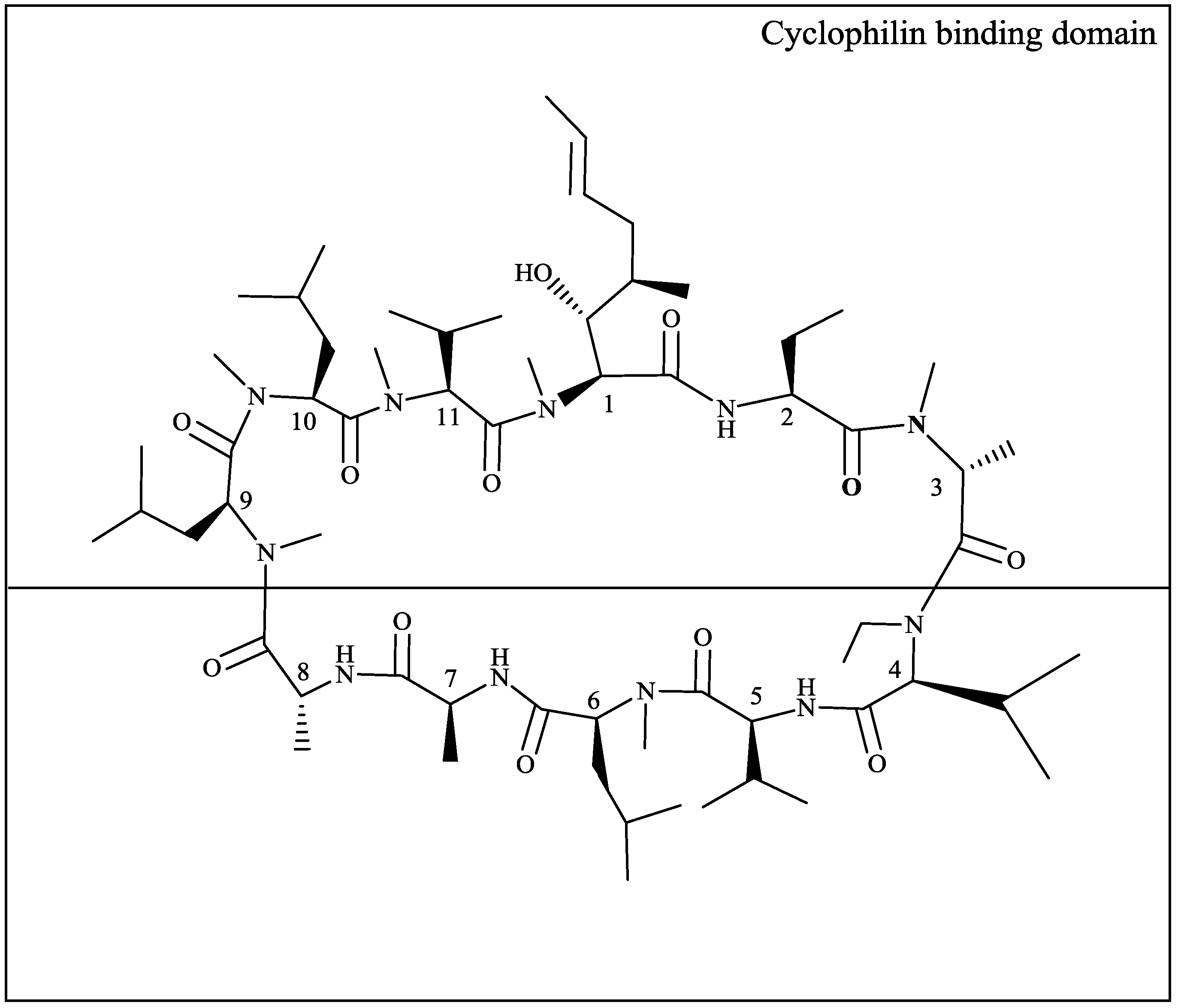{kind=link}
{kind=link}
 |
2.2 Restriction Factors, Cyclophilin A and the Loop around HIV-1 CA Amino Acid P122
2.3. Amino Acid Polymorphisms in CA of HIV-1 Isolates from the Outlier (O) Group
2.4. Amino Acid Polymorphisms in CA and In vitro Resistance to DEB025 of HIV-1 Isolates from Nonresponder Patients
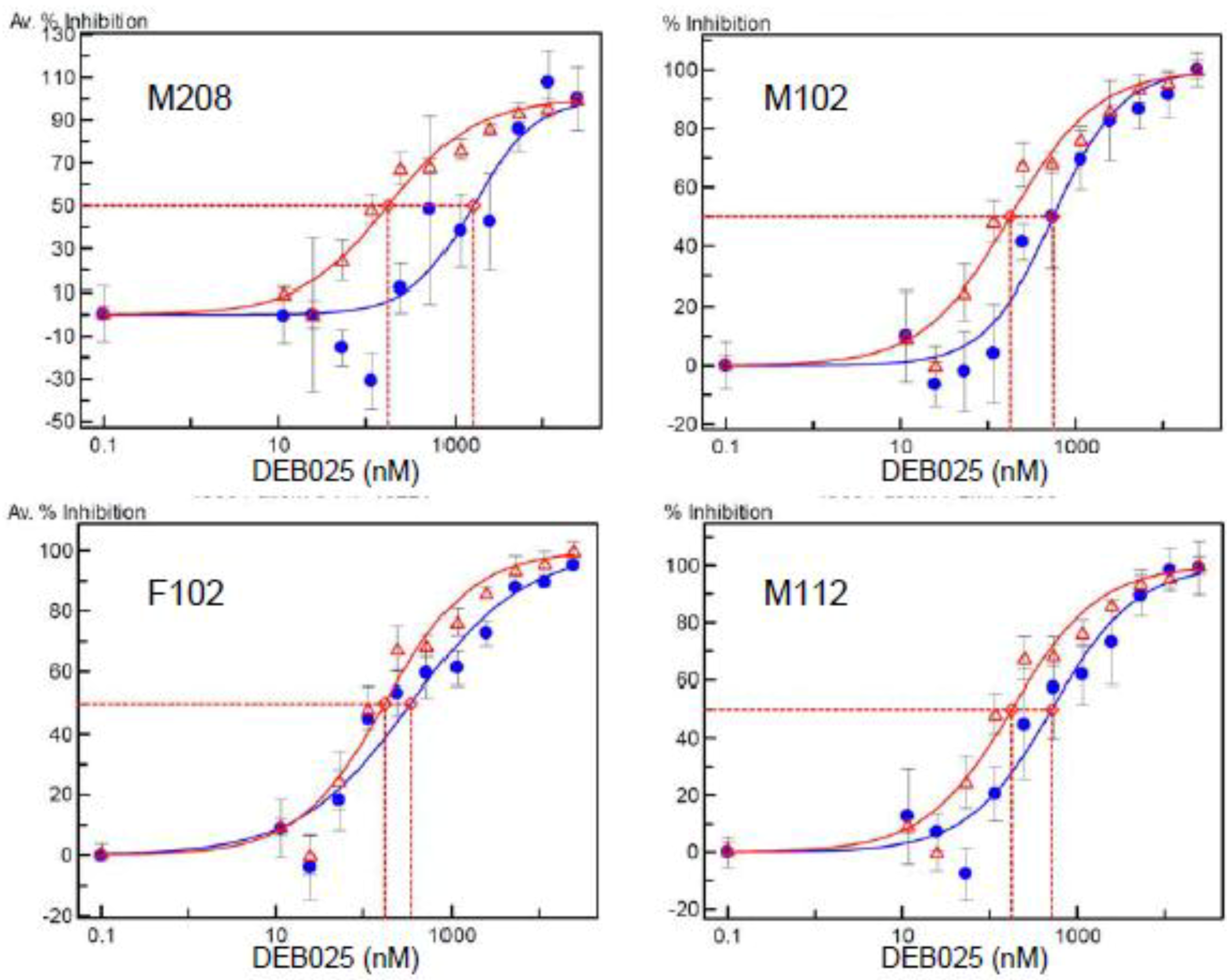
2.5. Levels of CypA Expression in PBMCs from Nonresponder Patients
2.6. Amino Acid Polymorphisms outside the CypA Binding Loop
2.7. Presumed Mode-of-Action of Cyclosporines in Inhibition of HIV-1
2.8. Presumed Mode-of-Action of Cyclosporines in Inhibition of HCV
 |
3. Experimental Section
4. Conclusions
Acknowledgements
Conflict of Interest
References
- Fischer, G.; Bang, H.; Mech, C. Determination of enzymatic catalysis for the cis-trans-isomerization of peptide binding in proline-containing peptides. Biomed. Biochim. Acta 1984, 43, 1101–1111. [Google Scholar]
- Snyder, S.H.; Sabatini, D.M. Immunophilins and the nervous system. Nat. Med. 1995, 1, 32–37. [Google Scholar] [CrossRef]
- Franke, E.K.; Yuan, H.E.; Luban, J. Specific incorporation of cyclophilin A into HIV-1 virions. Nature 1994, 372, 359–362. [Google Scholar]
- Rosenwirth, B.; Billich, A.; Steinkasserer, A.; Hammerschmid, F.; Peichl, P.; Göttlinger, H.; Traber, R.; Wenger, R. Cyclophilin A as a novel target in anti-HIV-I chemotherapy. Int. Antivir. News 1995, 3, 62–63. [Google Scholar]
- Thali, M.; Bukovsky, A.; Kondo, E.; Rosenwirth, B.; Walsh, C.T.; Sodroski, J.; Gottlinger, H.G. Functional association of cyclophilin A with HIV-1 virions. Nature 1994, 372, 363–365. [Google Scholar]
- Chatterji, U.; Bobardt, M.; Selvarajah, S.; Yang, F.; Tang, H.; Sakamoto, N.; Vuagniaux, G.; Parkinson, T.; Gallay, P. The isomerase active site of cyclophilin A is critical for hepatitis C virus replication. J. Biol. Chem. 2009, 284, 16998–17005. [Google Scholar] [CrossRef]
- Coelmont, L.; Hanoulle, X.; Chatterji, U.; Berger, C.; Snoeck, J.; Bobardt, M.; Lim, P.; Vliegen, I.; Paeshuyse, J.; Vuagniaux, G.; Vandamme, A.M.; Bartenschlager, R.; Gallay, P.; Lippens, G.; Neyts, J. DEB025 (Alisporivir) inhibits hepatitis C virus replication by preventing a cyclophilin A induced cis-trans isomerisation in domain II of NS5A. PLoS One 2010, 5, e13687. [Google Scholar] [CrossRef]
- Kaul, A.; Stauffer, S.; Berger, C.; Pertel, T.; Schmitt, J.; Kallis, S.; Lopez, M.Z.; Lohmann, V.; Luban, J.; Bartenschlager, R. Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS Pathog. 2009, 5, e1000546. [Google Scholar] [CrossRef]
- Liu, Z.; Yang, F.; Robotham, J.M.; Tang, H. Critical role of cyclophilin A and its prolyl-peptidyl isomerase activity in the structure and function of the hepatitis C virus replication complex. J. Virol. 2009, 83, 6554–6565. [Google Scholar] [CrossRef]
- Bukovsky, A.A.; Weimann, A.; Accola, M.A.; Gottlinger, H.G. Transfer of the HIV-1 cyclophilin-binding site to simian immunodeficiency virus from Macaca mulatta can confer both cyclosporin sensitivity and cyclosporin dependence. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 10943–10948. [Google Scholar] [CrossRef]
- Gamble, T.R.; Vajdos, F.F.; Yoo, S.; Worthylake, D.K.; Houseweart, M.; Sundquist, W.I.; Hill, C.P. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 1996, 87, 1285–1294. [Google Scholar] [CrossRef]
- Luban, J.; Bossolt, K.L.; Franke, E.K.; Kalpana, G.V.; Goff, S.P. Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell 1993, 73, 1067–1078. [Google Scholar] [CrossRef]
- Billich, A.; Hammerschmid, F.; Peichl, P.; Wenger, R.; Zenke, G.; Quesniaux, V.; Rosenwirth, B. Mode of action of SDZ NIM 811, a nonimmunosuppressive cyclosporin A analog with activity against human immunodeficiency virus (HIV) type 1: interference with HIV protein-cyclophilin A interactions. J. Virol. 1995, 69, 2451–2461. [Google Scholar]
- Braaten, D.; Franke, E.K.; Luban, J. Cyclophilin A is required for an early step in the life cycle of human immunodeficiency virus type 1 before the initiation of reverse transcription. J. Virol. 1996, 70, 3551–3560. [Google Scholar]
- Mlynar, E.; Bevec, D.; Billich, A.; Rosenwirth, B.; Steinkasserer, A. The non-immunosuppressive cyclosporin A analogue SDZ NIM 811 inhibits cyclophilin A incorporation into virions and virus replication in human immunodeficiency virus type 1-infected primary and growth-arrested T cells. J. Gen. Virol. 1997, 78, 825–835. [Google Scholar]
- Watashi, K.; Shimotohno, K. Cyclophilin and viruses: cyclophilin as a cofactor for viral infection and possible anti-viral target. Drug. Target Insights 2007, 2, 9–18. [Google Scholar]
- Yang, F.; Robotham, J.M.; Nelson, H.B.; Irsigler, A.; Kenworthy, R.; Tang, H. Cyclophilin A is an essential cofactor for Hepatitis C virus infection and the principal mediator of cyclosporine A resistance in vitro. J. Virol. 2008, 82, 5269–5278. [Google Scholar] [CrossRef]
- Dreyfuss, M.; Harri, H.; Hoffmann, H.; Kobel, H.; Pache, W.; Tscherter, H. Cyclosporin A and C. New Metabolites from Trichoderma ploysporum. Eur. J. Appl. Microbiol. 1976, 3, 125–133. [Google Scholar] [CrossRef]
- Handschumacher, R.E.; Harding, M.W.; Rice, J.; Drugge, R.J.; Speicher, D.W. Cyclophilin: a specific cytosolic binding protein for cyclosporin A. Science 1984, 226, 544–547. [Google Scholar]
- Ruegger, A.; Kuhn, M.; Lichti, H.; Loosli, H.R.; Huguenin, R.; Quiquerez, C.; von Wartburg, A. Cyclosporin A, a Peptide Metabolite from Trichoderma polysporum (Link ex Pers.) Rifai, with a remarkable immunosuppressive activity. Helv. Chim. Acta 1976, 59, 1075–1092. [Google Scholar] [CrossRef]
- Borel, J.F.; Di Padova, F.; Mason, J.; Quesniaux, V.; Ryffel, B.; Wenger, R. Pharmacology of cyclosporine (Sandimmune). Pharmacol. Rev. 1989, 41, 239–242. [Google Scholar]
- Baumann, G.; Zenke, G.; Wenger, R.; Hiestand, P.; Quesniaux, V.; Andersen, E.; Schreier, M.H. Molecular mechanisms of immunosuppression. J. Autoimmun. 1992, 5, 67–72. [Google Scholar] [CrossRef]
- Wenger, R.M.; Payne, T.G.; Schreier, M.H. Cyclosporine: Chemistry Structure-Activity Relationships and Mode of Action; Berger, W.R., Flueckiger, H.G., Koeppe, K.T., Holmes, C.E., Mountford, E.L., Nickoloff, T.G., Payne, M.H., Schreier, I.C.P., Smith, R., Wenger, M., Eds.; Progress in Clinical Biochemistry and Medicine, Vol. 3. Vii+191p; Springer-Verlag New York, Inc.: New York, N.Y., USA; Berlin, West Germany, Illus. Isbn 0-387-16249-6; Isbn 3-540-16249-6; 1986; pp. 157–191. [Google Scholar]
- Huai, Q.; Kim, H.Y.; Liu, Y.; Zhao, Y.; Mondragon, A.; Liu, J.O.; Ke, H. Crystal structure of calcineurin-cyclophilin-cyclosporin shows common but distinct recognition of immunophilin-drug complexes. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 12037–12042. [Google Scholar]
- Jin, L.; Harrison, S.C. Crystal structure of human calcineurin complexed with cyclosporin A and human cyclophilin. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 13522–13526. [Google Scholar] [CrossRef]
- Quesniaux, V.F.; Schreier, M.H.; Wenger, R.M.; Hiestand, P.C.; Harding, M.W.; Van Regenmortel, M.H. Cyclophilin binds to the region of cyclosporine involved in its immunosuppressive activity. Eur. J. Immunol. 1987, 17, 1359–1365. [Google Scholar] [CrossRef]
- Weber, C.; Wider, G.; von Freyberg, B.; Traber, R.; Braun, W.; Widmer, H.; Wuthrich, K. The NMR structure of cyclosporin A bound to cyclophilin in aqueous solution. Biochemistry 1991, 30, 6563–6574. [Google Scholar]
- Rosenwirth, B.; Billich, A.; Datema, R.; Donatsch, P.; Hammerschmid, F.; Harrison, R.; Hiestand, P.; Jaksche, H.; Mayer, P.; Peichl, P.; Quesniaux, V.; Schatz, F.; Schuurman, H.-J.; Traber, R.; Wenger, R.; Wolff, B.; Zenke, G.; Zurini, M. Inhibition of human immunodeficiency virus type 1 replication by SDZ NIM 811, a nonimmunosuppressive cyclosporine analog. Antimicrobial. Agents Chemother. 1994, 38, 1763–1772. [Google Scholar] [CrossRef]
- Bartz, S.R.; Hohenwalter, E.; Hu, M.K.; Rich, D.H.; Malkovsky, M. Inhibition of human immunodeficiency virus replication by nonimmunosuppressive analogs of cyclosporin A. Proc. Natl. Acad. Sci. U.S.A. 1995, 92((12)), 5381–5385. [Google Scholar]
- Ishii, N.; Watashi, K.; Hishiki, T.; Goto, K.; Inoue, D.; Hijikata, M.; Wakita, T.; Kato, N.; Shimotohno, K. Diverse effects of cyclosporine on hepatitis C virus strain replication. J. Virol. 2006, 80, 4510–4520. [Google Scholar] [CrossRef]
- Ma, S.; Boerner, J.E.; TiongYip, C.; Weidmann, B.; Ryder, N.S.; Cooreman, M.P.; Lin, K. NIM811, a cyclophilin inhibitor, exhibits potent in vitro activity against hepatitis C virus alone or in combination with alpha interferon. Antimicrob. Agents Chemother. 2006, 50, 2976–2982. [Google Scholar] [CrossRef]
- Nakagawa, M.; Sakamoto, N.; Tanabe, Y.; Koyama, T.; Itsui, Y.; Takeda, Y.; Chen, C.H.; Kakinuma, S.; Oooka, S.; Maekawa, S.; Enomoto, N.; Watanabe, M. Suppression of hepatitis C virus replication by cyclosporin A is mediated by blockade of cyclophilins. Gastroenterology 2005, 129, 1031–1041. [Google Scholar] [CrossRef]
- Traber, R.; Kobel, H.; Loosli, H.R.; Senn, H.; Rosenwirth, B.; Lawen, A. [Melle4] cyclosporin, a novel natural cylosporin with anti-HIV activity. Antivir. Chem. Chemother. 1994, 5, 331–339. [Google Scholar]
- Nakagawa, M.; Sakamoto, N.; Enomoto, N.; Tanabe, Y.; Kanazawa, N.; Koyama, T.; Kurosaki, M.; Maekawa, S.; Yamashiro, T.; Chen, C.H.; Itsui, Y.; Kakinuma, S.; Watanabe, M. Specific inhibition of hepatitis C virus replication by cyclosporin A. Biochem. Biophys. Res. Commun 2004, 313, 42–47. [Google Scholar] [CrossRef]
- Lawitz, E.; Godofsky, E.; Rouzier, R.; Marbury, T.; Nguyen, T.; Ke, J.; Huang, M.; Praestgaard, J.; Serra, D.; Evans, T.G. Safety, pharmacokinetics, and antiviral activity of the cyclophilin inhibitor NIM811 alone or in combination with pegylated interferon in HCV-infected patients receiving 14 days of therapy. Antiviral. Res. 2011, 89, 238–245. [Google Scholar] [CrossRef]
- Hubler, F.; Ruckle, T.; Patiny, L.; Muamba, T.; Guichou, J.F.; Mutter, M.; Wenger, R. Synthetic routes to NEtXaa4-cyclosporin A derivates as potential anti-HIV drugs. Tetrahedron Letters 2000, 41, 7193–7196. [Google Scholar] [CrossRef]
- Wenger, R.M. Cyclosporine and analogues--isolation and synthesis--mechanism of action and structural requirements for pharmacological activity. Fortschr. Chem. Org. Naturst. 1986, 50, 123–136. [Google Scholar] [CrossRef]
- Wenger, R.; Mutter, M.; Ruckle, T. Novel cyclosporin with improved activity profile. WO 00/01715, 2000. [Google Scholar]
- Coelmont, L.; Kaptein, S.; Paeshuyse, J.; Vliegen, I.; Dumont, J.M.; Vuagniaux, G.; Neyts, J. Debio 025, a cyclophilin binding molecule, is highly efficient in clearing hepatitis C virus (HCV) replicon-containing cells when used alone or in combination with specifically targeted antiviral therapy for HCV (STAT-C) inhibitors. Antimicrob. Agents Chemother. 2009, 53, 967–976. [Google Scholar] [CrossRef]
- Paeshuyse, J.; Kaul, A.; De Clercq, E.; Rosenwirth, B.; Dumont, J.M.; Scalfaro, P.; Bartenschlager, R.; Neyts, J. The non-immunosuppressive cyclosporin DEBIO-025 is a potent inhibitor of hepatitis C virus replication in vitro. Hepatology 2006, 43, 761–770. [Google Scholar] [CrossRef]
- Ptak, R.G.; Gallay, P.A.; Jochmans, D.; Halestrap, A.P.; Ruegg, U.T.; Pallansch, L.A.; Bobardt, M.D.; de Bethune, M.P.; Neyts, J.; De Clercq, E.; Dumont, J.M.; Scalfaro, P.; Besseghir, K.; Wenger, R.M.; Rosenwirth, B. Inhibition of Human Immunodeficiency Virus Type 1 Replication in Human Cells by Debio-025, a Novel Cyclophilin Binding Agent. Antimicrob. Agents Chemother. 2008, 52, 1302–1317. [Google Scholar] [CrossRef]
- Landrieu, I.; Hanoulle, X.; Bonachera, F.; Hamel, A.; Sibille, N.; Yin, Y.; Wieruszeski, J.M.; Horvath, D.; Wei, Q.; Vuagniaux, G.; Lippens, G. Structural basis for the non-immunosuppressive character of the cyclosporin A analogue Debio 025. Biochemistry 2010, 49, 4679–4986. [Google Scholar] [CrossRef]
- Chatterji, U.; Bobardt, M.D.; Stanfield, R.; Ptak, R.G.; Pallansch, L.A.; Ward, P.A.; Jones, M.J.; Stoddart, C.A.; Scalfaro, P.; Dumont, J.M.; Besseghir, K.; Rosenwirth, B.; Gallay, P.A. Naturally occurring capsid substitutions render HIV-1 cyclophilin A independent in human cells and TRIM-cyclophilin-resistant in Owl monkey cells. J. Biol. Chem. 2005, 280, 40293–40300. [Google Scholar] [CrossRef]
- Daelemans, D.; Dumont, J.M.; Rosenwirth, B.; De Clercq, E.; Pannecouque, C. Debio-025 inhibits HIV-1 by interfering with an early event in the replication cycle. Antiviral. Res. 2010, 85, 418–421. [Google Scholar] [CrossRef]
- Flisiak, R.; Dumont, J.M.; Crabbe, R. Cyclophilin inhibitors in hepatitis C viral infection. Expert Opin. Investig. Drugs 2007, 16, 1345–1354. [Google Scholar] [CrossRef]
- Gallay, P.A. Cyclophilin inhibitors. Clin. Liver. Dis. 2009, 13, 403–417. [Google Scholar] [CrossRef]
- Flisiak, R.; Horban, A.; Gallay, P.; Bobardt, M.; Selvarajah, S.; Wiercinska-Drapalo, A.; Siwak, E.; Cielniak, I.; Higersberger, J.; Kierkus, J.; Aeschlimann, C.; Grosgurin, P.; Nicolas-Métral, V.; Dumont, J.-M.; Porchet, H.; Crabbé, R.; Scalfaro, P. The cyclophilin inhibitor Debio-025 shows potent anti-hepatitis C effect in patients coinfected with hepatitis C and human immunodeficiency virus. Hepatology 2008, 47, 817–826. [Google Scholar] [CrossRef]
- Crabbe, R.; Vuagniaux, G.; Dumont, J.M.; Nicolas-Metral, V.; Marfurt, J.; Novaroli, L. An evaluation of the cyclophilin inhibitor Debio 025 and its potential as a treatment for chronic hepatitis C. Expert. Opin. Investig. Drugs 2009, 18, 211–220. [Google Scholar] [CrossRef]
- Flisiak, R.; Feinman, S.V.; Jablkowski, M.; Horban, A.; Kryczka, W.; Pawlowska, M.; Heathcote, J.E.; Mazzella, G.; Vandelli, C.; Nicolas-Metral, V.; Grosgurin, P.; Liz, J.S.; Scalfaro, P.; Porchet, H.; Crabbe, R. The cyclophilin inhibitor Debio 025 combined with PEG IFNalpha2a significantly reduces viral load in treatment-naive hepatitis C patients. Hepatology 2009, 49, 1460–1468. [Google Scholar] [CrossRef]
- Chatterji, U.; Lim, P.; Bobardt, M.D.; Wieland, S.; Cordek, D.G.; Vuagniaux, G.; Chisari, F.; Cameron, C.E.; Targett-Adams, P.; Parkinson, T.; Gallay, P.A. HCV resistance to cyclosporin A does not correlate with a resistance of the NS5A-cyclophilin A interaction to cyclophilin inhibitors. J. Hepatol. 2010, 53, 50–56. [Google Scholar] [CrossRef]
- Chatterji, U.; Bobardt, M.; Selvarajah, S.; Yang, F.; Tang, H.; Sakamoto, N.; Vuagniaux, G.; Parkinson, T.; Gallay, P. The isomerase active site of cyclophilin a is critical for HCV replication. J. Biol. Chem. 2009, 284, 16998–17005. [Google Scholar] [CrossRef]
- Aberham, C.; Weber, S.; Phares, W. Spontaneous mutations in the human immunodeficiency virus type 1 gag gene that affect viral replication in the presence of cyclosporins. J. Virol. 1996, 70, 3536–3544. [Google Scholar]
- Braaten, D.; Aberham, C.; Franke, E.K.; Yin, L.; Phares, W.; Luban, J. Cyclosporine A-resistant human immunodeficiency virus type 1 mutants demonstrate that Gag encodes the functional target of cyclophilin A. J. Virol. 1996, 70, 5170–5176. [Google Scholar]
- Yin, L.; Braaten, D.; Luban, J. Human immunodeficiency virus type 1 replication is modulated by host cyclophilin A expression levels. J. Virol. 1998, 72, 6430–6436. [Google Scholar]
- Berthoux, L.; Sebastian, S.; Sokolskaja, E.; Luban, J. Cyclophilin A is required for TRIM5{alpha}-mediated resistance to HIV-1 in Old World monkey cells. Proc. Natl. Acad. Sci. U.S.A 2005, 102, 14849–14853. [Google Scholar] [CrossRef]
- Luban, J. Cyclophilin A, TRIM5, and resistance to human immunodeficiency virus type 1 infection. J. Virol. 2007, 81, 1054–1061. [Google Scholar] [CrossRef]
- Owens, C.M.; Song, B.; Perron, M.J.; Yang, P.C.; Stremlau, M.; Sodroski, J. Binding and susceptibility to postentry restriction factors in monkey cells are specified by distinct regions of the human immunodeficiency virus type 1 capsid. J. Virol. 2004, 78, 5423–5437. [Google Scholar] [CrossRef]
- Sayah, D.M.; Sokolskaja, E.; Berthoux, L.; Luban, J. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 2004, 430, 569–573. [Google Scholar]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 2004, 427, 848–853. [Google Scholar]
- Chatterji, U.; Bobardt, M.D.; Gaskill, P.; Sheeter, D.; Fox, H.; Gallay, P.A. Trim5alpha accelerates degradation of cytosolic capsid associated with productive HIV-1 entry. J. Biol. Chem. 2006, 281, 37025–37033. [Google Scholar]
- Hatziioannou, T.; Perez-Caballero, D.; Cowan, S.; Bieniasz, P.D. Cyclophilin interactions with incoming human immunodeficiency virus type 1 capsids with opposing effects on infectivity in human cells. J. Virol. 2005, 79, 176–183. [Google Scholar] [CrossRef]
- Ikeda, Y.; Ylinen, L.M.; Kahar-Bador, M.; Towers, G.J. Influence of gag on human immunodeficiency virus type 1 species-specific tropism. J. Virol. 2004, 78, 11816–11822. [Google Scholar]
- Sayah, D.M.; Luban, J. Selection for loss of Ref1 activity in human cells releases human immunodeficiency virus type 1 from cyclophilin A dependence during infection. J. Virol. 2004, 78, 12066–12070. [Google Scholar] [CrossRef]
- Sokolskaja, E.; Berthoux, L.; Luban, J. Cyclophilin A and TRIM5alpha independently regulate human immunodeficiency virus type 1 infectivity in human cells. J. Virol. 2006, 80, 2855–2862. [Google Scholar] [CrossRef]
- Stremlau, M.; Song, B.; Javanbakht, H.; Perron, M.; Sodroski, J. Cyclophilin A: an auxiliary but not necessary cofactor for TRIM5alpha restriction of HIV-1. Virology 2006, 351, 112–120. [Google Scholar] [CrossRef]
- Hatziioannou, T.; Cowan, S.; Goff, S.P.; Bieniasz, P.D.; Towers, G.J. Restriction of multiple divergent retroviruses by Lv1 and Ref1. EMBO J. 2003, 22, 385–394. [Google Scholar] [CrossRef]
- Towers, G.J.; Hatziioannou, T.; Cowan, S.; Goff, S.P.; Luban, J.; Bieniasz, P.D. Cyclophilin A modulates the sensitivity of HIV-1 to host restriction factors. Nat. Med. 2003, 9, 1138–1143. [Google Scholar]
- Gitti, R.K.; Lee, B.M.; Walker, J.; Summers, M.F.; Yoo, S.; Sundquist, W.I. Structure of the amino-terminal core domain of the HIV-1 capsid protein. Science 1996, 273, 231–235. [Google Scholar]
- Wiegers, K.; Krausslich, H.G. Differential dependence of the infectivity of HIV-1 group O isolates on the cellular protein cyclophilin A. Virology 2002, 294, 289–295. [Google Scholar] [CrossRef]
- Braaten, D.; Franke, E.K.; Luban, J. Cyclophilin A is required for the replication of group M human immunodeficiency virus type 1 (HIV-1) and simian immunodeficiency virus SIV(CPZ)GAB but not group O HIV-1 or other primate immunodeficiency viruses. J. Virol. 1996, 70, 4220–4227. [Google Scholar]
- Matsuoka, S.; Dam, E.; Lecossier, D.; Clavel, F.; Hance, A.J. Modulation of HIV-1 infectivity and cyclophilin A-dependence by Gag sequence and target cell type. Retrovirology 2009, 6, 21. [Google Scholar] [CrossRef]
- Hatziioannou, T.; Cowan, S.; Von Schwedler, U.K.; Sundquist, W.I.; Bieniasz, P.D. Species-specific tropism determinants in the human immunodeficiency virus type 1 capsid. J. Virol. 2004, 78, 6005–6012. [Google Scholar] [CrossRef]
- Tang, C.; Ndassa, Y.; Summers, M.F. Structure of the N-terminal 283-residue fragment of the immature HIV-1 Gag polyprotein. Nat. Struct. Biol. 2002, 9, 537–543. [Google Scholar]
- Yang, R.; Aiken, C. A mutation in alpha helix 3 of CA renders human immunodeficiency virus type 1 cyclosporin A resistant and dependent: rescue by a second-site substitution in a distal region of CA. J. Virol. 2007, 81, 3749–3756. [Google Scholar] [CrossRef]
- Fassati, A.; Goff, S.P. Characterization of intracellular reverse transcription complexes of human immunodeficiency virus type 1. J. Virol. 2001, 75, 3626–3635. [Google Scholar] [CrossRef]
- Gatanaga, H.; Das, D.; Suzuki, Y.; Yeh, D.D.; Hussain, K.A.; Ghosh, A.K.; Mitsuya, H. Altered HIV-1 Gag protein interactions with cyclophilin A (CypA) on the acquisition of H219Q and H219P substitutions in the CypA binding loop. J. Biol. Chem. 2006, 281, 1241–1250. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gallay, P.A.; Ptak, R.G.; Bobardt, M.D.; Dumont, J.-M.; Vuagniaux, G.; Rosenwirth, B. Correlation of Naturally Occurring HIV-1 Resistance to DEB025 with Capsid Amino Acid Polymorphisms. Viruses 2013, 5, 981-997. https://doi.org/10.3390/v5030981
Gallay PA, Ptak RG, Bobardt MD, Dumont J-M, Vuagniaux G, Rosenwirth B. Correlation of Naturally Occurring HIV-1 Resistance to DEB025 with Capsid Amino Acid Polymorphisms. Viruses. 2013; 5(3):981-997. https://doi.org/10.3390/v5030981
Chicago/Turabian StyleGallay, Philippe A., Roger G. Ptak, Michael D. Bobardt, Jean-Maurice Dumont, Grégoire Vuagniaux, and Brigitte Rosenwirth. 2013. "Correlation of Naturally Occurring HIV-1 Resistance to DEB025 with Capsid Amino Acid Polymorphisms" Viruses 5, no. 3: 981-997. https://doi.org/10.3390/v5030981