
Two Novel Myoviruses from the North of Iraq Reveal Insights into Clostridium difficile Phage Diversity and Biology

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Phage Isolation
2.2. Phage Host Range Assay
2.3. Purification of Phage Genomic DNA
2.4. Phage Genome Sequencing
2.5. Phylogenetic Analyses and Comparative Genomics
2.6. Protein Analysis
3. Results
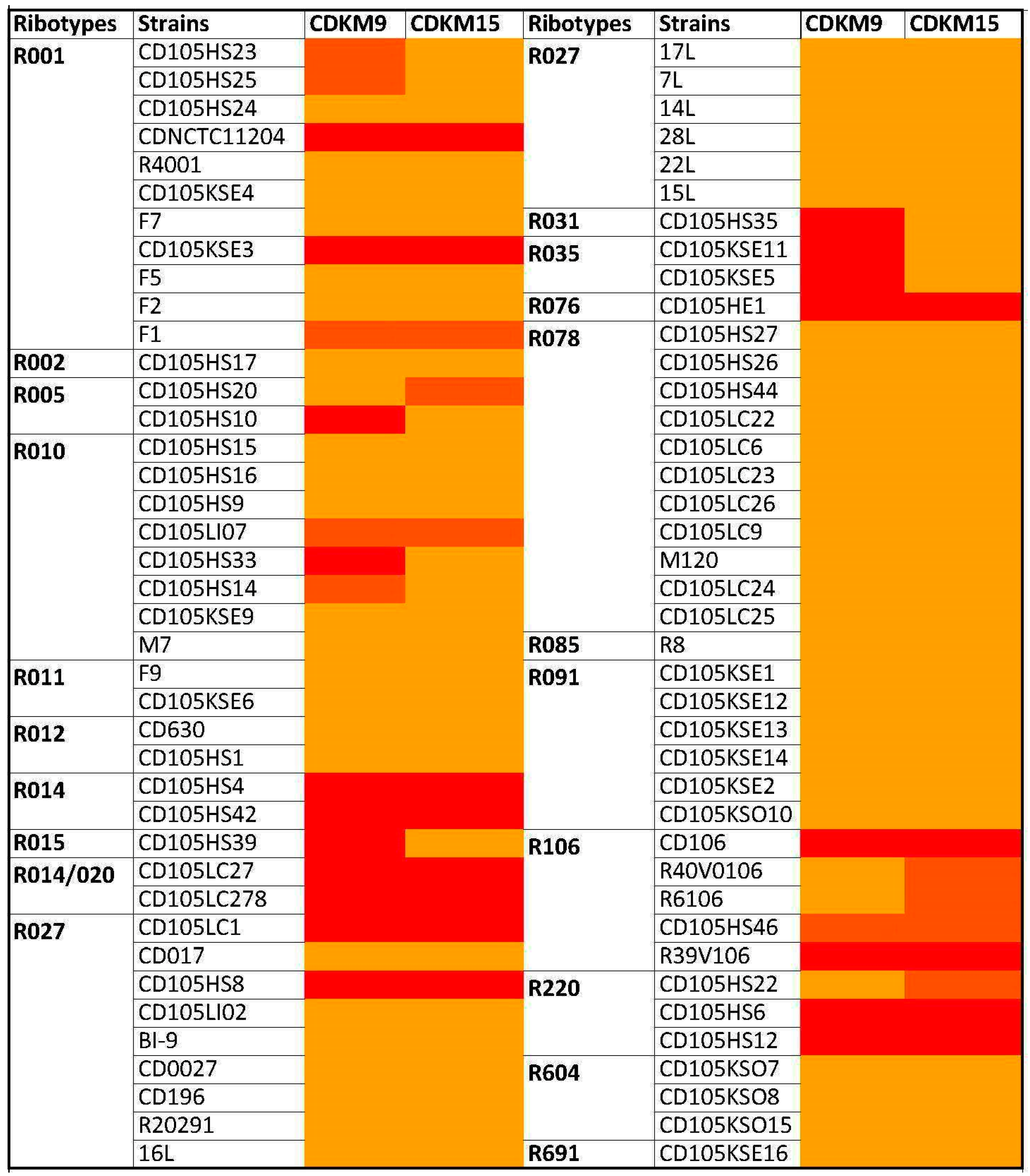
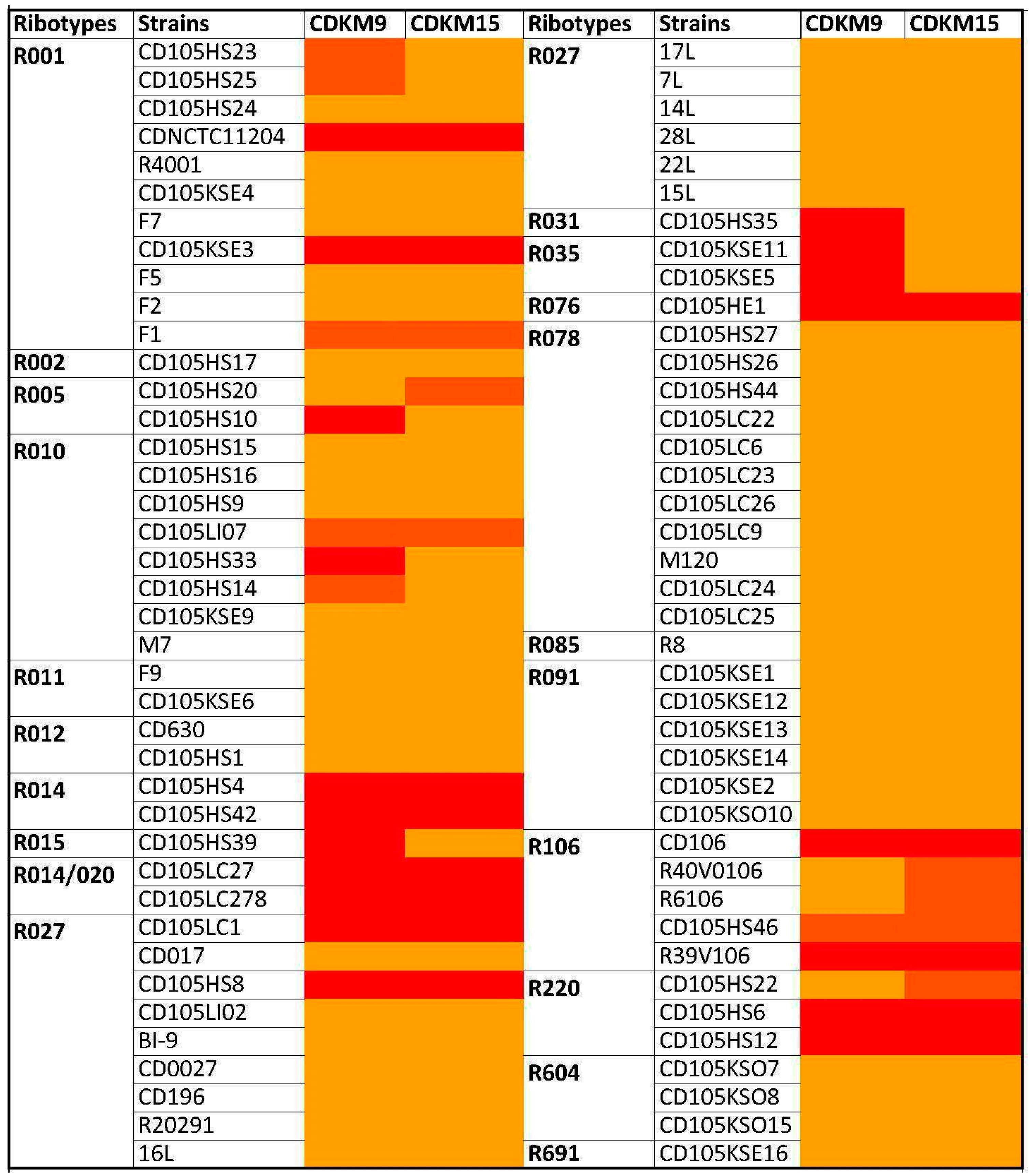
3.1. Phage Isolates and Host Range Analysis
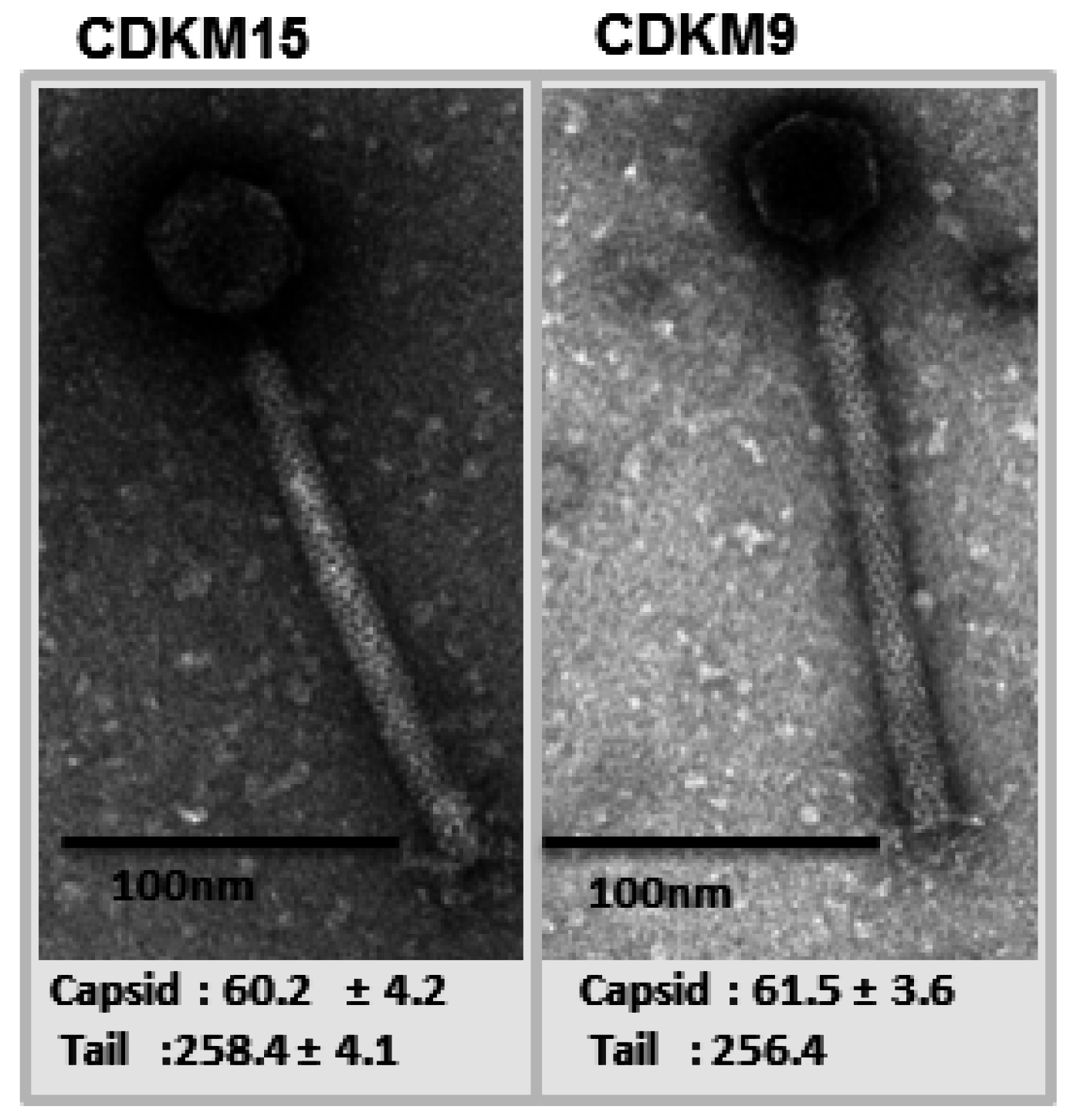
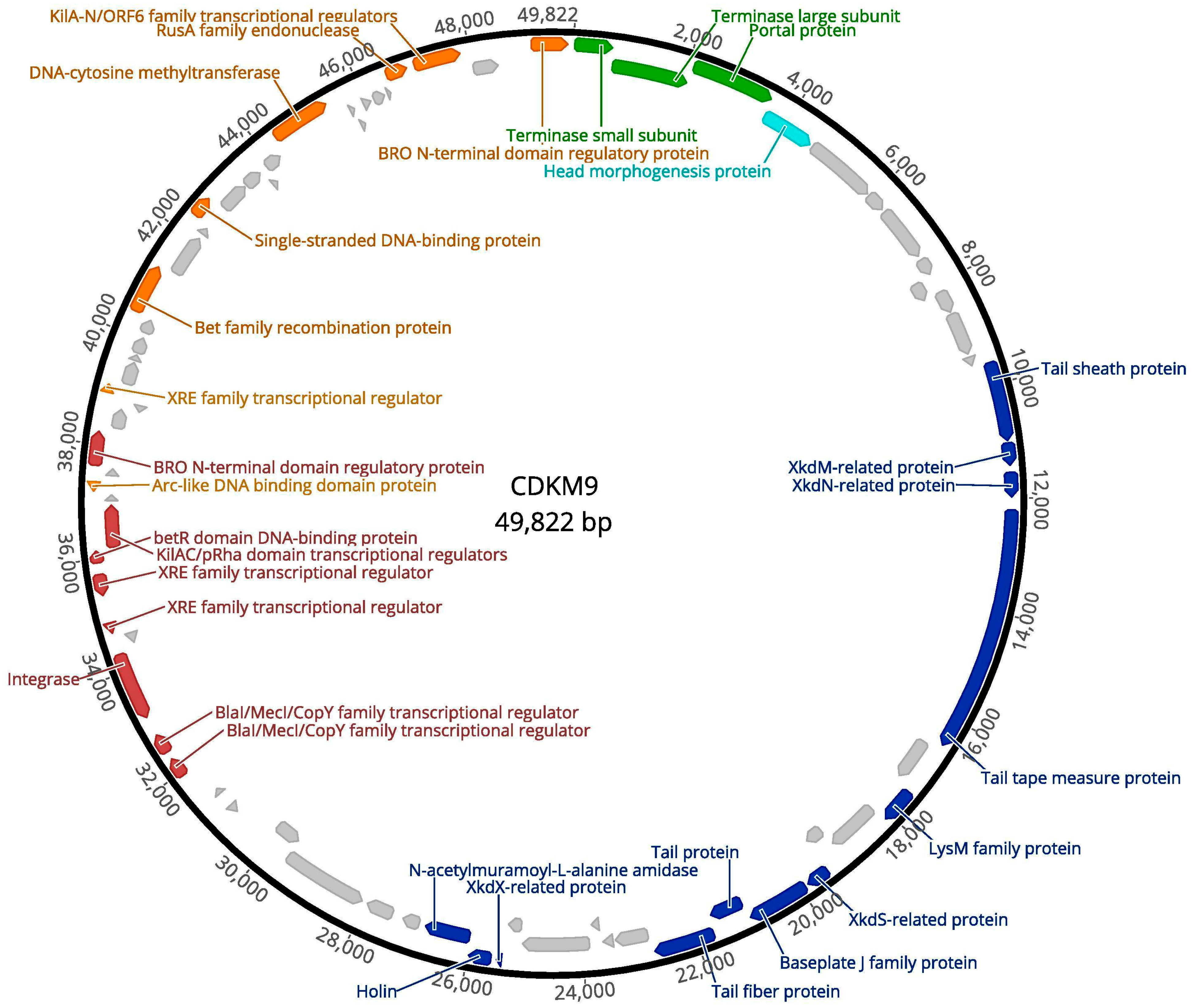
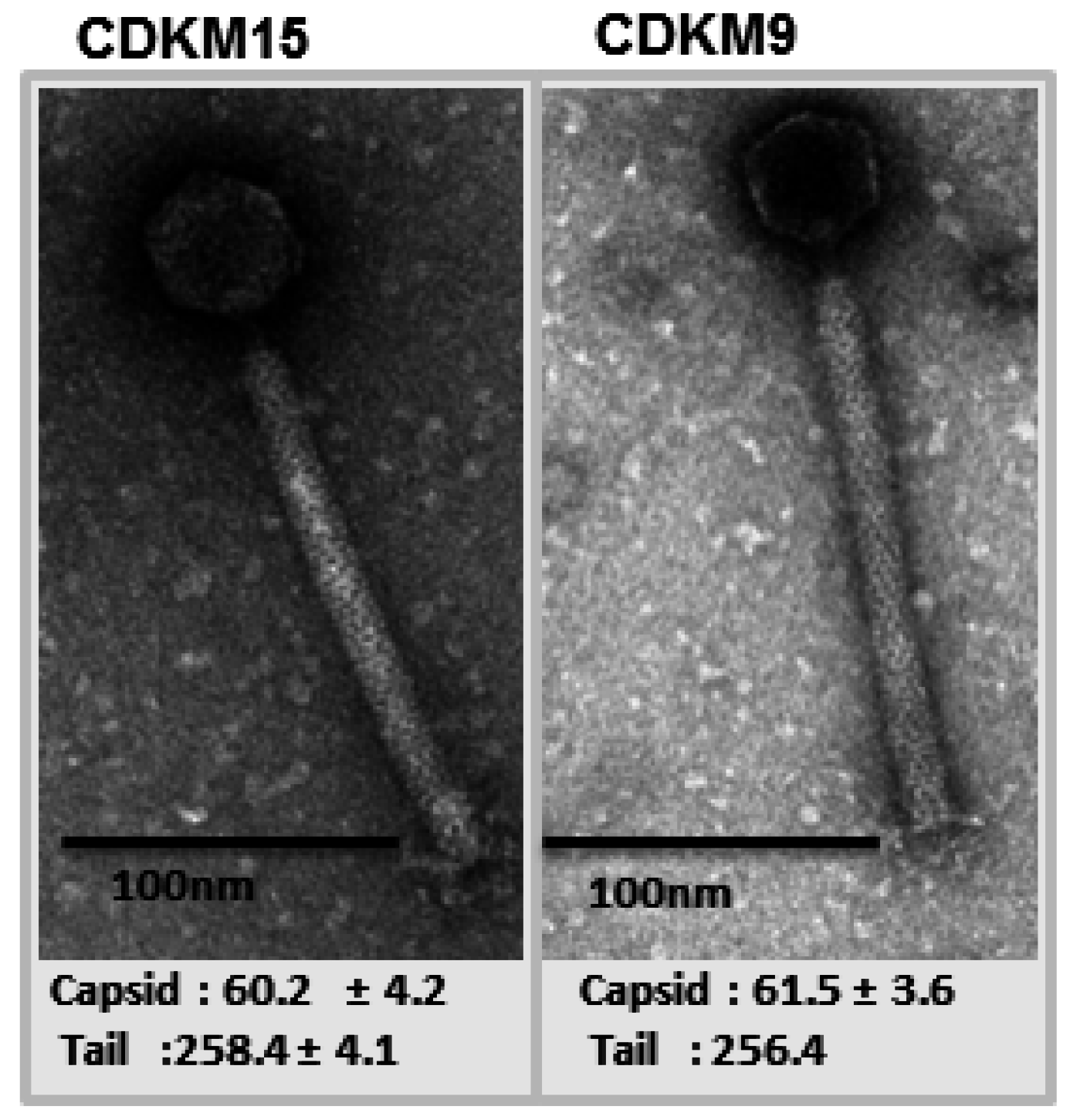
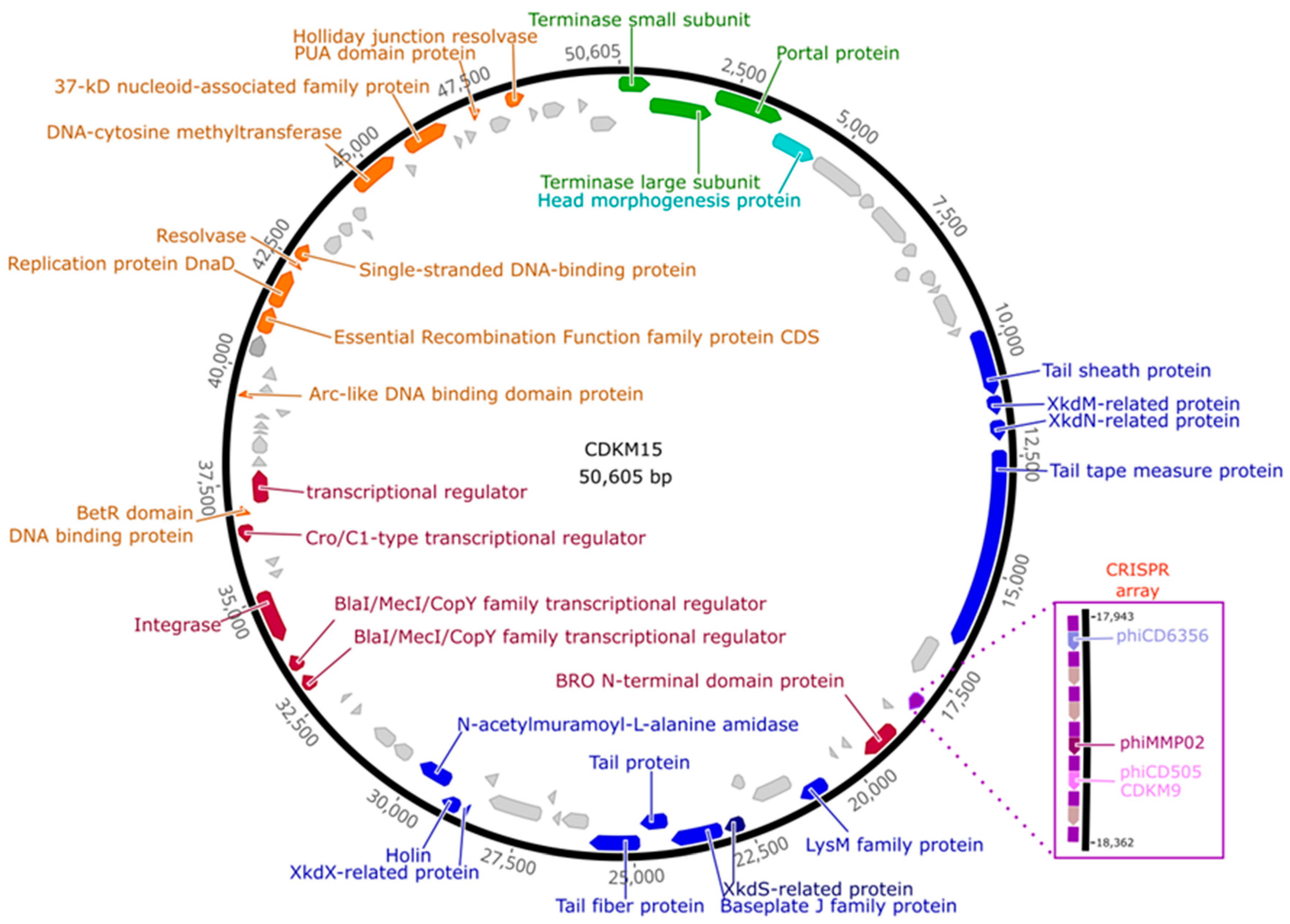
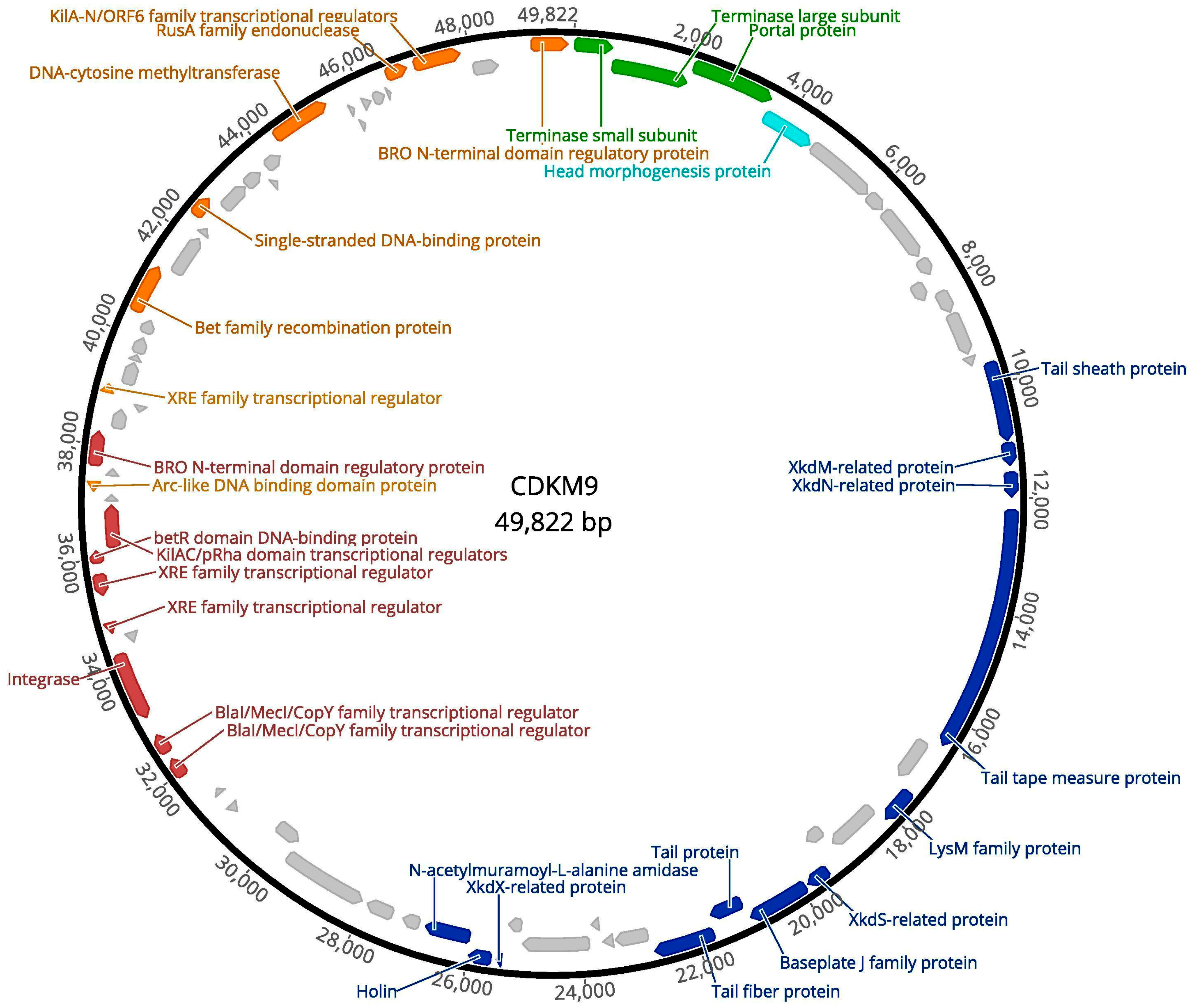
3.2. Genome Features of CDKM15 and CDKM9
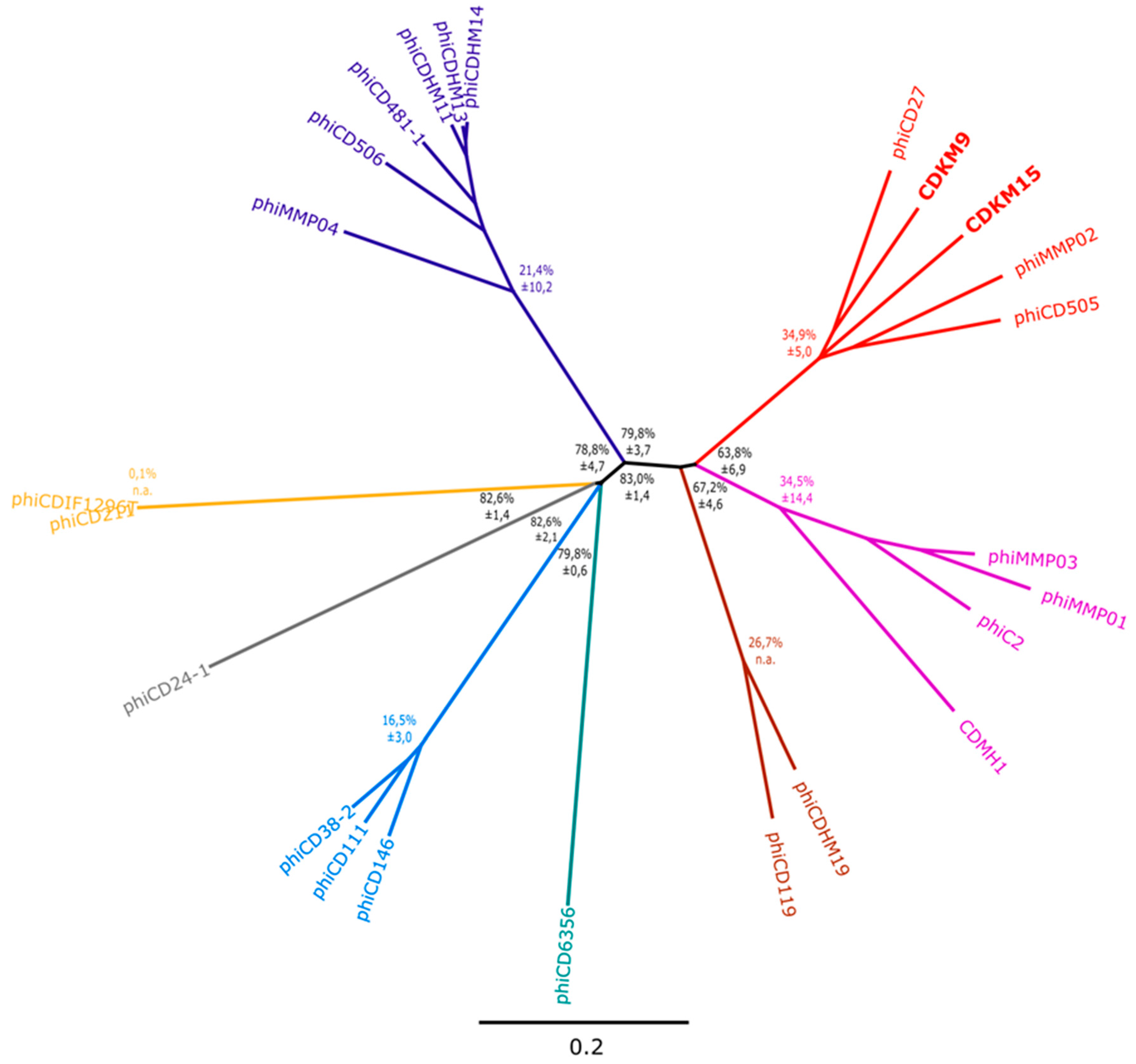
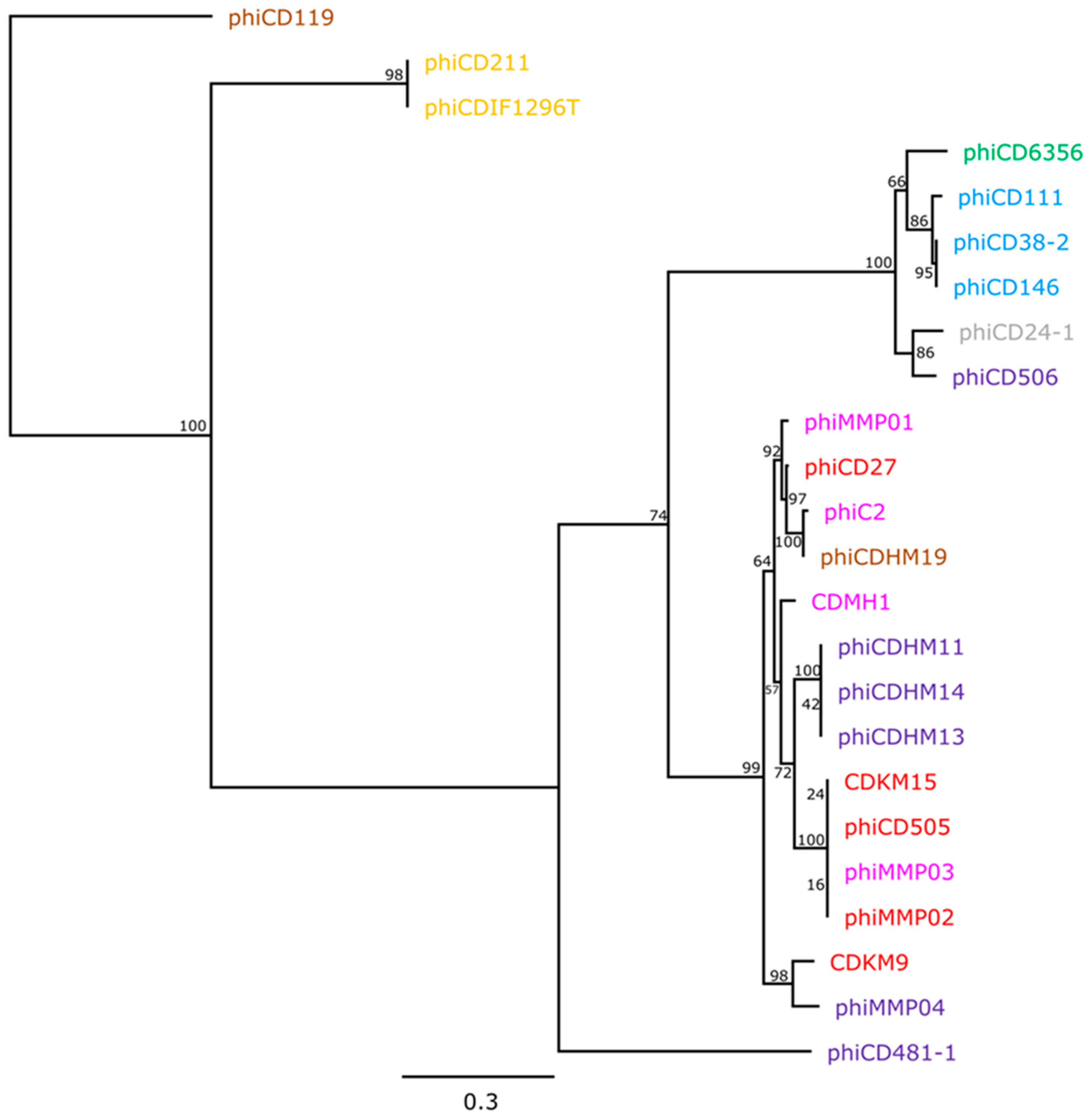
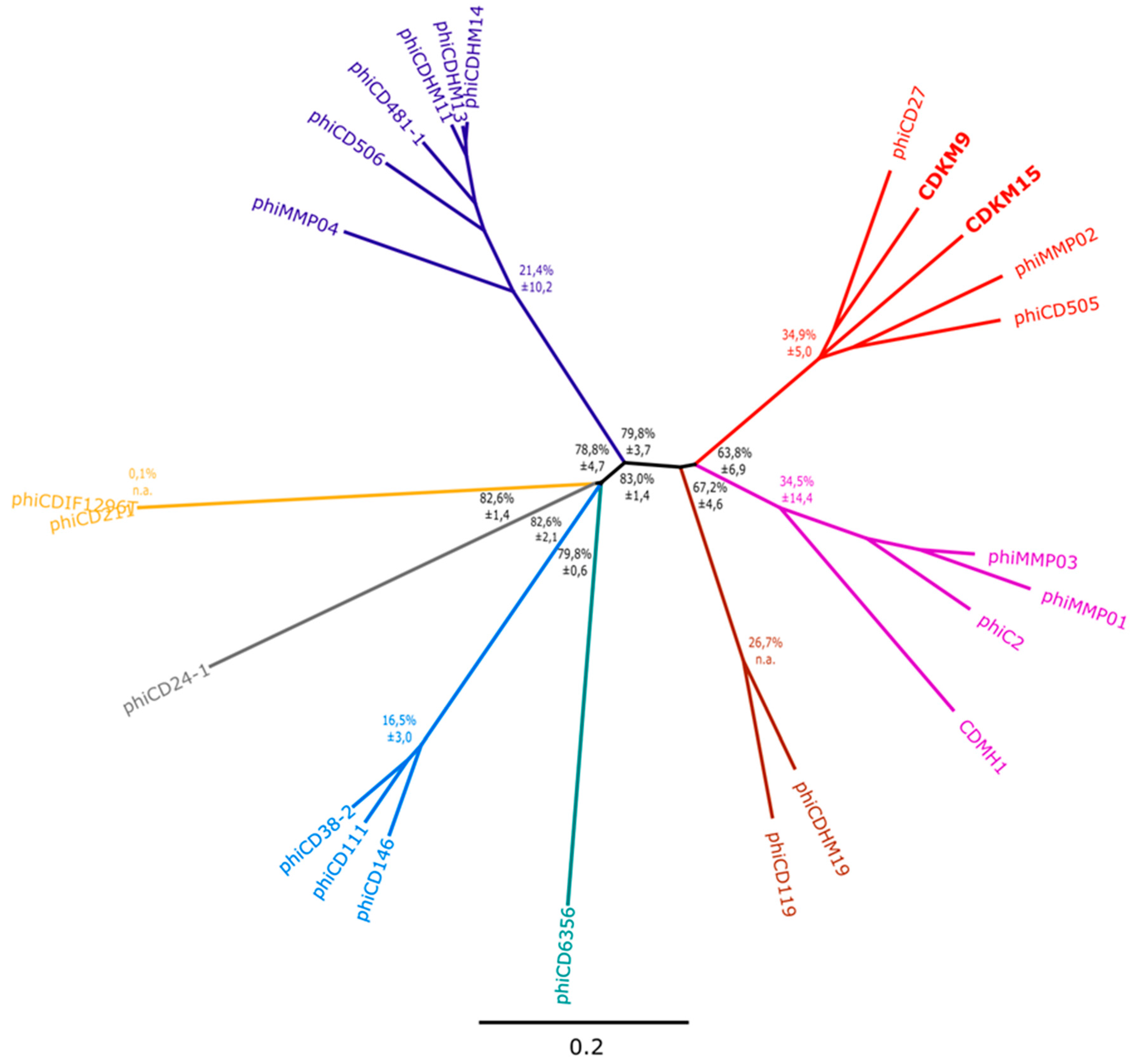
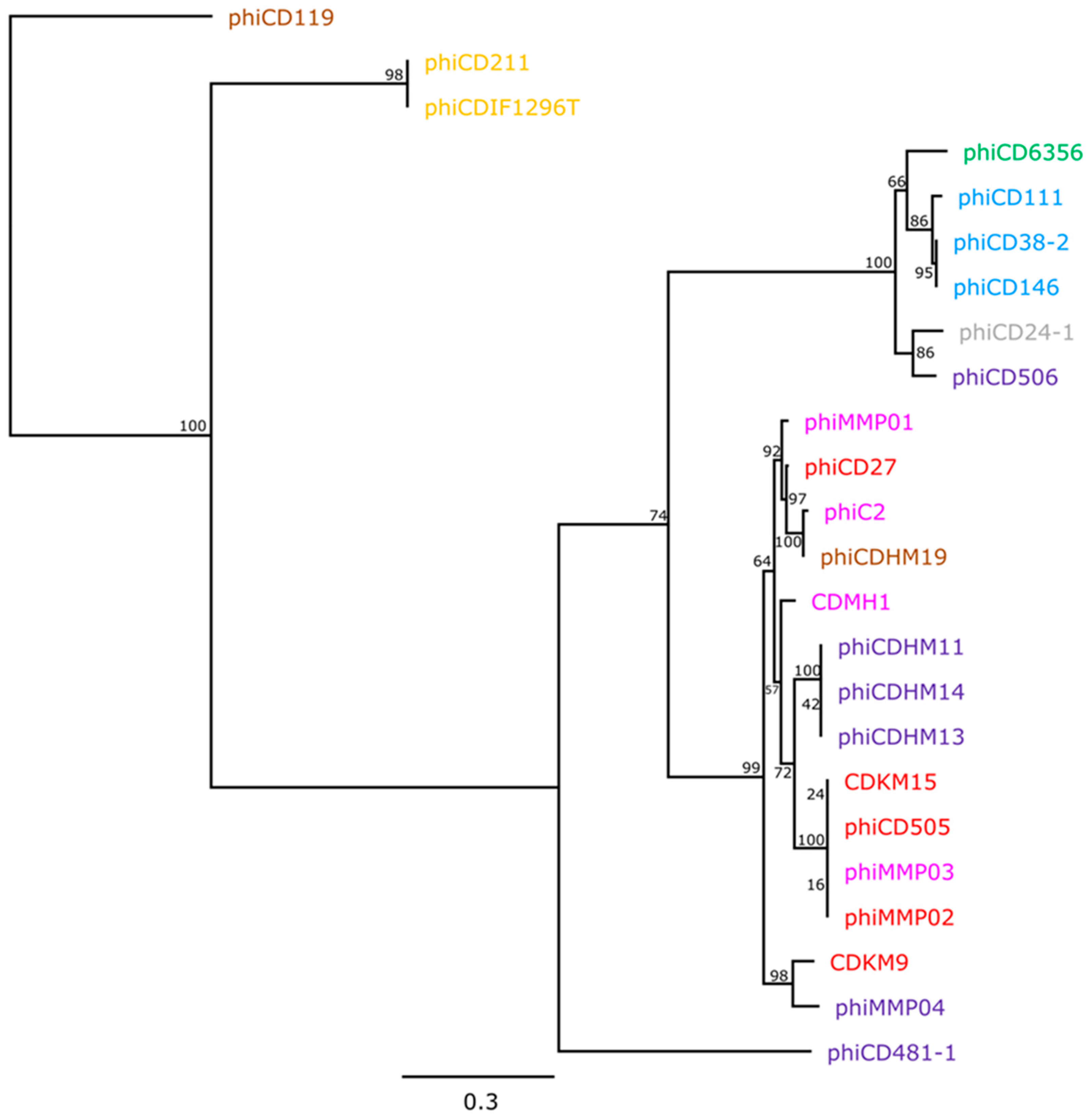
3.3. Phylogenetic Analyses
3.4. Phylogeny of the Endolysin Genes
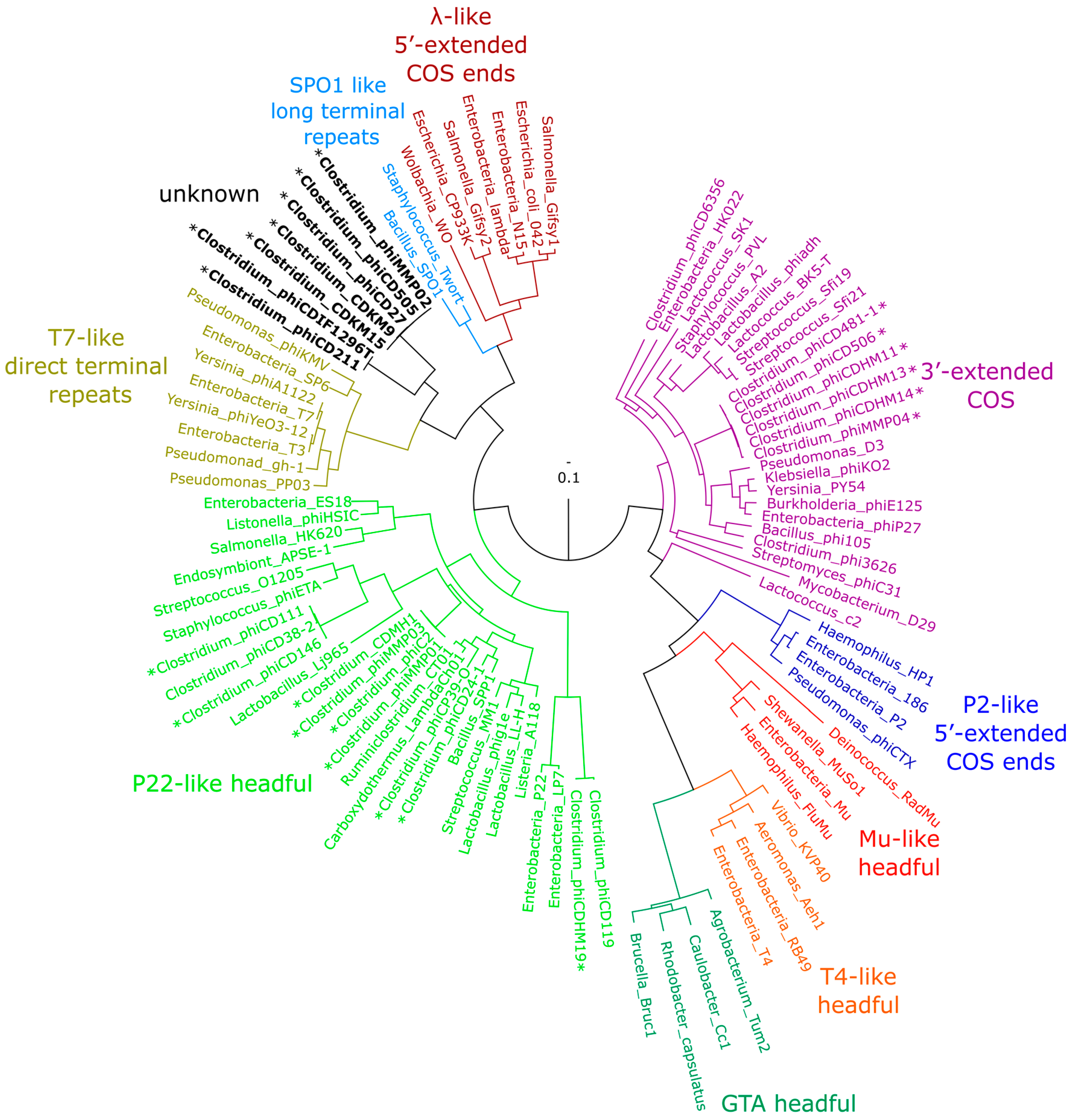
3.5. Phylogenetic Analysis of terL and the Packaging Strategy of the Isolated Phages
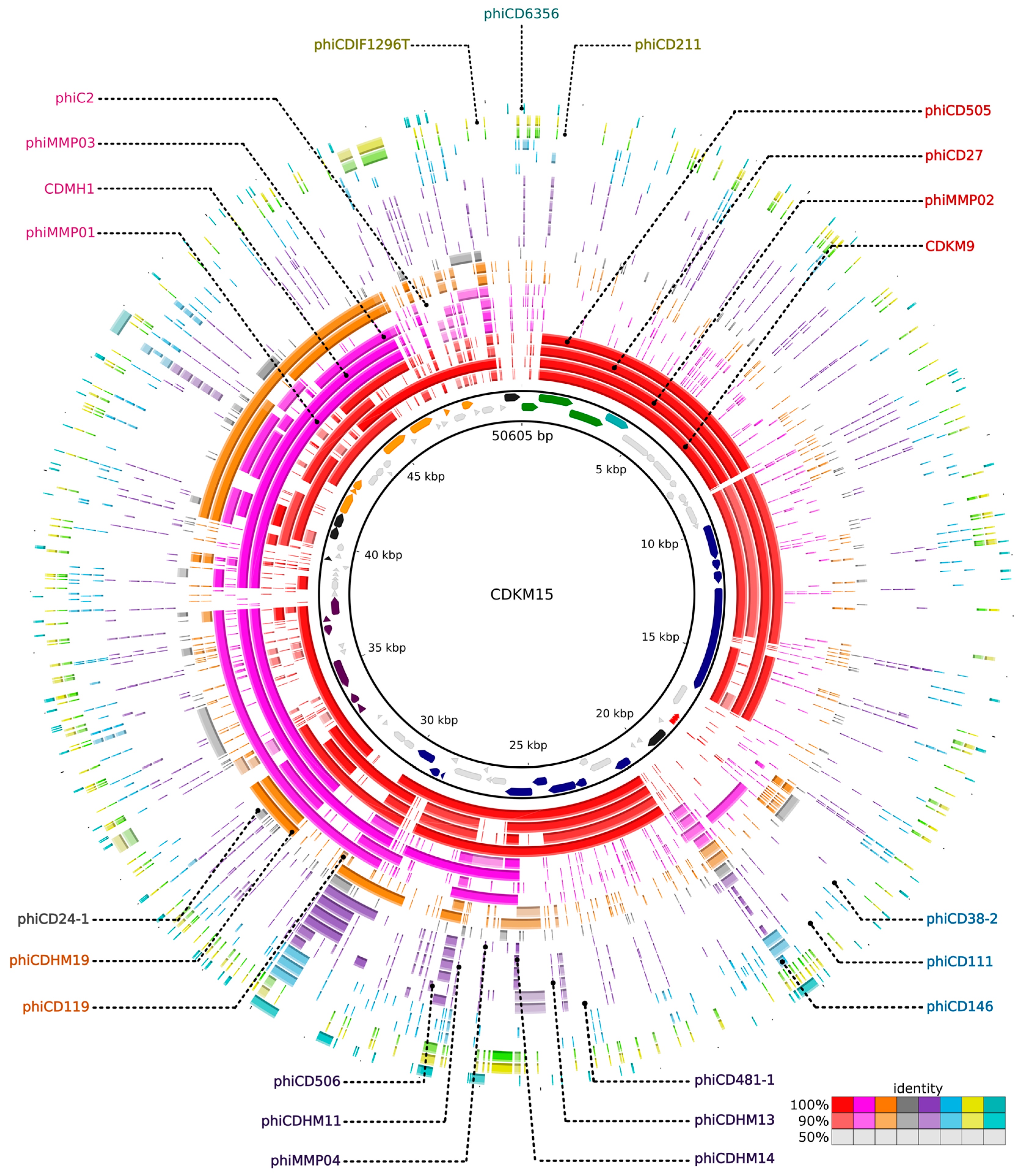
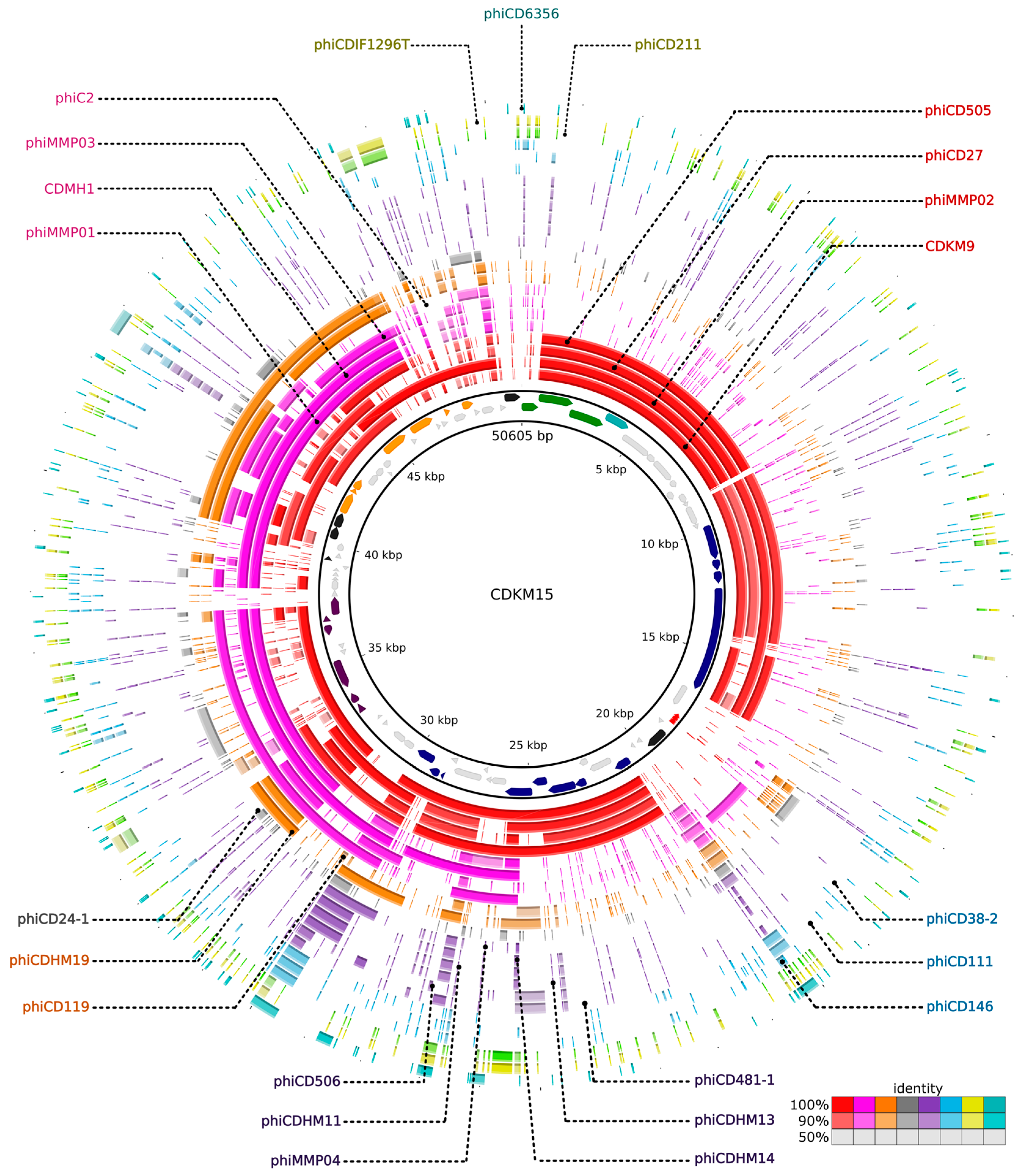
3.6. Comparative Genomics
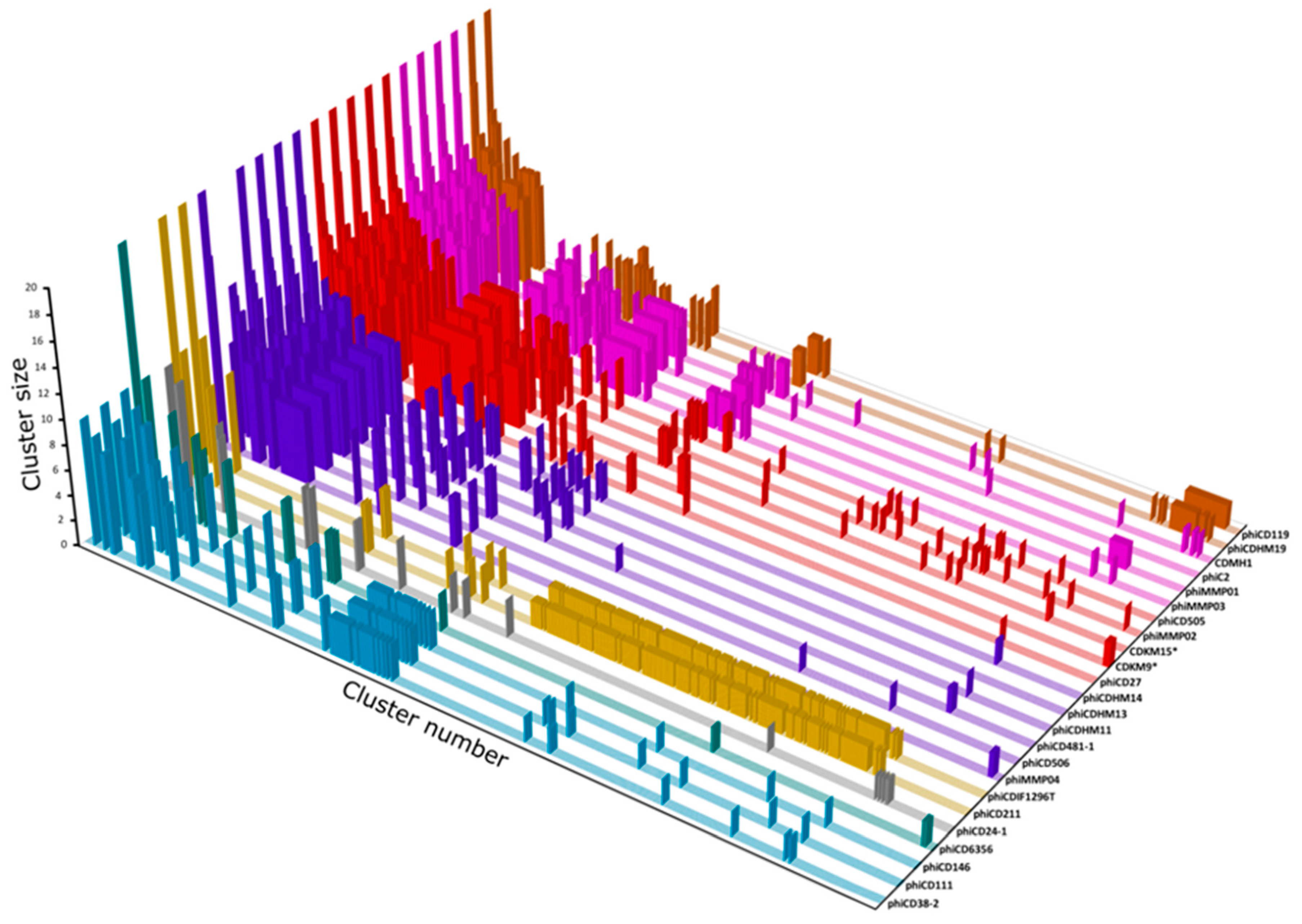
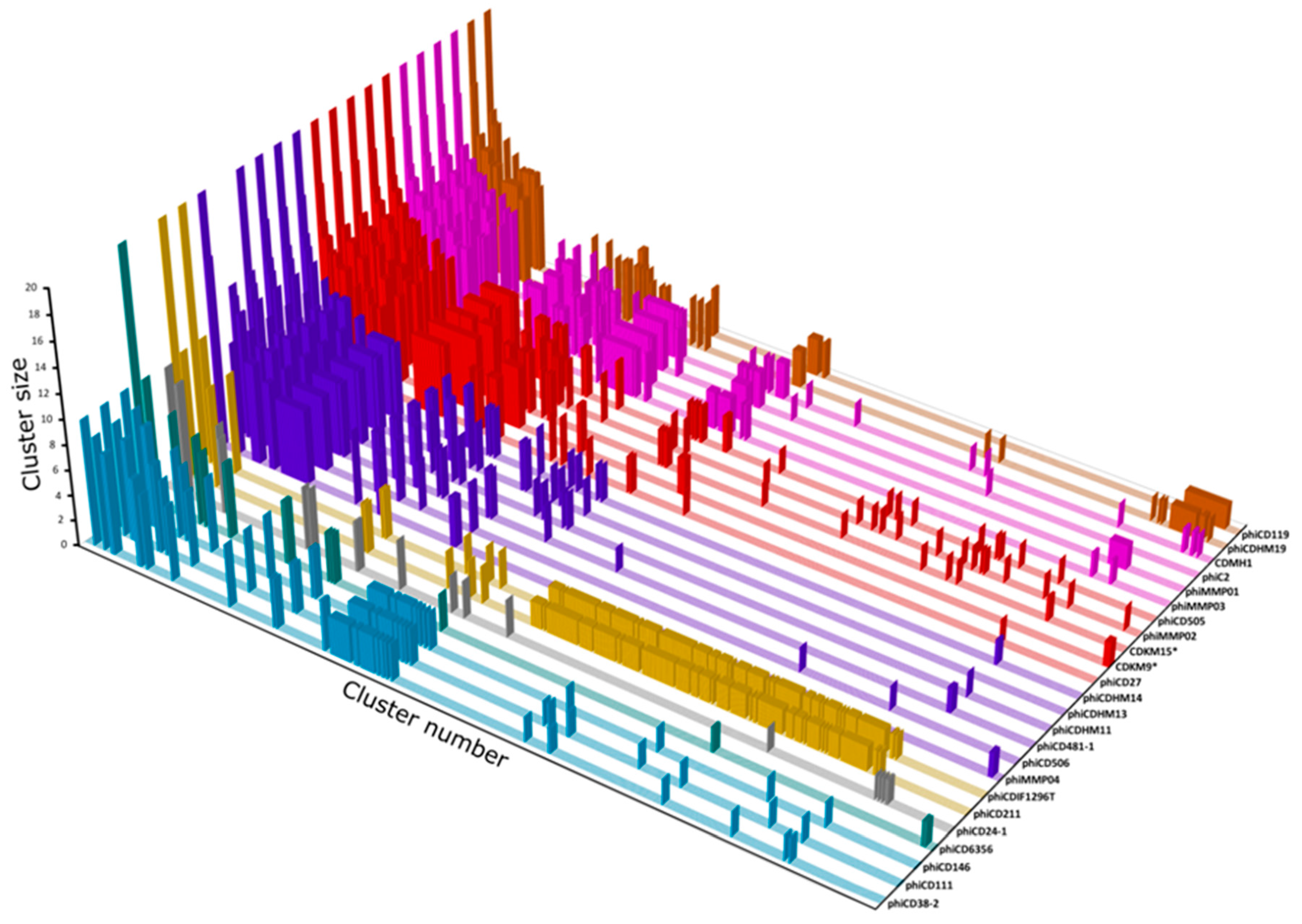
3.7. Protein Cluster Analysis
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Surawicz, C.M.; McFarland, L.V. Pseudomembranous colitis: Causes and cures. Digestion 1999, 60, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Goudarzi, M.; Seyedjavadi, S.S.; Goudarzi, H.; Mehdizadeh Aghdam, E.; Nazeri, S. Clostridium difficile infection: Epidemiology, pathogenesis, risk factors, and therapeutic options. Scientifica 2014, 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Buffie, C.G.; Jarchum, I.; Equinda, M.; Lipuma, L.; Gobourne, A.; Viale, A.; Ubeda, C.; Xavier, J.; Pamer, E.G. Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to Clostridium difficile-induced colitis. Infect. Immun. 2012, 80, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Knight, D.R.; Elliott, B.; Chang, B.J.; Perkins, T.T.; Riley, T.V. Diversity and evolution in the genome of Clostridium difficile. Clin. Microbiol. Rev. 2015, 28, 721–741. [Google Scholar] [CrossRef] [PubMed]
- Hunt, J.J.; Ballard, J.D. Variations in virulence and molecular biology among emerging strains of Clostridium difficile. Microbiol. Mol. Biol. Rev. 2013, 77, 567–581. [Google Scholar] [CrossRef] [PubMed]
- Nale, J.Y.; Spencer, J.; Hargreaves, K.R.; Buckley, A.M.; Trzepinski, P.; Douce, G.R.; Clokie, M.R. Better together: Bacteriophage combinations significantly reduce Clostridium difficile growth in vitro and proliferation in vivo. Antimicrob. Agents Chemother. 2015, 60, 968–981. [Google Scholar] [CrossRef] [PubMed]
- Meader, E.; Mayer, M.J.; Gasson, M.J.; Steverding, D.; Carding, S.R.; Narbad, A. Bacteriophage treatment significantly reduces viable Clostridium difficile and prevents toxin production in an in vitro model system. Anaerobe 2010, 16, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, V.; Fralick, J.A.; Rolfe, R.D. Prevention of Clostridium difficile-induced ileocecitis with bacteriophage. Anaerobe 1999, 5, 69–78. [Google Scholar] [CrossRef]
- Goh, S.; Ong, P.F.; Song, K.P.; Riley, T.V.; Chang, B.J. The complete genome sequence of Clostridium difficile phage ΦC2 and comparisons to ΦCD119 and inducible prophages of CD630. Microbiology 2007, 153, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Govind, R.; Fralick, J.A.; Rolfe, R.D. Genomic organization and molecular characterization of Clostridium difficile bacteriophage phicd119. J. Bacteriol. 2006, 188, 2568–2577. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, K.R.; Flores, C.O.; Lawley, T.D.; Clokie, M.R. Abundant and diverse clustered regularly interspaced short palindromic repeat spacers in Clostridium difficile strains and prophages target multiple phage types within this pathogen. mBio 2014, 5, e01045–e01013. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, K.R.; Kropinski, A.M.; Clokie, M.R.J. What does the talking? Quorum sensing signalling genes discovered in a bacteriophage genome. PLoS ONE 2014, 9, e85131. [Google Scholar] [CrossRef] [PubMed]
- Horgan, M.; O’Sullivan, O.; Coffey, A.; Fitzgerald, G.F.; van Sinderen, D.; McAuliffe, O.; Ross, R.P. Genome analysis of the Clostridium difficile phage ΦCD6356, a temperate phage of the Siphoviridae family. Gene 2010, 462, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.J.; Narbad, A.; Gasson, M.J. Molecular characterization of a Clostridium difficile bacteriophage and its cloned biologically active endolysin. J. Bacteriol. 2008, 190, 6734–6740. [Google Scholar] [CrossRef] [PubMed]
- Meessen-Pinard, M.; Sekulovic, O.; Fortier, L.-C. Evidence of in vivo prophage induction during Clostridium difficile infection. Appl. Environ. Microbiol. 2012, 78, 7662–7670. [Google Scholar] [CrossRef] [PubMed]
- Sekulovic, O.; Garneau, J.R.; Neron, A.; Fortier, L.C. Characterization of temperate phages infecting Clostridium difficile isolates of human and animal origins. Appl. Environ. Microbiol. 2014, 80, 2555–2563. [Google Scholar] [CrossRef] [PubMed]
- Sekulovic, O.; Meessen-Pinard, M.; Fortier, L.C. Prophage-stimulated toxin production in Clostridium difficile NAP1/027 lysogens. J. Bacteriol. 2011, 193, 2726–2734. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, R.; Darius, P.; Summer, E.J.; Seto, D.; Mahadevan, P.; Nilsson, A.S.; Ackermann, H.W.; Kropinski, A.M. Classification of myoviridae bacteriophages using protein sequence similarity. BMC Microbiol. 2009, 9, 224. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, K.R.; Clokie, M.R.J. A taxonomic review of Clostridium difficile phages and proposal of a novel genus, “Phimmp04likevirus”. Viruses 2015, 7, 2534–2541. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, K.R. Isolation and Characterisation of Bacteriophages Infecting Environmental Strains of Clostridium difficile. Ph.D. Thesis, University of Leicester, Leicester, UK, 2012. [Google Scholar]
- Hargreaves, K.R.; Colvin, H.V.; Patel, K.V.; Clokie, J.J.; Clokie, M.R. Genetically diverse Clostridium difficile strains harboring abundant prophages in an estuarine environment. Appl. Environ. Microbiol. 2013, 79, 6236–6243. [Google Scholar] [CrossRef] [PubMed]
- Goh, S.; Riley, T.V.; Chang, B.J. Isolation and characterization of temperate bacteriophages of Clostridium difficile. Appl. Environ. Microbiol. 2005, 71, 1079–1083. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001. [Google Scholar]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 13 November 2016).
- Joshi, N.A.; Fass, J.N. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for Fastq Files. 2011. Available online: https://github.com/najoshi/sickle (accessed on 1 October 2015).
- Li, R.; Zhu, H.; Ruan, J.; Qian, W.; Fang, X.; Shi, Z.; Li, Y.; Li, S.; Shan, G.; Kristiansen, K.; et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010, 20, 265–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Li, Y.; Kristiansen, K.; Wang, J. SOAP: Short oligonucleotide alignment program. Bioinformatics 2008, 24, 713–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. Spades: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Besemer, J.; Borodovsky, M. Genemark: Web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 2005, 33, W451–W454. [Google Scholar] [CrossRef] [PubMed]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bryant, S.H. CD-Search: Protein domain annotations on the fly. Nucleic Acids Res. 2004, 32, W327–W331. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. Interproscan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. PILER-CR: Fast and accurate identification of CRISPR repeats. BMC Bioinformatics 2007, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef] [PubMed]
- Biswas, A.; Gagnon, J.N.; Brouns, S.J.J.; Fineran, P.C.; Brown, C.M. Crisprtarget. RNA Biol. 2013, 10, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Ågren, J.; Sundström, A.; Håfström, T.; Segerman, B. Gegenees: Fragmented alignment of multiple genomes for determining phylogenomic distances and genetic signatures unique for specified target groups. PLoS ONE 2012, 7, e39107. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evolut. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Casjens, S.R.; Gilcrease, E.B. Determining DNA packaging strategy by analysis of the termini of the chromosomes in tailed-bacteriophage virions. Methods Mol. Biol. 2009, 502, 91–111. [Google Scholar] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Linder, C.R.; Warnow, T. RAxML and FastTree: Comparing two methods for large-scale maximum likelihood phylogeny estimation. PLoS ONE 2011, 6, e27731. [Google Scholar] [CrossRef] [PubMed]
- Alikhan, N.F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 2011, 12, 402. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Sebaihia, M.; Wren, B.W.; Mullany, P.; Fairweather, N.F.; Minton, N.; Stabler, R.; Thomson, N.R.; Roberts, A.P.; Cerdeno-Tarraga, A.M.; Wang, H.; et al. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat. Genet. 2006, 38, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Boudry, P.; Semenova, E.; Monot, M.; Datsenko, K.A.; Lopatina, A.; Sekulovic, O.; Ospina-Bedoya, M.; Fortier, L.C.; Severinov, K.; Dupuy, B.; et al. Function of the CRISPR-Cas System of the Human Pathogen Clostridium difficile. mBio 2015, 6, e01112–e01115. [Google Scholar] [PubMed]
- Soutourina, O.A.; Monot, M.; Boudry, P.; Saujet, L.; Pichon, C.; Sismeiro, O.; Semenova, E.; Severinov, K.; Le Bouguenec, C.; Coppee, J.Y.; et al. Genome-wide identification of regulatory RNAs in the human pathogen Clostridium difficile. PLoS Genet. 2013, 9, e1003493. [Google Scholar] [CrossRef] [PubMed]
- Nafissi, N.; Slavcev, R. Bacteriophage recombination systems and biotechnical applications. Appl. Microbiol. Biotechnol. 2014, 98, 2841–2851. [Google Scholar] [CrossRef] [PubMed]
- Hillyar, C.R. Genetic recombination in bacteriophage lambda. Biosci. Horiz. 2012, 5, hzs001. [Google Scholar] [CrossRef]
- McDonald, L.; Killgore, G.; Thompson, A.; Owens, R.; Kazakova, S.; Sambol, S.; Johnson, S.; Gerding, D. An epidemic, toxin gene-variant strain of Clostridium difficile. N. Engl. J. Med. 2005, 353, 2433–2441. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.D.; Auchtung, J.M.; Collins, J.; Britton, R.A. Epidemic Clostridium difficile strains demonstrate increased competitive fitness compared to nonepidemic isolates. Infect. Immun. 2014, 82, 2815–2825. [Google Scholar] [CrossRef] [PubMed]
- Tickler, I.A.; Goering, R.V.; Whitmore, J.D.; Lynn, A.N.; Persing, D.H.; Tenover, F.C. Strain types and antimicrobial resistance patterns of Clostridium difficile isolates from the United States, 2011 to 2013. Antimicrob. Agents Chemother. 2014, 58, 4214–4218. [Google Scholar] [CrossRef] [PubMed]
- Fortier, L.-C.; Moineau, S. Morphological and genetic diversity of temperate phages in Clostridium difficile. Appl. Environ. Microbiol. 2007, 73, 7358–7366. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.M.; Shoup, M.; Robinson, C.; Britton, R.; Olsen, K.E.; Barrangou, R. CRISPR diversity and microevolution in Clostridium difficile. Genome Biol. Evol. 2016, 8, 2841–2855. [Google Scholar] [CrossRef] [PubMed]
- Minot, S.; Sinha, R.; Chen, J.; Li, H.; Keilbaugh, S.A.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. The human gut virome: Inter-individual variation and dynamic response to diet. Genome Res. 2011, 21, 1616–1625. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, H.; Melo, L.D.; Santos, S.B.; Nobrega, F.L.; Ferreira, E.C.; Cerca, N.; Azeredo, J.; Kluskens, L.D. Molecular aspects and comparative genomics of bacteriophage endolysins. J. Virol. 2013, 87, 4558–4570. [Google Scholar] [CrossRef] [PubMed]
- Nagy, E.; Foldes, J. Electron microscopic investigation of lysogeny of clostridium difficile strains isolated from antibiotic-associated diarrhea cases and from healthy carriers. Acta Pathol. Microbiol. Immunol. Scand. 1991, 99, 321–326. [Google Scholar] [CrossRef]
- Nale, J.Y. Isolation and Characterisation of Temperate Bacteriophages of the Hypervirulent Clostridium difficile 027 Strains. Ph.D. Thesis, University of Leicester, Leicester, UK, 2013. [Google Scholar]
- Shan, J.; Patel, K.V.; Hickenbotham, P.T.; Nale, J.Y.; Hargreaves, K.R.; Clokie, M.R. Prophage carriage and diversity within clinically relevant strains of Clostridium difficile. Appl. Environ. Microbiol. 2012, 78, 6027–6034. [Google Scholar] [CrossRef] [PubMed]
- Casjens, S.R.; Gilcrease, E.B.; Winn-Stapley, D.A.; Schicklmaier, P.; Schmieger, H.; Pedulla, M.L.; Ford, M.E.; Houtz, J.M.; Hatfull, G.F.; Hendrix, R.W. The generalized transducing Salmonella bacteriophage ES18: Complete genome sequence and DNA packaging strategy. J. Bacteriol. 2005, 187, 1091–1104. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Carpena, N.; Quiles-Puchalt, N.; Ram, G.; Novick, R.P.; Penades, J.R. Intra- and inter-generic transfer of pathogenicity island-encoded virulence genes by cos phages. ISME J. 2015, 9, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.B.; Feiss, M. Mechanisms of DNA packaging by large double-stranded DNA viruses. Annu. Rev. Virol. 2015, 2, 351–378. [Google Scholar] [CrossRef] [PubMed]
- Goh, S.; Hussain, H.; Chang, B.J.; Emmett, W.; Riley, T.V.; Mullany, P. Phage ΦC2 mediates transduction of Tn6215, encoding erythromycin resistance, between Clostridium difficile strains. mBio 2013, 4, e00840-13. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rashid, S.J.; Barylski, J.; Hargreaves, K.R.; Millard, A.A.; Vinner, G.K.; Clokie, M.R.J. Two Novel Myoviruses from the North of Iraq Reveal Insights into Clostridium difficile Phage Diversity and Biology. Viruses 2016, 8, 310. https://doi.org/10.3390/v8110310
Rashid SJ, Barylski J, Hargreaves KR, Millard AA, Vinner GK, Clokie MRJ. Two Novel Myoviruses from the North of Iraq Reveal Insights into Clostridium difficile Phage Diversity and Biology. Viruses. 2016; 8(11):310. https://doi.org/10.3390/v8110310
Chicago/Turabian StyleRashid, Srwa J., Jakub Barylski, Katherine R. Hargreaves, Andrew A. Millard, Gurinder K. Vinner, and Martha R. J. Clokie. 2016. "Two Novel Myoviruses from the North of Iraq Reveal Insights into Clostridium difficile Phage Diversity and Biology" Viruses 8, no. 11: 310. https://doi.org/10.3390/v8110310