
Sequence and Structure Analysis of Distantly-Related Viruses Reveals Extensive Gene Transfer between Viruses and Hosts and among Viruses

Abstract
:1. Introduction
2. Materials and Methods
3. Results
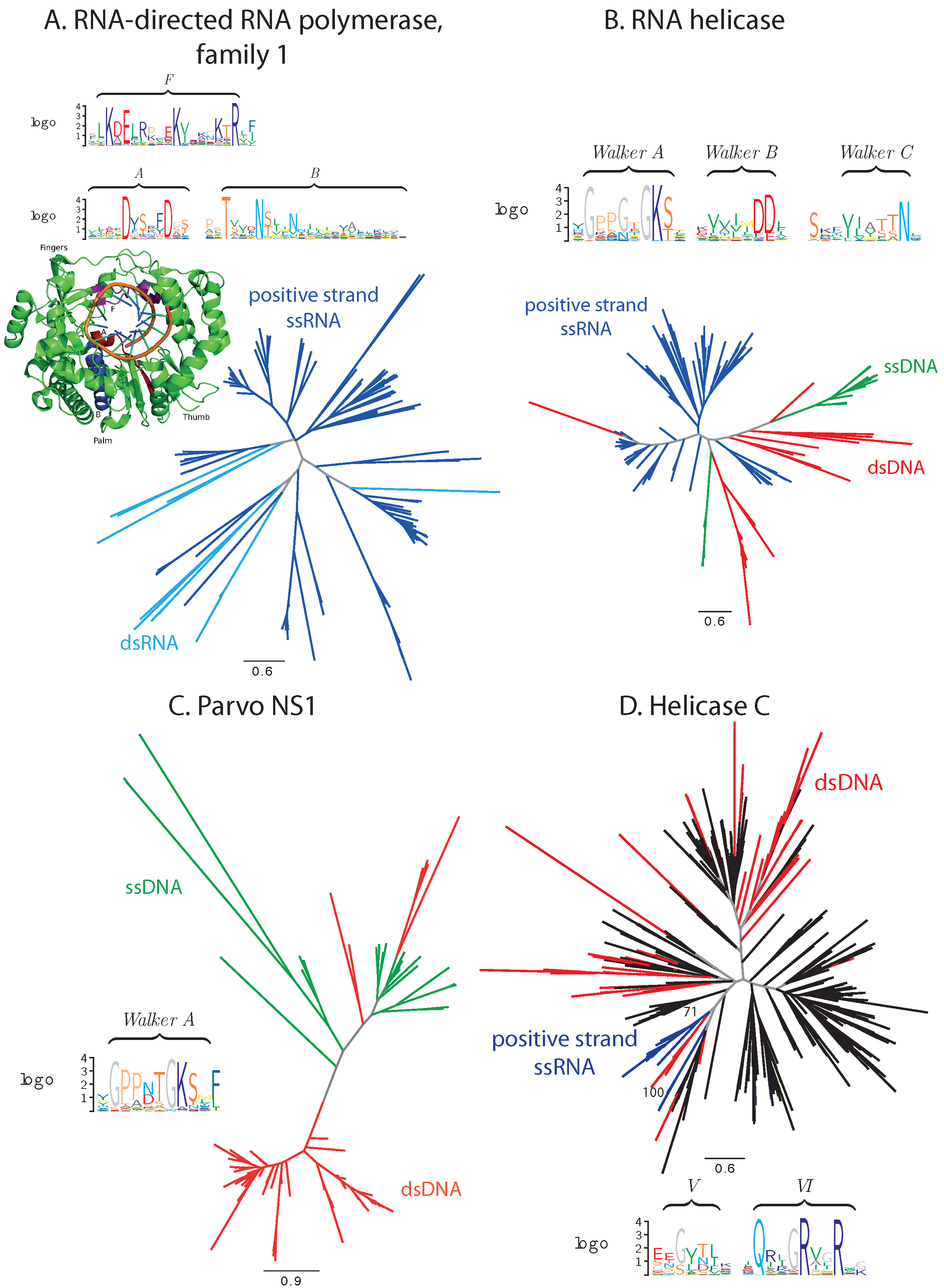
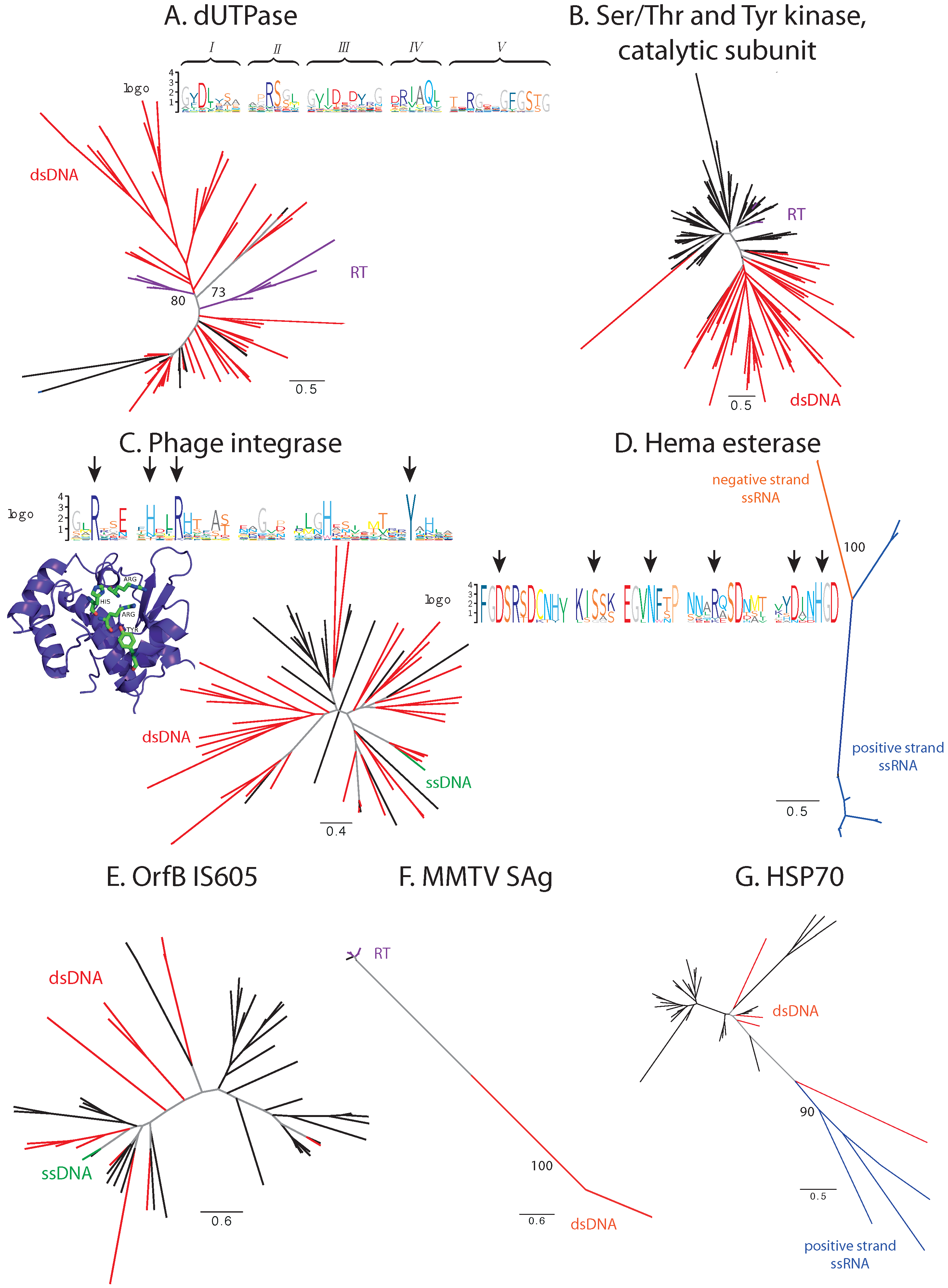
3.1. Sequence Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pfam Family | Structure | Number of | Matched Viruses and Their Types |
|---|---|---|---|
| Representative | Similar Pairs | ||
| PDB ID | |||
| Balanced sequence distribution | |||
| Helicase C | 4C9B | 3 | Molluscipoxvirus (dsDNA) |
| – Pestivirus (positive-strand ssRNA) | |||
| Parvo NS1 | 1U0J | 5 | Bocavirus (ssDNA) |
| – Fowl adenovirus A (dsDNA) | |||
| RdRP 1 | 2EC0 | 2 | Cryspovirus (dsRNA) |
| – Potyviridae (positive-strand ssRNA) | |||
| RNA helicase | – | 4 | Norovirus (positive-strand ssRNA) |
| – Circovirus (ssDNA) | |||
| Unbalanced sequence distribution | |||
| dUTPase | 1SYL | 106 | Betaretrovirus (retro-transcribing ssRNA) |
| – several dsDNA viruses | |||
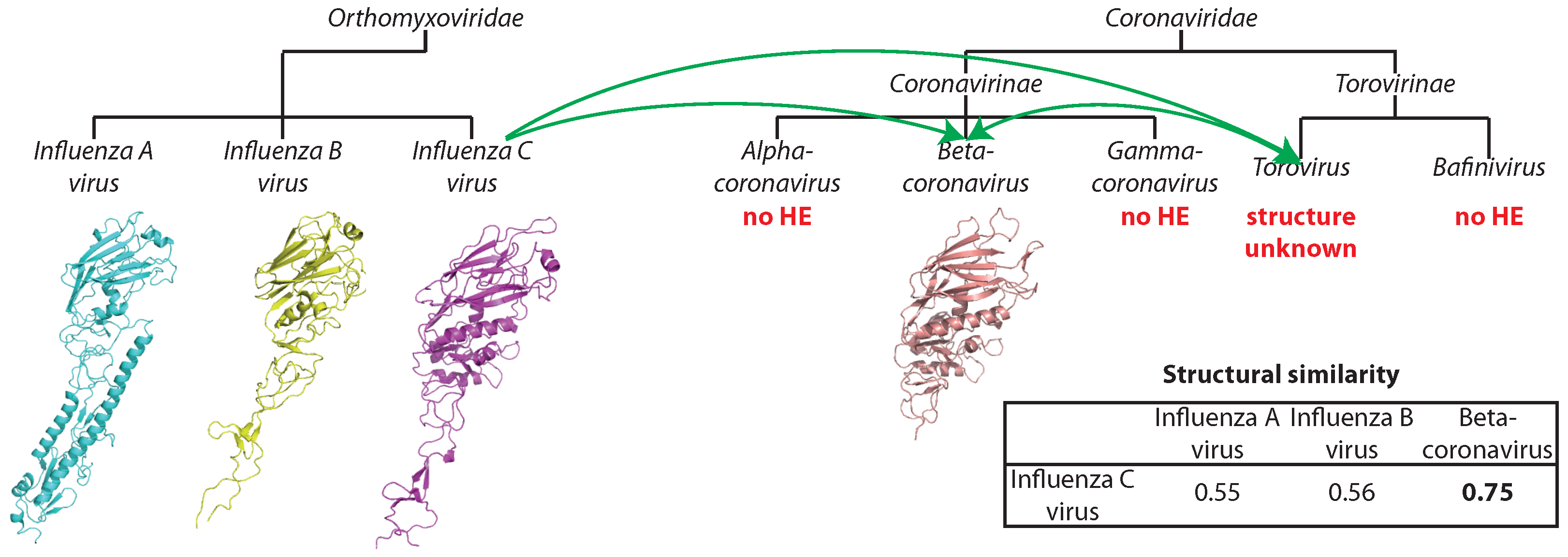
| Hema esterase | 3I27 | 4 | Influenza C virus (negative-strand ssRNA) |
| – Coronaviridae (positive-strand ssRNA) | |||
| HSP70 | 2V7Y | 1 | Cafeteriavirus (dsDNA) |
| – Velarivirus (positive-strand ssRNA) | |||
| MMTV SAg | – | 2 | Betaretrovirus (retro-transcribing ssRNA) |
| – Rhadinovirus (dsDNA) | |||
| OrfB IS605 | – | 4 | Inovirus (ssDNA) |
| – Myoviridae, Bicaudavirus (dsDNA) | |||
| Phage integrase | 1AIH | 4 | Inovirus (ssDNA) |
| – Caudovirales (dsDNA) | |||
| Pkinase | 2IVS | 5 | Alpharetrovirus (retro-transcribing ssRNA) |
| – Mimivirus (dsDNA) | |||
| Not considered in detail | |||
| Parvo coat N | – | 2 | Gammabaculovirus (dsDNA) |
| – Densovirinae (ssDNA) | |||
| – | – | 1 | Alphanudivirus (dsDNA) |
| – Ambidensovirus (ssDNA) | |||
3.1.1. Balanced Viral Sequence Distribution

3.1.2. Unbalanced Viral Sequence Distribution

3.2. Structural Analysis

3.2.1. Widely-Populated Folds
| Dataset | Groups | PDB ID and Chain | Description | Classes of Viral Genomes |
|---|---|---|---|---|
| Widely Populated Folds | ||||
| Capsid | Group 1 | 3NAPB, 1B35B, 4QPIB, | Capsid subunits with jelly roll fold | positive-strand ssRNA, ssDNA, dsDNA, dsRNA |
| 3CJIC, 4GMP0, 3CJIB, | ||||
| 1BEV3, 4QPIC, 1B35C, | ||||
| 3NAPC, 1F2NA, 1X35A, | ||||
| 2IZWA, 2FZ2A, 3ZXAC, | ||||
| 1OPOA, 2TBVA, 2WS91, | ||||
| 3CJIA, 4Q4W1, 4QPIA, | ||||
| 2MEV1, 1B35A, 3NAPA, | ||||
| 3IYOA, 2GH8A, 4NWVA, | ||||
| 1IHMA, 3J1PA, 1CWPA, | ||||
| 1F15A, 4V4MA, 3J4UH, | ||||
| 3J40H, 1YQ5A, 1A6CA, | ||||
| 1S58A, 3J1QA, 4G0RA, | ||||
| 3N7XA, 1DNVA, 3P0SA, | ||||
| 3R0RA, 2BBVA, 2WUYA, | ||||
| 2W0CL, 1SVA1, 1DZLA, | ||||
| 3FBMA, 1OHFA | ||||
| Group 2 | 1HX6A, 2BBDA, 2W0CA, | Capsid subunits with double jelly roll fold | dsDNA, unclassified virophage (Sputnik) | |
| 1M3YA, 3J26A | ||||
| Envelope | Group 1 | 4HJ1A, 3J0CA, 4ADIA, | Envelope glycoproteins | positive-strand ssRNA, |
| 3J27A, 2GG1A | negative-strand ssRNA | |||
| Poly- | 4GZKA, 4A8OA, 2R7RA, | RNA-dependent | dsRNA, | |
| merases | 1N35A, 3ZEDA, 4WSBB, | RNA polymerases | positive-strand ssRNA | |
| 4OBCA, 2CJQA, 4HDHA, | negative-strand ssRNA | |||
| 2EC0A, 3UQSA, 1KHVA | ||||
| Helicases | Group 1 | 2V9PA, 4GDFA, 1U0JA | Superfamily 3 helicases | dsDNA, ssDNA |
| Group 2 | 3VKWA, 4N0NA, 3UPUA, | Superfamily 1 and 2 helicases | positive-strand ssRNA, dsDNA | |
| 2OCAA, 3O8BA, 2WHXA | ||||
| Confined folds | ||||
| Proteases | 4QBBA, 4M0WA, 2J7QA, | papain-like | positive-strand ssRNA, | |
| 4IUMA, 3MTVA | cysteine proteases | dsDNA | ||
| Methyl- | 3MAGA, 2XYQA, 3EMDA | Methyltransferases | dsDNA | |
| transferases | positive-strand ssRNA | |||
| Envelope | Group 2 | 2WR1A, 3BT6A, 1FLCA, | Hemagglutinin, | negative-strand ssRNA, |
| 3CL5A | Hemagglutinin esterases | positive-strand ssRNA | ||
3.2.2. Confined Folds


4. Discussion
| Baltimore Classes | Family | Fold | Baltimore Classes | |
|---|---|---|---|---|
| Families with balanced sequence distribution/widely-populated folds | ||||
| dsRNA (22) | RdRP 1 | ↔ | Polymerases | dsRNA (5) |
| positive-strand ssRNA (205) | positive-strand ssRNA (6) | |||
| negative-strand ssRNA (1) | ||||
| dsDNA (31) | RNA helicase | ↔ | Helicases | dsDNA (2) |
| ssDNA (24) | Group 1 | ssDNA (1) | ||
| positive-strand ssRNA (124) | ||||
| dsDNA (101) | Helicase C | ↔ | Helicases | dsDNA (2) |
| positive-strand ssRNA (44) | Group 2 | positive-strand ssRNA (4) | ||
| dsDNA (76) | Parvo NS1 | |||
| ssDNA (34) | ||||
| Families with unbalanced sequence distribution / confined folds | ||||
| positive-strand ssRNA (16) | Hema esterase | ↔ | Envelope | positive-strand ssRNA (1) |
| negative-strand ssRNA (3) | Group 2 | negative-strand ssRNA (3) | ||
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dimmock, N.; Easton, A.; Leppard, K. Introduction to Modern Virology, 6th ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2009. [Google Scholar]
- Raoult, D.; Audic, S.; Robert, C.; Abergel, C.; Renesto, P.; Ogata, H.; la Scola, B.; Suzan, M.; Claverie, J. The 1.2-megabase genome sequence of Mimivirus. Science 2004, 306, 1344–1350. [Google Scholar] [CrossRef] [PubMed]
- Philippe, N.; Legendre, M.; Doutre, G.; Couté, Y.; Poirot, O.; Lescot, M.; Arslan, D.; Seltzer, V.; Bertaux, L.; Bruley, C.; et al. Pandoraviruses: Amoeba viruses with genomes up to 2.5 Mb reaching that of parasitic eukaryotes. Science 2013, 341, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Nasir, A.; Kim, K.; Caetano-Anolles, G. Giant viruses coexisted with the cellular ancestors and represent a distinct supergroup along with superkingdoms Archaea, Bacteria and Eukarya. BMC Evol. Biol. 2012, 12. [Google Scholar] [CrossRef] [PubMed]
- King, A.; Adams, M.; Carstens, E.; Lefkowitz, E. Virus Taxonomy: Classification and Nomenclature of Viruses. Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier: San Diego, CA, USA, 2012. [Google Scholar]
- Baltimore, D. Expression of animal virus genomes. Bacteriol. Rev. 1971, 35, 235–241. [Google Scholar] [PubMed]
- Koonin, E.; Senkevich, T.; Dolja, V. The ancient Virus World and evolution of cells. Biol. Direct. 2006, 1. [Google Scholar] [CrossRef]
- Krupovic, M.; Bamford, D. Protein Conservation in Virus Evolution; eLS: Chicago, IL, USA, 2011. [Google Scholar]
- Forterre, P. Defining life: The virus viewpoint. Orig. Life Evol. Biosph. 2010, 40, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Rossmann, M.; Johnson, J. Icosahedral RNA virus structure. Annu. Rev. Biochem. 1989, 58, 533–573. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Ravantti, J.; Bamford, D. Geminiviruses: A tale of a plasmid becoming a virus. BMC Evol. Biol. 2009, 9. [Google Scholar] [CrossRef] [PubMed]
- Raoult, D.; Forterre, P. Redefining viruses: Lessons from Mimivirus. Nat. Rev. Microbiol. 2008, 6, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Bamford, D.; Grimes, J.; Stuart, D. What does structure tell us about virus evolution? Curr. Opin. Struct. Biol. 2005, 15, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Bamford, D. Virus evolution: How far does the double β-barrel viral lineage extend? Nat. Rev. Microbiol. 2008, 6, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.; Dolya, V. Virus world as an evolutionary network of viruses and capsidless selfish elements. Microbiol. Mol. Biol. Rev. 2014, 78, 278–303. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Koonin, E. Polintons: A hotbed of eukaryotic virus, transposon and plasmid evolution. Nat. Rev. Microbiol. 2015, 13, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Ochman, H.; Lawrence, J.; Groisman, E.A. Lateral gene transfer and the nature of bacterial innovation. Nature 2000, 405, 299–304. [Google Scholar] [CrossRef] [PubMed]
- De la Cruz, F.; Davies, J. Horizontal gene transfer and the origin of species: Lessons from bacteria. Trends Microbiol. 2000, 8, 128–133. [Google Scholar] [CrossRef]
- Liu, H.; Fu, Y.; Li, B.; Yu, X.; Xie, J.; Cheng, J.; Ghabrial, S.; Li, G.; Yi, X.; Jiang, D. Widespread horizontal gene transfer from circular single-stranded DNA viruses to eukaryotic genomes. BMC Evol. Biol. 2011, 11. [Google Scholar] [CrossRef] [PubMed]
- Boto, L. Horizontal gene transfer in the acquisition of novel traits by metazoans. Proc. Biol. Sci. 2014, 281. [Google Scholar] [CrossRef] [PubMed]
- Filée, J.; Pouget, N.; Chandler, M. Phylogenetic evidence for extensive lateral acquisition of cellular genes by Nucleocytoplasmic large DNA viruses. BMC Evol. Biol. 2008, 8. [Google Scholar] [CrossRef] [PubMed]
- Monier, A.; Pagarete, A.; de Vargas, C.; Allen, M.; Read, B.; Claverie, J.M.; Ogata, H. Horizontal gene transfer of an entire metabolic pathway between a eukaryotic alga and its DNA virus. Genome Res. 2009, 19, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Odom, M.; Hendrickson, R.; Lefkowitz, E. Poxvirus protein evolution: Family wide assessment of possible horizontal gene transfer events. Virus Res. 2009, 144, 233–249. [Google Scholar] [CrossRef] [PubMed]
- La Scola, B.; Desnues, C.; Pagnier, I.; Robert, C.; Barrassi, L.; Fournous, G.; Merchat, M.; Suzan-Monti, M.; Forterre, P.; Koonin, E.; et al. The virophage as a unique parasite of the giant mimivirus. Nature 2008, 455, 100–104. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res. 2014, 42, D191–D198. [Google Scholar]
- Altschul, S.; Gish, W.; Miller, W.; Myers, E.; Lipman, D. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Punta, M.; Coggill, P.; Eberhardt, R.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J.; et al. The Pfam protein families database. Nucleic Acids Res. 2012, 40, D290–D301. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.; Clements, J.; Eddy, S. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML Version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.; Scornavacca, C. Dendroscope 3: An interactive tool for rooted phylogenetic trees and networks. Syst. Biol. 2012, 61, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, R.; Akhtar, R.; Birney, E.; Bower, L.; Cerdeno-Tárraga, A.; Cheng, Y.; Cleland, I.; Faruque, N.; Goodgame, N.; Gibson, R.; et al. The European nucleotide archive. Nucleic Acids Res. 2011, 39, D28–D31. [Google Scholar] [CrossRef] [PubMed]
- Peden, J. CodonW. Available online: http://codonw.sourceforge.net/ (accessed on 1 April 2013).
- Bernstein, F.; Koetzle, T.; Williams, G.; Meyer, E., Jr.; Brice, M.; Rogers, J.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The Protein Data Bank: A computer-based archival file for macromolecular structures. J. Mol. Biol. 1977, 112, 535–542. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. CD-HIT: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins 2004, 57, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics 2010, 26, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Rosenström, P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010, 38, W545–W549. [Google Scholar] [CrossRef]
- Pettersen, E.; Goddard, T.; Huang, C.; Couch, G.; Greenblatt, D.; Meng, E.; Ferrin, T. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.; Saraste, M.; Runswick, M.; Gay, N. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP requiring enzymes and a common nucleotide binding fold. EMBO J. 1982, 1, 945–951. [Google Scholar] [PubMed]
- Sharma, D.; Say, A.; Ledford, L.; Hughes, A.; Sehorn, H.; Dwyer, D.; Sehorn, M. Role of the conserved lysine within the Walker A motif of human DMC1. DNA Repair 2013, 12, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Orta, C.; Arias, A.; Perez-Luque, R.; Escarmís, C.; Domingo, E.; Verdaguer, N. Structure of foot-and-mouth disease virus RNA-dependent RNA polymerase and its complex with a template-primer RNA. J. Biol. Chem. 2004, 279, 47212–47221. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, M.; Weiller, G. Evidence that a plant virus switched hosts to infect a vertebrate and then recombined with a vertebrate-infecting virus. Proc. Natl. Acad. Sci. USA 1999, 96, 8022–8027. [Google Scholar] [CrossRef] [PubMed]
- Saraste, M.; Sibbald, P.; Wittinghofer, A. The P-loop—A common motif in ATP- and GTP-binding proteins. Trends Biochem. Sci. 1990, 15, 430–434. [Google Scholar] [CrossRef]
- Caruthers, J.; Johnson, E.; McKay, D. Crystal structure of yeast initiation factor 4A, a DEAD-box RNA helicase. Proc. Natl. Acad. Sci. USA 2000, 97, 13080–13085. [Google Scholar] [CrossRef]
- Vértessy, B.; Tóth, J. Keeping uracil out of DNA: Physiological role, structure and catalytic mechanism of dUTPases. Acc. Chem. Res. 2009, 42, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Barabás, O.; Nemeth-Pongracz, V.; Magyar, A.; Hudecz, F.; Vértessy, B. Role of the C-terminal conserved sequence motif in the function of dUTPase from Mason-Pfizer monkey retrovirus. J. Pept. Sci. 2006, 12, 206. [Google Scholar]
- Leonard, C.; Aravind, L.; Koonin, E. Novel families of putative protein kinases in bacteria and Archaea: evolution of the “eukaryotic” protein kinase superfamily. Genome Res. 1998, 8, 1038–1047. [Google Scholar] [PubMed]
- Hickman, A.; Waninger, S.; Scocca, J.; Dyda, F. Molecular organization in site-specific recombination: The catalytic domain of bacteriophage HP1 integrase at 2.7 Å resolution. Cell 1997, 89, 227–237. [Google Scholar] [CrossRef]
- De Groot, R. Structure, function and evolution of the hemagglutininesterase proteins of corona- and toroviruses. Glycoconj. J. 2006, 23, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Acha-Orbea, H.; MacDonald, H. Superantigens of mouse mammary tumor virus. Annu. Rev. Immunol. 1995, 13, 459–486. [Google Scholar] [CrossRef] [PubMed]
- Knappe, A.; Hiller, C.; Thurau, M.; Wittmann, S.; Hofmann, H.; Fleckenstein, B.; Fickenscher, H. The superantigen-homologous viral immediateearly gene ie14/vsag in herpesvirus saimiri-transformed human T cells. J. Virol. 1997, 71, 9124–9133. [Google Scholar] [PubMed]
- Peremyslov, V.; Andreev, I.; Prokhnevsky, A.; Duncan, G.; Taliansky, M.; Dolja, V. Complex molecular architecture of beet yellow virus particles. Proc. Natl. Acad. Sci. USA 2004, 101, 5030–5035. [Google Scholar] [CrossRef] [PubMed]
- Lange, M.; Lehle, J. The genome of Cryptophlebia leucotreta granulovirus. Virology 2003, 317, 220–236. [Google Scholar] [CrossRef]
- Morse, M.; Marriott, A.; Nuttall, P. The glycoprotein of Thogoto virus (a tick-borne othomyxo-like virus) is related to the baculovirus glycoprotein GP64. Virology 1992, 186, 640–646. [Google Scholar] [CrossRef]
- Van Beurden, S.; Leroy, B.; Wattiez, R.; Haenen, O.; Boeren, S.; Vervoort, J.; Peeters, B.; Rottier, P.; Engelsma, M.; Vanderplasschen, A. Identification and localization of the structural proteins of anguillid herpesvirus 1. Vet. Res. 2011, 42. [Google Scholar] [CrossRef] [PubMed]
- Mushegian, A.; Elena, S. Evolution of plant virus movement proteins from the 30K superfamily and their homologs integrated in plant genomes. Virology 2015, 476, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Rost, B. Twilight zone of protein sequence alignments. Protein Eng. 1999, 12, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Irigoyen, N.; Garriga, D.; Navarro, A.; Verdaguer, N.; Rodriguez, J.; Caston, J. Autoproteolytic activity derived from the infectious bursal disease virus capsid protein. J. Biol. Chem. 2009, 284, 8064–8072. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sun, S.; Xiang, Y.; Wong, J.; Klose, T.; Raoult, D.; Rossmann, M. Structure of Sputnik, a virophage, at 3.5 Å resolution. Proc. Natl. Acad. Sci. USA 2012, 109, 18431–18436. [Google Scholar] [CrossRef] [PubMed]
- Benson, S.; Bamford, J.; Bamford, D.; Burnett, R. The X-ray crystal structure of P3, the major coat protein of the lipid-containing bacteriophage PRD1,at 1.65 Å resolution. Acta Crystallogr. Sect. D 2002, 39, 39–59. [Google Scholar] [CrossRef]
- Khayat, R.; Tang, L.; Larson, E.; Lawrence, M.; Young, M.; Johnson, J. Structure of an Archaeal virus capsid protein reveals a common ancestry to eukaryotic and bacterial viruses. Proc. Natl. Acad. Sci. USA 2005, 102, 18944–18949. [Google Scholar] [CrossRef] [PubMed]
- Abrescia, N.; Grimes, J.; Kivela, H.; Assenberg, R.; Sutton, G.; Butcher, S.; Bamford, J.K.H.; Bamford, D.H.; Stuart, D.I. Insights into virus evolution and membrane biogenesis from the structure of the marine lipid-containing bacteriophage PM2. Mol. Cell 2008, 31, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Nandhagopal, N.; Simpson, A.; Gurnon, J.; Yan, X.; Baker, T.; Graves, M.; van Etten, J.; Rossmann, M. The structure and evolution of the major capsid protein of a large, lipid-containing DNA virus. Proc. Natl. Acad. Sci. USA 2002, 99, 14758–14763. [Google Scholar] [CrossRef] [PubMed]
- Dessau, M.; Modis, Y. Crystal structure of glycoprotein C from Rift Valley fever virus. Proc. Natl. Acad. Sci. USA 2013, 110, 1696–1701. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Vargas, J.; Krey, T.; Valansi, C.; Avinoam, O.; Haouz, A.; Jamin, M.; Raveh-Barak, H.; Podbilewicz, B.; Rey, F. Structural basis of eukaryotic cell-cell fusion. Cell 2014, 157, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Gibrat, J.F.; Mariadassou, M.; Boudinot, P.; Delmas, B. Analyses of the radiation of birnaviruses from diverse host phyla and of their evolutionary affinities with other double-stranded RNA and positive strand RNA viruses using robust structure-based multiple sequence alignments and advanced phylogenetic methods. BMC Evol. Biol. 2013, 13. [Google Scholar] [CrossRef]
- Reich, S.; Guilligay, D.; Pflug, A.; Malet, H.; Berger, I.; Crépin, T.; Hart, D.; Lunardi, T.; Nanao, M.; Ruigrok, R.; et al. Structural insight into cap-snatching and RNA synthesis by influenza polymerase. Nature 2014, 516, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Salgado, P.S.; Koivunen, M.R.L.; Makeyev, E.V.; Bamford, D.H.; Stuart, D.I.; Grimes, J.M. The structure of an RNAi polymerase links RNA silencing and transcription. PLoS Biol. 2006, 4, e434. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Byrd, A.; Yun, M.; Pemble, C.; Harrison, D.; Yeruva, L.; Dahl, C.; Kreuzer, K.; Raney, K.; White, S. The T4 phage SF1B helicase Dda is structurally optimized to perform DNA strand separation. Structure 2012, 20, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.; Koonin, E. Helicases: Amino acid sequence comparisons and structure-function relationships. Curr. Opin. Struct. Biol. 1993, 3, 419–429. [Google Scholar] [CrossRef]
- Zeng, Q.; Langereis, M.; van Viet, A.; Huizinga, E.; de Groot, R. Structure of coronavirus hemagglutinin-esterase offers insight into Corona- and Influenza virus evolution. Proc. Natl. Acad. Sci. USA 2008, 105, 9065–9069. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Cheng, F.; Lu, M.; Tian, X.; Ma, J. Crystal structure of unliganded influenza B virus hemagglutinin. J. Virol. 2008, 82, 3011–3020. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.; Dolja, V. Evolution and taxonomy of positive-strand RNA viruses: Implications of comparative analysis of amino acid sequences. Crit. Rev. Biochem. Mol. Biol. 1993, 28, 375–430. [Google Scholar] [CrossRef] [PubMed]
- Abrescia, N.G.; Bamford, D.H.; Grimes, J.M.; Stuart, D.I. Structure unifies the viral universe. Ann. Rev. Biochem. 2012, 81, 795–822. [Google Scholar] [CrossRef] [PubMed]
- Sillitoe, I.; Lewis, T.E.; Cuff, A.; Das, S.; Ashford, P.; Dawson, N.L.; Furnham, N.; Laskowski, R.A.; Lee, D.; Lees, J.G.; et al. CATH: Comprehensive structural and functional annotations for genome sequences. Nucleic Acids Res. 2015, 43, D376–D381. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, A.; Howorth, D.; Chandonia, J.M.; Brenner, S.E.; Hubbard, T.J.P.; Chothia, C.; Murzin, A.G. Data growth and its impact on the SCOP database: New developments. Nucleic Acids Res. 2007, 36, D419–D425. [Google Scholar] [CrossRef] [PubMed]
- Moreira, D. Multiple independent horizontal transfers of informational genes from bacteria to plasmids and phages: implications for the origin of bacterial replication machinery. Mol. Microbiol. 2000, 35, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.; Makarova, K.; Aravind, L. Horizontal gene transfer in prokaryotes: quantification and classification. Ann. Rev. Microbiol. 2001, 55, 709–742. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, P.; Bize, L.; Muri, F.; Hoebeke, M.; Rodolphe, F.; Ehrlich, S.D.; Prum, B.; Bessières, P. Mining Bacillus subtilis chromosome heterogeneities using hidden Markov models. Nucleic Acids Res. 2002, 30, 1418–1426. [Google Scholar] [CrossRef] [PubMed]
- Tsirigos, A.; Rigoutsos, I. A sensitive, support-vector-machine method for the detection of horizontal gene transfers in viral, Archaeal and bacterial genomes. Nucleic Acids Res. 2005, 33, 3699–3707. [Google Scholar] [CrossRef] [PubMed]
- Metzler, S.; Kalinina, O.V. Detection of atypical genes in virus families using a one-class SVM. BMC Genomics 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Yutin, N. Origin and evolution of eukaryotic large nucleo-cytoplasmic DNA viruses. Intervirology 2010, 53, 284–292. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caprari, S.; Metzler, S.; Lengauer, T.; Kalinina, O.V. Sequence and Structure Analysis of Distantly-Related Viruses Reveals Extensive Gene Transfer between Viruses and Hosts and among Viruses. Viruses 2015, 7, 5388-5409. https://doi.org/10.3390/v7102882
Caprari S, Metzler S, Lengauer T, Kalinina OV. Sequence and Structure Analysis of Distantly-Related Viruses Reveals Extensive Gene Transfer between Viruses and Hosts and among Viruses. Viruses. 2015; 7(10):5388-5409. https://doi.org/10.3390/v7102882
Chicago/Turabian StyleCaprari, Silvia, Saskia Metzler, Thomas Lengauer, and Olga V. Kalinina. 2015. "Sequence and Structure Analysis of Distantly-Related Viruses Reveals Extensive Gene Transfer between Viruses and Hosts and among Viruses" Viruses 7, no. 10: 5388-5409. https://doi.org/10.3390/v7102882