Gene Therapy Strategies to Exploit TRIM Derived Restriction Factors against HIV-1

Abstract
:1. Introduction

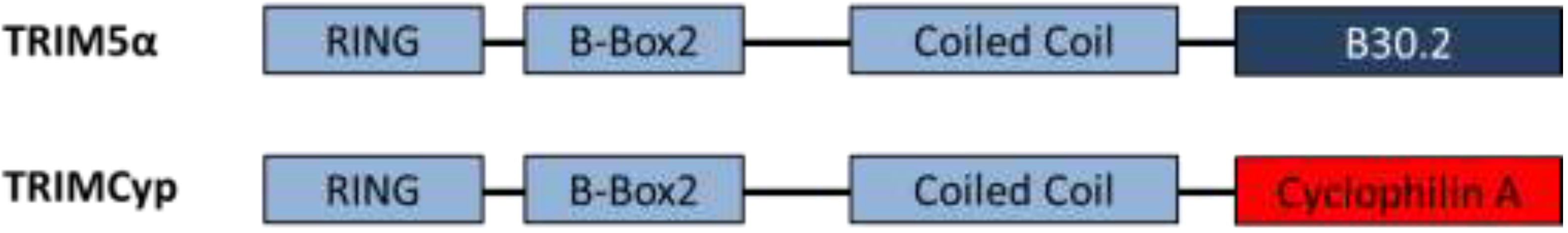{kind=link}
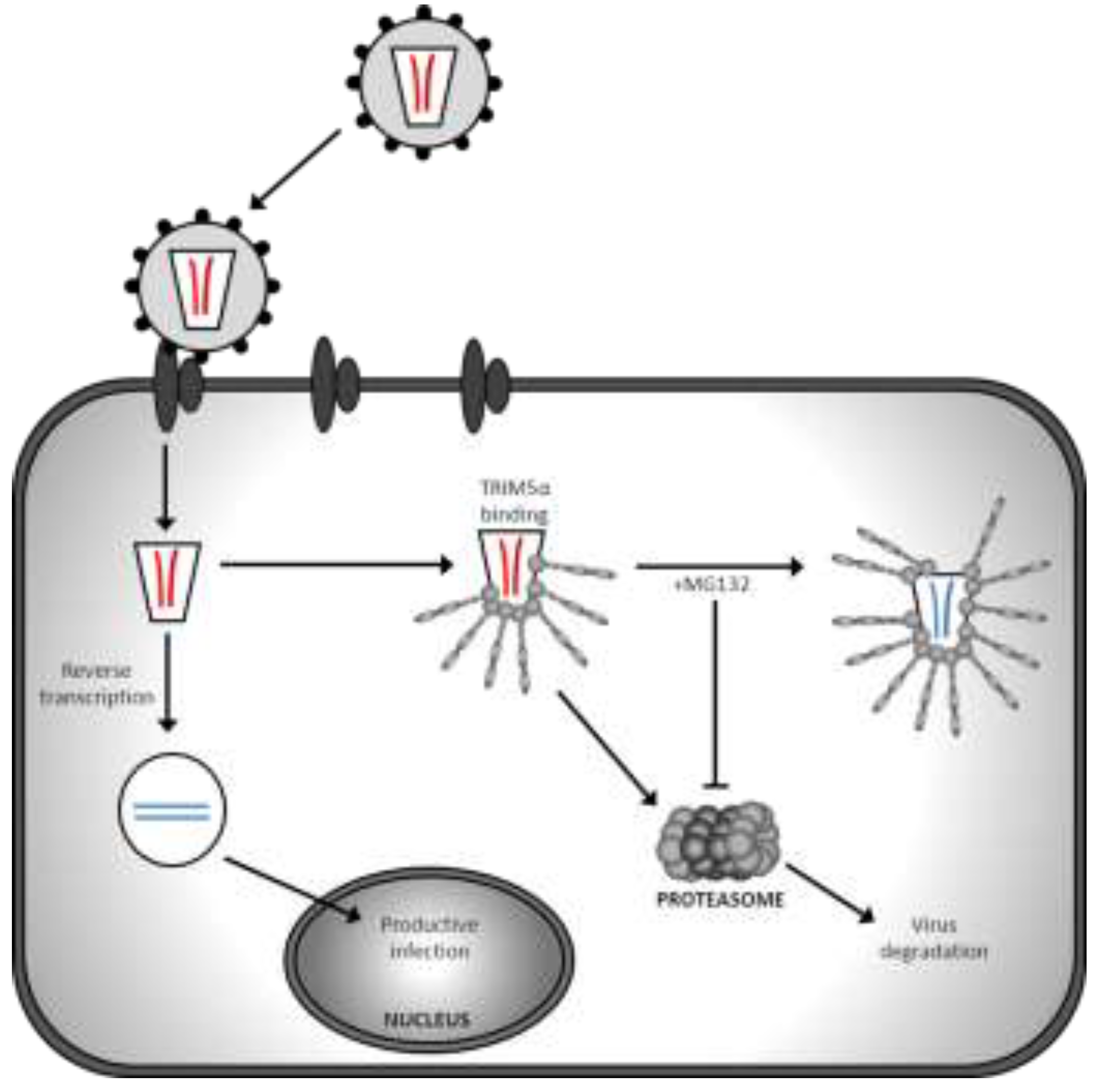{kind=link}
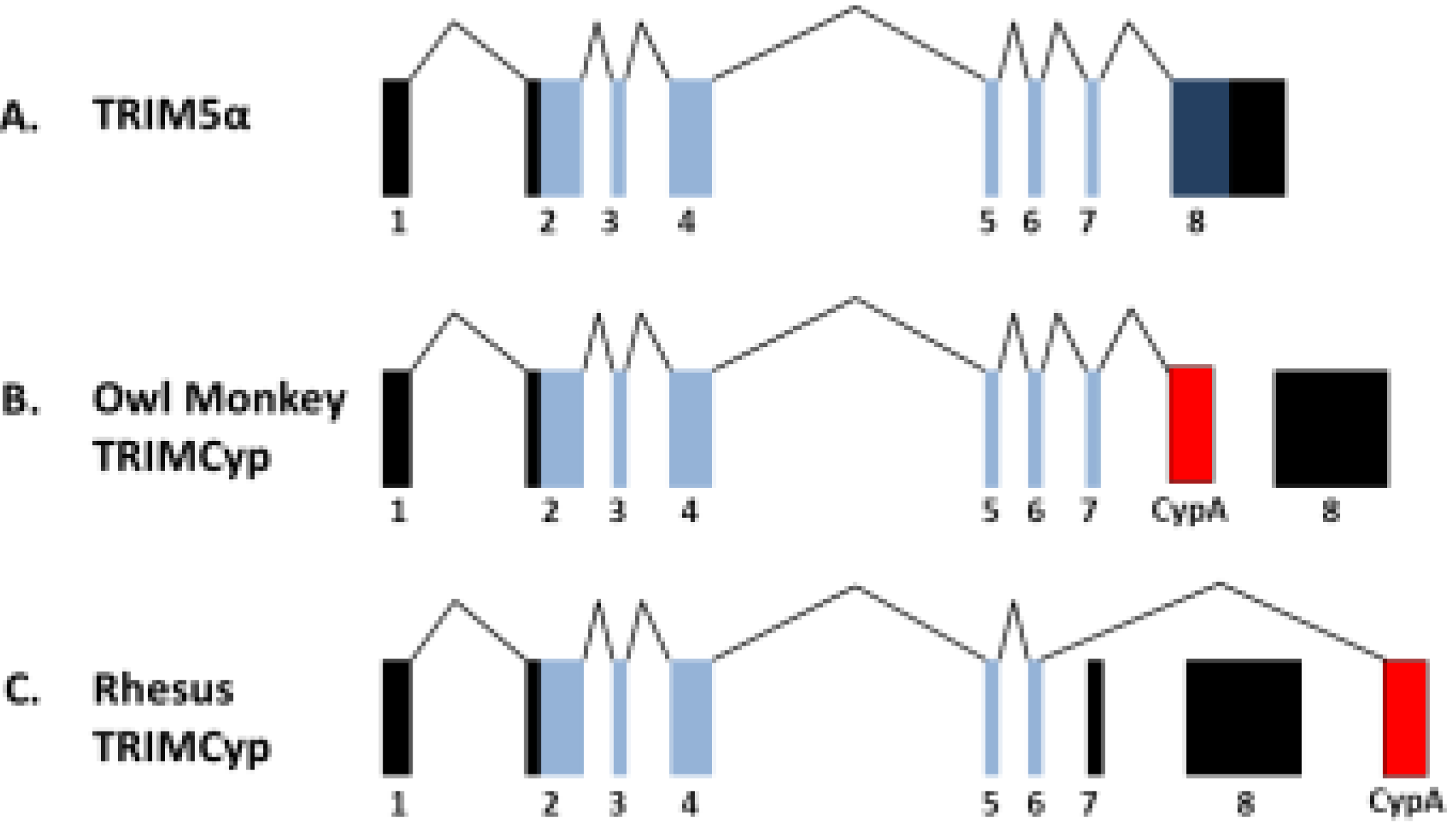{kind=link}
| Restriction Factor | Mechanism | Viral evasion | Disadvantages | Exploitation |
|---|---|---|---|---|
| APOBEC | Cytidine deamination of viral genome | Vif mediated proteasomal degradation | APOBEC3G induced mutations may promote evolution of HIV | Modified APOBEC3G restricts HIV in T cells and macrophages [11] |
| SAMHD1 | dNTP triphosphohydrolase activity depletes dNTPs and prevents reverse transcription | HIV-2 Vpx causes proteasomal degradation | Antiviral function limited to quiescent cells | Undefined |
| Tetherin | Prevents HIV release by anchoring budding virus particles | Lysosomal degradation is promoted by Vpu | Reduces, but does not abolish spread of virus.or establishment of reservoirs | Vpu resistant tetherin in cell lines [12] |
| TRIM5α/TRIMCyp | Targets virus for proteasomal degradation and disrupts uncoating | HIV-1 accessory proteins are unable to counteract TRIM restriction | Species specific restriction; human TRIM5α does not restrict HIV-1 | Chimeric TRIM5α and humanised TRIMCyp restriction demonstrated in humanised mice [13,14] |
2. Innate Immunity against HIV



3. Gene Therapy Exploitation of TRIMCyp Factors
4. Conclusions
Acknowledgments
Conflicts of Interest
References and Notes
- Mitsuyasu, R.T.; Anton, P.A.; Deeks, S.G.; Scadden, D.T.; Connick, E.; Downs, M.T.; Bakker, A.; Roberts, M.R.; June, C.H.; Jalali, S.; et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4+ and CD8+ T cells in human immunodeficiency virus-infected subjects. Blood 2000, 96, 785–793. [Google Scholar]
- Deeks, S.G.; Wagner, B.; Anton, P.A.; Mitsuyasu, R.T.; Scadden, D.T.; Huang, C.; Macken, C.; Richman, D.D.; Christopherson, C.; June, C.H.; et al. A Phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Mol. Ther. 2002, 5, 788–797. [Google Scholar] [CrossRef]
- Maier, D.A.; Brennan, A.L.; Jiang, S.; Binder-Scholl, G.K.; Lee, G.; Plesa, G.; Zheng, Z.; Cotte, J.; Carpenito, C.; Wood, T.; et al. Efficient clinical scale gene modification via zinc finger nuclease-targeted disruption of the hiv co-receptor CCR5. Hum. Gene Ther. 2013, 24, 245–258. [Google Scholar] [CrossRef]
- Holt, N.; Wang, J.; Kim, K.; Friedman, G.; Wang, X.; Taupin, V.; Crooks, G.M.; Kohn, D.B.; Gregory, P.D.; Holmes, M.C.; et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat. Biotechnol. 2010, 28, 839–847. [Google Scholar] [CrossRef]
- Podsakoff, G.M.; Engel, B.C.; Carbonaro, D.A.; Choi, C.; Smogorzewska, E.M.; Bauer, G.; Selander, D.; Csik, S.; Wilson, K.; Betts, M.R.; et al. Selective survival of peripheral blood lymphocytes in children with HIV-1 following delivery of an anti-HIV gene to bone marrow CD34+ cells. Mol. Ther. 2005, 12, 77–86. [Google Scholar] [CrossRef]
- Von Laer, D.; Hasselmann, S.; Hasselmann, K. Gene therapy for HIV infection: What does it need to make it work? J. Gene Med. 2006, 8, 658–667. [Google Scholar] [CrossRef]
- Macpherson, J.L.; Boyd, M.P.; Arndt, A.J.; Todd, A.V.; Fanning, G.C.; Ely, J.A.; Elliott, F.; Knop, A.; Raponi, M.; Murray, J.; et al. Long-term survival and concomitant gene expression of ribozyme-transduced CD4+ T-lymphocytes in HIV-infected patients. J. Gene Med. 2005, 7, 552–564. [Google Scholar] [CrossRef]
- Tebas, P.; Stein, D.; Binder-Scholl, G.; Mukherjee, R.; Brady, T.; Rebello, T.; Humeau, L.; Kalos, M.; Papasavvas, E.; Montaner, L.J.; et al. Antiviral effects of autologous CD4 T cells genetically modified with a conditionally replicating lentiviral vector expressing long antisense to HIV. Blood 2013, 121, 1524–1533. [Google Scholar] [CrossRef]
- Van Lunzen, J.; Glaunsinger, T.; Stahmer, I.; von Baehr, V.; Baum, C.; Schilz, A.; Kuehlcke, K.; Naundorf, S.; Martinius, H.; Hermann, F.; et al. Transfer of autologous gene-modified T cells in HIV-infected patients with advanced immunodeficiency and drug-resistant virus. Mol. Ther. 2007, 15, 1024–1033. [Google Scholar]
- Morgan, R.A.; Walker, R.; Carter, C.S.; Natarajan, V.; Tavel, J.A.; Bechtel, C.; Herpin, B.; Muul, L.; Zheng, Z.; Jagannatha, S.; et al. Preferential survival of CD4+ T lymphocytes engineered with anti-human immunodeficiency virus (HIV) genes in HIV-infected individuals. Hum. Gene Ther. 2005, 16, 1065–1074. [Google Scholar] [CrossRef]
- Ao, Z.; Wang, X.; Bello, A.; Jayappa, K.D.; Yu, Z.; Fowke, K.; He, X.; Chen, X.; Li, J.; Kobinger, G.; et al. Characterization of anti-HIV activity mediated by R88-APOBEC3G mutant fusion proteins in CD4+ T cells, peripheral blood mononuclear cells, and macrophages. Hum. Gene Ther. 2011, 22, 1225–1237. [Google Scholar]
- Gupta, R.K.; Hué, S.; Schaller, T.; Verschoor, E.; Pillay, D.; Towers, G.J. Mutation of a single residue renders human tetherin resistant to HIV-1 Vpu-mediated depletion. PLoS Pathog. 2009, 5. [Google Scholar] [CrossRef]
- Neagu, M.R.; Ziegler, P.; Pertel, T.; Strambio-De-Castillia, C.; Grütter, C.; Martinetti, G.; Mazzucchelli, L.; Grütter, M.; Manz, M.G.; Luban, J. Potent inhibition of HIV-1 by TRIM5-cyclophilin fusion proteins engineered from human components. J. Clin. Invest. 2009, 119, 3035–3047. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.E.; Chen, R.X.; McGee, J.; Nacey, C.; Pollard, R.B.; Abedi, M.; Bauer, G.; Nolta, J.A.; Anderson, J.S. Generation of an HIV-1 resistant immune system with CD34+ HSCS transduced with a triple combination anti-HIV lentiviral vector. J. Virol. 2012. [Google Scholar] [CrossRef]
- Bieniasz, P.D. Intrinsic immunity: A front-line defense against viral attack. Nat. Immunol. 2004, 5, 1109–1115. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Venkatesh, S.; Bieniasz, P.D. Intrinsic cellular defenses against human immunodeficiency viruses. Immunity 2012, 37, 399–411. [Google Scholar] [CrossRef]
- Hultquist, J.F.; Lengyel, J.A.; Refsland, E.W.; LaRue, R.S.; Lackey, L.; Brown, W.L.; Harris, R.S. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J. Virol. 2011, 85, 11220–11234. [Google Scholar]
- Le Tortorec, A.; Willey, S.; Neil, S.J. Antiviral inhibition of enveloped virus release by tetherin/bst-2: Action and counteraction. Viruses 2011, 3, 520–540. [Google Scholar] [CrossRef]
- Si, Z.; Vandegraaff, N.; O’Huigin, C.; Song, B.; Yuan, W.; Xu, C.; Perron, M.; Li, X.; Marasco, W.A.; Engelman, A.; et al. Evolution of a cytoplasmic tripartite motif (TRIM) protein in cows that restricts retroviral infection. Proc. Natl. Acad. Sci. USA 2006, 103, 7454–7459. [Google Scholar] [CrossRef]
- Ylinen, L.M.J.; Keckesova, Z.; Webb, B.L.J.; Gifford, R.J.M.; Smith, T.P.L.; Towers, G.J. Isolation of an active Lv1 gene from cattle indicates that tripartite motif protein-mediated innate immunity to retroviral infection is widespread among mammals. J. Virol. 2006, 80, 7332–7338. [Google Scholar] [CrossRef]
- Gabuzda, D.H.; Lawrence, K.; Langhoff, E.; Terwilliger, E.; Dorfman, T.; Haseltine, W.A.; Sodroski, J. Role of Vif in replication of human immunodeficiency virus type 1 in CD4+ T lymphocytes. J. Virol. 1992, 66, 6489–6495. [Google Scholar]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef]
- Harris, R.S.; Petersen-Mahrt, S.K.; Neuberger, M.S. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol. Cell 2002, 10, 1247–1253. [Google Scholar] [CrossRef]
- Mangeat, B.; Turelli, P.; Caron, G.; Friedli, M.; Perrin, L.; Trono, D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 2003, 424, 99–103. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, B.; Pomerantz, R.J.; Zhang, C.; Arunachalam, S.C.; Gao, L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 2003, 424, 94–98. [Google Scholar] [CrossRef]
- Zennou, V.; Perez-Caballero, D.; Gottlinger, H.; Bieniasz, P.D. APOBEC3G incorporation into human immunodeficiency virus type 1 particles. J. Virol. 2004, 78, 12058–12061. [Google Scholar] [CrossRef]
- Khan, M.A.; Kao, S.; Miyagi, E.; Takeuchi, H.; Goila-Gaur, R.; Opi, S.; Gipson, C.L.; Parslow, T.G.; Ly, H.; Strebel, K. Viral rna is required for the association of APOBEC3G with human immunodeficiency virus type 1 nucleoprotein complexes. J. Virol. 2005, 79, 5870–5874. [Google Scholar] [CrossRef]
- Newman, E.N.C.; Holmes, R.K.; Craig, H.M.; Klein, K.C.; Lingappa, J.R.; Malim, M.H.; Sheehy, A.M. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr. Biol. 2005, 15, 166–170. [Google Scholar] [CrossRef]
- Schrofelbauer, B.; Chen, D.; Landau, N.R. A single amino acid of APOBEC3G controls its species-specific interaction with virion infectivity factor (Vif). Proc. Natl. Acad. Sci. USA 2004, 101, 3927–3932. [Google Scholar] [CrossRef]
- Xu, H.; Svarovskaia, E.S.; Barr, R.; Zhang, Y.; Khan, M.A.; Strebel, K.; Pathak, V.K. A single amino acid substitution in human APOBEC3G antiretroviral enzyme confers resistance to HIV-1 virion infectivity factor-induced depletion. Proc. Natl. Acad. Sci. USA 2004, 101, 5652–5657. [Google Scholar] [CrossRef]
- Ao, Z.; Yu, Z.; Wang, L.; Zheng, Y.; Yao, X. Vpr14-88-Apobec3G fusion protein is efficiently incorporated into Vif-positive HIV-1 particles and inhibits viral infection. PLoS One 2008, 3. [Google Scholar] [CrossRef]
- Kim, E.-Y.; Bhattacharya, T.; Kunstman, K.; Swantek, P.; Koning, F.A.; Malim, M.H.; Wolinsky, S.M. Human Apobec3G-mediated editing can promote HIV-1 sequence diversification and accelerate adaptation to selective pressure. J. Virol. 2010, 84, 10402–10405. [Google Scholar] [CrossRef]
- Fourati, S.; Malet, I.; Binka, M.; Boukobza, S.; Wirden, M.; Sayon, S.; Simon, A.; Katlama, C.; Simon, V.; Calvez, V.; et al. Partially active HIV-1 Vif alleles facilitate viral escape from specific antiretrovirals. AIDS 2010, 24, 2313–2321. [Google Scholar]
- Neil, S.J.D.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef]
- Kupzig, S.; Korolchuk, V.; Rollason, R.; Sugden, A.; Wilde, A.; Banting, G. BST-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic 2003, 4, 694–709. [Google Scholar] [CrossRef]
- Erikson, E.; Adam, T.; Schmidt, S.; Lehmann-Koch, J.; Over, B.; Goffinet, C.; Harter, C.; Bekeredjian-Ding, I.; Sertel, S.; Lasitschka, F.; et al. In vivo expression profile of the antiviral restriction factor and tumor-targeting antigen CD317/BST-2/HM1.24/tetherin in humans. Proc. Natl. Acad. Sci. USA 2011, 108, 13688–13693. [Google Scholar] [CrossRef]
- Douglas, J.L.; Viswanathan, K.; McCarroll, M.N.; Gustin, J.K.; Früh, K.; Moses, A.V. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/tetherin via a βTrcP-dependent mechanism. J. Virol. 2009, 83, 7931–7947. [Google Scholar] [CrossRef]
- Mitchell, R.S.; Katsura, C.; Skasko, M.A.; Fitzpatrick, K.; Lau, D.; Ruiz, A.; Stephens, E.B.; Margottin-Goguet, F.; Benarous, R.; Guatelli, J.C. Vpu antagonizes BST-2-mediated restriction of HIV-1 release via beta-TrcP and endo-lysosomal trafficking. PLoS Pathog. 2009, 5, e1000450. [Google Scholar] [CrossRef]
- Lv, M.; Wang, J.; Zhu, Y.; Wang, X.; Zuo, T.; Liu, D.; Zhang, J.; Wu, J.; Zhang, H.; Kong, W.; et al. Overexpression of inactive tetherin delGPI mutant inhibits HIV-1 Vpu-mediated antagonism of endogenous tetherin. FEBS Lett. 2013, 587, 37–43. [Google Scholar] [CrossRef]
- Jia, B.; Serra-Moreno, R.; Neidermyer, W.; Rahmberg, A.; Mackey, J.; Fofana, I.B.; Johnson, W.E.; Westmoreland, S.; Evans, D.T. Species-specific activity of SIV nef and HIV-1 Vpu in overcoming restriction by tetherin/BST2. PLoS Pathog. 2009, 5, e1000429. [Google Scholar] [CrossRef]
- Gupta, R.K.; Mlcochova, P.; Pelchen-Matthews, A.; Petit, S.J.; Mattiuzzo, G.; Pillay, D.; Takeuchi, Y.; Marsh, M.; Towers, G.J. Simian immunodeficiency virus envelope glycoprotein counteracts tetherin/BST-2/CD317 by intracellular sequestration. Proc. Natl. Acad. Sci. USA 2009, 106, 20889–20894. [Google Scholar] [CrossRef]
- Le Tortorec, A.; Neil, S.J. Antagonism to and intracellular sequestration of human tetherin by the human immunodeficiency virus type 2 envelope glycoprotein. J. Virol. 2009, 83, 11966–11978. [Google Scholar] [CrossRef]
- Rice, G.I.; Bond, J.; Asipu, A.; Brunette, R.L.; Manfield, I.W.; Carr, I.M.; Fuller, J.C.; Jackson, R.M.; Lamb, T.; Briggs, T.A.; et al. Mutations involved in Aicardi-Goutières syndrome implicate SAMHD1 as regulator of the innate immune response. Nat. Genet. 2009, 41, 829–832. [Google Scholar] [CrossRef]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the samhd1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Ségéral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef]
- Baldauf, H.-M.; Pan, X.; Erikson, E.; Schmidt, S.; Daddacha, W.; Burggraf, M.; Schenkova, K.; Ambiel, I.; Wabnitz, G.; Gramberg, T.; et al. Samhd1 restricts HIV-1 infection in resting CD4+ T cells. Nat. Med. 2012, 18, 1682–1687. [Google Scholar] [CrossRef]
- Descours, B.; Cribier, A.; Chable-Bessia, C.; Ayinde, D.; Rice, G.; Crow, Y.; Yatim, A.; Schwartz, O.; Laguette, N.; Benkirane, M. SAMHD1 restricts HIV-1 reverse transcription in quiescent CD4+ T-cells. Retrovirology 2012, 9. [Google Scholar] [CrossRef]
- Goldstone, D.C.; Ennis-Adeniran, V.; Hedden, J.J.; Groom, H.C.T.; Rice, G.I.; Christodoulou, E.; Walker, P.A.; Kelly, G.; Haire, L.F.; Yap, M.W.; et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 2011, 480, 379–382. [Google Scholar] [CrossRef]
- Lahouassa, H.; Daddacha, W.; Hofmann, H.; Ayinde, D.; Logue, E.C.; Dragin, L.; Bloch, N.; Maudet, C.; Bertrand, M.; Gramberg, T.; et al. Samhd1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 2012, 13, 223–228. [Google Scholar] [CrossRef]
- Powell, R.D.; Holland, P.J.; Hollis, T.; Perrino, F.W. Aicardi-Goutieres syndrome gene and HIV-1 restriction factor SAMHD1 is a dGTP-regulated deoxynucleotide triphosphohydrolase. J. Biol. Chem. 2011, 286, 43596–43600. [Google Scholar] [CrossRef]
- Beloglazova, N.; Flick, R.; Tchigvintsev, A.; Brown, G.; Popovic, A.; Nocek, B.; Yakunin, A.F. Nuclease activity of the human SAMHD1 protein implicated in the Aicardi-Goutieres syndrome and HIV-1 restriction. J. Biol. Chem. 2013, 288, 8101–8110. [Google Scholar] [CrossRef]
- Cribier, A.; Descours, B.; Valadão, A.L.C.; Laguette, N.; Benkirane, M. Phosphorylation of SAMHD1 by cyclin A2/Cdk1 regulates its restriction activity toward HIV-1. Cell Rep. 2013, 3, 1036–1043. [Google Scholar] [CrossRef]
- White, T.E.; Brandariz-Nuñez, A.; Valle-Casuso, J.C.; Amie, S.; Nguyen, L.A.; Kim, B.; Tuzova, M.; Diaz-Griffero, F. The retroviral restriction ability of SAMHD1, but not its deoxynucleotide triphosphohydrolase activity, is regulated by phosphorylation. Cell Host Microbe 2013, 13, 441–451. [Google Scholar] [CrossRef]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The cytoplasmic body component TRIM5α restricts HIV-1 infection in old world monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef]
- Hatziioannou, T.; Perez-Caballero, D.; Yang, A.; Cowan, S.; Bieniasz, P.D. Retrovirus resistance factors Ref1 and Lv1 are species-specific variants of TRIM5α. Proc. Natl. Acad. Sci. USA 2004, 101, 10774–10779. [Google Scholar] [CrossRef]
- Keckesova, Z.; Ylinen, L.M.J.; Towers, G.J. The human and African green monkey TRIM5α genes encode Ref1 and Lv1 retroviral restriction factor activities. Proc. Natl. Acad. Sci. USA 2004, 101, 10780–10785. [Google Scholar] [CrossRef]
- Towers, G.; Bock, M.; Martin, S.; Takeuchi, Y.; Stoye, J.P.; Danos, O. A conserved mechanism of retrovirus restriction in mammals. Proc. Natl. Acad. Sci. USA 2000, 97, 12295–12299. [Google Scholar] [CrossRef]
- Towers, G.; Collins, M.; Takeuchi, Y. Abrogation of Ref1 retrovirus restriction in human cells. J. Virol. 2002, 76, 2548–2550. [Google Scholar] [CrossRef]
- Hatziioannou, T.; Cowan, S.; Goff, S.P.; Bieniasz, P.D.; Towers, G.J. Restriction of multiple divergent retroviruses by Lv1 and Ref1. EMBO J. 2003, 22, 385–394. [Google Scholar] [CrossRef]
- Besnier, C.; Takeuchi, Y.; Towers, G. Restriction of lentivirus in monkeys. Proc. Natl. Acad. Sci. USA 2002, 99, 11920–11925. [Google Scholar] [CrossRef]
- Cowan, S.; Hatziioannou, T.; Cunningham, T.; Muesing, M.A.; Gottlinger, H.G.; Bieniasz, P.D. Cellular inhibitors with Fv1-like activity restrict human and simian immunodeficiency virus tropism. Proc. Natl. Acad. Sci. USA 2002, 99, 11914–11919. [Google Scholar] [CrossRef]
- Munk, C.; Brandt, S.M.; Lucero, G.; Landau, N.R. A dominant block to HIV-1 replication at reverse transcription in simian cells. Proc. Natl. Acad. Sci. USA 2002, 99, 13843–13848. [Google Scholar] [CrossRef]
- Nakayama, E.E.; Miyoshi, H.; Nagai, Y.; Shioda, T. A specific region of 37 amino acid residues in the SPRY (B30.2) domain of african green monkey TRIM5α determines species-specific restriction of simian immunodeficiency virus SIVmac infection. J. Virol. 2005, 79, 8870–8877. [Google Scholar] [CrossRef]
- Perez-Caballero, D.; Hatziioannou, T.; Yang, A.; Cowan, S.; Bieniasz, P.D. Human tripartite motif 5α domains responsible for retrovirus restriction activity and specificity. J. Virol. 2005, 79, 8969–8978. [Google Scholar] [CrossRef]
- Stremlau, M.; Perron, M.; Welikala, S.; Sodroski, J. Species-specific variation in the B30.2 (SPRY) domain of TRIM5α determines the potency of human immunodeficiency virus restriction. J. Virol. 2005, 79, 3139–3145. [Google Scholar] [CrossRef]
- Yap, M.W.; Nisole, S.; Stoye, J.P. A single amino acid change in the spry domain of human TRIM5α leads to HIV-1 restriction. Curr. Biol. 2005, 15, 73–78. [Google Scholar] [CrossRef]
- Perez-Caballero, D.; Hatziioannou, T.; Yang, A.; Cowan, S.; Bieniasz, P.D. Human tripartite motif 5α domains responsible for retrovirus restriction activity and specificity. J. Virol. 2005, 79, 8969–8978. [Google Scholar] [CrossRef]
- Sawyer, S.L.; Wu, L.I.; Emerman, M.; Malik, H.S. Positive selection of primate TRIM5α identifies a critical species-specific retroviral restriction domain. Proc. Natl. Acad. Sci. USA 2005, 102, 2832–2837. [Google Scholar] [CrossRef]
- Johnson, W.E.; Sawyer, S.L. Molecular evolution of the antiretroviral TRIM5 gene. Immunogenetics 2009, 61, 163–176. [Google Scholar] [CrossRef]
- Schaller, T.; Hué, S.p.; Towers, G.J. An active TRIM5 protein in rabbits indicates a common antiviral ancestor for mammalian TRIM5 proteins. J. Virol. 2007, 81, 11713–11721. [Google Scholar] [CrossRef]
- Fletcher, A.J.; Hué, S.; Schaller, T.; Pillay, D.; Towers, G.J. Hare TRIM5α restricts divergent retroviruses and exhibits significant sequence variation from closely related lagomorpha TRIM5 genes. J. Virol. 2010, 84, 12463–12468. [Google Scholar] [CrossRef]
- Reymond, A.; Meroni, G.; Fantozzi, A.; Merla, G.; Cairo, S.; Luzi, L.; Riganelli, D.; Zanaria, E.; Messali, S.; Cainarca, S.; et al. The tripartite motif family identifies cell compartments. EMBO J. 2001, 20, 2140–2151. [Google Scholar] [CrossRef]
- Sawyer, S.L.; Emerman, M.; Malik, H.S. Discordant evolution of the adjacent antiretroviral genes TRIM22 and TRIM5 in mammals. PLoS Pathog. 2007, 3, e197. [Google Scholar] [CrossRef]
- Asaoka, K.; Ikeda, K.; Hishinuma, T.; Horie-Inoue, K.; Takeda, S.; Inoue, S. A retrovirus restriction factor TRIM5α is transcriptionally regulated by interferons. Biochem. Biophys. Res. Commun. 2005, 338, 1950–1956. [Google Scholar] [CrossRef]
- Ylinen, L.M.; Keckesova, Z.; Wilson, S.J.; Ranasinghe, S.; Towers, G.J. Differential restriction of human immunodeficiency virus type 2 and simian immunodeficiency virus SIVmac by trim5α alleles. J. Virol. 2005, 79, 11580–11587. [Google Scholar] [CrossRef]
- Perron, M.J.; Stremlau, M.; Song, B.; Ulm, W.; Mulligan, R.C.; Sodroski, J. TRIM5α mediates the postentry block to N-tropic murine leukemia viruses in human cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11827–11832. [Google Scholar] [CrossRef]
- Newman, R.M.; Hall, L.; Connole, M.; Chen, G.L.; Sato, S.; Yuste, E.; Diehl, W.; Hunter, E.; Kaur, A.; Miller, G.M.; et al. Balancing selection and the evolution of functional polymorphism in old world monkey TRIM5α. Proc. Natl. Acad. Sci. USA 2006, 103, 19134–19139. [Google Scholar] [CrossRef]
- Wilson, S.J.; Webb, B.L.J.; Maplanka, C.; Newman, R.M.; Verschoor, E.J.; Heeney, J.L.; Towers, G.J. Rhesus macaque TRIM5 alleles have divergent antiretroviral specificities. J. Virol. 2008, 82, 7243–7247. [Google Scholar] [CrossRef]
- Speelmon, E.C.; Livingston-Rosanoff, D.; Li, S.S.; Vu, Q.; Bui, J.; Geraghty, D.E.; Zhao, L.P.; McElrath, M.J. Genetic association of the antiviral restriction factor TRIM5α with human immunodeficiency virus type 1 infection. J. Virol. 2006, 80, 2463–2471. [Google Scholar] [CrossRef]
- Van Manen, D.L.; Rits, M.A.N.; Beugeling, C.; van Dort, K.; Schuitemaker, H.; Kootstra, N.A. The effect of TRIM5 polymorphisms on the clinical course of HIV-1 infection. PLoS Pathog. 2008, 4, e18. [Google Scholar] [CrossRef]
- Sewram, S.; Singh, R.; Kormuth, E.; Werner, L.; Mlisana, K.; Karim, S.S.A.; Ndung’u, T. Human TRIM5α expression levels and reduced susceptibility to HIV-1 infection. J. Infect. Dis. 2009, 199, 1657–1663. [Google Scholar] [CrossRef]
- Anderson, J.; Akkina, R. Human immunodeficiency virus type 1 restriction by human-rhesus chimeric tripartite motif 5α (TRIM5α) in CD34+ cell-derived macrophages in vitro and in T cells in vivo in severe combined immunodeficient (SCID-hu) mice transplanted with human fetal tissue. Hum. Gene Ther. 2008, 19, 217–228. [Google Scholar] [CrossRef]
- Li, Y.; Li, X.; Stremlau, M.; Lee, M.; Sodroski, J. Removal of arginine 332 allows human TRIM5α to bind human immunodeficiency virus capsids and to restrict infection. J. Virol. 2006, 80, 6738–6744. [Google Scholar] [CrossRef]
- Luban, J. Cyclophilin a, TRIM5, and resistance to human immunodeficiency virus type 1 infection. J. Virol. 2007, 81, 1054–1061. [Google Scholar] [CrossRef]
- Ganser-Pornillos, B.K.; Chandrasekaran, V.; Pornillos, O.; Sodroski, J.G.; Sundquist, W.I.; Yeager, M. Hexagonal assembly of a restricting TRIM5α protein. Proc. Natl. Acad. Sci. USA 2011, 108, 534–539. [Google Scholar] [CrossRef]
- Stremlau, M.; Perron, M.; Lee, M.; Li, Y.; Song, B.; Javanbakht, H.; az-Griffero, F.; Anderson, D.J.; Sundquist, W.I.; Sodroski, J. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5α restriction factor. Proc. Natl. Acad. Sci. USA 2006, 103, 5514–5519. [Google Scholar] [CrossRef]
- Perron, M.J.; Stremlau, M.; Lee, M.; Javanbakht, H.; Song, B.; Sodroski, J. The human trim5alpha restriction factor mediates accelerated uncoating of the N-tropic murine leukemia virus capsid. J. Virol. 2007, 81, 2138–2148. [Google Scholar] [CrossRef]
- Black, L.R.; Aiken, C. TRIM5α disrupts the structure of assembled HIV-1 capsid complexes in vitro. J. Virol. 2010, 84, 6564–6569. [Google Scholar] [CrossRef]
- Zhao, G.; Ke, D.; Vu, T.; Ahn, J.; Shah, V.B.; Yang, R.; Aiken, C.; Charlton, L.M.; Gronenborn, A.M.; Zhang, P. Rhesus TRIM5α disrupts the HIV-1 capsid at the inter-hexamer interfaces. PLoS Pathog. 2011, 7, e1002009. [Google Scholar] [CrossRef] [Green Version]
- Rold, C.J.; Aiken, C. Proteasomal degradation of TRIM5α during retrovirus restriction. PLoS Pathog. 2008, 4, e1000074. [Google Scholar] [CrossRef]
- Anderson, J.L.; Campbell, E.M.; Wu, X.; Vandegraaff, N.; Engelman, A.; Hope, T.J. Proteasome inhibition reveals that a functional preintegration complex intermediate can be generated during restriction by diverse TRIM5 proteins. J. Virol. 2006, 80, 9754–9760. [Google Scholar] [CrossRef]
- Wu, X.; Anderson, J.L.; Campbell, E.M.; Joseph, A.M.; Hope, T.J. Proteasome inhibitors uncouple rhesus TRIM5α restriction of HIV-1 reverse transcription and infection. Proc. Natl. Acad. Sci. USA 2006, 103, 7465–7470. [Google Scholar] [CrossRef]
- Campbell, E.M.; Perez, O.; Anderson, J.L.; Hope, T.J. Visualization of a proteasome-independent intermediate during restriction of HIV-1 by rhesus TRIM5α. J. Cell Biol. 2008, 180, 549–561. [Google Scholar] [CrossRef]
- Campbell, E.M.; Dodding, M.P.; Yap, M.W.; Wu, X.; Gallois-Montbrun, S.; Malim, M.H.; Stoye, J.P.; Hope, T.J. TRIM5α cytoplasmic bodies are highly dynamic structures. Mol. Biol. Cell 2007, 18, 2102–2111. [Google Scholar] [CrossRef]
- Perez-Caballero, D.; Hatziioannou, T.; Zhang, F.; Cowan, S.; Bieniasz, P.D. Restriction of human immunodeficiency virus type 1 by TRIM-CypA occurs with rapid kinetics and independently of cytoplasmic bodies, ubiquitin, and proteasome activity. J. Virol. 2005, 79, 15567–15572. [Google Scholar] [CrossRef]
- Song, B.; Diaz-Griffero, F.; Park, D.H.; Rogers, T.; Stremlau, M.; Sodroski, J. TRIM5α association with cytoplasmic bodies is not required for antiretroviral activity. Virology 2005, 343, 201–211. [Google Scholar] [CrossRef]
- Diaz-Griffero, F.; Gallo, D.E.; Hope, T.J.; Sodroski, J. Trafficking of some old world primate TRIM5α proteins through the nucleus. Retrovirology 2011, 8, 38–38. [Google Scholar] [CrossRef]
- Tareen, S.U.; Emerman, M. Human TRIM5α has additional activities that are uncoupled from retroviral capsid recognition. Virology 2011, 409, 113–120. [Google Scholar] [CrossRef]
- Pertel, T.; Hausmann, S.P.; Morger, D.; Züger, S.; Guerra, J.; Lascano, J.; Reinhard, C.; Santoni, F.A.; Uchil, P.D.; Chatel, L.; et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 2011, 472, 361–365. [Google Scholar] [CrossRef]
- Gamble, T.R.; Vajdos, F.F.; Yoo, S.; Worthylake, D.K.; Houseweart, M.; Sundquist, W.I.; Hill, C.P. Crystal structure of human cyclophilin a bound to the amino-terminal domain of HIV-1 capsid. Cell 1996, 87, 1285–1294. [Google Scholar] [CrossRef]
- Bosco, D.A.; Eisenmesser, E.Z.; Pochapsky, S.; Sundquist, W.I.; Kern, D. Catalysis of cis/trans isomerization in native HIV-1 capsid by human cyclophilin A. Proc. Natl. Acad. Sci. USA 2002, 99, 5247–5252. [Google Scholar]
- Gitti, R.K.; Lee, B.M.; Walker, J.; Summers, M.F.; Yoo, S.; Sundquist, W.I. Structure of the amino-terminal core domain of the HIV-1 capsid protein. Science 1996, 273, 231–235. [Google Scholar]
- Yoo, S.; Myszka, D.G.; Yeh, C.; McMurray, M.; Hill, C.P.; Sundquist, W.I. Molecular recognition in the HIV-1 capsid/cyclophilin a complex. J. Mol. Biol. 1997, 269, 780–795. [Google Scholar] [CrossRef]
- Franke, E.K.; Yuan, H.E.; Luban, J. Specific incorporation of cyclophilin a into HIV-1 virions. Nature 1994, 372, 359–362. [Google Scholar] [CrossRef]
- Thali, M.; Bukovsky, A.; Kondo, E.; Rosenwirth, B.; Walsh, C.T.; Sodroski, J.; Ýttlinger, H.G. Functional association of cyclophilin a with HIV-1 virions. Nature 1994, 372, 363–365. [Google Scholar] [CrossRef]
- Braaten, D.; Luban, J. Cyclophilin a regulates HIV-1 infectivity, as demonstrated by gene targeting in human T cells. EMBO J. 2001, 20, 1300–1309. [Google Scholar] [CrossRef]
- Sokolskaja, E.; Sayah, D.M.; Luban, J. Target cell cyclophilin a modulates human immunodeficiency virus type 1 infectivity. J. Virol. 2004, 78, 12800–12808. [Google Scholar] [CrossRef]
- Hatziioannou, T.; Perez-Caballero, D.; Cowan, S.; Bieniasz, P.D. Cyclophilin interactions with incoming human immunodeficiency virus type 1 capsids with opposing effects on infectivity in human cells. J. Virol. 2005, 79, 176–183. [Google Scholar] [CrossRef]
- Diaz-Griffero, F.; Vandegraaff, N.; Li, Y.; McGee-Estrada, K.; Stremlau, M.; Welikala, S.; Si, Z.; Engelman, A.; Sodroski, J. Requirements for capsid-binding and an effector function in trimcyp-mediated restriction of HIV-1. Virology 2006, 351, 404–419. [Google Scholar] [CrossRef]
- Lin, T.Y.; Emerman, M. Cyclophilin a interacts with diverse lentiviral capsids. Retrovirology 2006, 3. [Google Scholar] [CrossRef]
- Zhang, F.; Hatziioannou, T.; Perez-Caballero, D.; Derse, D.; Bieniasz, P.D. Antiretroviral potential of human tripartite motif-5 and related proteins. Virology 2006, 353, 396–409. [Google Scholar] [CrossRef]
- Braaten, D.; Franke, E.K.; Luban, J. Cyclophilin a is required for an early step in the life cycle of human immunodeficiency virus type 1 before the initiation of reverse transcription. J. Virol. 1996, 70, 3551–3560. [Google Scholar]
- Li, Y.; Kar, A.K.; Sodroski, J. Target cell type-dependent modulation of human immunodeficiency virus type 1 capsid disassembly by cyclophilin A. J. Virol. 2009, 83, 10951–10962. [Google Scholar] [CrossRef]
- Towers, G.J. The control of viral infection by tripartite motif proteins and cyclophilin A. Retrovirology 2007, 4. [Google Scholar] [CrossRef]
- Sokolskaja, E.; Berthoux, L.; Luban, J. Cyclophilin A and TRIM5α independently regulate human immunodeficiency virus type 1 infectivity in human cells. J. Virol. 2006, 80, 2855–2862. [Google Scholar] [CrossRef]
- Battivelli, E.; Lecossier, D.; Matsuoka, S.; Migraine, J.; Clavel, F.; Hance, A.J. Strain-specific differences in the impact of human TRIM5α, different TRIM5α alleles, and the inhibition of capsid-cyclophilin a interactions on the infectivity of HIV-1. J. Virol. 2010, 84, 11010–11019. [Google Scholar] [CrossRef]
- Keckesova, Z.; Ylinen, L.M.; Towers, G.J. Cyclophilin A renders human immunodeficiency virus type 1 sensitive to old world monkey but not human TRIM5α antiviral activity. J. Virol. 2006, 80, 4683–4690. [Google Scholar] [CrossRef]
- Towers, G.J.; Hatziioannou, T.; Cowan, S.; Goff, S.P.; Luban, J.; Bieniasz, P.D. Cyclophilin A modulates the sensitivity of HIV-1 to host restriction factors. Nat. Med. 2003, 9, 1138–1143. [Google Scholar] [CrossRef]
- Rasaiyaah, J.T.; Tan, C.P.; Fletcher, A.J.; Price, A.J.; Blondeau, C.; Hilditch, L.; Jacques, D.A.; Selwood, D.L.; James, L.C.; Noursadeghi, M.; Towers, G.J. HIV-1 evades innate immune recognition through specific co-factor recruitment. Nature 2013. [Google Scholar] [CrossRef]
- Schaller, T.; Ocwieja, K.E.; Rasaiyaah, J.; Price, A.J.; Brady, T.L.; Roth, S.L.; Hué, S.; Fletcher, A.J.; Lee, K.; KewalRamani, V.N.; et al. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 2011, 7, e1002439. [Google Scholar] [CrossRef]
- Matreyek, K.A.; Yucel, S.S.; Li, X.; Engelman, A. Nucleoporin NUP153 phenylalanine-glycine motifs engage a common binding pocket within the HIV-1 capsid protein to mediate lentiviral infectivity. PLoS Pathog 2013, 9, e1003693. [Google Scholar] [CrossRef]
- Nisole, S.; Lynch, C.; Stoye, J.P.; Yap, M.W. A TRIM5-cyclophilin a fusion protein found in owl monkey kidney cells can restrict HIV-1. Proc. Natl. Acad. Sci. USA 2004, 101, 13324–13328. [Google Scholar] [CrossRef]
- Sayah, D.M.; Sokolskaja, E.; Berthoux, L.; Luban, J. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 2004, 430, 569–573. [Google Scholar] [CrossRef]
- Brennan, G.; Kozyrev, Y.; Hu, S.L. Trimcyp expression in old world primates macaca nemestrina and macaca fascicularis. Proc. Natl. Acad. Sci. USA 2008, 105, 3569–3574. [Google Scholar] [CrossRef]
- Newman, R.M.; Hall, L.; Kirmaier, A.; Pozzi, L.A.; Pery, E.; Farzan, M.; O’Neil, S.P.; Johnson, W. Evolution of a TRIM5-CypA splice isoform in old world monkeys. PLoS Pathog. 2008, 4, e1000003. [Google Scholar] [CrossRef]
- Langelier, C.R.; Sandrin, V.; Eckert, D.M.; Christensen, D.E.; Chandrasekaran, V.; Alam, S.L.; Aiken, C.; Olsen, J.C.; Kar, A.K.; Sodroski, J.G.; et al. Biochemical characterization of a recombinant TRIM5α protein that restricts human immunodeficiency virus type 1 replication. J. Virol. 2008, 82, 11682–11694. [Google Scholar] [CrossRef]
- Wilson, S.J.; Webb, B.L.; Ylinen, L.M.; Verschoor, E.; Heeney, J.L.; Towers, G.J. Independent evolution of an antiviral TRIMCyp in rhesus macaques. Proc. Natl. Acad. Sci. USA 2008, 105, 3557–3562. [Google Scholar] [CrossRef]
- Price, A.J.; Marzetta, F.; Lammers, M.; Ylinen, L.M.J.; Schaller, T.; Wilson, S.J.; Towers, G.J.; James, L.C. Active site remodeling switches HIV specificity of antiretroviral TRIMCyp. Nat. Struct. Mol. Biol. 2009, 16, 1036–1042. [Google Scholar] [CrossRef]
- Ylinen, L.M.J.; Price, A.J.; Rasaiyaah, J.; Hué, S.; Rose, N.J.; Marzetta, F.; James, L.C.; Towers, G.J. Conformational adaptation of asian macaque TRIMCyp directs lineage specific antiviral activity. PLoS Pathog. 2010, 6. [Google Scholar] [CrossRef]
- Malfavon-Borja, R.; Wu, L.I.; Emerman, M.; Malik, H.S. Birth, decay, and reconstruction of an ancient TRIMCyp gene fusion in primate genomes. Proc. Natl. Acad. Sci. USA 2013, 110, E583–E592. [Google Scholar] [CrossRef]
- Walker, J.E.; Chen, R.X.; McGee, J.; Nacey, C.; Pollard, R.B.; Abedi, M.; Bauer, G.; Nolta, J.A.; Anderson, J.S. Generation of an HIV-1-resistant immune system with CD34+ hematopoietic stem cells transduced with a triple-combination anti-HIV lentiviral vector. J. Virol. 2012, 86, 5719–5729. [Google Scholar] [CrossRef]
- Chan, E.; Schaller, T.; Eddaoudi, A.; Zhan, H.; Tan, C.P.; Jacobsen, M.; Thrasher, A.J.; Towers, G.J.; Qasim, W. Lentiviral gene therapy against human immunodeficiency virus type 1, using a novel human TRIM21-cyclophilin a restriction factor. Hum. Gene Ther. 2012, 23, 1176–1185. [Google Scholar] [CrossRef]
- Mallery, D.L.; McEwan, W.A.; Bidgood, S.R.; Towers, G.J.; Johnson, C.M.; James, L.C. Antibodies mediate intracellular immunity through tripartite motif-containing 21 (TRIM21). Proc. Natl. Acad. Sci. USA 2010, 107, 19985–19990. [Google Scholar]
- McEwan, W.A.; Tam, J.C.; Watkinson, R.E.; Bidgood, S.R.; Mallery, D.L.; James, L.C. Intracellular antibody-bound pathogens stimulate immune signaling via the Fc receptor TRIM21. Nat. Immunol. 2013, 14, 327–336. [Google Scholar] [CrossRef]
- Yap, M.W.; Dodding, M.P.; Stoye, J.P. TRIM-cyclophilin a fusion proteins can restrict human immunodeficiency virus type 1 infection at two distinct phases in the viral life cycle. J. Virol. 2006, 80, 4061–4067. [Google Scholar] [CrossRef]
- Schaller, T.; Ylinen, L.M.; Webb, B.L.; Singh, S.; Towers, G.J. Fusion of cyclophilin a to Fv1 enables cyclosporine-sensitive restriction of human and feline immunodeficiency viruses. J. Virol. 2007, 81, 10055–10063. [Google Scholar] [CrossRef]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 2013, 341. [Google Scholar] [CrossRef]
- Aiuti, A.; Biasco, L.; Scaramuzza, S.; Ferrua, F.; Cicalese, M.P.; Baricordi, C.; Dionisio, F.; Calabria, A.; Giannelli, S.; Castiello, M.C.; et al. Lentiviral hematopoietic stem cell gene therapy in patients with wiskott-aldrich syndrome. Science 2013, 341. [Google Scholar] [CrossRef]
- Hütter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Müssig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kücherer, C.; Blau, O.; et al. Long-term control of HIV by CCR5 delta32/delta32 stem-cell transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chan, E.; Towers, G.J.; Qasim, W. Gene Therapy Strategies to Exploit TRIM Derived Restriction Factors against HIV-1. Viruses 2014, 6, 243-263. https://doi.org/10.3390/v6010243
Chan E, Towers GJ, Qasim W. Gene Therapy Strategies to Exploit TRIM Derived Restriction Factors against HIV-1. Viruses. 2014; 6(1):243-263. https://doi.org/10.3390/v6010243
Chicago/Turabian StyleChan, Emma, Greg J. Towers, and Waseem Qasim. 2014. "Gene Therapy Strategies to Exploit TRIM Derived Restriction Factors against HIV-1" Viruses 6, no. 1: 243-263. https://doi.org/10.3390/v6010243