Biochemical and Functional Interactions of Human Papillomavirus Proteins with Polycomb Group Proteins

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Human Papillomaviruses
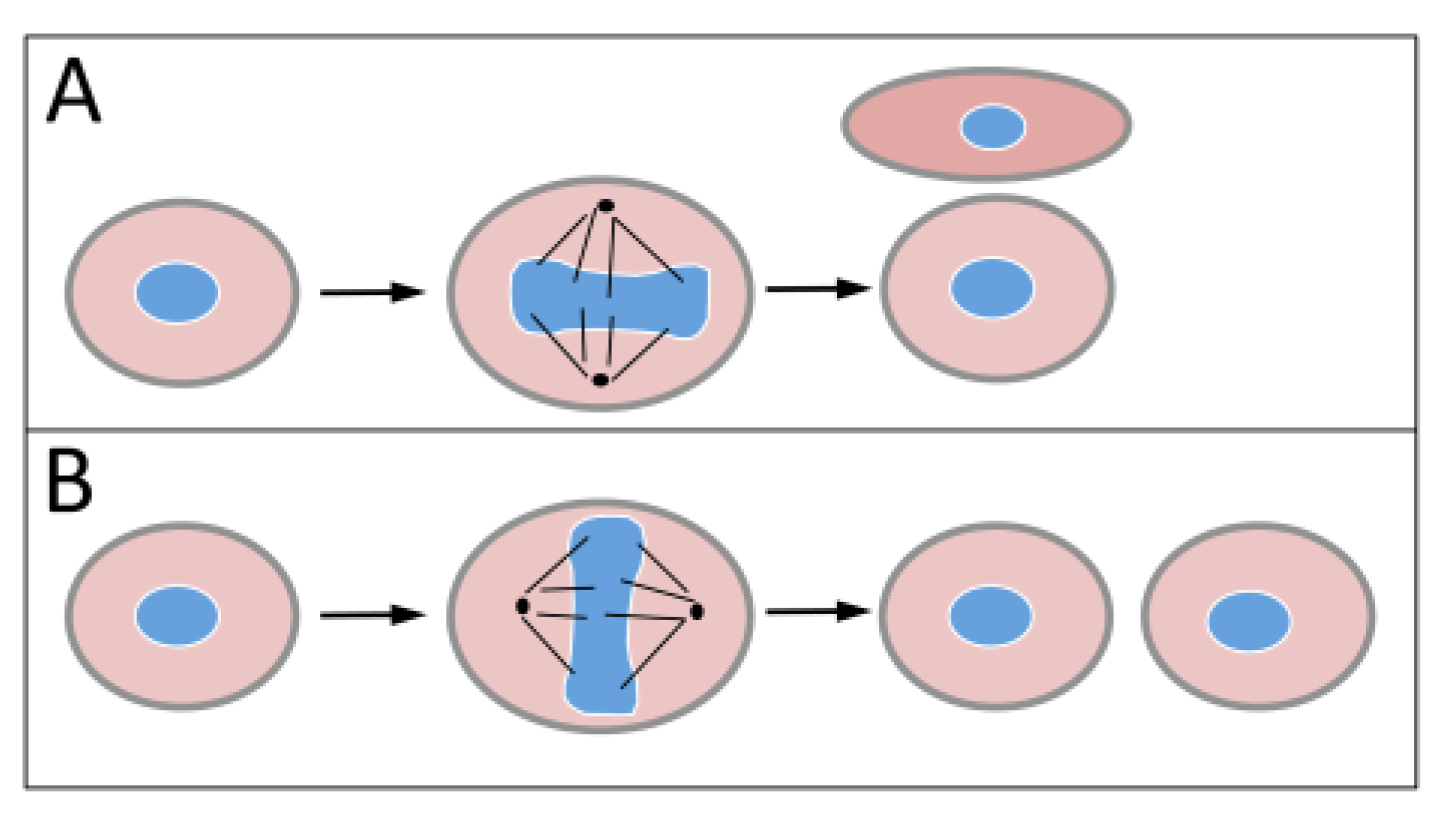
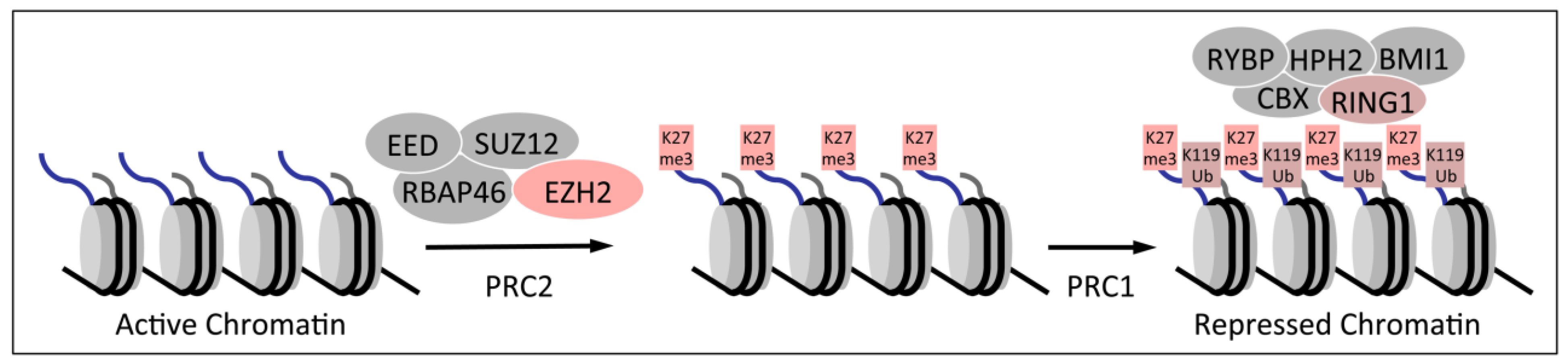
1.2. Polycomb Group Proteins and Homeobox Genes
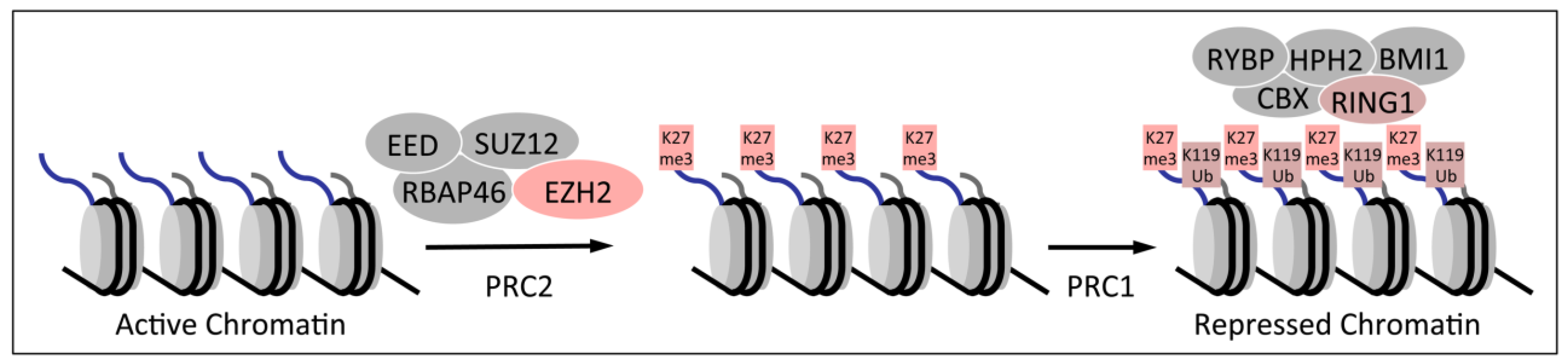
2. Dysregulation of Homeobox Gene Expression in Cervical Cancers
3. Association of HPV16 E7 with Polycomb Group Proteins
3.1. HPV16 E7
3.2. Association of HPV16 E7 with E2F6 Containing Polycomb Repressive Complexes
3.3. Decreased Trimethylation of Lysine 27 of Histone H3 in HPV16 E7 Expressing Cells
4. Modulation of Polycomb Group Protein Expression by HPV16 Oncoproteins
4.1. Increased EZH2 Expression in HPV E7 Expressing Cells
4.2. Modulation of BMI1 Expression by HPV E6 and E7
5. Modulation of PRC Regulators by HPV16 E7
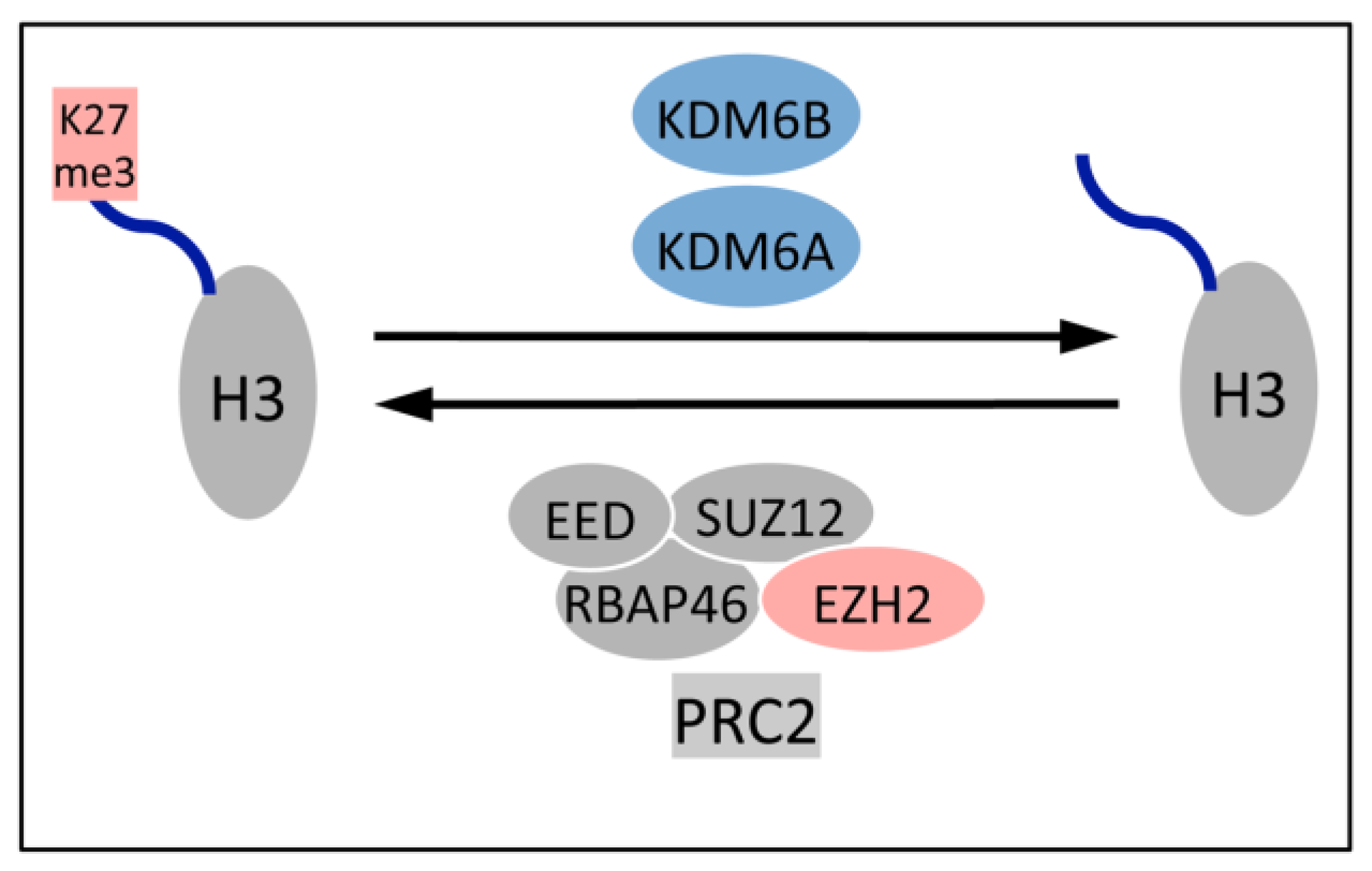
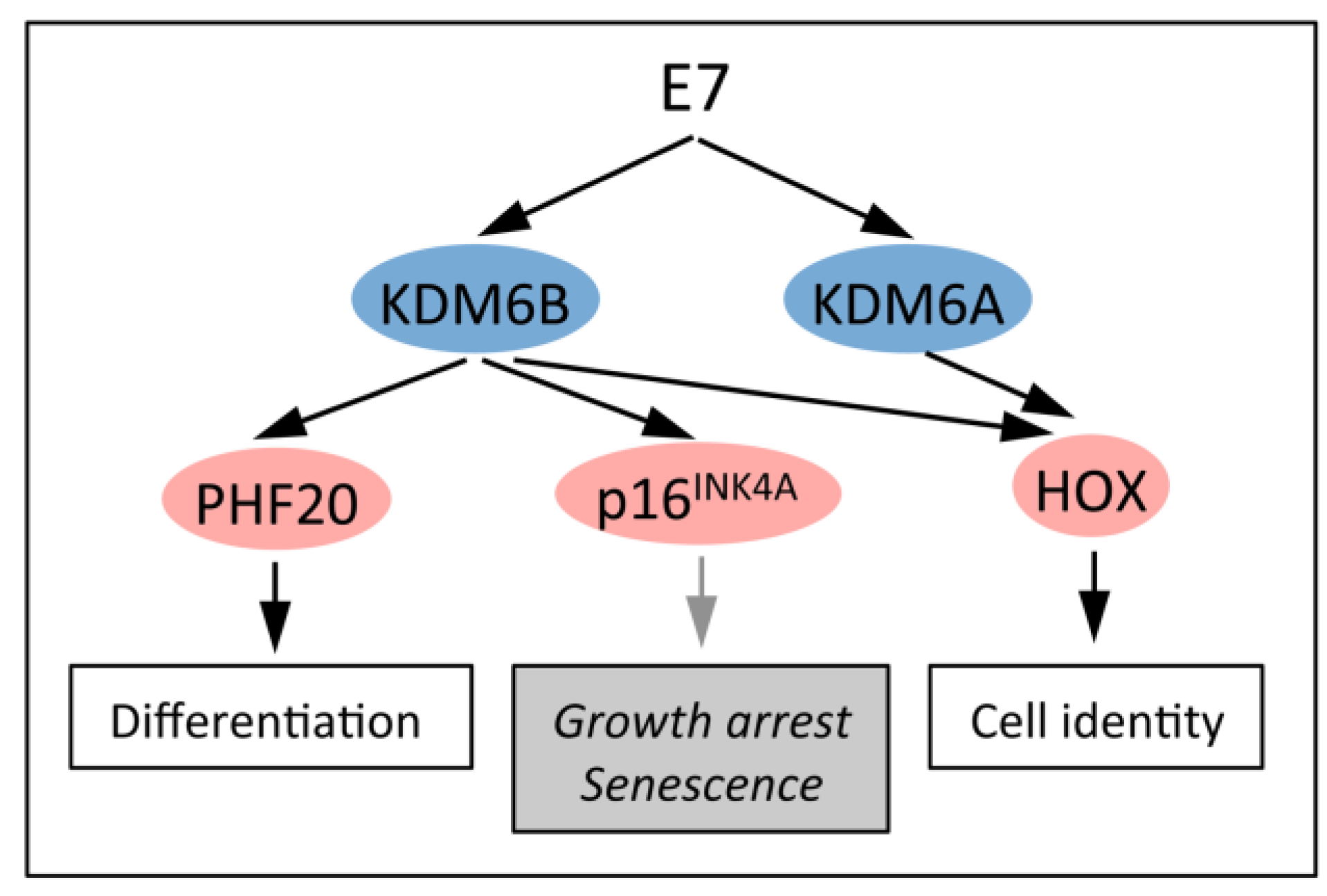
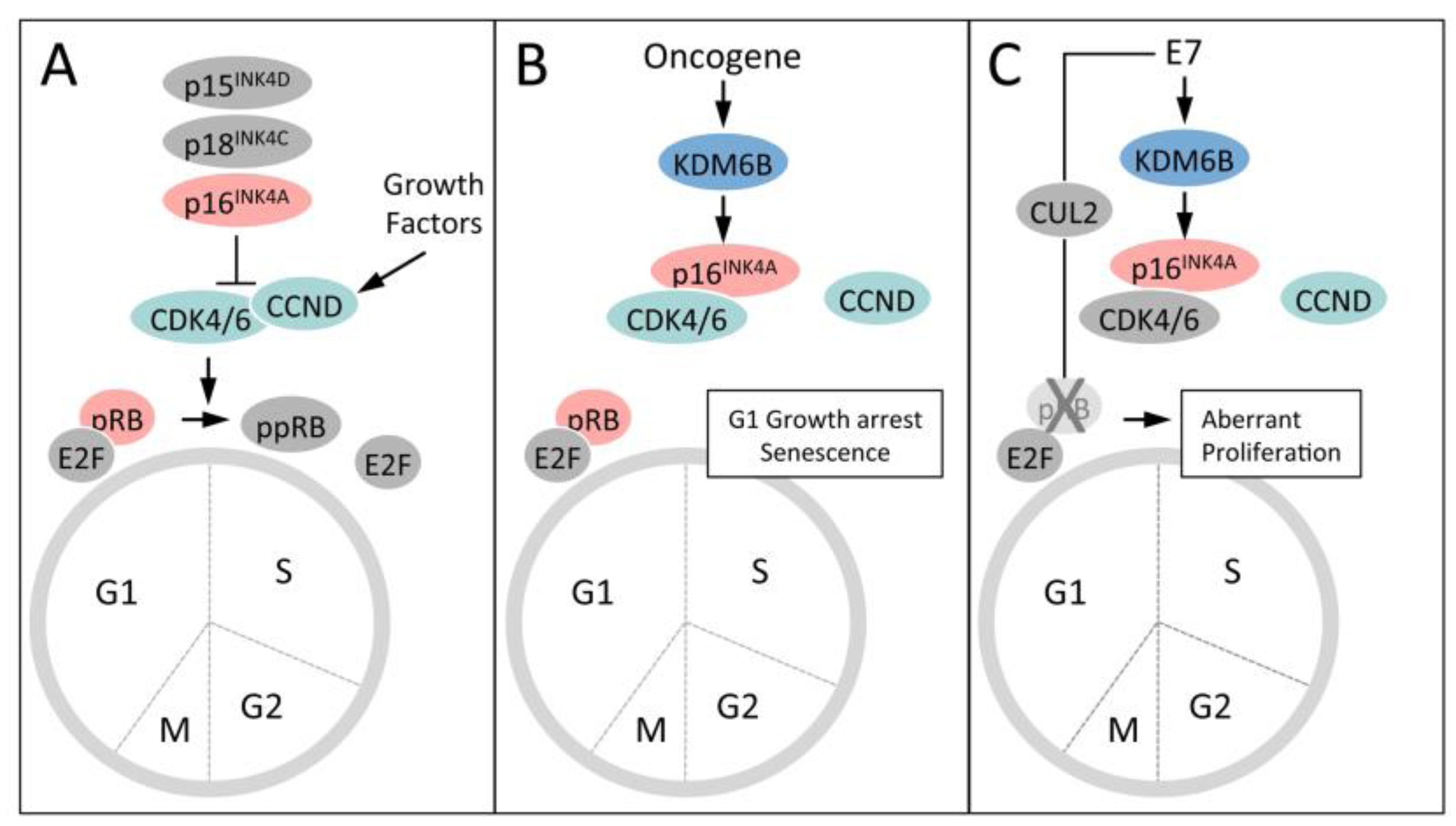
5.1. Upregulation of the H3K27 Demethylases KDM6A and KDM6B
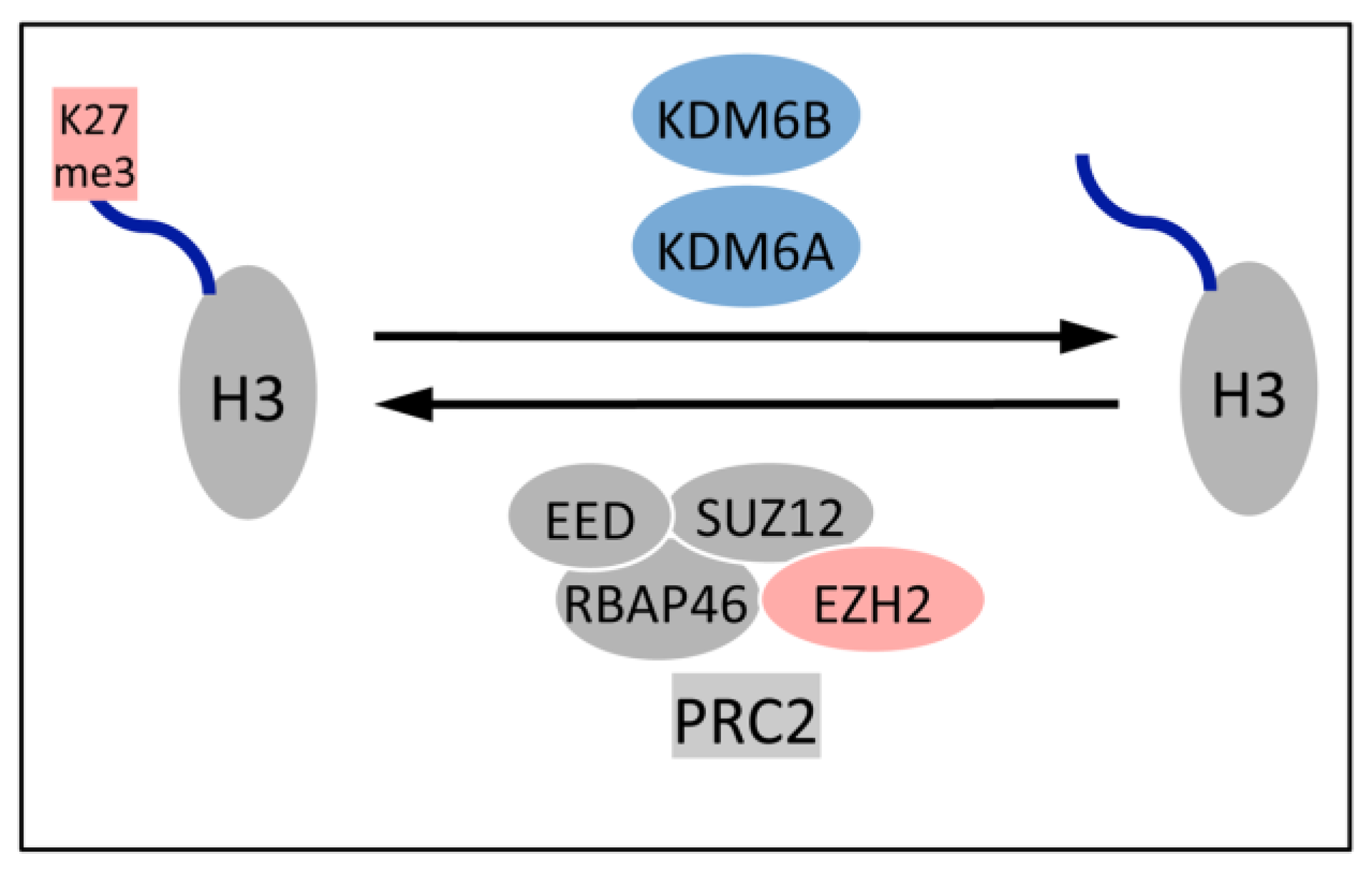
5.2. KDM6B Controls Epithelial Differentiation
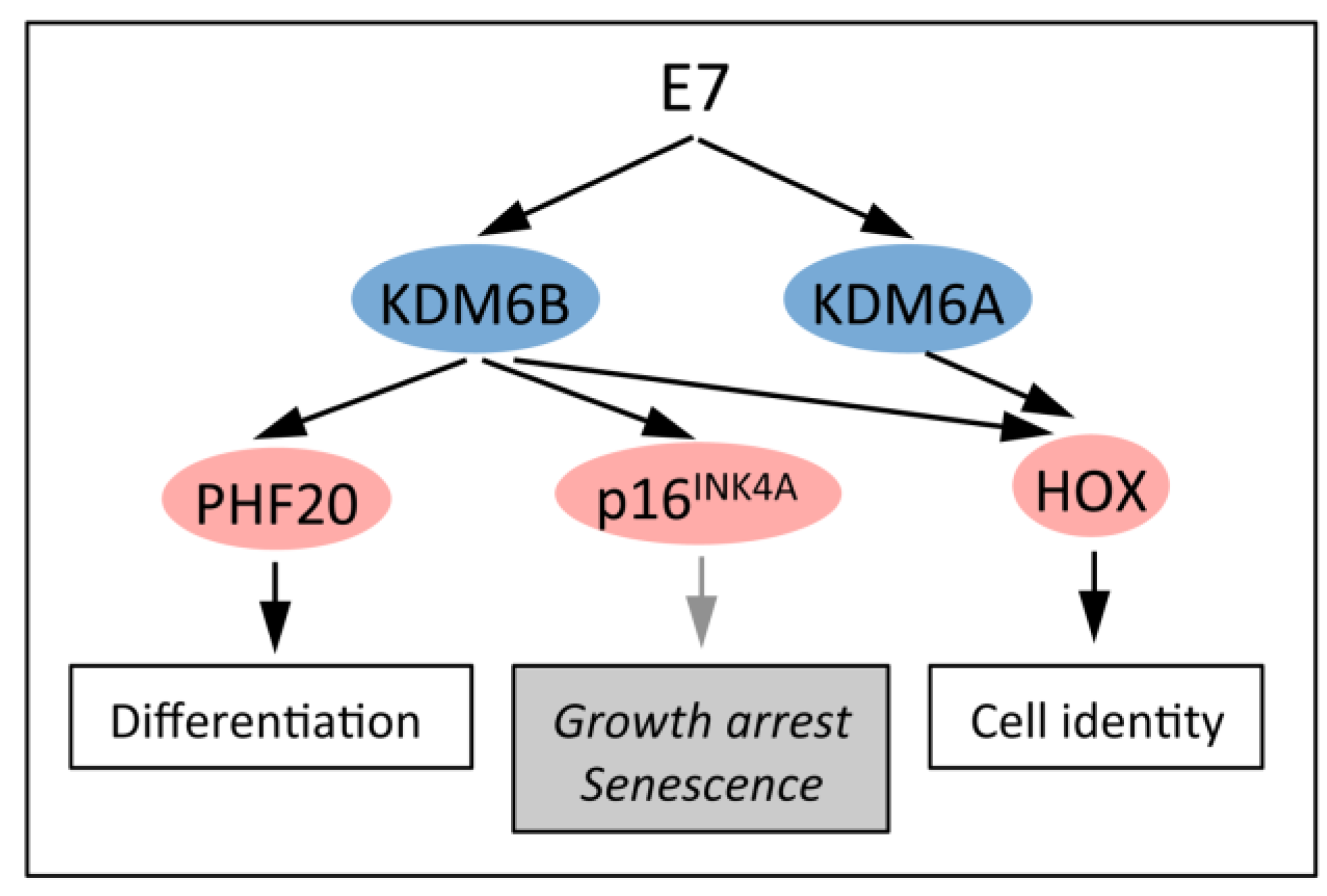
5.3. KDM6B Mediates the Oncogene-Induced Senescence (OIS) Response to RAS/RAF

5.4. HPV16 E7 Induces the Cervical Cancer Biomarker p16INK4A through KDM6B
5.5. Dysregulated Homeobox Gene Expression in HPV16 E7 Expressing Cells
5.6. KDM6A and KDM6B are Essential in Cervical Carcinoma Cells
6. Concluding Remarks
Acknowledgments
Conflict of Interest
References
- Bernard, H.U.; Burk, R.D.; Chen, Z.; van Doorslaer, K.; Hausen, H.; de Villiers, E.M. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 2010, 401, 70–79. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef]
- Munoz, N.; Bosch, F.X.; de Sanjose, S.; Herrero, R.; Castellsague, X.; Shah, K.V.; Snijders, P.J.; Meijer, C.J. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 2003, 348, 518–527. [Google Scholar] [CrossRef]
- Jemal, A.; Simard, E.P.; Dorell, C.; Noone, A.M.; Markowitz, L.E.; Kohler, B.; Eheman, C.; Saraiya, M.; Bandi, P.; Saslow, D.; et al. Annual report to the nation on the status of cancer, 1975–2009, Featuring the burden and trends in human papillomavirus(HPV)-associated cancers and HPV vaccination coverage levels. J. Nat. Cancer Inst. 2013, 105, 175–201. [Google Scholar] [CrossRef]
- Kane, M.A.; Serrano, B.; de Sanjose, S.; Wittet, S. Implementation of human papillomavirus immunization in the developing world. Vaccine 2012, 30 Suppl. 5, F192–F200. [Google Scholar] [CrossRef]
- Frazer, I.H. Prevention of cervical cancer through papillomavirus vaccination. Nat. Rev. Immunol. 2004, 4, 46–54. [Google Scholar] [CrossRef]
- Hebner, C.M.; Laimins, L.A. Human papillomaviruses: Basic mechanisms of pathogenesis and oncogenicity. Rev. Med. Virol. 2006, 16, 83–97. [Google Scholar] [CrossRef]
- Bodily, J.; Laimins, L.A. Persistence of human papillomavirus infection: Keys to malignant progression. Trends Microbiol. 2011, 19, 33–39. [Google Scholar] [CrossRef]
- Baker, C.C.; Phelps, W.C.; Lindgren, V.; Braun, M.J.; Gonda, M.A.; Howley, P.M. Structural and translational analysis of human papillomavirus type 16 sequences in cervical carcinoma cell lines. J. Virol. 1987, 61, 962–971. [Google Scholar]
- Schwarz, E.; Freese, U.K.; Gissmann, L.; Mayer, W.; Roggenbuck, B.; Stremlau, A.; zur Hausen, H. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature 1985, 314, 111–114. [Google Scholar] [CrossRef]
- McLaughlin-Drubin, M.E.; Munger, K. Oncogenic activities of human papillomaviruses. Virus Res. 2009, 143, 195–208. [Google Scholar] [CrossRef]
- Riley, R.R.; Duensing, S.; Brake, T.; Munger, K.; Lambert, P.F.; Arbeit, J.M. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res. 2003, 63, 4862–4871. [Google Scholar]
- Francis, D.A.; Schmid, S.I.; Howley, P.M. Repression of the integrated papillomavirus E6/E7 promoter is required for growth suppression of cervical cancer cells. J. Virol. 2000, 74, 2679–2686. [Google Scholar] [CrossRef]
- Goodwin, E.C.; DiMaio, D. Repression of human papillomavirus oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant tumor suppressor pathways. Proc. Natl. Acad. Sci. USA 2000, 97, 12513–81251. [Google Scholar] [CrossRef]
- Thierry, F.; Yaniv, M. The BPV-1 E2 trans-acting protein can be either an activator or repressor of the HPV-18 regulatory region. EMBO 1987, 6, 3391–3397. [Google Scholar]
- Bracken, A.P.; Helin, K. Polycomb group proteins: Navigators of lineage pathways led astray in cancer. Nat. Rev. Cancer 2009, 9, 773–784. [Google Scholar] [CrossRef]
- Trojer, P.; Cao, A.R.; Gao, Z.; Li, Y.; Zhang, J.; Xu, X.; Li, G.; Losson, R.; Erdjument-Bromage, H.; Tempst, P.; et al. L3MBTL2 protein acts in concert with PcG protein-mediated monoubiquitination of H2A to establish a repressive chromatin structure. Mol. Cell 2011, 42, 438–450. [Google Scholar] [CrossRef]
- Qin, J.; Whyte, W.A.; Anderssen, E.; Apostolou, E.; Chen, H.H.; Akbarian, S.; Bronson, R.T.; Hochedlinger, K.; Ramaswamy, S.; Young, R.A.; et al. The polycomb group protein L3mbtl2 assembles an atypical PRC1-family complex that is essential in pluripotent stem cells and early development. Cell Stem Cell 2012, 11, 319–332. [Google Scholar] [CrossRef]
- Zhang, J.; Bardot, E.; Ezhkova, E. Epigenetic regulation of skin: Focus on the Polycomb complex. Cell Mol. Life Sci. 2012, 69, 2161–2172. [Google Scholar] [CrossRef]
- Eckert, R.L.; Adhikary, G.; Rorke, E.A.; Chew, Y.C.; Balasubramanian, S. Polycomb group proteins are key regulators of keratinocyte function. J. Invest. Dermatol. 2011, 131, 295–301. [Google Scholar] [CrossRef]
- Shaw, T.; Martin, P. Epigenetic reprogramming during wound healing: Loss of polycomb-mediated silencing may enable upregulation of repair genes. EMBO Rep. 2009, 10, 881–886. [Google Scholar] [CrossRef]
- Eklund, E. The role of Hox proteins in leukemogenesis: Insights into key regulatory events in hematopoiesis. Crit. Rev. Oncog. 2011, 16, 65–76. [Google Scholar] [CrossRef]
- Kelly, Z.L.; Michael, A.; Butler-Manuel, S.; Pandha, H.S.; Morgan, R.G. HOX genes in ovarian cancer. J. Ovarian. Res. 2011, 4, e16. [Google Scholar] [CrossRef] [Green Version]
- Ewing, C.M.; Ray, A.M.; Lange, E.M.; Zuhlke, K.A.; Robbins, C.M.; Tembe, W.D.; Wiley, K.E.; Isaacs, S.D.; Johng, D.; Wang, Y.; et al. Germline mutations in HOXB13 and prostate-cancer risk. N. Engl. J. Med. 2012, 366, 141–149. [Google Scholar] [CrossRef]
- Martin, N.; Popov, N.; Aguilo, F.; O'Loghlen, A.; Raguz, S.; Snijders, A.P.; Dharmalingam, G.; Li, S.; Thymiakou, E.; Carroll, T.; et al. Interplay between homeobox proteins and polycomb repressive complexes in p16 regulation. EMBO 2013, 32, 982–995. [Google Scholar] [CrossRef]
- Hung, Y.C.; Ueda, M.; Terai, Y.; Kumagai, K.; Ueki, K.; Kanda, K.; Yamaguchi, H.; Akise, D.; Ueki, M. Homeobox gene expression and mutation in cervical carcinoma cells. Cancer Sci. 2003, 94, 437–441. [Google Scholar] [CrossRef]
- Alami, Y.; Castronovo, V.; Belotti, D.; Flagiello, D.; Clausse, N. HOXC5 and HOXC8 expression are selectively turned on in human cervical cancer cells compared to normal keratinocytes. Biochem. Biophys. Res. Commun. 1999, 257, 738–745. [Google Scholar] [CrossRef]
- Barba-de la Rosa, A.P.; Briones-Cerecero, E.; Lugo-Melchor, O.; de Leon-Rodriguez, A.; Santos, L.; Castelo-Ruelas, J.; Valdivia, A.; Pina, P.; Chagolla-Lopez, A.; Hernandez-Cueto, D.; et al. Hox B4 as potential marker of non-differentiated cells in human cervical cancer cells. J. Cancer Res. Clin. Oncol. 2012, 138, 293–300. [Google Scholar] [CrossRef]
- Zhai, Y.; Kuick, R.; Nan, B.; Ota, I.; Weiss, S.J.; Trimble, C.L.; Fearon, E.R.; Cho, K.R. Gene expression analysis of preinvasive and invasive cervical squamous cell carcinomas identifies HOXC10 as a key mediator of invasion. Cancer Res. 2007, 67, 10163–10172. [Google Scholar] [CrossRef]
- White, E.A.; Howley, P.M. Proteomic approaches to the study of papillomavirus-host interactions. Virology 2013, 435, 57–69. [Google Scholar] [CrossRef]
- Huh, K.W.; DeMasi, J.; Ogawa, H.; Nakatani, Y.; Howley, P.M.; Munger, K. Association of the human papillomavirus type 16 E7 oncoprotein with the 600-kDa retinoblastoma protein-associated factor, p600. Proc. Natl. Acad. Sci. USA 2005, 102, 11492–11497. [Google Scholar] [CrossRef]
- McLaughlin-Drubin, M.E.; Huh, K.W.; Munger, K. Human papillomavirus type 16 E7 oncoprotein associates with E2F6. J. Virol. 2008, 82, 8695–8705. [Google Scholar] [CrossRef]
- Cartwright, P.; Muller, H.; Wagener, C.; Holm, K.; Helin, K. E2F-6: A novel member of the E2F family is an inhibitor of E2F-dependent transcription. Oncogene 1998, 17, 611–623. [Google Scholar]
- Gaubatz, S.; Wood, J.G.; Livingston, D.M. Unusual proliferation arrest and transcriptional control properties of a newly discovered E2F family member, E2F-6. Proc. Natl. Acad. Sci. USA 1998, 95, 9190–9195. [Google Scholar] [CrossRef]
- Trimarchi, J.M.; Fairchild, B.; Wen, J.; Lees, J.A. The E2F6 transcription factor is a component of the mammalian Bmi1-containing polycomb complex. Proc. Natl. Acad. Sci. USA 2001, 98, 1519–1524. [Google Scholar] [CrossRef]
- Ogawa, H.; Ishiguro, K.; Gaubatz, S.; Livingston, D.M.; Nakatani, Y. A complex with chromatin modifiers that occupies E2F- and Myc-responsive genes in G0 cells. Science 2002, 296, 1132–1136. [Google Scholar] [CrossRef]
- Attwooll, C.; Oddi, S.; Cartwright, P.; Prosperini, E.; Agger, K.; Steensgaard, P.; Wagener, C.; Sardet, C.; Moroni, M.C.; Helin, K. A novel repressive E2F6 complex containing the polycomb group protein, EPC1, that interacts with EZH2 in a proliferation-specific manner. J. Biol. Chem. 2005, 280, 1199–1208. [Google Scholar]
- Deshpande, A.M.; Akunowicz, J.D.; Reveles, X.T.; Patel, B.B.; Saria, E.A.; Gorlick, R.G.; Naylor, S.L.; Leach, R.J.; Hansen, M.F. PHC3, a component of the hPRC-H complex, associates with E2F6 during G0 and is lost in osteosarcoma tumors. Oncogene 2007, 26, 1714–1722. [Google Scholar] [CrossRef]
- McLaughlin-Drubin, M.E.; Crum, C.P.; Munger, K. Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B histone demethylase expression and causes epigenetic reprogramming. Proc. Natl. Acad. Sci. USA 2011, 108, 2130–2135. [Google Scholar] [CrossRef]
- Hyland, P.L.; McDade, S.S.; McCloskey, R.; Dickson, G.J.; Arthur, K.; McCance, D.J.; Patel, D. Evidence for alteration of EZH2, BMI1, and KDM6A and epigenetic reprogramming in human papillomavirus type 16 E6/E7-expressing keratinocytes. J. Virol. 2011, 85, 10999–11006. [Google Scholar] [CrossRef]
- Bracken, A.P.; Pasini, D.; Capra, M.; Prosperini, E.; Colli, E.; Helin, K. EZH2 is downstream of the pRB-E2F pathway, Essential for proliferation and amplified in cancer. EMBO J. 2003, 22, 5323–5335. [Google Scholar] [CrossRef]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185. [Google Scholar] [CrossRef]
- Chase, A.; Cross, N.C. Aberrations of EZH2 in cancer. Clin. Cancer Res. 2011, 17, 2613–2618. [Google Scholar] [CrossRef]
- Holland, D.; Hoppe-Seyler, K.; Schuller, B.; Lohrey, C.; Maroldt, J.; Durst, M.; Hoppe-Seyler, F. Activation of the enhancer of zeste homologue 2 gene by the human papillomavirus E7 oncoprotein. Cancer Res. 2008, 68, 9964–9972. [Google Scholar] [CrossRef]
- Cha, T.L.; Zhou, B.P.; Xia, W.; Wu, Y.; Yang, C.C.; Chen, C.T.; Ping, B.; Otte, A.P.; Hung, M.C. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science 2005, 310, 306–310. [Google Scholar] [CrossRef]
- Menges, C.W.; Baglia, L.A.; Lapoint, R.; McCance, D.J. Human papillomavirus type 16 E7 up-regulates AKT activity through the retinoblastoma protein. Cancer Res. 2006, 66, 5555–5559. [Google Scholar] [CrossRef]
- Spangle, J.M.; Munger, K. The human papillomavirus type 16 E6 oncoprotein activates mTORC1 signaling and increases protein synthesis. J. Virol. 2010, 84, 9398–9407. [Google Scholar] [CrossRef]
- Xu, K.; Wu, Z.J.; Groner, A.C.; He, H.H.; Cai, C.; Lis, R.T.; Wu, X.; Stack, E.C.; Loda, M.; Liu, T.; et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 2012, 338, 1465–1469. [Google Scholar] [CrossRef]
- Kuzmichev, A.; Margueron, R.; Vaquero, A.; Preissner, T.S.; Scher, M.; Kirmizis, A.; Ouyang, X.; Brockdorff, N.; Abate-Shen, C.; Farnham, P.; et al. Composition and histone substrates of polycomb repressive group complexes change during cellular differentiation. Proc. Natl. Acad. Sci. USA 2005, 102, 1859–1864. [Google Scholar]
- Honig, A.; Weidler, C.; Hausler, S.; Krockenberger, M.; Buchholz, S.; Koster, F.; Segerer, S.E.; Dietl, J.; Engel, J.B. Overexpression of polycomb protein BMI-1 in human specimens of breast, Ovarian, endometrial and cervical cancer. Anticancer Res. 2010, 30, 1559–1564. [Google Scholar]
- Min, L.; Dong-Xiang, S.; Xiao-Tong, G.; Ting, G.; Xiao-Dong, C. Clinicopathological and prognostic significance of Bmi-1 expression in human cervical cancer. Acta Obstet. Gynecol. Scand. 2011, 90, 737–745. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, C.X.; Zhu, C.B.; Zhang, J.; Kan, S.F.; Du, L.T.; Li, W.; Wang, L.L.; Wang, S. Overexpression of Bmi-1 in uterine cervical cancer: Correlation with clinicopathology and prognosis. Int. J. Gynecol. Cancer 2010, 20, 1597–1603. [Google Scholar]
- Zhang, X.; Wang, C.; Wang, L.; Du, L.; Wang, S.; Zheng, G.; Li, W.; Zhuang, X.; Dong, Z. Detection of circulating Bmi-1 mRNA in plasma and its potential diagnostic and prognostic value for uterine cervical cancer. Int. J. Cancer 2012, 131, 165–172. [Google Scholar] [CrossRef]
- Tong, Y.Q.; Liu, B.; Zheng, H.Y.; He, Y.J.; Gu, J.; Li, F.; Li, Y. BMI-1 autoantibody as a new potential biomarker for cervical carcinoma. PLoS One 2011, 6, e27804. [Google Scholar]
- Miller, J.; Dakic, A.; Chen, R.; Palechor-Ceron, N.; Dai, Y.; Kallakury, B.; Schlegel, R.; Liu, X. HPV16 E7 Protein and hTERT proteins defective for telomere maintenance cooperate to immortalize human keratinocytes. PLoS Pathog. 2013, 9, e1003284. [Google Scholar] [CrossRef]
- Chen, F.; Li, Y.; Wang, L.; Hu, L. Knockdown of BMI-1 causes cell-cycle arrest and derepresses p16INK4a, HOXA9 and HOXC13 mRNA expression in HeLa cells. Med. Oncol. 2011, 28, 1201–1209. [Google Scholar] [CrossRef]
- Greenfield, A.; Carrel, L.; Pennisi, D.; Philippe, C.; Quaderi, N.; Siggers, P.; Steiner, K.; Tam, P.P.; Monaco, A.P.; Willard, H.F.; et al. The UTX gene escapes X inactivation in mice and humans. Hum. Mol. Genet. 1998, 7, 737–742. [Google Scholar] [CrossRef]
- Shpargel, K.B.; Sengoku, T.; Yokoyama, S.; Magnuson, T. UTX and UTY demonstrate histone demethylase-independent function in mouse embryonic development. PLoS Genet. 2012, 8, e1002964. [Google Scholar] [CrossRef]
- Lee, S.; Lee, J.W.; Lee, S.K. UTX, a histone H3-lysine 27 demethylase, acts as a critical switch to activate the cardiac developmental program. Dev. Cell 2012, 22, 25–37. [Google Scholar] [CrossRef]
- Welstead, G.G.; Creyghton, M.P.; Bilodeau, S.; Cheng, A.W.; Markoulaki, S.; Young, R.A.; Jaenisch, R. X-linked H3K27me3 demethylase Utx is required for embryonic development in a sex-specific manner. Proc. Natl. Acad. Sci. USA 2012, 109, 13004–13009. [Google Scholar] [CrossRef]
- Wang, C.; Lee, J.E.; Cho, Y.W.; Xiao, Y.; Jin, Q.; Liu, C.; Ge, K. UTX regulates mesoderm differentiation of embryonic stem cells independent of H3K27 demethylase activity. Proc. Natl. Acad. Sci. USA 2012, 109, 15324–15329. [Google Scholar]
- Burgold, T.; Voituron, N.; Caganova, M.; Tripathi, P.P.; Menuet, C.; Tusi, B.K.; Spreafico, F.; Bevengut, M.; Gestreau, C.; Buontempo, S.; et al. The H3K27 demethylase JMJD3 is required for maintenance of the embryonic respiratory neuronal network, Neonatal breathing, and survival. Cell Rep. 2012, 2, 1244–1258. [Google Scholar] [CrossRef]
- De Santa, F.; Totaro, M.G.; Prosperini, E.; Notarbartolo, S.; Testa, G.; Natoli, G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 2007, 130, 1083–1094. [Google Scholar] [CrossRef]
- De Santa, F.; Narang, V.; Yap, Z.H.; Tusi, B.K.; Burgold, T.; Austenaa, L.; Bucci, G.; Caganova, M.; Notarbartolo, S.; Casola, S.; et al. Jmjd3 contributes to the control of gene expression in LPS-activated macrophages. EMBO 2009, 28, 3341–3352. [Google Scholar] [CrossRef]
- Lee, M.G.; Villa, R.; Trojer, P.; Norman, J.; Yan, K.P.; Reinberg, D.; Di Croce, L.; Shiekhattar, R. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science 2007, 318, 447–450. [Google Scholar] [CrossRef]
- Issaeva, I.; Zonis, Y.; Rozovskaia, T.; Orlovsky, K.; Croce, C.M.; Nakamura, T.; Mazo, A.; Eisenbach, L.; Canaani, E. Knockdown of ALR (MLL2) reveals ALR target genes and leads to alterations in cell adhesion and growth. Mol. Cell Biol. 2007, 27, 1889–1903. [Google Scholar] [CrossRef]
- van Haaften, G.; Dalgliesh, G.L.; Davies, H.; Chen, L.; Bignell, G.; Greenman, C.; Edkins, S.; Hardy, C.; O'Meara, S.; Teague, J.; et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat. Genet. 2009, 41, 521–523. [Google Scholar]
- Xiang, Y.; Zhu, Z.; Han, G.; Lin, H.; Xu, L.; Chen, C.D. JMJD3 is a histone H3K27 demethylase. Cell Res. 2007, 17, 850–857. [Google Scholar] [CrossRef]
- Sen, G.L.; Webster, D.E.; Barragan, D.I.; Chang, H.Y.; Khavari, P.A. Control of differentiation in a self-renewing mammalian tissue by the histone demethylase JMJD3. Gene. Develop. 2008, 22, 1865–1870. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Barradas, M.; Anderton, E.; Acosta, J.C.; Li, S.; Banito, A.; Rodriguez-Niedenfuhr, M.; Maertens, G.; Banck, M.; Zhou, M.M.; Walsh, M.J.; et al. Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Gene. Develop. 2009, 23, 1177–1182. [Google Scholar] [CrossRef]
- Agger, K.; Cloos, P.A.; Rudkjaer, L.; Williams, K.; Andersen, G.; Christensen, J.; Helin, K. The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene- and stress-induced senescence. Gene. Develop. 2009, 23, 1171–1176. [Google Scholar] [CrossRef]
- Sano, T.; Oyama, T.; Kashiwabara, K.; Fukuda, T.; Nakajima, T. Expression status of p16 protein is associated with human papillomavirus oncogenic potential in cervical and genital lesions. Amer. J. Pathol. 1998, 153, 1741–1748. [Google Scholar] [CrossRef]
- Klaes, R.; Friedrich, T.; Spitkovsky, D.; Ridder, R.; Rudy, W.; Petry, U.; Dallenbach-Hellweg, G.; Schmidt, D.; von Knebel Doeberitz, M. Overexpression of p16(INK4A) as a specific marker for dysplastic and neoplastic epithelial cells of the cervix uteri. Int. J. Cancer 2001, 92, 276–284. [Google Scholar] [CrossRef]
- Khleif, S.N.; DeGregori, J.; Yee, C.L.; Otterson, G.A.; Kaye, F.J.; Nevins, J.R.; Howley, P.M. Inhibition of cyclin D-CDK4/CDK6 activity is associated with an E2F-mediated induction of cyclin kinase inhibitor activity. Proc. Natl. Acad. Sci. USA 1996, 93, 4350–4354. [Google Scholar]
- Gonzalez, S.L.; Stremlau, M.; He, X.; Basile, J.R.; Münger, K. Degradation of the retinoblastoma tumor suppressor by the human papillomavirus type 16 E7 oncoprotein is important for functional inactivation and is separable from proteasomal degradation of E7. J. Virol. 2001, 75, 7583–7591. [Google Scholar] [CrossRef]
- Skalska, L.; White, R.E.; Parker, G.A.; Sinclair, A.J.; Paschos, K.; Allday, M.J. Induction of p16(INK4a) is the major barrier to proliferation when Epstein-Barr Virus (EBV) transforms primary B cells into lymphoblastoid cell lines. PLoS Pathog. 2013, 9, e1003187. [Google Scholar] [CrossRef]
- Skalska, L.; White, R.E.; Franz, M.; Ruhmann, M.; Allday, M.J. Epigenetic repression of p16(INK4A) by latent Epstein-Barr virus requires the interaction of EBNA3A and EBNA3C with CtBP. PLoS Pathog. 2010, 6, e1000951. [Google Scholar] [CrossRef]
- White, R.E.; Groves, I.J.; Turro, E.; Yee, J.; Kremmer, E.; Allday, M.J. Extensive co-operation between the Epstein-Barr virus EBNA3 proteins in the manipulation of host gene expression and epigenetic chromatin modification. PLoS One 2010, 5, e13979. [Google Scholar]
- Paschos, K.; Parker, G.A.; Watanatanasup, E.; White, R.E.; Allday, M.J. BIM promoter directly targeted by EBNA3C in polycomb-mediated repression by EBV. Nucleic Acids Res. 2012, 40, 7233–7246. [Google Scholar] [CrossRef]
- Andrisani, O.M. Deregulation of epigenetic mechanisms by the hepatitis B virus x protein in hepatocarcinogenesis. Viruses 2013, 5, 858–872. [Google Scholar] [CrossRef]
- Herschkowitz, J.I.; He, X.; Fan, C.; Perou, C.M. The functional loss of the retinoblastoma tumour suppressor is a common event in basal-like and luminal B breast carcinomas. Breast Cancer Res. 2008, 10, R75. [Google Scholar] [CrossRef]
- Jarrard, D.F.; Modder, J.; Fadden, P.; Fu, V.; Sebree, L.; Heisey, D.; Schwarze, S.R.; Friedl, A. Alterations in the p16/pRb cell cycle checkpoint occur commonly in primary and metastatic human prostate cancer. Cancer Lett. 2002, 185, 191–199. [Google Scholar] [CrossRef]
- Kommoss, S.; du Bois, A.; Ridder, R.; Trunk, M.J.; Schmidt, D.; Pfisterer, J.; Kommoss, F. Independent prognostic significance of cell cycle regulator proteins p16(INK4a) and pRb in advanced-stage ovarian carcinoma including optimally debulked patients: a translational research subprotocol of a randomised study of the Arbeitsgemeinschaft Gynaekologische Onkologie Ovarian Cancer Study Group. Br. J. Cancer 2007, 96, 306–313. [Google Scholar] [CrossRef]
- Andujar, P.; Wang, J.; Descatha, A.; Galateau-Salle, F.; Abd-Alsamad, I.; Billon-Galland, M.A.; Blons, H.; Clin, B.; Danel, C.; Housset, B.; et al. p16INK4A inactivation mechanisms in non-small-cell lung cancer patients occupationally exposed to asbestos. Lung Cancer 2010, 67, 23–30. [Google Scholar] [Green Version]
- Malumbres, M.; Barbacid, M. To cycle or not to cycle: a critical decision in cancer. Nat. Rev. Cancer 2001, 1, 222–231. [Google Scholar] [CrossRef]
- Horner, S.M.; DeFilippis, R.A.; Manuelidis, L.; DiMaio, D. Repression of the human papillomavirus E6 gene initiates p53-dependent, Telomerase-independent senescence and apoptosis in HeLa cervical carcinoma cells. J. Virol. 2004, 78, 4063–4073. [Google Scholar] [CrossRef]
- Psyrri, A.; DeFilippis, R.A.; Edwards, A.P.; Yates, K.E.; Manuelidis, L.; DiMaio, D. Role of the retinoblastoma pathway in senescence triggered by repression of the human papillomavirus E7 protein in cervical carcinoma cells. Cancer Res. 2004, 64, 3079–3086. [Google Scholar] [CrossRef]
- DeFilippis, R.A.; Goodwin, E.C.; Wu, L.; DiMaio, D. Endogenous human papillomavirus E6 and E7 proteins differentially regulate proliferation, senescence, and apoptosis in HeLa cervical carcinoma cells. J. Virol. 2003, 77, 1551–1563. [Google Scholar] [CrossRef]
- Westbrook, T.F.; Martin, E.S.; Schlabach, M.R.; Leng, Y.; Liang, A.C.; Feng, B.; Zhao, J.J.; Roberts, T.M.; Mandel, G.; Hannon, G.J.; et al. A genetic screen for candidate tumor suppressors identifies REST. Cell 2005, 121, 837–848. [Google Scholar] [CrossRef]
- Coulson, J.M.; Edgson, J.L.; Woll, P.J.; Quinn, J.P. A splice variant of the neuron-restrictive silencer factor repressor is expressed in small cell lung cancer: A potential role in derepression of neuroendocrine genes and a useful clinical marker. Cancer Res. 2000, 60, 1840–1844. [Google Scholar]
- Westbrook, T.F.; Hu, G.; Ang, X.L.; Mulligan, P.; Pavlova, N.N.; Liang, A.; Leng, Y.; Maehr, R.; Shi, Y.; Harper, J.W.; et al. SCFbeta-TRCP controls oncogenic transformation and neural differentiation through REST degradation. Nature 2008, 452, 370–374. [Google Scholar] [CrossRef]
- Kruidenier, L.; Chung, C.W.; Cheng, Z.; Liddle, J.; Che, K.; Joberty, G.; Bantscheff, M.; Bountra, C.; Bridges, A.; Diallo, H.; et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 2012, 488, 404–408. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
McLaughlin-Drubin, M.E.; Munger, K. Biochemical and Functional Interactions of Human Papillomavirus Proteins with Polycomb Group Proteins. Viruses 2013, 5, 1231-1249. https://doi.org/10.3390/v5051231
McLaughlin-Drubin ME, Munger K. Biochemical and Functional Interactions of Human Papillomavirus Proteins with Polycomb Group Proteins. Viruses. 2013; 5(5):1231-1249. https://doi.org/10.3390/v5051231
Chicago/Turabian StyleMcLaughlin-Drubin, Margaret E., and Karl Munger. 2013. "Biochemical and Functional Interactions of Human Papillomavirus Proteins with Polycomb Group Proteins" Viruses 5, no. 5: 1231-1249. https://doi.org/10.3390/v5051231