
Adenoviral Vectors Armed with Cell Fusion-Inducing Proteins as Anti-Cancer Agents

Abstract
:1. Introduction
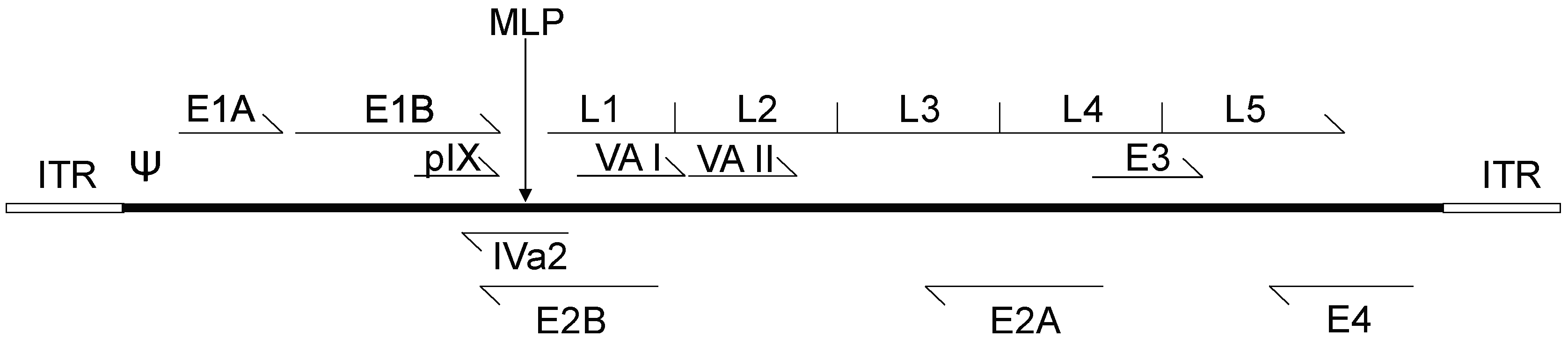
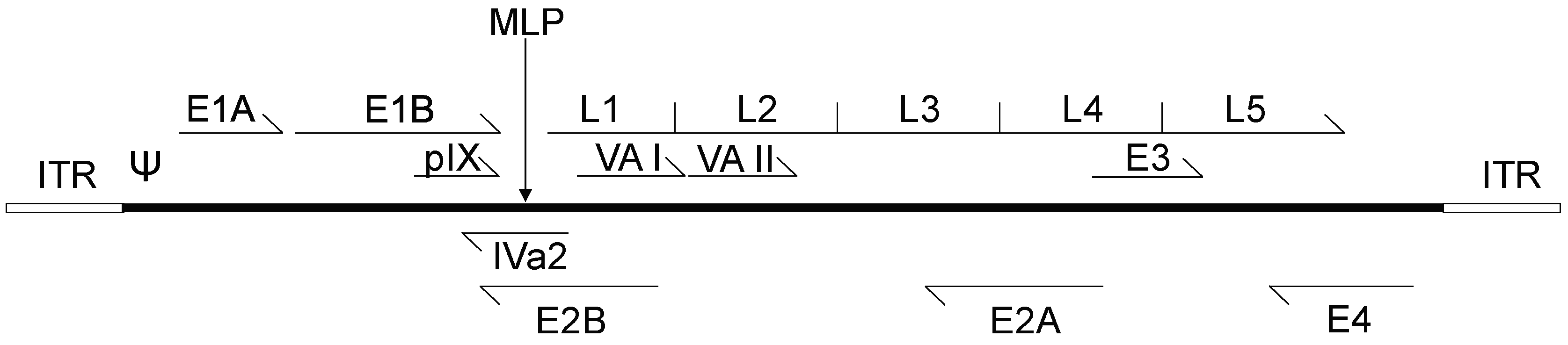
2. Adenovirus Biology
3. Fusogenic Proteins
4. Expression of Fusogenic Proteins from Replication-Defective Adenovirus Vectors
4.1. Gibbon-Ape Leukemia Virus Fusogenic Membrane Glycoprotein
4.2. Measles Virus Fusion Proteins
4.3. Other Fusogenic Envelope Proteins
4.4. Fusion Associated Small Transmembrane Proteins
5. Trans-Complementing Adenovirus Systems for Expression of Fusion Proteins
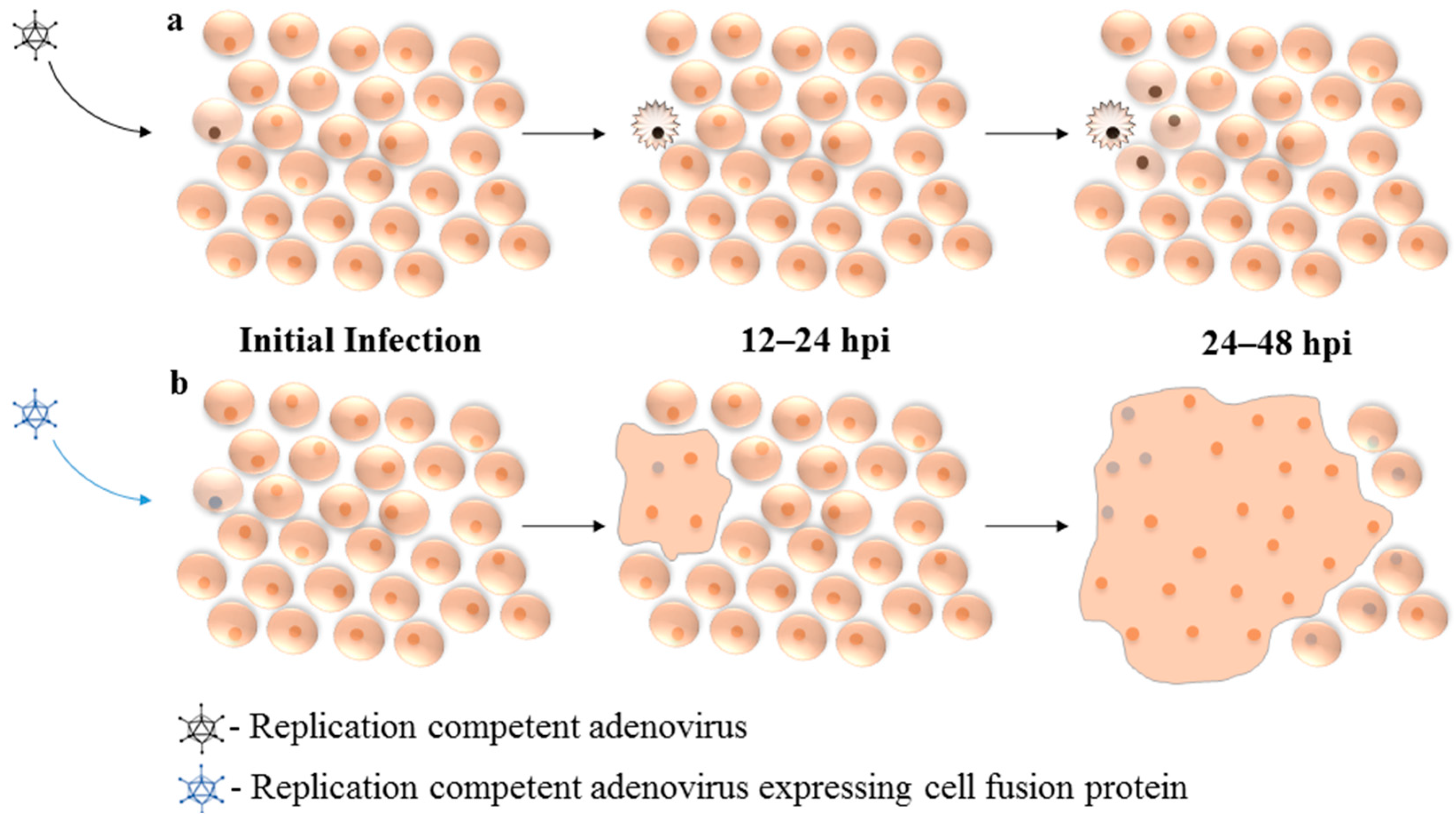
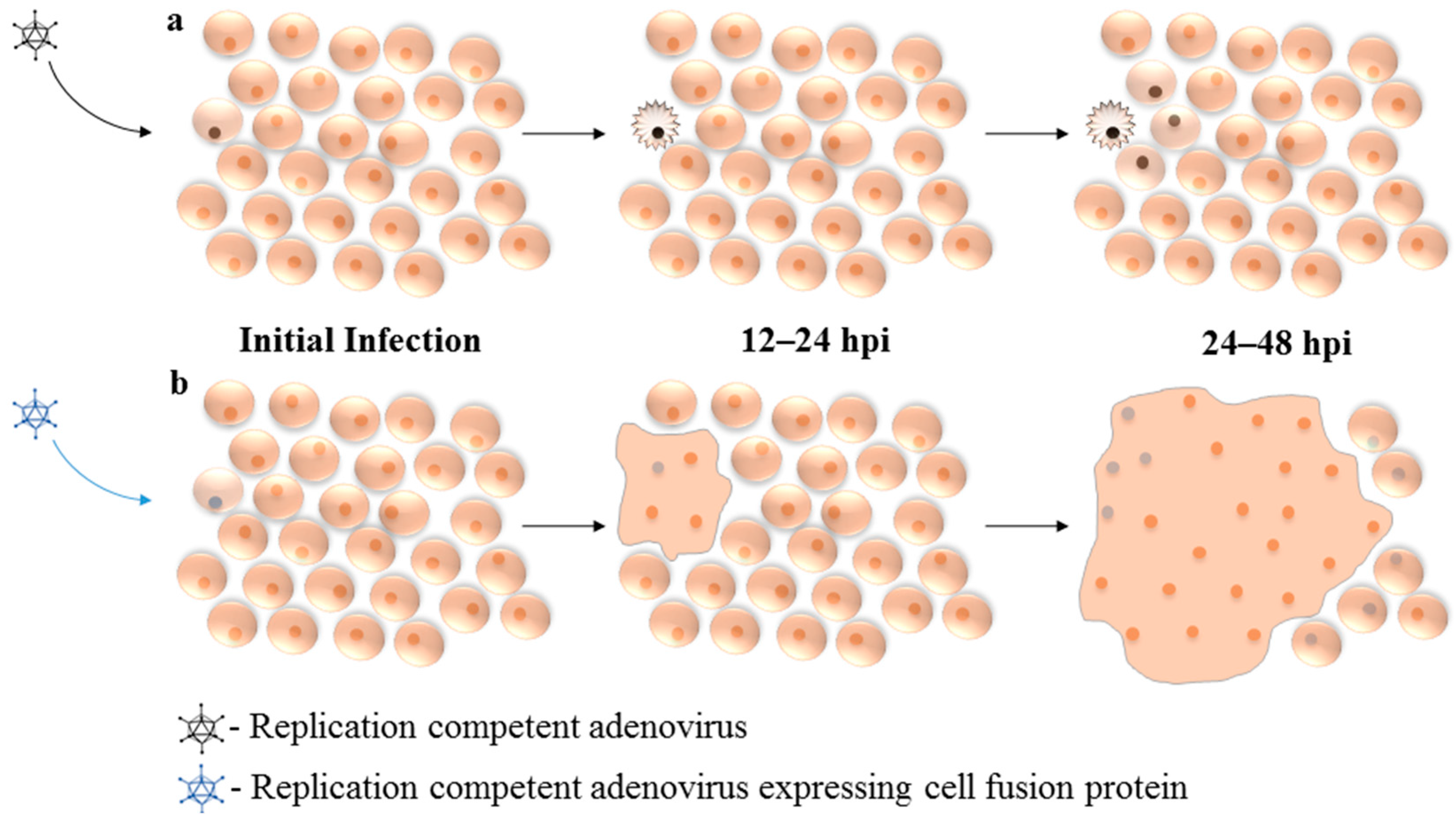
6. Expression of Fusogenic Proteins from Replication Competent Adenovirus
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Garcia, M.; Jemal, A.; Ward, E.; Center, M.; Hao, Y.; Siegel, R.; Thun, M. Global cancer facts & figures 2007. Atlanta GA Am. Cancer Soc. 2007, 1, 1–52. [Google Scholar]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M. Encyclopedia of Cancer; Schwab, M., Ed.; Springer: Heidelberg, Germany, 2008. [Google Scholar]
- Green, D.M.; Kun, L.E.; Matthay, K.K.; Meadows, A.T.; Meyer, W.H.; Meyers, P.A.; Spunt, S.L.; Robison, L.L.; Hudson, M.M. Relevance of historical therapeutic approaches to the contemporary treatment of pediatric solid tumors. Pediatr. Blood Cancer 2013, 60, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed]
- García-Cao, I.; García-Cao, M.; Martín-Caballero, J.; Criado, L.M.; Klatt, P.; Flores, J.M.; Weill, J.C.; Blasco, M.A.; Serrano, M. ‘Super p53’ mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J. 2002, 21, 6225–6235. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Bauerschmitz, G.J.; Barker, S.D.; Hemminki, A. Adenoviral gene therapy for cancer: From vectors to targeted and replication competent agents (review). Int. J. Oncol. 2002, 21, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Heise, C.; Kirn, D.H. Replication-selective adenoviruses as oncolytic agents. J. Clin. Investig. 2000, 105, 847–851. [Google Scholar] [CrossRef] [PubMed]
- Kirn, D.H.; Wang, Y.; Le Boeuf, F.; Bell, J.; Thorne, S.H. Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PLoS Med 2007, 4, e353. [Google Scholar] [CrossRef] [PubMed]
- Senzer, N.N.; Kaufman, H.L.; Amatruda, T.; Nemunaitis, M.; Reid, T.; Daniels, G.; Gonzalez, R.; Glaspy, J.; Whitman, E.; Harrington, K. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor–encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J. Clin. Oncol. 2009, 27, 5763–5771. [Google Scholar] [CrossRef] [PubMed]
- Lichty, B.D.; Power, A.T.; Stojdl, D.F.; Bell, J.C. Vesicular stomatitis virus: Re-inventing the bullet. Trends Mol. Med. 2004, 10, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Khuri, F.R.; Nemunaitis, J.; Ganly, I.; Arseneau, J.; Tannock, I.F.; Romel, L.; Gore, M.; Ironside, J.; MacDougall, R.; Heise, C. A controlled trial of intratumoral ONYX-015, a selectively-replicating adenovirus, in combination with cisplatin and 5-fluorouracil in patients with recurrent head and neck cancer. Nat. Med. 2000, 6, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Greig, S.L. Talimogene laherparepvec: First global approval. Drugs 2016, 76, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z. Current status of gendicine in China: Recombinant human Ad-p53 agent for treatment of cancers. Hum. Gene Ther. 2005, 16, 1016–1027. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Chang, J.; Zhang, L.; Jiang, W.; Guan, Z.; Liu, J.; Zhang, Y.; Hu, X.; Wu, G.; Wang, H. Phase III randomized clinical trial of intratumoral injection of E1B gene-deleted adenovirus (H101) combined with cisplatin-based chemotherapy in treating squamous cell cancer of head and neck or esophagus. Chin. J. Cancer 2004, 23, 1666–1670. [Google Scholar]
- Edelstein, M. Gene Therapy Clinical Trials Worldwide. Available online: http://www.abedia.com/wiley/ (accessed on 25 July 2016).
- DeWeese, T.L.; van der Poel, H.; Li, S.; Mikhak, B.; Drew, R.; Goemann, M.; Hamper, U.; DeJong, R.; Detorie, N.; Rodriguez, R. A phase I trial of CV706, a replication-competent, PSA selective oncolytic adenovirus, for the treatment of locally recurrent prostate cancer following radiation therapy. Cancer Res. 2001, 61, 7464–7472. [Google Scholar] [PubMed]
- Vecil, G.G.; Lang, F.F. Clinical trials of adenoviruses in brain tumors: A review of Ad-p53 and oncolytic adenoviruses. J. Neuro-Oncol. 2003, 65, 237–246. [Google Scholar] [CrossRef]
- Sauthoff, H.; Hu, J.; Maca, C.; Goldman, M.; Heitner, S.; Yee, H.; Pipiya, T.; Rom, W.N.; Hay, J.G. Intratumoral spread of wild-type adenovirus is limited after local injection of human xenograft tumors: Virus persists and spreads systemically at late time points. Hum. Gene Ther. 2003, 14, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.; Sauthoff, H.; Heitner, S.; Jagirdar, J.; Rom, W.N.; Hay, J.G. Wild-type adenovirus decreases tumor xenograft growth, but despite viral persistence complete tumor responses are rarely achieved-deletion of the viral E1b-19-kD gene increases the viral oncolytic effect. Hum. Gene Ther. 2001, 12, 1323–1332. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Rojas, J.J.; Gros, A.; Mercade, E.; Cascallo, M.; Alemany, R. Hyaluronidase expression by an oncolytic adenovirus enhances its intratumoral spread and suppresses tumor growth. Mol. Ther. 2010, 18, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Toth, K.; Djeha, H.; Ying, B.; Tollefson, A.E.; Kuppuswamy, M.; Doronin, K.; Krajcsi, P.; Lipinski, K.; Wrighton, C.J.; Wold, W.S. An oncolytic adenovirus vector combining enhanced cell-to-cell spreading, mediated by the ADP cytolytic protein, with selective replication in cancer cells with deregulated Wnt signaling. Cancer Res. 2004, 64, 3638–3644. [Google Scholar] [CrossRef] [PubMed]
- Yumul, R.; Richter, M.; Lu, Z.Z.; Saydaminova, K.; Wang, H.; Wang, C.H.; Carter, D.; Lieber, A. Epithelial Junction Opener Improves Oncolytic Adenovirus Therapy in Mouse Tumor Models. Hum. Gene Ther. 2016, 27, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Dmitriev, I.; Krasnykh, V.; Miller, C.R.; Wang, M.; Kashentseva, E.; Mikheeva, G.; Belousova, N.; Curiel, D.T. An adenovirus vector with genetically modified fibers demonstrates expanded tropism via utilization of a coxsackievirus and adenovirus receptor-independent cell entry mechanism. J. Virol. 1998, 72, 9706–9713. [Google Scholar] [PubMed]
- Li, Y.; Pong, R.-C.; Bergelson, J.M.; Hall, M.C.; Sagalowsky, A.I.; Tseng, C.-P.; Wang, Z.; Hsieh, J.-T. Loss of adenoviral receptor expression in human bladder cancer cells: A potential impact on the efficacy of gene therapy. Cancer Res. 1999, 59, 325–330. [Google Scholar] [PubMed]
- Vigne, E.; Mahfouz, I.; Dedieu, J.-F.; Brie, A.; Perricaudet, M.; Yeh, P. RGD inclusion in the hexon monomer provides adenovirus type 5-based vectors with a fiber knob-independent pathway for infection. J. Virol. 1999, 73, 5156–5161. [Google Scholar] [PubMed]
- Poulin, K.L.; Lanthier, R.M.; Smith, A.C.; Christou, C.; Quiroz, M.R.; Powell, K.L.; O’Meara, R.W.; Kothary, R.; Lorimer, I.A.; Parks, R.J. Retargeting of adenovirus vectors through genetic fusion of a single-chain or single-domain antibody to capsid protein IX. J. Virol. 2010, 84, 10074–10086. [Google Scholar] [CrossRef] [PubMed]
- Poulin, K.L.; Tong, G.; Vorobyova, O.; Pool, M.; Kothary, R.; Parks, R.J. Use of Cre/loxP recombination to swap cell binding motifs on the adenoviral capsid protein IX. Virology 2011, 420, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Nwanegbo, E.; Vardas, E.; Gao, W.; Whittle, H.; Sun, H.; Rowe, D.; Robbins, P.D.; Gambotto, A. Prevalence of neutralizing antibodies to adenoviral serotypes 5 and 35 in the adult populations of The Gambia, South Africa, and the United States. Clin. Diagn. Lab. Immunol. 2004, 11, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Mast, T.C.; Kierstead, L.; Gupta, S.B.; Nikas, A.A.; Kallas, E.G.; Novitsky, V.; Mbewe, B.; Pitisuttithum, P.; Schechter, M.; Vardas, E. International epidemiology of human pre-existing adenovirus (Ad) type-5, type-6, type-26 and type-36 neutralizing antibodies: Correlates of high Ad5 titers and implications for potential HIV vaccine trials. Vaccine 2010, 28, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Tsai, V.; Johnson, D.E.; Rahman, A.; Wen, S.F.; LaFace, D.; Philopena, J.; Nery, J.; Zepeda, M.; Maneval, D.C.; Demers, G.W. Impact of human neutralizing antibodies on antitumor efficacy of an oncolytic adenovirus in a murine model. Clin. Cancer Res. 2004, 10, 7199–7206. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Knipe, D.; Fields, B.; Howley, P.; Griffin, D.; Lamb, R. Adenoviridae: The viruses and their replication. Fields’ Virology 2007, 2, 2355–2395. [Google Scholar]
- Kojaoghlanian, T.; Flomenberg, P.; Horwitz, M.S. The impact of adenovirus infection on the immunocompromised host. Rev. Med. Virol. 2003, 13, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Giberson, A.N.; Davidson, A.R.; Parks, R.J. Chromatin structure of adenovirus DNA throughout infection. Nucleic Acids Res. 2012, 40, 2369–2376. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.J.; Benkő, M.; Harrach, B. Genetic content and evolution of adenoviruses. J. Gen. Virol. 2003, 84, 2895–2908. [Google Scholar] [CrossRef] [PubMed]
- Nash, L.A.; Parks, R.J. Adenovirus Biology and Development as a Gene Delivery Vector. In Gene Therapy with Adenoviral Based Vectors; Ng, P., Brunetti-Pierri, N., Eds.; Taylor and Francis: New York, NY, USA, 2016. [Google Scholar]
- Smith, A.C.; Poulin, K.L.; Parks, R.J. DNA genome size affects the stability of the adenovirus virion. J. Virol. 2009, 83, 2025–2028. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.B.; Byrd, S.A.; Walker, A.K.; Strahler, J.R.; Andrews, P.C.; Imperiale, M.J. Presence of the adenovirus IVa2 protein at a single vertex of the mature virion. J. Virol. 2008, 82, 9086–9093. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, R.P.; Mariano, T.M.; Siekierka, J.; Mathews, M.B. A mechanism for the control of protein synthesis by adenovirus VA RNA I. Cell 1986, 44, 391–400. [Google Scholar] [CrossRef]
- Aparicio, O.; Razquin, N.; Zaratiegui, M.; Narvaiza, I.; Fortes, P. Adenovirus virus-associated RNA is processed to functional interfering RNAs involved in virus production. J. Virol. 2006, 80, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Wong, C.M.; Parks, R.J. The adenovirus genome contributes to the structural stability of the virion. Viruses 2014, 6, 3563–3583. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.E.; Kay, M.A. Persistence of recombinant adenovirus in vivo is not dependent on vector DNA replication. J. Virol. 1997, 71, 8902–8907. [Google Scholar] [PubMed]
- Wolf, J.K.; Bodurka, D.C.; Gano, J.B.; Deavers, M.; Ramondetta, L.; Ramirez, P.T.; Levenback, C.; Gershenson, D.M. A phase I study of Adp53 (INGN 201; ADVEXIN) for patients with platinum-and paclitaxel-resistant epithelial ovarian cancer. Gynecol. Oncol. 2004, 94, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.; Lahrs, S.; DeMatteo, R.P. Overexpression of interleukin-12 enables dendritic cells to activate NK cells and confer systemic antitumor immunity. FASEB J. 2003, 17, 728–730. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.; Schuur, E.R.; Lim, H.Y.; Henderson, G.A.; Simons, J.W.; Henderson, D.R. Prostate attenuated replication competent adenovirus (ARCA) CN706: A selective cytotoxic for prostate-specific antigen-positive prostate cancer cells. Cancer Res. 1997, 57, 2559–2563. [Google Scholar] [PubMed]
- Ding, M.; Cao, X.; Xu, H.-N.; Fan, J.-K.; Huang, H.-L.; Yang, D.-Q.; Li, Y.-H.; Wang, J.; Li, R.; Liu, X.-Y. Prostate cancer-specific and potent antitumor effect of a DD3-controlled oncolytic virus harboring the PTEN gene. PLoS ONE 2012, 7, e35153. [Google Scholar] [CrossRef] [PubMed]
- Bandara, L.R.; La Thangue, N.B. Adenovirus E1a prevents the retinoblastoma gene product from complexing with a cellular transcription factor. Nature 1991, 351, 494–497. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Alemany, R.; Gomez-Manzano, C.; Medrano, D.R.; Lemoine, M.G.; Olson, M.V.; Alonso, M.M.; Lee, O.-H.; Conrad, C.C.; Yung, W.A. Downmodulation of El A Protein Expression as a Novel Strategy to Design Cancer-Selective Adenoviruses. Neoplasia 2005, 7, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Nevins, J.R. The Rb/E2F pathway and cancer. Hum. Mol. Genet. 2001, 10, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Zheng, S.; Li, X.-F.; Huang, J.-J.; Zheng, X.; Li, Z. Intra-tumor injection of H101, a recombinant adenovirus, in combination with chemotherapy in patients with advanced cancers: A pilot phase II clinical trial. World J. Gastroenterol. 2004, 10, 3634–3638. [Google Scholar] [CrossRef] [PubMed]
- Heise, C.C.; Williams, A.; Olesch, J.; Kirn, D.H. Efficacy of a replication-competent adenovirus (ONYX-015) following intratumoral injection: Intratumoral spread and distribution effects. Cancer Gene Ther. 1999, 6, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.F.; Bruner, J.M.; Fuller, G.N.; Aldape, K.; Prados, M.D.; Chang, S.; Berger, M.S.; McDermott, M.W.; Kunwar, S.M.; Junck, L.R. Phase I trial of adenovirus-mediated p53 gene therapy for recurrent glioma: Biological and clinical results. J. Clin. Oncol. 2003, 21, 2508–2518. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.; Breznik, J.; Lichty, B.D. Strategies to enhance viral penetration of solid tumors. Hum. Gene Ther. 2011, 22, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Delassus, S.; Sonigo, P.; Wain-Hobson, S. Genetic organization of gibbon ape leukemia virus. Virology 1989, 173, 205–213. [Google Scholar] [CrossRef]
- Plattet, P.; Alves, L.; Herren, M.; Aguilar, H.C. Measles Virus Fusion Protein: Structure, Function and Inhibition. Viruses 2016, 8, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Singh, M.; Malashkevich, V.N.; Kim, P.S. Structural characterization of the human respiratory syncytial virus fusion protein core. Proc. Natl. Acad. Sci. USA 2000, 97, 14172–14177. [Google Scholar] [CrossRef] [PubMed]
- Shmulevitz, M.; Duncan, R. A new class of fusion-associated small transmembrane (FAST) proteins encoded by the non-enveloped fusogenic reoviruses. EMBO J. 2000, 19, 902–912. [Google Scholar] [CrossRef] [PubMed]
- Chernomordik, L.V.; Kozlov, M.M. Mechanics of membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Bullough, F.; Murphy, S.; Emiliusen, L.; Lavillette, D.; Cosset, F.L.; Cattaneo, R.; Russell, S.J.; Vile, R.G. Fusogenic membrane glycoproteins as a novel class of genes for the local and immune-mediated control of tumor growth. Cancer Res. 2000, 60, 1492–1497. [Google Scholar] [PubMed]
- Bateman, A.R.; Harrington, K.J.; Kottke, T.; Ahmed, A.; Melcher, A.A.; Gough, M.J.; Linardakis, E.; Riddle, D.; Dietz, A.; Lohse, C.M.; et al. Viral fusogenic membrane glycoproteins kill solid tumor cells by nonapoptotic mechanisms that promote cross presentation of tumor antigens by dendritic cells. Cancer Res. 2002, 62, 6566–6578. [Google Scholar] [PubMed]
- Wong, C.M.; Poulin, K.L.; Tong, G.; Christou, C.; Kennedy, M.A.; Falls, T.; Bell, J.C.; Parks, R.J. Adenovirus-Mediated Expression of the p14 Fusion-Associated Small Transmembrane Protein Promotes Cancer Cell Fusion and Apoptosis In Vitro but Does Not Provide Therapeutic Efficacy in a Xenograft Mouse Model of Cancer. PLoS ONE 2016, 11, e0151516. [Google Scholar] [CrossRef] [PubMed]
- Salsman, J.; Top, D.; Boutilier, J.; Duncan, R. Extensive syncytium formation mediated by the reovirus FAST proteins triggers apoptosis-induced membrane instability. J. Virol. 2005, 79, 8090–8100. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Jia, H.; Liu, R.; Wu, J.; Han, H.; Zuo, Y.; Yang, S.; Huang, W. Inhibition of NF-κB in fusogenic membrane glycoprotein causing HL-60 cell death: Implications for acute myeloid leukemia. Cancer Lett. 2009, 273, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; McDonald, C.; Giannini, C.; Peng, K.W.; Rosales, G.; Russell, S.J.; Galanis, E. Adenoviral vectors expressing fusogenic membrane glycoproteins activated via matrix metalloproteinase cleavable linkers have significant antitumor potential in the gene therapy of gliomas. J. Gene Med. 2004, 6, 1216–1227. [Google Scholar] [CrossRef] [PubMed]
- Brade, A.M.; Szmitko, P.; Ngo, D.; Liu, F.F.; Klamut, H.J. Heat-directed tumor cell fusion. Hum. Gene Ther. 2003, 14, 447–461. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.; Cawood, R.; El-Sherbini, Y.; Purdie, L.; Bazan-Peregrino, M.; Seymour, L.W.; Carlisle, R.C. Active adenoviral vascular penetration by targeted formation of heterocellular endothelial-epithelial syncytia. Mol. Ther. 2011, 19, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Jevremovic, D.; Suzuki, K.; Kottke, T.; Thompson, J.; Emery, S.; Harrington, K.; Bateman, A.; Vile, R. Intratumoral expression of a fusogenic membrane glycoprotein enhances the efficacy of replicating adenovirus therapy. Gene Ther. 2003, 10, 1663–1671. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Gros, A.; Cascallo, M.; Vile, R.; Mercade, E.; Alemany, R. Syncytia formation affects the yield and cytotoxicity of an adenovirus expressing a fusogenic glycoprotein at a late stage of replication. Gene Ther. 2008, 15, 1240–1245. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Grases, D.; Rojas, J.J.; Gros, A.; Vilardell, F.; Vile, R.; Mercade, E.; Cascallo, M.; Alemany, R. GALV expression enhances the therapeutic efficacy of an oncolytic adenovirus by inducing cell fusion and enhancing virus distribution. Gene Ther. 2012, 19, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Alkhatib, G.; Richardson, C.; Shen, S.H. Intracellular processing, glycosylation, and cell-surface expression of the measles virus fusion protein (F) encoded by a recombinant adenovirus. Virology 1990, 175, 262–270. [Google Scholar] [CrossRef]
- Galanis, E.; Bateman, A.; Johnson, K.; Diaz, R.M.; James, C.D.; Vile, R.; Russell, S.J. Use of viral fusogenic membrane glycoproteins as novel therapeutic transgenes in gliomas. Hum. Gene Ther. 2001, 12, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, D.; Bayer, W.; Wildner, O. In situ tumor vaccination with adenovirus vectors encoding measles virus fusogenic membrane proteins and cytokines. World J. Gastroenterol. 2007, 13, 3063–3070. [Google Scholar] [PubMed]
- Hoffmann, D.; Wildner, O. Enhanced killing of pancreatic cancer cells by expression of fusogenic membrane glycoproteins in combination with chemotherapy. Mol. Cancer Ther. 2006, 5, 2013–2022. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, D.; Bangen, J.M.; Bayer, W.; Wildner, O. Synergy between expression of fusogenic membrane proteins, chemotherapy and facultative virotherapy in colorectal cancer. Gene Ther. 2006, 13, 1534–1544. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, D.; Bayer, W.; Heim, A.; Potthoff, A.; Nettelbeck, D.M.; Wildner, O. Evaluation of twenty-one human adenovirus types and one infectivity-enhanced adenovirus for the treatment of malignant melanoma. J. Investig. Dermatol. 2008, 128, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, D.; Bayer, W.; Grunwald, T.; Wildner, O. Intratumoral expression of respiratory syncytial virus fusion protein in combination with cytokines encoded by adenoviral vectors as in situ tumor vaccine for colorectal cancer. Mol. Cancer Ther. 2007, 6, 1942–1950. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, D.; Bayer, W.; Wildner, O. Therapeutic immune response induced by intratumoral expression of the fusogenic membrane protein of vesicular stomatitis virus and cytokines encoded by adenoviral vectors. Int. J. Mol. Med. 2007, 20, 673–681. [Google Scholar] [PubMed]
- Gomez-Trevino, A.; Castel, S.; Lopez-Iglesias, C.; Cortadellas, N.; Comas-Riu, J.; Mercade, E. Effects of adenovirus-mediated SV5 fusogenic glycoprotein expression on tumor cells. J. Gene Med. 2003, 5, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.M.; Nash, L.A.; Del Papa, J.; Poulin, K.L.; Falls, T.; Bell, J.C.; Parks, R.J. Expression of the fusogenic p14 FAST protein from a replication-defective adenovirus vector does not provide a therapeutic benefit in an immunocompetent mouse model of cancer. Cancer Gene Ther. 2016, 23, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Dewar, R.L.; Vasudevachari, M.; Natarajan, V.; Salzman, N. Biosynthesis and processing of human immunodeficiency virus type 1 envelope glycoproteins: Effects of monensin on glycosylation and transport. J. Virol. 1989, 63, 2452–2456. [Google Scholar] [PubMed]
- Li, H.; Haviv, Y.S.; Derdeyn, C.A.; Lam, J.; Coolidge, C.; Hunter, E.; Curiel, D.T.; Blackwell, J.L. Human immunodeficiency virus type 1-mediated syncytium formation is compatible with adenovirus replication and facilitates efficient dispersion of viral gene products and de novo-synthesized virus particles. Hum. Gene Ther. 2001, 12, 2155–2165. [Google Scholar] [CrossRef] [PubMed]
- Van Zeijl, M.; Johann, S.V.; Closs, E.; Cunningham, J.; Eddy, R.; Shows, T.B.; O’Hara, B. A human amphotropic retrovirus receptor is a second member of the gibbon ape leukemia virus receptor family. Proc. Natl. Acad. Sci. USA 1994, 91, 1168–1172. [Google Scholar] [CrossRef] [PubMed]
- Kavanaugh, M.P.; Kabat, D. Identification and characterization of a widely expressed phosphate transporter/retrovirus receptor family. Kidney Int. 1996, 49, 959–963. [Google Scholar] [CrossRef] [PubMed]
- Forestell, S.P.; Dando, J.S.; Chen, J.; de Vries, P.; Bohnlein, E.; Rigg, R.J. Novel retroviral packaging cell lines: Complementary tropisms and improved vector production for efficient gene transfer. Gene Ther. 1997, 4, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Fielding, A.K.; Chapel-Fernandes, S.; Chadwick, M.P.; Bullough, F.J.; Cosset, F.L.; Russell, S.J. A hyperfusogenic gibbon ape leukemia envelope glycoprotein: Targeting of a cytotoxic gene by ligand display. Hum. Gene Ther. 2000, 11, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.J.; Peng, K.W.; Allen, C.; Russell, S.J.; Galanis, E. Targeting the cytotoxicity of fusogenic membrane glycoproteins in gliomas through protease-substrate interaction. Gene Ther. 2003, 10, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Wust, P.; Hildebrandt, B.; Sreenivasa, G.; Rau, B.; Gellermann, J.; Riess, H.; Felix, R.; Schlag, P.M. Hyperthermia in combined treatment of cancer. Lancet Oncol. 2002, 3, 487–497. [Google Scholar] [CrossRef]
- Katz, S.L.; Yanagi, Y.; Kemper, C.; Navaratnarajah, C.K.; Rima, B.K. Measles: History and Basic Biology, 1st ed.; Griffin, D.E., Oldstone, M.B., Eds.; Springer: Heidelberg, Germany, 2008. [Google Scholar]
- Nakamura, T.; Peng, K.W.; Vongpunsawad, S.; Harvey, M.; Mizuguchi, H.; Hayakawa, T.; Cattaneo, R.; Russell, S.J. Antibody-targeted cell fusion. Nat. Biotechnol. 2004, 22, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Hammond, A.L.; Plemper, R.K.; Zhang, J.; Schneider, U.; Russell, S.J.; Cattaneo, R. Single-chain antibody displayed on a recombinant measles virus confers entry through the tumor-associated carcinoembryonic antigen. J. Virol. 2001, 75, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.W.; Donovan, K.A.; Schneider, U.; Cattaneo, R.; Lust, J.A.; Russell, S.J. Oncolytic measles viruses displaying a single-chain antibody against CD38, a myeloma cell marker. Blood 2003, 101, 2557–2562. [Google Scholar] [CrossRef] [PubMed]
- Linardakis, E.; Bateman, A.; Phan, V.; Ahmed, A.; Gough, M.; Olivier, K.; Kennedy, R.; Errington, F.; Harrington, K.J.; Melcher, A.; et al. Enhancing the efficacy of a weak allogeneic melanoma vaccine by viral fusogenic membrane glycoprotein-mediated tumor cell-tumor cell fusion. Cancer Res. 2002, 62, 5495–5504. [Google Scholar] [PubMed]
- Brown, C.W.; Stephenson, K.B.; Hanson, S.; Kucharczyk, M.; Duncan, R.; Bell, J.C.; Lichty, B.D. The p14 FAST protein of reptilian reovirus increases vesicular stomatitis virus neuropathogenesis. J. Virol. 2009, 83, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Stojdl, D.F.; Lichty, B.D.; Paterson, J.M.; Power, A.T.; Knowles, S.; Marius, R.; Reynard, J.; Poliquin, L.; Atkins, H.; Brown, E.G. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 2003, 4, 263–275. [Google Scholar] [CrossRef]
- Le Boeuf, F.; Diallo, J.-S.; McCart, J.A.; Thorne, S.; Falls, T.; Stanford, M.; Kanji, F.; Auer, R.; Brown, C.W.; Lichty, B.D. Synergistic interaction between oncolytic viruses augments tumor killing. Mol. Ther. 2010, 18, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Symons, J.A.; Alcamí, A.; Smith, G.L. Vaccinia virus encodes a soluble type I interferon receptor of novel structure and broad species soecificity. Cell 1995, 81, 551–560. [Google Scholar] [CrossRef]
- Bett, A.; Prevec, L.; Graham, F. Packaging capacity and stability of human adenovirus type 5 vectors. J. Virol. 1993, 67, 5911–5921. [Google Scholar] [PubMed]
- Kennedy, M.A.; Parks, R.J. Adenovirus virion stability and the viral genome: Size matters. Mol. Ther. 2009, 17, 1664–1666. [Google Scholar] [CrossRef] [PubMed]
- Imler, J.-L.; Bout, A.; Dreyer, D.; Dieterlé, A.; Schultz, H.; Valerio, D.; Mehtali, M.; Pavirani, A. Trans-complementation of E1-deleted adenovirus: A new vector to reduce the possibility of codissemination of wild-type and recombinant adenoviruses. Hum. Gene Ther. 1995, 6, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Tietge, U.J.; Kozarsky, K.F.; Donahee, M.H.; Rader, D.J. A tetracycline-regulated adenoviral expression system for in vivo delivery of transgenes to lung and liver. J. Gene Med. 2003, 5, 567–575. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Origin of Fusion Protein | Replication-Defective Ad Vector | Replication-Competent Ad Vector | Reference |
|---|---|---|---|
| Gibbon-Ape Leukemia Virus | ✓ | - | [65,66] |
| - | ✓ | [67] 1, [68] 2, [69,70] | |
| Measles Virus | ✓ | - | [71,72,73] |
| - | ✓ | [74,75,76] 3 | |
| Respiratory Syncytial Virus | ✓ | - | [77] |
| Vesicular Stomatitis Virus | ✓ | - | [78] |
| Simian Virus 5 | ✓ | - | [79] |
| Reptilian Reovirus | ✓ | - | [62,80] |
| Human Immunodeficiency Virus | - | ✓ | [81,82] |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Del Papa, J.; Parks, R.J. Adenoviral Vectors Armed with Cell Fusion-Inducing Proteins as Anti-Cancer Agents. Viruses 2017, 9, 13. https://doi.org/10.3390/v9010013
Del Papa J, Parks RJ. Adenoviral Vectors Armed with Cell Fusion-Inducing Proteins as Anti-Cancer Agents. Viruses. 2017; 9(1):13. https://doi.org/10.3390/v9010013
Chicago/Turabian StyleDel Papa, Joshua, and Robin J. Parks. 2017. "Adenoviral Vectors Armed with Cell Fusion-Inducing Proteins as Anti-Cancer Agents" Viruses 9, no. 1: 13. https://doi.org/10.3390/v9010013