Genomic and Functional Characteristics of Human Cytomegalovirus Revealed by Next-Generation Sequencing

Abstract
:1. Introduction

2. HCMV Genomics before the Introduction of NGS
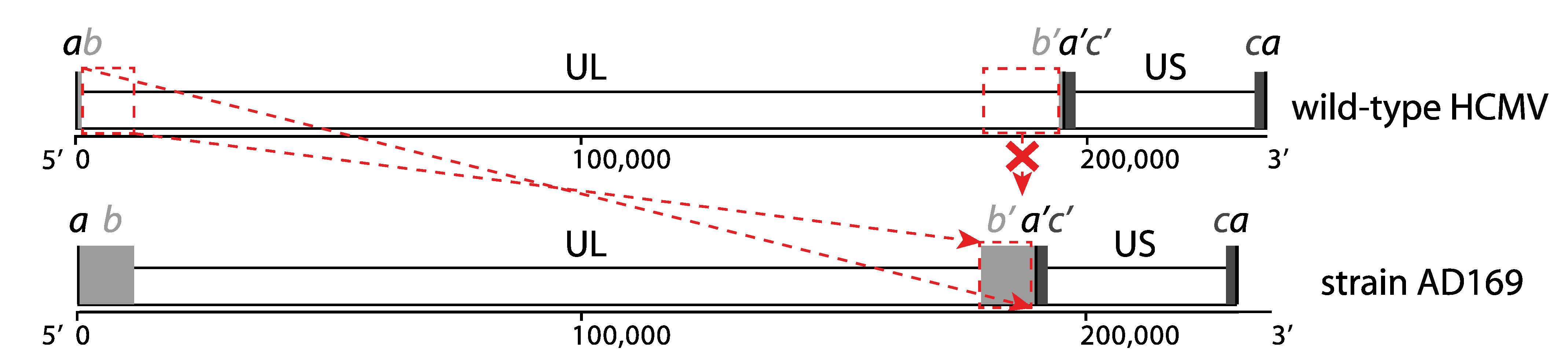
2.1. Genome Alterations during Cell Culture Adaptation

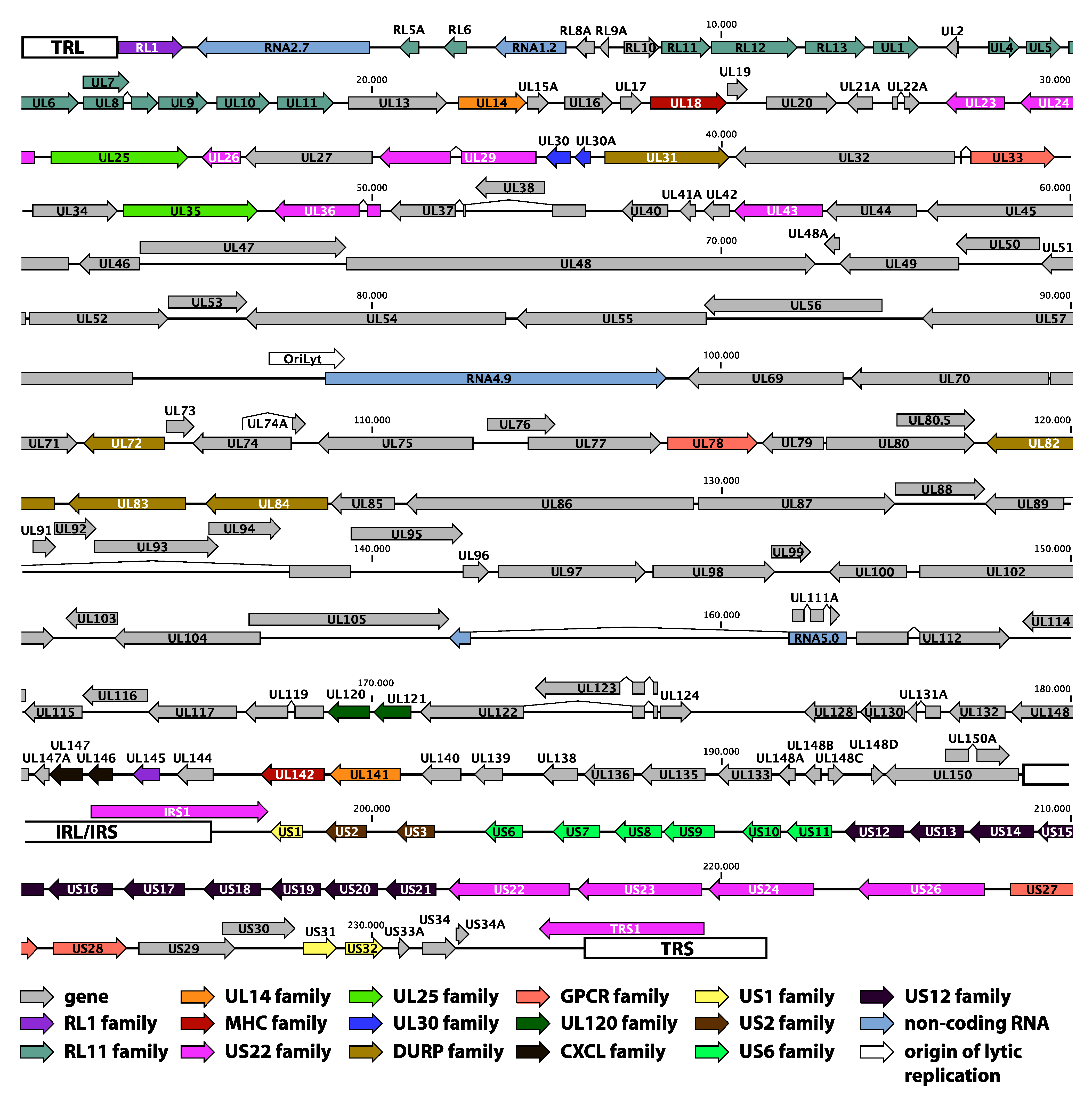
2.2. Genome Annotation

{kind=link}
{kind=link}
| GenBank accession | Strain name | Clinical source | Passage history | Ref. | Submission date |
|---|---|---|---|---|---|
| X17403 | AD169 | Adenoids of a 7-year old girl | Passaged extensively in human fibroblasts | [12] | December 6, 1989 |
| BK00039’ | AD169 varUK | Adenoids of a 7-year old girl | Passaged extensively in human fibroblasts | [40] | May 1, 2002 |
| NC_006273*GU179001 | Merlin | Urine from a congenitally infected infant | Passaged 3 times in human fibroblasts | [25] | September 27, 2002 |
| AY315197 | Towne varS | Urine of a 2-month-old infant with microcephaly and hepatosplenomegaly | Passaged extensively in human fibroblasts | [30] | June 6, 2003 |
| AC146851 | Towne-BAC | Urine of a 2-month-old infant with microcephaly and hepatosplenomegaly | BAC clone from a plaque purified Towne derivative (varS) | [39] | October 14, 2003 |
| AC146904 | PH-BAC | Transplant patient with HCMV disease | BAC clone from isolate PH (passaged less than 12 times) | [39] | October 21, 2003 |
| AC146905 | Toledo-BAC | Urine from a congenitally infected infant | BAC clone from a plaque purified Toledo derivative | [39] | October 21, 2013 |
| AC146906 | TR-BAC | AIDS patient with CMV retinitis | BAC clone from isolate TR | [39] | October 21, 2013 |
| AC146907 | FIX-BAC | Cervical secretions of a pregnant woman with a primary HCMV infection | BAC clone from isolate VR1814 | [39] | October 21, 2013 |
| AC146999 | AD169-BAC | Adenoids of a 7-year old girl | BAC clone from a plaque purified AD169 derivative (varATCC) | [39] | October 31, 2013 |
| EF999921 | TB40/E clone TB40-BAC4 | Throat wash of a bone marrow transplant recipient | BAC clone from TB40/E passaged 5 times in human fibroblasts and 22 times in human endothelial cells | [41] | June 25, 2007 |
| FJ527563 | AD169 varUC | Adenoids of a 7-year old girl | Passaged extensively in human fibroblasts | [20] | December 1, 2008 |
| FJ616285 | Towne varL | Urine of a 2-month-old infant with microcephaly and hepatosplenomegaly | Passaged extensively in human fibroblasts | [20] | January 9, 2009 |
| GQ221973 | HAN13 | Bronchoalveolar lavage | Passaged 3 times in human fibroblasts | [42] | May 28, 2009 |
| GQ221974 | 3157 | Urine from a congenitally infected infant | Passaged 3 times in human fibroblasts | [42] | May 28, 2009 |
| GQ221975 | JP | Post mortem prostate tissue from an AIDS patient | Unpassaged | [42] | May 28, 2009 |
| GQ396662 | HAN38 | Bronchoalveolar lavage | Passaged 2 times in human fibroblasts | [42] | July 17, 2009 |
| GQ396663 | HAN20 | Bronchoalveolar lavage | Passaged 2 times in human fibroblasts | [42] | July 17, 2009 |
| GQ466044 | 3301 | Urine from a congenitally infected infant | Unpassaged | [42] | August 7, 2009 |
| GU179288 | U8 | Urine from a congenitally infected infant | Unpassaged | [42] | November 5, 2009 |
| GU179289 | VR1814 | Cervical secretions of a pregnant woman with a primary HCMV infection | Unpassaged | [42] | November 5, 2009 |
| GU179290 | U11 | Urine from a congenitally infected infant | Unpassaged | [42] | November 5, 2009 |
| GU179291 | AF1 | Amniotic fluid | Unpassaged | [42] | November 5, 2009 |
| GU937742 | Toledo | Urine from a congenitally infected infant | Passaged several times in human fibroblasts | [25] | February 26, 2010 |
| HQ380895 | JHC | Blood from a bone marrow transplant patient | Plaque purified and passaged 3 times in human fibroblasts | [43] | October 7, 2010 |
| JX512197 | 6397 | Urine from a congenitally infected infant | Passaged 3 times in human fibroblasts | - | August 21, 2012 |
| JX512198 | Davis | Liver biopsy from a congenitally infected infant | Passaged many times in human fibroblasts | - | August 21, 2012 |
| JX512199 | HAN1 | Bronchoalveolar lavage | No information | - | August 21, 2012 |
| JX512200 | HAN2 | Bronchoalveolar lavage | Passaged 3 times in human fibroblasts | - | August 21, 2012 |
| JX512201 | HAN3 | Bronchoalveolar lavage | Passaged 3 times in human fibroblasts | - | August 21, 2012 |
| JX512202 | HAN8 | Bronchoalveolar lavage | Passaged 3 times in human fibroblasts | - | August 21, 2012 |
| JX512203 | HAN12 | Bronchoalveolar lavage | Passaged 3 times in human fibroblasts | - | August 21, 2012 |
| JX512204 | HAN16 | Urine from an infant | Passaged 2 times in human fibroblasts | - | August 21, 2012 |
| JX512205 | HAN19 | Bronchoalveolar lavage | Passaged 2 times in human fibroblasts | - | August 21, 2012 |
| JX512206 | HAN22 | Bronchoalveolar lavage | Passaged 2 times in human fibroblasts | - | August 21, 2012 |
| JX512207 | HAN28 | Bronchoalveolar lavage | Passaged 3 times in human fibroblasts | - | August 21, 2012 |
| JX512208 | HAN31 | Bronchoalveolar lavage | Passaged 2 times in human fibroblasts | - | August 21, 2012 |
| KC519319 | BE/9/2010 | Urine from an infant | Passaged 2 times in human fibroblasts | - | January 23, 2013 |
| KC519320 | BE/10/2010 | Urine from a congenitally infected infant | Passaged 2 times in human fibroblasts | - | January 23, 2013 |
| KC519321 | BE/11/2010 | Urine from an infant | Passaged 2 times in human fibroblasts | - | January 23, 2013 |
| KC519322 | BE/21/2010 | Urine from a pulmonary transplant recipient | Unpassaged | - | January 23, 2013 |
| KC519323 | BE/27/2010 | Urine from a renal transplant recipient | Passaged 4 times in human fibroblasts | - | January 23, 2013 |
| KF021605 | TR | Vitreous humor from eye of HIV-positive male | Passaged several times in human fibroblasts | [44] | May 9, 2013 |
| KF297339 | TB40/E clone Lisa | Throat wash of a bone marrow transplant recipient | Generated on human fibroblasts by passaging strain TB40/E once, plaque purifying 3 times and passaging once more | [45] | June 26, 2013 |
2.3. Genetic Diversity
3. Characterization of Complete HCMV Genomes Using NGS
| First author | Title | Journal | NGS technology | Ref. | Publication date |
|---|---|---|---|---|---|
| Bradley et al. | High-throughput sequence analysis of variants of human cytomegalovirus strains Towne and AD169. | J. Gen. Virol. | IGA° | [20] | June 24, 2009 |
| Cunningham et al. | Sequences of complete human cytomegalovirus genomes from infected cell cultures and clinical specimens. | J. Gen. Virol. | IGA° | [42] | November 11, 2009 |
| Görzer et al. | Deep sequencing reveals highly complex dynamics of human cytomegalovirus genotypes in transplant patients over time. | J. Virol. | GSF* | [77] | May 12, 2010 |
| Stanton et al. | Reconstruction of the complete human cytomegalovirus genome in a BAC reveals RL13 to be a potent inhibitor of replication. | J. Clin. Invest. | IGA° | [35] | August 2, 2010 |
| Görzer et al. | The impact of PCR-generated recombination on diversity estimation of mixed viral populations by deep sequencing. | J. Virol. Methods | GSF* | [78] | August 4, 2010 |
| Jung et al. | Full genome sequencing and analysis of human cytomegalovirus strain JHC isolated from a Korean patient. | Virus Res. | GSF* | [43] | January 19, 2011 |
| Renzette et al. | Extensive genome-wide variability of human cytomegalovirus in congenitally infected infants. | PLoS Pathog. | IGA° | [79] | May 19, 2011 |
| James et al. | Cyclopropavir inhibits the normal function of the human cytomegalovirus UL97 kinase. | Antimicrob. Agents Chemother. | IGA° | [80] | July 25, 2011 |
| Stark et al. | High-resolution profiling and analysis of viral and host small RNAs during human cytomegalovirus infection. | J. Virol. | IGA° | [81] | October 19, 2011 |
| Gatherer et al. | High-resolution human cytomegalovirus transcriptome. | Proc. Natl. Acad. Sci. U. S. A. | IGA° | [82] | November 22, 2011 |
| Bhattacharjee et al. | Genetic analysis of cytomegalovirus in malignant gliomas. | J. Virol. | IGA° | [83] | April 11, 2012 |
| Meshesha et al. | The microRNA Transcriptome of Human Cytomegalovirus (HCMV). | Open Virol. J. | IGA° | [84] | April 11, 2012 |
| Stern-Ginossar et al. | Decoding human cytomegalovirus. | Science | IGA°, HiSeq^ | [85] | November 23, 2012 |
| Rossetto et al. | Cis and trans acting factors involved in human cytomegalovirus experimental and natural latent infection of CD14 (+) monocytes and CD34 (+) cells. | PLoS Pathog. | MiSeq’ | [86] | May 23, 2013 |
| Sahoo et al. | Detection of cytomegalovirus drug resistance mutations by next-generation sequencing. | J. Clin. Microbiol. | GSJ” | [87] | August 28, 2013 |
| Renzette et al. | Rapid intrahost evolution of human cytomegalovirus is shaped by demography and positive selection. | PLoS Genet. | IGA° | [88] | September 26, 2013 |
| Brechtel et al. | Complete Genome Sequence of a Cytomegalovirus Towne-BAC (Bacterial Artificial Chromosome) Isolate Maintained in Escherichia coli for 10 Years and Then Serially Passaged in Human Fibroblasts. | Genome Announc. | MiSeq’ | [75] | September 26, 2013 |
| Brechtel et al. | Complete Genome Sequence of a UL96 Mutant Cytomegalovirus Towne-BAC (Bacterial Artificial Chromosome) Isolate Passaged in Fibroblasts To Allow Accumulation of Compensatory Mutations. | Genome Announc. | MiSeq’ | [76] | October 24, 2013 |
4. Deep Sequencing of Intrahost HCMV Populations
5. NGS in HCMV Transcriptome Studies
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Cannon, M.J.; Schmid, D.S.; Hyde, T.B. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev. Med. Virol. 2010, 20, 202–213. [Google Scholar] [CrossRef]
- Reeves, M.; Sinclair, J. Aspects of human cytomegalovirus latency and reactivation. Curr. Top. Microbiol. Immunol. 2008, 325, 297–313. [Google Scholar]
- Boeckh, M.; Geballe, A.P. Cytomegalovirus: Pathogen, paradigm, and puzzle. J. Clin. Investig. 2011, 121, 1673–1680. [Google Scholar] [CrossRef]
- Manicklal, S.; Emery, V.C.; Lazzarotto, T.; Boppana, S.B.; Gupta, R.K. The “silent” global burden of congenital cytomegalovirus. Clin. Microbiol. Rev. 2013, 26, 86–102. [Google Scholar] [CrossRef]
- Murphy, E.; Shenk, T. Human cytomegalovirus genome. Curr. Top. Microbiol. Immunol. 2008, 325, 1–19. [Google Scholar]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef]
- Zhang, J.; Chiodini, R.; Badr, A.; Zhang, G. The impact of next-generation sequencing on genomics. J. Genet. Genomics 2011, 38, 95–109. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Steinberg, K.M.; Larson, D.E.; Wilson, R.K.; Mardis, E.R. The next-generation sequencing revolution and its impact on genomics. Cell 2013, 155, 27–38. [Google Scholar] [CrossRef]
- Radford, A.D.; Chapman, D.; Dixon, L.; Chantrey, J.; Darby, A.C.; Hall, N. Application of next-generation sequencing technologies in virology. J. Gen. Virol. 2012, 93, 1853–1868. [Google Scholar]
- Capobianchi, M.R.; Giombini, E.; Rozera, G. Next-generation sequencing technology in clinical virology. Clin. Microbiol. Infect. 2013, 19, 15–22. [Google Scholar] [CrossRef]
- Barzon, L.; Lavezzo, E.; Costanzi, G.; Franchin, E.; Toppo, S.; Palu, G. Next-generation sequencing technologies in diagnostic virology. J. Clin. Virol. 2013, 58, 346–350. [Google Scholar] [CrossRef]
- Chee, M.S.; Bankier, A.T.; Beck, S.; Bohni, R.; Brown, C.M.; Cerny, R.; Horsnell, T.; Hutchison, C.A., 3rd.; Kouzarides, T.; Martignetti, J.A.; et al. Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr. Top. Microbiol. Immunol. 1990, 154, 125–169. [Google Scholar]
- Bankier, A.T.; Beck, S.; Bohni, R.; Brown, C.M.; Cerny, R.; Chee, M.S.; Hutchison, C.A., 3rd.; Kouzarides, T.; Martignetti, J.A.; Preddie, E.; et al. The DNA sequence of the human cytomegalovirus genome. DNA Seq. 1991, 2, 1–12. [Google Scholar]
- Just, M.; Buergin-Wolff, A.; Emoedi, G.; Hernandez, R. Immunisation trials with live attenuated cytomegalovirus TOWNE 125. Infection 1975, 3, 111–114. [Google Scholar] [CrossRef]
- Plotkin, S.A.; Farquhar, J.; Horberger, E. Clinical trials of immunization with the Towne 125 strain of human cytomegalovirus. J. Infect. Dis. 1976, 134, 470–475. [Google Scholar]
- Neff, B.J.; Weibel, R.E.; Buynak, E.B.; McLean, A.A.; Hilleman, M.R. Clinical and laboratory studies of live cytomegalovirus vaccine Ad-169. Proc. Soc. Exp. Biol. Med. 1979, 160, 32–37. [Google Scholar] [CrossRef]
- Quinnan, G.V., Jr.; Delery, M.; Rook, A.H.; Frederick, W.R.; Epstein, J.S.; Manischewitz, J.F.; Jackson, L.; Ramsey, K.M.; Mittal, K.; Plotkin, S.A.; et al. Comparative virulence and immunogenicity of the Towne strain and a nonattenuated strain of cytomegalovirus. Ann. Intern. Med. 1984, 101, 478–483. [Google Scholar] [CrossRef]
- Cha, T.A.; Tom, E.; Kemble, G.W.; Duke, G.M.; Mocarski, E.S.; Spaete, R.R. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J. Virol. 1996, 70, 78–83. [Google Scholar]
- Prichard, M.N.; Penfold, M.E.; Duke, G.M.; Spaete, R.R.; Kemble, G.W. A review of genetic differences between limited and extensively passaged human cytomegalovirus strains. Rev. Med. Virol. 2001, 11, 191–200. [Google Scholar] [CrossRef]
- Bradley, A.J.; Lurain, N.S.; Ghazal, P.; Trivedi, U.; Cunningham, C.; Baluchova, K.; Gatherer, D.; Wilkinson, G.W.; Dargan, D.J.; Davison, A.J. High-throughput sequence analysis of variants of human cytomegalovirus strains Towne and AD169. J. Gen. Virol. 2009, 90, 2375–2380. [Google Scholar] [CrossRef]
- Skaletskaya, A.; Bartle, L.M.; Chittenden, T.; McCormick, A.L.; Mocarski, E.S.; Goldmacher, V.S. A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc. Natl. Acad. Sci. USA 2001, 98, 7829–7834. [Google Scholar]
- Yu, D.; Smith, G.A.; Enquist, L.W.; Shenk, T. Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J. Virol. 2002, 76, 2316–2328. [Google Scholar] [CrossRef]
- Davison, A.J.; Akter, P.; Cunningham, C.; Dolan, A.; Addison, C.; Dargan, D.J.; Hassan-Walker, A.F.; Emery, V.C.; Griffiths, P.D.; Wilkinson, G.W. Homology between the human cytomegalovirus RL11 gene family and human adenovirus E3 genes. J. Gen. Virol. 2003, 84, 657–663. [Google Scholar] [CrossRef]
- Akter, P.; Cunningham, C.; McSharry, B.P.; Dolan, A.; Addison, C.; Dargan, D.J.; Hassan-Walker, A.F.; Emery, V.C.; Griffiths, P.D.; Wilkinson, G.W.; et al. Two novel spliced genes in human cytomegalovirus. J. Gen. Virol. 2003, 84, 1117–1122. [Google Scholar] [CrossRef]
- Dolan, A.; Cunningham, C.; Hector, R.D.; Hassan-Walker, A.F.; Lee, L.; Addison, C.; Dargan, D.J.; McGeoch, D.J.; Gatherer, D.; Emery, V.C.; et al. Genetic content of wild-type human cytomegalovirus. J. Gen. Virol. 2004, 85, 1301–1312. [Google Scholar] [CrossRef]
- Hahn, G.; Revello, M.G.; Patrone, M.; Percivalle, E.; Campanini, G.; Sarasini, A.; Wagner, M.; Gallina, A.; Milanesi, G.; Koszinowski, U.; et al. Human cytomegalovirus UL131–128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J. Virol. 2004, 78, 10023–10033. [Google Scholar] [CrossRef]
- Wang, D.; Shenk, T. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc. Natl. Acad. Sci. USA 2005, 102, 18153–18158. [Google Scholar] [CrossRef]
- Revello, M.G.; Gerna, G. Human cytomegalovirus tropism for endothelial/epithelial cells: Scientific background and clinical implications. Rev. Med. Virol. 2010, 20, 136–155. [Google Scholar] [CrossRef]
- Yu, D.; Silva, M.C.; Shenk, T. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 12396–12401. [Google Scholar] [CrossRef]
- Dunn, W.; Chou, C.; Li, H.; Hai, R.; Patterson, D.; Stolc, V.; Zhu, H.; Liu, F. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. USA 2003, 100, 14223–14228. [Google Scholar] [CrossRef]
- Sekulin, K.; Gorzer, I.; Heiss-Czedik, D.; Puchhammer-Stockl, E. Analysis of the variability of CMV strains in the RL11D domain of the RL11 multigene family. Virus Genes 2007, 35, 577–583. [Google Scholar] [CrossRef]
- Engel, P.; Perez-Carmona, N.; Alba, M.M.; Robertson, K.; Ghazal, P.; Angulo, A. Human cytomegalovirus UL7, a homologue of the SLAM-family receptor CD229, impairs cytokine production. Immunol. Cell Biol. 2011, 89, 753–766. [Google Scholar] [CrossRef]
- Gabaev, I.; Steinbruck, L.; Pokoyski, C.; Pich, A.; Stanton, R.J.; Schwinzer, R.; Schulz, T.F.; Jacobs, R.; Messerle, M.; Kay-Fedorov, P.C. The human cytomegalovirus UL11 protein interacts with the receptor tyrosine phosphatase CD45, resulting in functional paralysis of T cells. PLoS Pathog. 2011, 7, e1002432. [Google Scholar] [CrossRef]
- Cortese, M.; Calo, S.; D’Aurizio, R.; Lilja, A.; Pacchiani, N.; Merola, M. Recombinant Human Cytomegalovirus (HCMV) RL13 Binds Human Immunoglobulin G Fc. PloS One 2012, 7, e50166. [Google Scholar]
- Stanton, R.J.; Baluchova, K.; Dargan, D.J.; Cunningham, C.; Sheehy, O.; Seirafian, S.; McSharry, B.P.; Neale, M.L.; Davies, J.A.; Tomasec, P.; et al. Reconstruction of the complete human cytomegalovirus genome in a BAC reveals RL13 to be a potent inhibitor of replication. J. Clin. Investig. 2010, 120, 3191–3208. [Google Scholar]
- Dargan, D.J.; Douglas, E.; Cunningham, C.; Jamieson, F.; Stanton, R.J.; Baluchova, K.; McSharry, B.P.; Tomasec, P.; Emery, V.C.; Percivalle, E.; et al. Sequential mutations associated with adaptation of human cytomegalovirus to growth in cell culture. J. Gen. Virol. 2010, 91, 1535–1546. [Google Scholar] [CrossRef]
- Davison, A.J.; Dolan, A.; Akter, P.; Addison, C.; Dargan, D.J.; Alcendor, D.J.; McGeoch, D.J.; Hayward, G.S. The human cytomegalovirus genome revisited: comparison with the chimpanzee cytomegalovirus genome. J. Gen. Virol. 2003, 84, 17–28. [Google Scholar] [CrossRef]
- Murphy, E.; Rigoutsos, I.; Shibuya, T.; Shenk, T.E. Reevaluation of human cytomegalovirus coding potential. Proc. Natl. Acad. Sci. USA 2003, 100, 13585–13590. [Google Scholar] [CrossRef]
- Murphy, E.; Yu, D.; Grimwood, J.; Schmutz, J.; Dickson, M.; Jarvis, M.A.; Hahn, G.; Nelson, J.A.; Myers, R.M.; Shenk, T.E. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc. Natl. Acad. Sci. USA 2003, 100, 14976–14981. [Google Scholar] [CrossRef]
- Dargan, D.J.; Jamieson, F.E.; MacLean, J.; Dolan, A.; Addison, C.; McGeoch, D.J. The published DNA sequence of human cytomegalovirus strain AD169 lacks 929 base pairs affecting genes UL42 and UL43. J. Virol. 1997, 71, 9833–9836. [Google Scholar]
- Sinzger, C.; Hahn, G.; Digel, M.; Katona, R.; Sampaio, K.L.; Messerle, M.; Hengel, H.; Koszinowski, U.; Brune, W.; Adler, B. Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J. Gen. Virol. 2008, 89, 359–368. [Google Scholar] [CrossRef]
- Cunningham, C.; Gatherer, D.; Hilfrich, B.; Baluchova, K.; Dargan, D.J.; Thomson, M.; Griffiths, P.D.; Wilkinson, G.W.; Schulz, T.F.; Davison, A.J. Sequences of complete human cytomegalovirus genomes from infected cell cultures and clinical specimens. J. Gen. Virol. 2010, 91, 605–615. [Google Scholar] [CrossRef]
- Jung, G.S.; Kim, Y.Y.; Kim, J.I.; Ji, G.Y.; Jeon, J.S.; Yoon, H.W.; Lee, G.C.; Ahn, J.H.; Lee, K.M.; Lee, C.H. Full genome sequencing and analysis of human cytomegalovirus strain JHC isolated from a Korean patient. Virus Res. 2011, 156, 113–120. [Google Scholar] [CrossRef]
- Murrell, I.; Tomasec, P.; Wilkie, G.S.; Dargan, D.J.; Davison, A.J.; Stanton, R.J. Impact of sequence variation in the UL128 locus on production of human cytomegalovirus in fibroblast and epithelial cells. J. Virol. 2013, 87, 10489–10500. [Google Scholar] [CrossRef]
- Tomasec, P.; Wang, E.C.; Davison, A.J.; Vojtesek, B.; Armstrong, M.; Griffin, C.; McSharry, B.P.; Morris, R.J.; Llewellyn-Lacey, S.; Rickards, C.; et al. Downregulation of natural killer cell-activating ligand CD155 by human cytomegalovirus UL141. Nat. Immunol. 2005, 6, 181–188. [Google Scholar]
- Chou, S.W.; Dennison, K.M. Analysis of interstrain variation in cytomegalovirus glycoprotein B sequences encoding neutralization-related epitopes. J. Infect. Dis. 1991, 163, 1229–1234. [Google Scholar] [CrossRef]
- Pignatelli, S.; Dal Monte, P.; Landini, M.P. gpUL73 (gN) genomic variants of human cytomegalovirus isolates are clustered into four distinct genotypes. J. Gen. Virol. 2001, 82, 2777–2784. [Google Scholar]
- Rasmussen, L.; Geissler, A.; Cowan, C.; Chase, A.; Winters, M. The genes encoding the gCIII complex of human cytomegalovirus exist in highly diverse combinations in clinical isolates. J. Virol. 2002, 76, 10841–10848. [Google Scholar] [CrossRef]
- Lurain, N.S.; Kapell, K.S.; Huang, D.D.; Short, J.A.; Paintsil, J.; Winkfield, E.; Benedict, C.A.; Ware, C.F.; Bremer, J.W. Human cytomegalovirus UL144 open reading frame: Sequence hypervariability in low-passage clinical isolates. J. Virol. 1999, 73, 10040–10050. [Google Scholar]
- Lurain, N.S.; Fox, A.M.; Lichy, H.M.; Bhorade, S.M.; Ware, C.F.; Huang, D.D.; Kwan, S.P.; Garrity, E.R.; Chou, S. Analysis of the human cytomegalovirus genomic region from UL146 through UL147A reveals sequence hypervariability, genotypic stability, and overlapping transcripts. Virol. J. 2006, 3, 4. [Google Scholar] [CrossRef]
- Qi, Y.; Mao, Z.Q.; Ruan, Q.; He, R.; Ma, Y.P.; Sun, Z.R.; Ji, Y.H.; Huang, Y. Human cytomegalovirus (HCMV) UL139 open reading frame: Sequence variants are clustered into three major genotypes. J. Med. Virol. 2006, 78, 517–522. [Google Scholar] [CrossRef]
- Hitomi, S.; Kozuka-Hata, H.; Chen, Z.; Sugano, S.; Yamaguchi, N.; Watanabe, S. Human cytomegalovirus open reading frame UL11 encodes a highly polymorphic protein expressed on the infected cell surface. Arch. Virol. 1997, 142, 1407–1427. [Google Scholar] [CrossRef]
- Bar, M.; Shannon-Lowe, C.; Geballe, A.P. Differentiation of human cytomegalovirus genotypes in immunocompromised patients on the basis of UL4 gene polymorphisms. J. Infect. Dis. 2001, 183, 218–225. [Google Scholar] [CrossRef]
- Pignatelli, S.; Dal Monte, P.; Rossini, G.; Landini, M.P. Genetic polymorphisms among human cytomegalovirus (HCMV) wild-type strains. Rev. Med. Virol. 2004, 14, 383–410. [Google Scholar] [CrossRef]
- Puchhammer-Stockl, E.; Gorzer, I. Cytomegalovirus and Epstein-Barr virus subtypes—The search for clinical significance. J. Clin. Virol. 2006, 36, 239–248. [Google Scholar] [CrossRef]
- Puchhammer-Stockl, E.; Gorzer, I. Human cytomegalovirus: An enormous variety of strains and their possible clinical significance in the human host. Future Virol. 2011, 6, 259–271. [Google Scholar] [CrossRef]
- Stanton, R.; Westmoreland, D.; Fox, J.D.; Davison, A.J.; Wilkinson, G.W. Stability of human cytomegalovirus genotypes in persistently infected renal transplant recipients. J. Med. Virol. 2005, 75, 42–46. [Google Scholar] [CrossRef]
- Pignatelli, S.; Dal Monte, P.; Rossini, G.; Chou, S.; Gojobori, T.; Hanada, K.; Guo, J.J.; Rawlinson, W.; Britt, W.; Mach, M.; et al. Human cytomegalovirus glycoprotein N (gpUL73-gN) genomic variants: Identification of a novel subgroup, geographical distribution and evidence of positive selective pressure. J. Gen. Virol. 2003, 84, 647–655. [Google Scholar] [CrossRef]
- Bradley, A.J.; Kovacs, I.J.; Gatherer, D.; Dargan, D.J.; Alkharsah, K.R.; Chan, P.K.; Carman, W.F.; Dedicoat, M.; Emery, V.C.; Geddes, C.C.; et al. Genotypic analysis of two hypervariable human cytomegalovirus genes. J. Med. Virol. 2008, 80, 1615–1623. [Google Scholar] [CrossRef]
- Bates, M.; Monze, M.; Bima, H.; Kapambwe, M.; Kasolo, F.C.; Gompels, U.A. High human cytomegalovirus loads and diverse linked variable genotypes in both HIV-1 infected and exposed, but uninfected, children in Africa. Virology 2008, 382, 28–36. [Google Scholar] [CrossRef]
- McGeoch, D.J.; Rixon, F.J.; Davison, A.J. Topics in herpes virus genomics and evolution. Virus Res. 2006, 117, 90–104. [Google Scholar] [CrossRef]
- Chou, S.W. Reactivation and recombination of multiple cytomegalovirus strains from individual organ donors. J. Infect. Dis. 1989, 160, 11–15. [Google Scholar] [CrossRef]
- Rasmussen, L.; Geissler, A.; Winters, M. Inter- and intragenic variations complicate the molecular epidemiology of human cytomegalovirus. J. Infect. Dis. 2003, 187, 809–819. [Google Scholar] [CrossRef]
- Faure-Della Corte, M.; Samot, J.; Garrigue, I.; Magnin, N.; Reigadas, S.; Couzi, L.; Dromer, C.; Velly, J.F.; Dechanet-Merville, J.; Fleury, H.J.; et al. Variability and recombination of clinical human cytomegalovirus strains from transplantation recipients. J. Clin. Virol. 2010, 47, 161–169. [Google Scholar] [CrossRef]
- Shepp, D.H.; Match, M.E.; Ashraf, A.B.; Lipson, S.M.; Millan, C.; Pergolizzi, R. Cytomegalovirus glycoprotein B groups associated with retinitis in AIDS. J. Infect. Dis. 1996, 174, 184–187. [Google Scholar] [CrossRef]
- Torok-Storb, B.; Boeckh, M.; Hoy, C.; Leisenring, W.; Myerson, D.; Gooley, T. Association of specific cytomegalovirus genotypes with death from myelosuppression after marrow transplantation. Blood 1997, 90, 2097–2102. [Google Scholar]
- Rossini, G.; Pignatelli, S.; Dal Monte, P.; Camozzi, D.; Lazzarotto, T.; Gabrielli, L.; Gatto, M.R.; Landini, M.P. Monitoring for human cytomegalovirus infection in solid organ transplant recipients through antigenemia and glycoprotein N (gN) variants: Evidence of correlation and potential prognostic value of gN genotypes. Microbes Infect. 2005, 7, 890–896. [Google Scholar] [CrossRef]
- Pignatelli, S.; Lazzarotto, T.; Gatto, M.R.; Dal Monte, P.; Landini, M.P.; Faldella, G.; Lanari, M. Cytomegalovirus gN genotypes distribution among congenitally infected newborns and their relationship with symptoms at birth and sequelae. Clin. Infect. Dis. 2010, 51, 33–41. [Google Scholar] [CrossRef]
- Arav-Boger, R.; Willoughby, R.E.; Pass, R.F.; Zong, J.C.; Jang, W.J.; Alcendor, D.; Hayward, G.S. Polymorphisms of the cytomegalovirus (CMV)-encoded tumor necrosis factor-alpha and beta-chemokine receptors in congenital CMV disease. J. Infect. Dis. 2002, 186, 1057–1064. [Google Scholar] [CrossRef]
- Arav-Boger, R.; Battaglia, C.A.; Lazzarotto, T.; Gabrielli, L.; Zong, J.C.; Hayward, G.S.; Diener-West, M.; Landini, M.P. Cytomegalovirus (CMV)-encoded UL144 (truncated tumor necrosis factor receptor) and outcome of congenital CMV infection. J. Infect. Dis. 2006, 194, 464–473. [Google Scholar] [CrossRef]
- Waters, A.; Hassan, J.; de Gascun, C.; Kissoon, G.; Knowles, S.; Molloy, E.; Connell, J.; Hall, W.W. Human cytomegalovirus UL144 is associated with viremia and infant development sequelae in congenital infection. J. Clin. Microbiol. 2010, 48, 3956–3962. [Google Scholar] [CrossRef]
- Arav-Boger, R.; Boger, Y.S.; Foster, C.B.; Boger, Z. The use of artificial neural networks in prediction of congenital CMV outcome from sequence data. Bioinform. Biol. Insights 2008, 2, 281–289. [Google Scholar]
- Kemble, G.; Duke, G.; Winter, R.; Spaete, R. Defined large-scale alterations of the human cytomegalovirus genome constructed by cotransfection of overlapping cosmids. J. Virol. 1996, 70, 2044–2048. [Google Scholar]
- Hahn, G.; Rose, D.; Wagner, M.; Rhiel, S.; McVoy, M.A. Cloning of the genomes of human cytomegalovirus strains Toledo, TownevarRIT3, and Towne long as BACs and site-directed mutagenesis using a PCR-based technique. Virology 2003, 307, 164–177. [Google Scholar] [CrossRef]
- Brechtel, T.; Tyner, M.; Tandon, R. Complete Genome Sequence of a Cytomegalovirus Towne-BAC (Bacterial Artificial Chromosome) Isolate Maintained in Escherichia coli for 10 Years and Then Serially Passaged in Human Fibroblasts. Genome Announc. 2013, 1, e00693-13. [Google Scholar]
- Brechtel, T.M.; Tyner, M.; Tandon, R. Complete Genome Sequence of a UL96 Mutant Cytomegalovirus Towne-BAC (Bacterial Artificial Chromosome) Isolate Passaged in Fibroblasts To Allow Accumulation of Compensatory Mutations. Genome Announc. 2013, 1, e00901-13. [Google Scholar]
- Gorzer, I.; Guelly, C.; Trajanoski, S.; Puchhammer-Stockl, E. Deep sequencing reveals highly complex dynamics of human cytomegalovirus genotypes in transplant patients over time. J. Virol. 2010, 84, 7195–7203. [Google Scholar] [CrossRef]
- Gorzer, I.; Guelly, C.; Trajanoski, S.; Puchhammer-Stockl, E. The impact of PCR-generated recombination on diversity estimation of mixed viral populations by deep sequencing. J. Virol. Methods 2010, 169, 248–252. [Google Scholar] [CrossRef]
- Renzette, N.; Bhattacharjee, B.; Jensen, J.D.; Gibson, L.; Kowalik, T.F. Extensive genome-wide variability of human cytomegalovirus in congenitally infected infants. PLoS Pathog. 2011, 7, e1001344. [Google Scholar] [CrossRef]
- James, S.H.; Hartline, C.B.; Harden, E.A.; Driebe, E.M.; Schupp, J.M.; Engelthaler, D.M.; Keim, P.S.; Bowlin, T.L.; Kern, E.R.; Prichard, M.N. Cyclopropavir inhibits the normal function of the human cytomegalovirus UL97 kinase. Antimicrob. Agents Chemother. 2011, 55, 4682–4691. [Google Scholar] [CrossRef]
- Stark, T.J.; Arnold, J.D.; Spector, D.H.; Yeo, G.W. High-resolution profiling and analysis of viral and host small RNAs during human cytomegalovirus infection. J. Virol. 2012, 86, 226–235. [Google Scholar] [CrossRef]
- Gatherer, D.; Seirafian, S.; Cunningham, C.; Holton, M.; Dargan, D.J.; Baluchova, K.; Hector, R.D.; Galbraith, J.; Herzyk, P.; Wilkinson, G.W.; et al. High-resolution human cytomegalovirus transcriptome. Proc. Natl. Acad. Sci. USA 2011, 108, 19755–19760. [Google Scholar] [CrossRef]
- Bhattacharjee, B.; Renzette, N.; Kowalik, T.F. Genetic analysis of cytomegalovirus in malignant gliomas. J. Virol. 2012, 86, 6815–6824. [Google Scholar] [CrossRef]
- Meshesha, M.K.; Veksler-Lublinsky, I.; Isakov, O.; Reichenstein, I.; Shomron, N.; Kedem, K.; Ziv-Ukelson, M.; Bentwich, Z.; Avni, Y.S. The microRNA Transcriptome of Human Cytomegalovirus (HCMV). Open Virol. J. 2012, 6, 38–48. [Google Scholar] [CrossRef]
- Stern-Ginossar, N.; Weisburd, B.; Michalski, A.; Le, V.T.; Hein, M.Y.; Huang, S.X.; Ma, M.; Shen, B.; Qian, S.B.; Hengel, H.; et al. Decoding human cytomegalovirus. Science 2012, 338, 1088–1093. [Google Scholar] [CrossRef]
- Rossetto, C.C.; Tarrant-Elorza, M.; Pari, G.S. Cis and trans acting factors involved in human cytomegalovirus experimental and natural latent infection of CD14 (+) monocytes and CD34 (+) cells. PLoS Pathog. 2013, 9, e1003366. [Google Scholar] [CrossRef]
- Sahoo, M.K.; Lefterova, M.I.; Yamamoto, F.; Waggoner, J.J.; Chou, S.; Holmes, S.P.; Anderson, M.W.; Pinsky, B.A. Detection of cytomegalovirus drug resistance mutations by next-generation sequencing. J. Clin. Microbiol. 2013, 51, 3700–3710. [Google Scholar] [CrossRef]
- Renzette, N.; Gibson, L.; Bhattacharjee, B.; Fisher, D.; Schleiss, M.R.; Jensen, J.D.; Kowalik, T.F. Rapid intrahost evolution of human cytomegalovirus is shaped by demography and positive selection. PLoS Genet. 2013, 9, e1003735. [Google Scholar] [CrossRef]
- Gorzer, I.; Kerschner, H.; Redlberger-Fritz, M.; Puchhammer-Stockl, E. Human cytomegalovirus (HCMV) genotype populations in immunocompetent individuals during primary HCMV infection. J. Clin. Virol. 2010, 48, 100–103. [Google Scholar] [CrossRef]
- Ross, S.A.; Novak, Z.; Pati, S.; Patro, R.K.; Blumenthal, J.; Danthuluri, V.R.; Ahmed, A.; Michaels, M.G.; Sanchez, P.J.; Bernstein, D.I.; et al. Mixed infection and strain diversity in congenital cytomegalovirus infection. J. Infect. Dis. 2011, 204, 1003–1007. [Google Scholar] [CrossRef]
- Cicin-Sain, L.; Podlech, J.; Messerle, M.; Reddehase, M.J.; Koszinowski, U.H. Frequent coinfection of cells explains functional in vivo complementation between cytomegalovirus variants in the multiply infected host. J. Virol. 2005, 79, 9492–9502. [Google Scholar] [CrossRef]
- Humar, A.; Kumar, D.; Gilbert, C.; Boivin, G. Cytomegalovirus (CMV) glycoprotein B genotypes and response to antiviral therapy, in solid-organ-transplant recipients with CMV disease. J. Infect. Dis. 2003, 188, 581–584. [Google Scholar] [CrossRef]
- Coaquette, A.; Bourgeois, A.; Dirand, C.; Varin, A.; Chen, W.; Herbein, G. Mixed cytomegalovirus glycoprotein B genotypes in immunocompromised patients. Clin. Infect. Dis. 2004, 39, 155–161. [Google Scholar] [CrossRef]
- Puchhammer-Stockl, E.; Gorzer, I.; Zoufaly, A.; Jaksch, P.; Bauer, C.C.; Klepetko, W.; Popow-Kraupp, T. Emergence of multiple cytomegalovirus strains in blood and lung of lung transplant recipients. Transplantation 2006, 81, 187–194. [Google Scholar] [CrossRef]
- Manuel, O.; Asberg, A.; Pang, X.; Rollag, H.; Emery, V.C.; Preiksaitis, J.K.; Kumar, D.; Pescovitz, M.D.; Bignamini, A.A.; Hartmann, A.; et al. Impact of genetic polymorphisms in cytomegalovirus glycoprotein B on outcomes in solid-organ transplant recipients with cytomegalovirus disease. Clin. Infect. Dis. 2009, 49, 1160–1166. [Google Scholar] [CrossRef]
- Beerenwinkel, N.; Zagordi, O. Ultra-deep sequencing for the analysis of viral populations. Curr. Opin. Virol. 2011, 1, 413–418. [Google Scholar] [CrossRef]
- Beerenwinkel, N.; Gunthard, H.F.; Roth, V.; Metzner, K.J. Challenges and opportunities in estimating viral genetic diversity from next-generation sequencing data. Front. Microbiol. 2012, 3, 329. [Google Scholar] [Green Version]
- McGettigan, P.A. Transcriptomics in the RNA-seq era. Curr. Opin. Chem. Biol. 2013, 17, 4–11. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, N.; Li, M.; Gao, S.; Wang, L.; Zheng, B.; Qi, Y.; Ruan, Q. Human CMV transcripts: An overview. Future Microbiol. 2012, 7, 577–593. [Google Scholar] [CrossRef]
- Chinen, M.; Tani, T. Diverse functions of nuclear non-coding RNAs in eukaryotic gene expression. Front. Biosci. 2012, 17, 1402–1417. [Google Scholar] [CrossRef]
- Reeves, M.B.; Davies, A.A.; McSharry, B.P.; Wilkinson, G.W.; Sinclair, J.H. Complex I binding by a virally encoded RNA regulates mitochondria-induced cell death. Science 2007, 316, 1345–1348. [Google Scholar] [CrossRef]
- Su, W.Y.; Xiong, H.; Fang, J.Y. Natural antisense transcripts regulate gene expression in an epigenetic manner. Biochem. Biophys. Res. Commun. 2010, 396, 177–181. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Pfeffer, S.; Zavolan, M.; Grasser, F.A.; Chien, M.; Russo, J.J.; Ju, J.; John, B.; Enright, A.J.; Marks, D.; Sander, C.; et al. Identification of virus-encoded microRNAs. Science 2004, 304, 734–736. [Google Scholar] [CrossRef]
- Dunn, W.; Trang, P.; Zhong, Q.; Yang, E.; van Belle, C.; Liu, F. Human cytomegalovirus expresses novel microRNAs during productive viral infection. Cell. Microbiol. 2005, 7, 1684–1695. [Google Scholar] [CrossRef]
- Grey, F.; Antoniewicz, A.; Allen, E.; Saugstad, J.; McShea, A.; Carrington, J.C.; Nelson, J. Identification and characterization of human cytomegalovirus-encoded microRNAs. J. Virol. 2005, 79, 12095–12099. [Google Scholar]
- Tuddenham, L.; Pfeffer, S. Roles and regulation of microRNAs in cytomegalovirus infection. Biochim. Biophys. Acta 2011, 1809, 613–622. [Google Scholar] [CrossRef]
- Marcinowski, L.; Lidschreiber, M.; Windhager, L.; Rieder, M.; Bosse, J.B.; Radle, B.; Bonfert, T.; Gyory, I.; de Graaf, M.; Prazeres da Costa, O.; et al. Real-time transcriptional profiling of cellular and viral gene expression during lytic cytomegalovirus infection. PLoS Pathog. 2012, 8, e1002908. [Google Scholar] [CrossRef] [Green Version]
- Juranic Lisnic, V.; Babic Cac, M.; Lisnic, B.; Trsan, T.; Mefferd, A.; das Mukhopadhyay, C.; Cook, C.H.; Jonjic, S.; Trgovcich, J. Dual analysis of the murine cytomegalovirus and host cell transcriptomes reveal new aspects of the virus-host cell interface. PLoS Pathog. 2013, 9, e1003611. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, R.; Xie, H.; Hui, Y.; Jiao, R.; Gong, Y.; Zhang, Y. Advances in nanopore sequencing technology. J. Nanosci. Nanotechnol. 2013, 13, 4521–4538. [Google Scholar] [CrossRef]
- Coupland, P.; Chandra, T.; Quail, M.; Reik, W.; Swerdlow, H. Direct sequencing of small genomes on the Pacific Biosciences RS without library preparation. BioTechniques 2012, 53, 365–372. [Google Scholar]
- Thompson, J.F.; Milos, P.M. The properties and applications of single-molecule DNA sequencing. Genome Biol. 2011, 12, 217. [Google Scholar]
- Korlach, J.; Turner, S.W. Going beyond five bases in DNA sequencing. Curr. Opin. Struct. Biol. 2012, 22, 251–261. [Google Scholar] [CrossRef]
- Davis, B.M.; Chao, M.C.; Waldor, M.K. Entering the era of bacterial epigenomics with single molecule real time DNA sequencing. Curr. Opin. Microbiol. 2013, 16, 192–198. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sijmons, S.; Van Ranst, M.; Maes, P. Genomic and Functional Characteristics of Human Cytomegalovirus Revealed by Next-Generation Sequencing. Viruses 2014, 6, 1049-1072. https://doi.org/10.3390/v6031049
Sijmons S, Van Ranst M, Maes P. Genomic and Functional Characteristics of Human Cytomegalovirus Revealed by Next-Generation Sequencing. Viruses. 2014; 6(3):1049-1072. https://doi.org/10.3390/v6031049
Chicago/Turabian StyleSijmons, Steven, Marc Van Ranst, and Piet Maes. 2014. "Genomic and Functional Characteristics of Human Cytomegalovirus Revealed by Next-Generation Sequencing" Viruses 6, no. 3: 1049-1072. https://doi.org/10.3390/v6031049