Characterization of a Novel Bat Adenovirus Isolated from Straw-Colored Fruit Bat (Eidolon helvum)

 ,
, 
 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Samples
2.2. Virus Isolation
2.3. Transmission Electron Microscopy
2.4. Indirect Immunofluorescence Assay
2.5. Virus Purification and DNA Extraction for Next-Generation Sequencing
2.6. Library Preparation and Full-Length Genomic Sequencing
2.7. Phylogenetic Analysis and Other Bioinformatics Analysis
2.8. Analysis of In Vitro Cell Tropism of EhAdV 06-106
2.9. Nested PCR for Molecular Epizootiology and Direct Sequencing
2.10. Analysis of the 5′ End of the Pol mRNA
3. Results
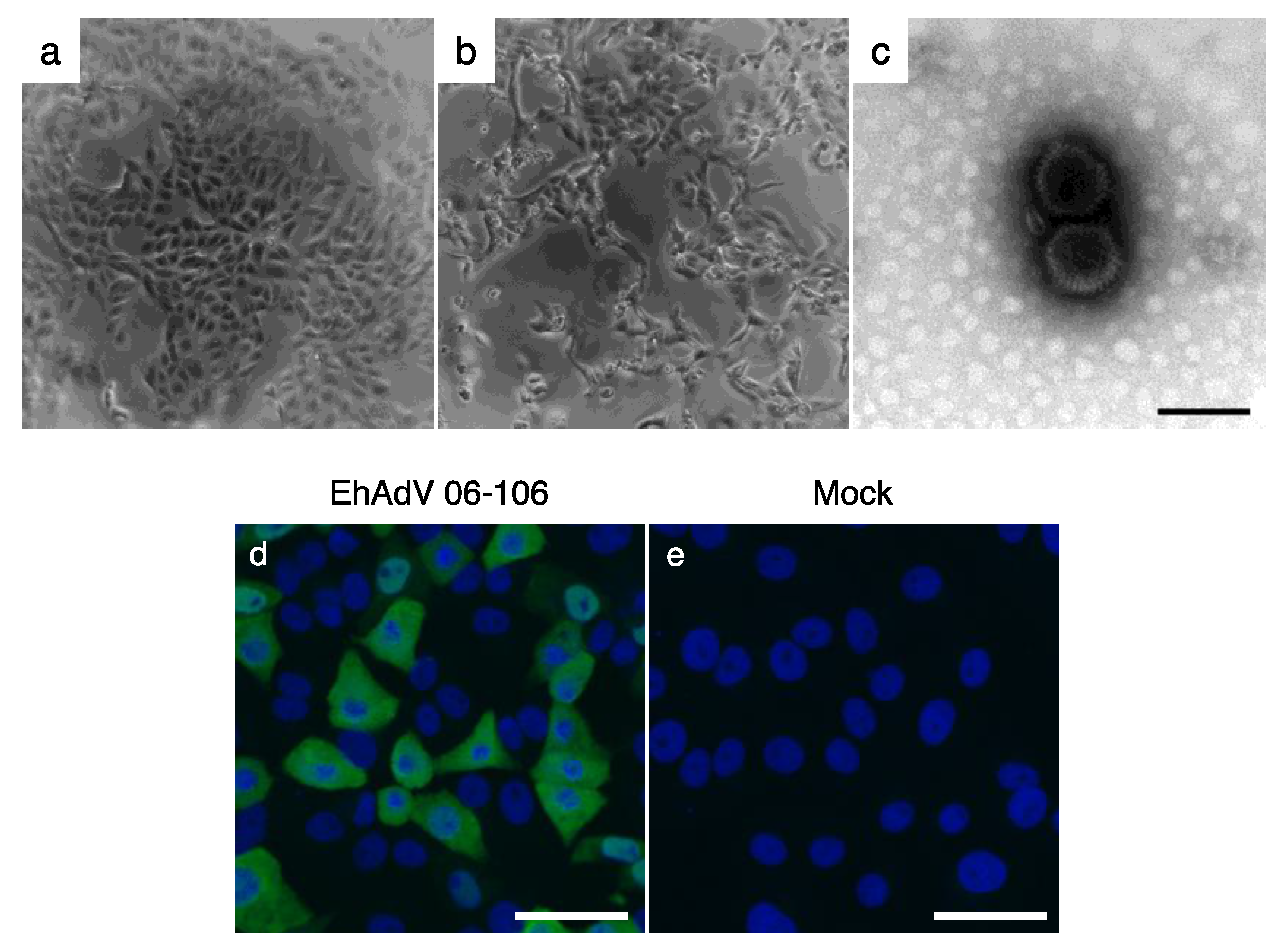
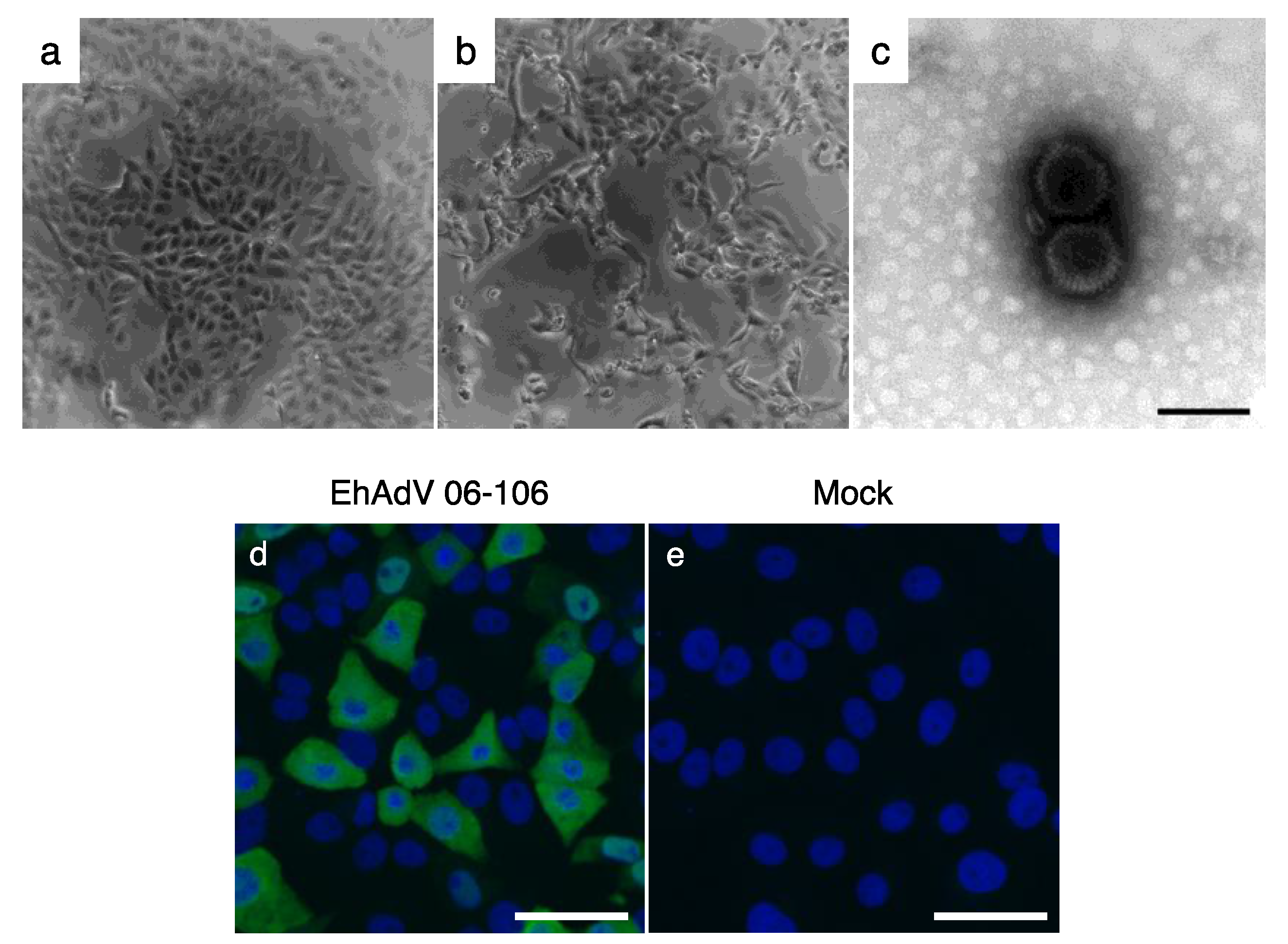
3.1. Isolation of a Novel Adenovirus from E. helvum Captured in Zambia
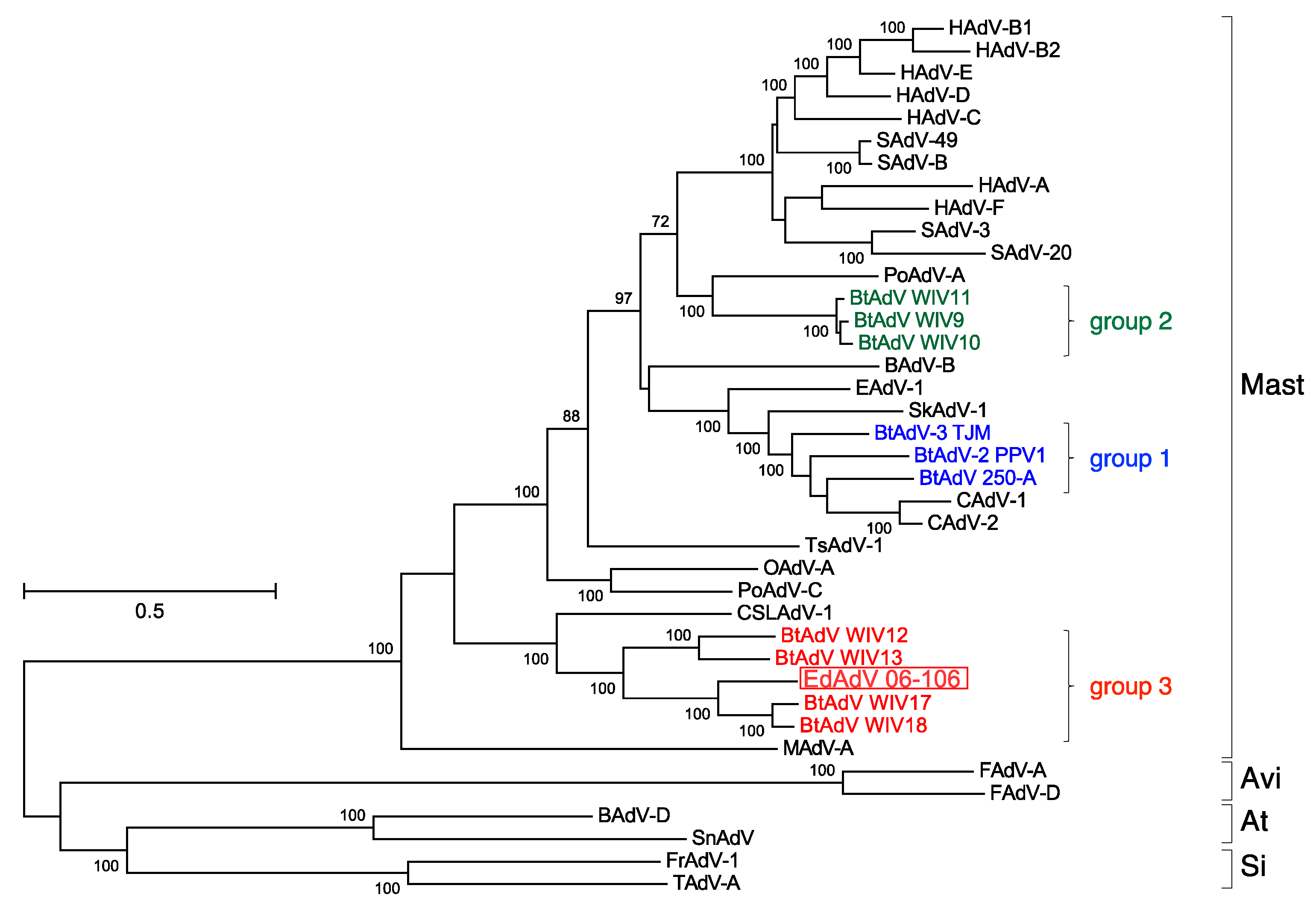
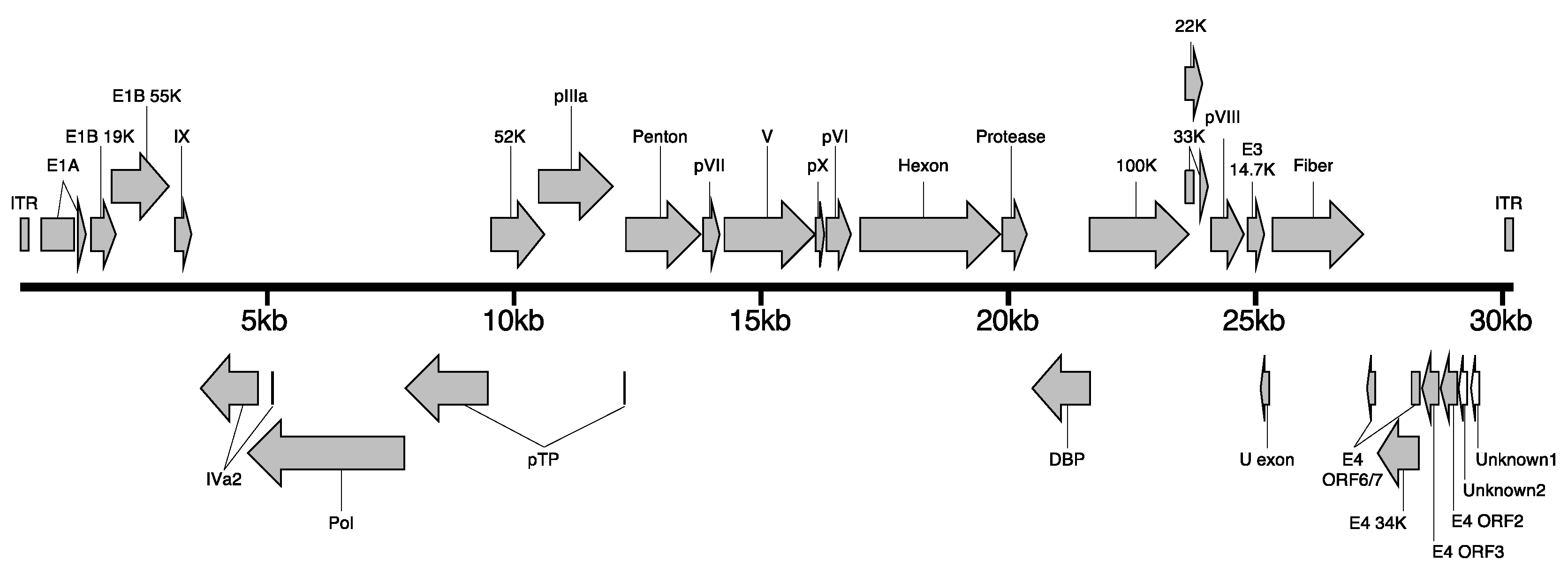
3.2. Whole Genome Analysis of EhAdV 06-106
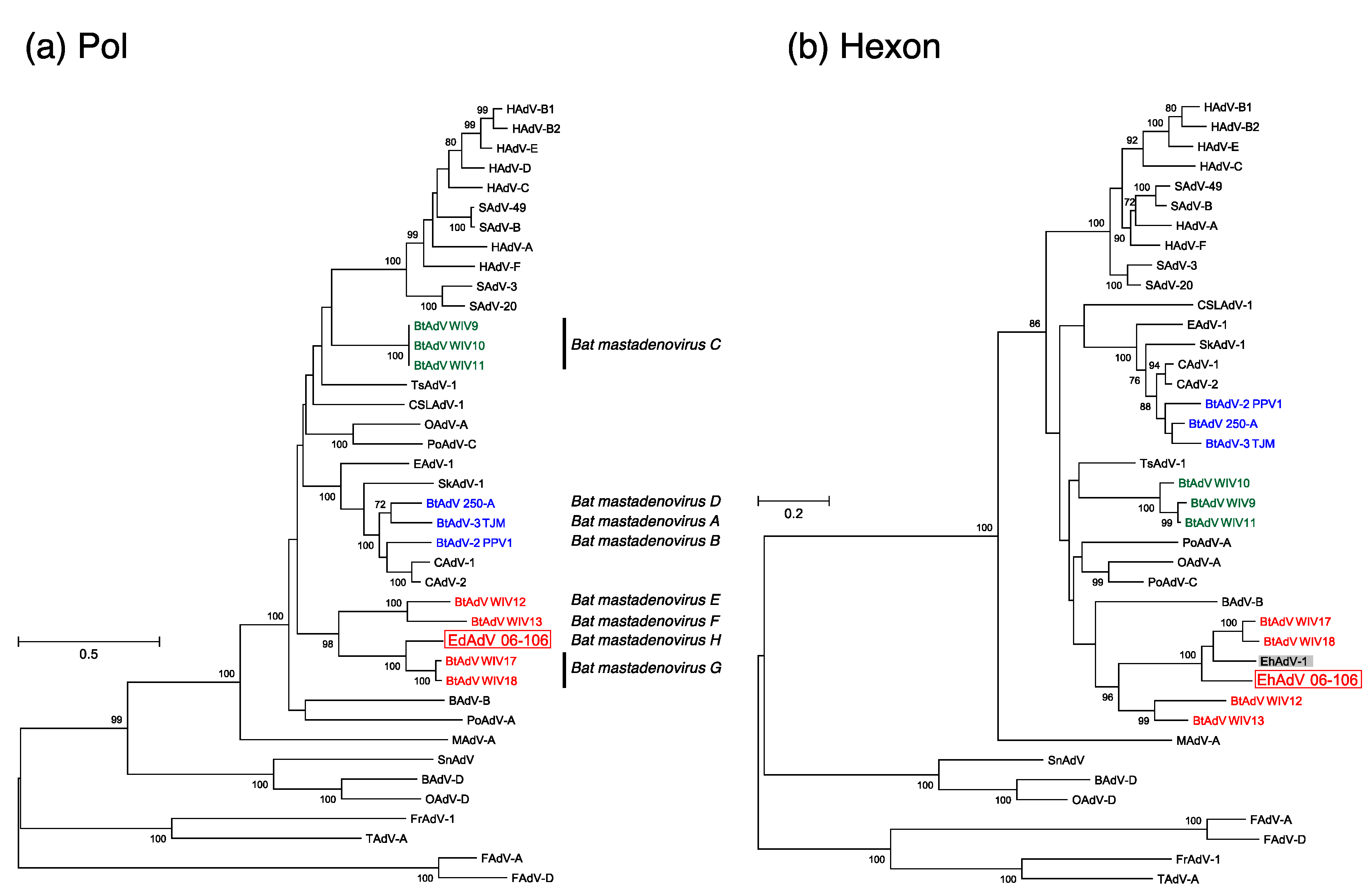
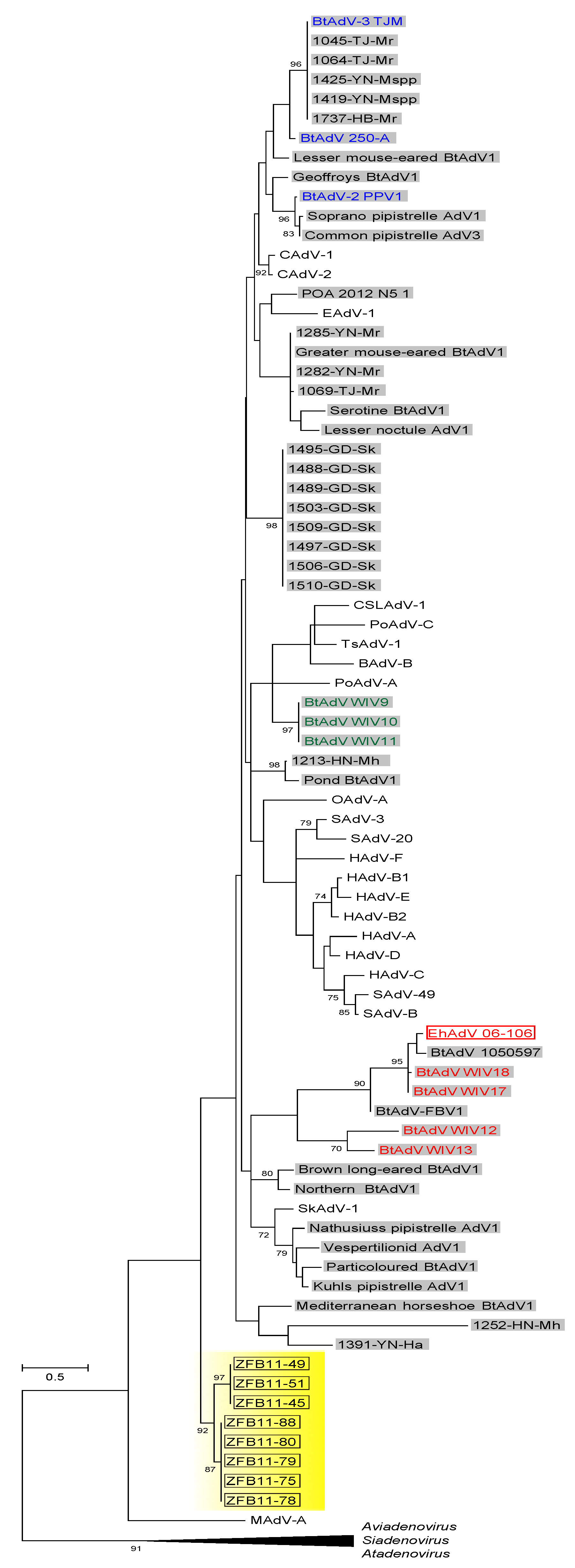
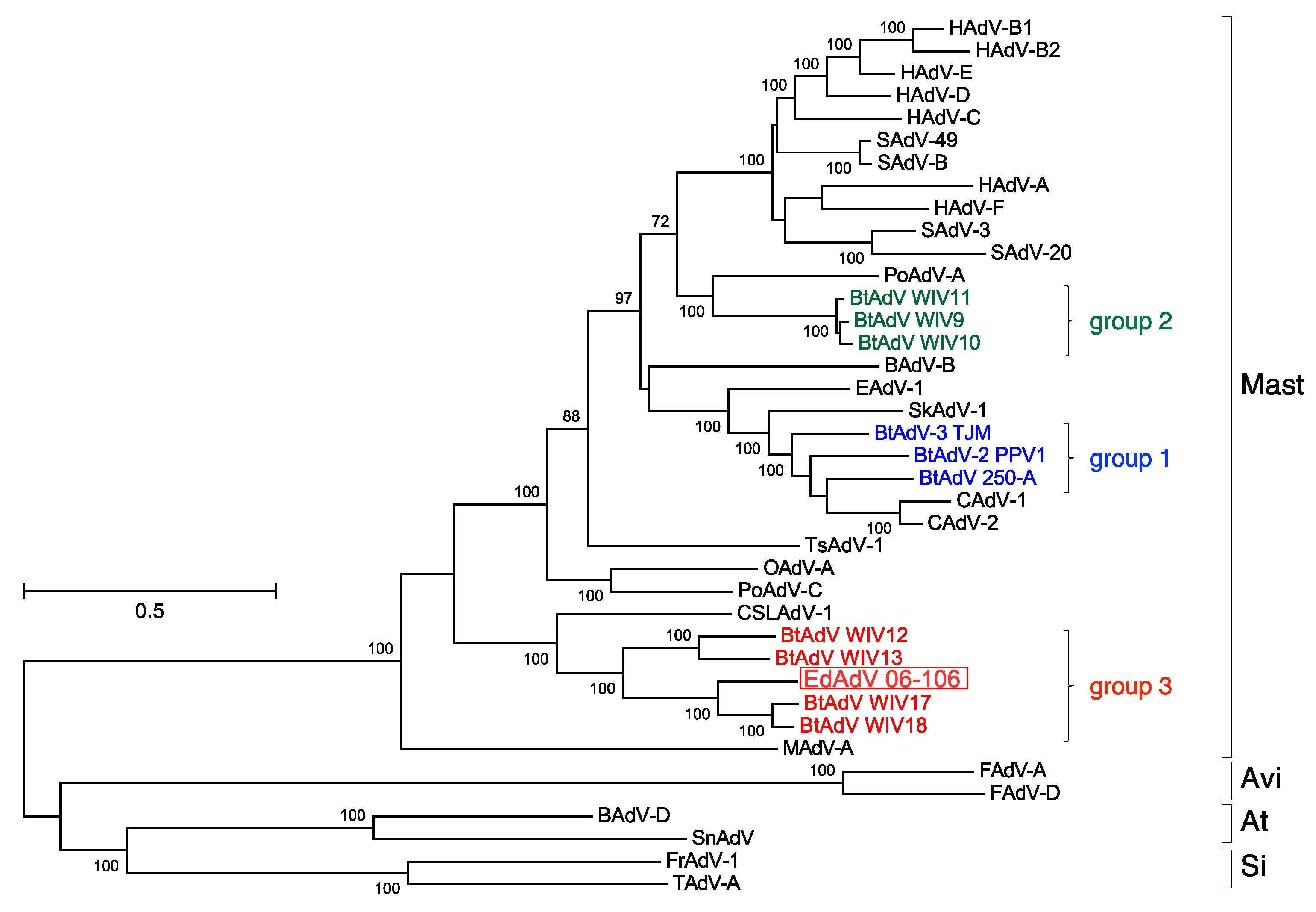
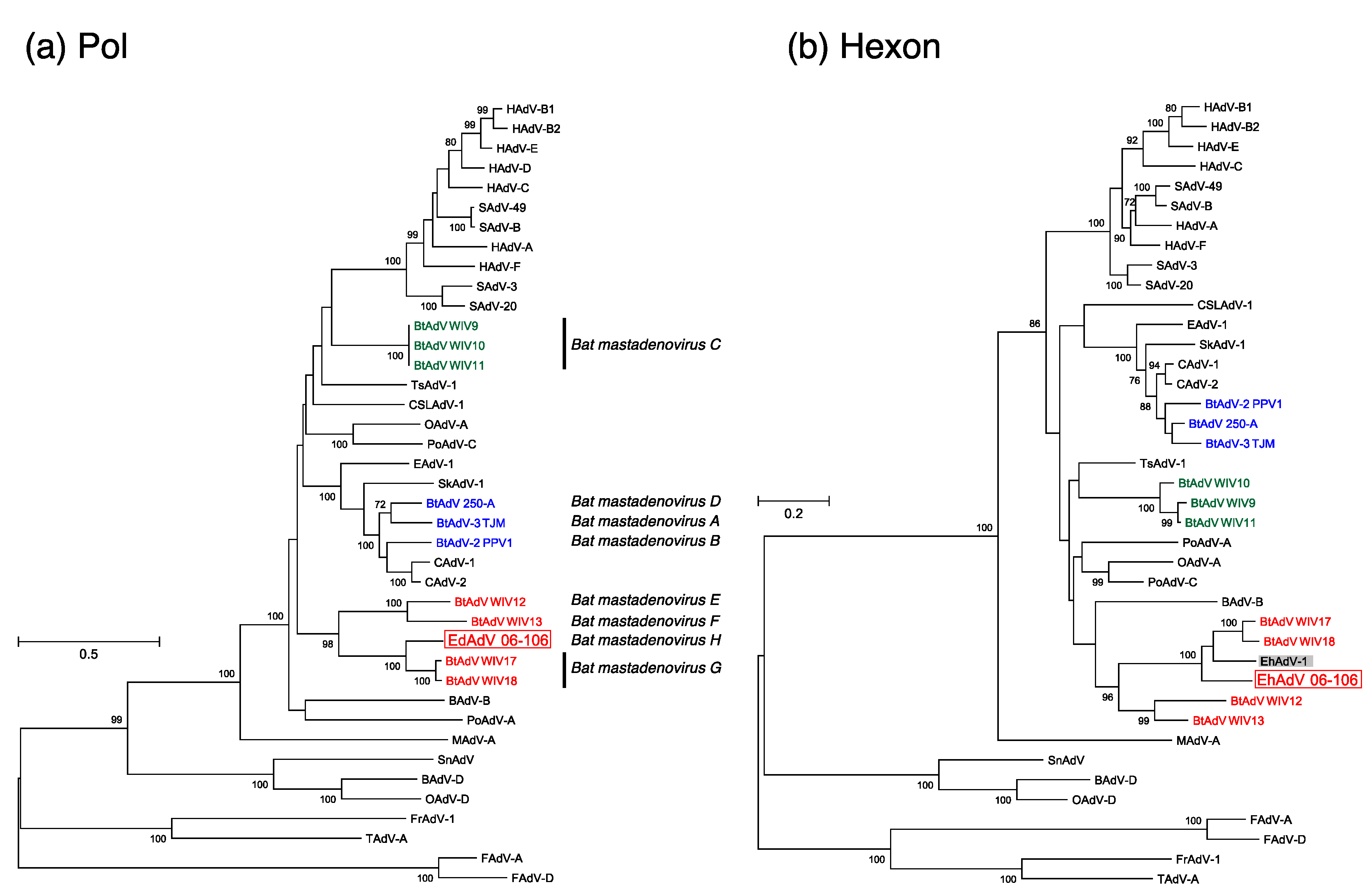
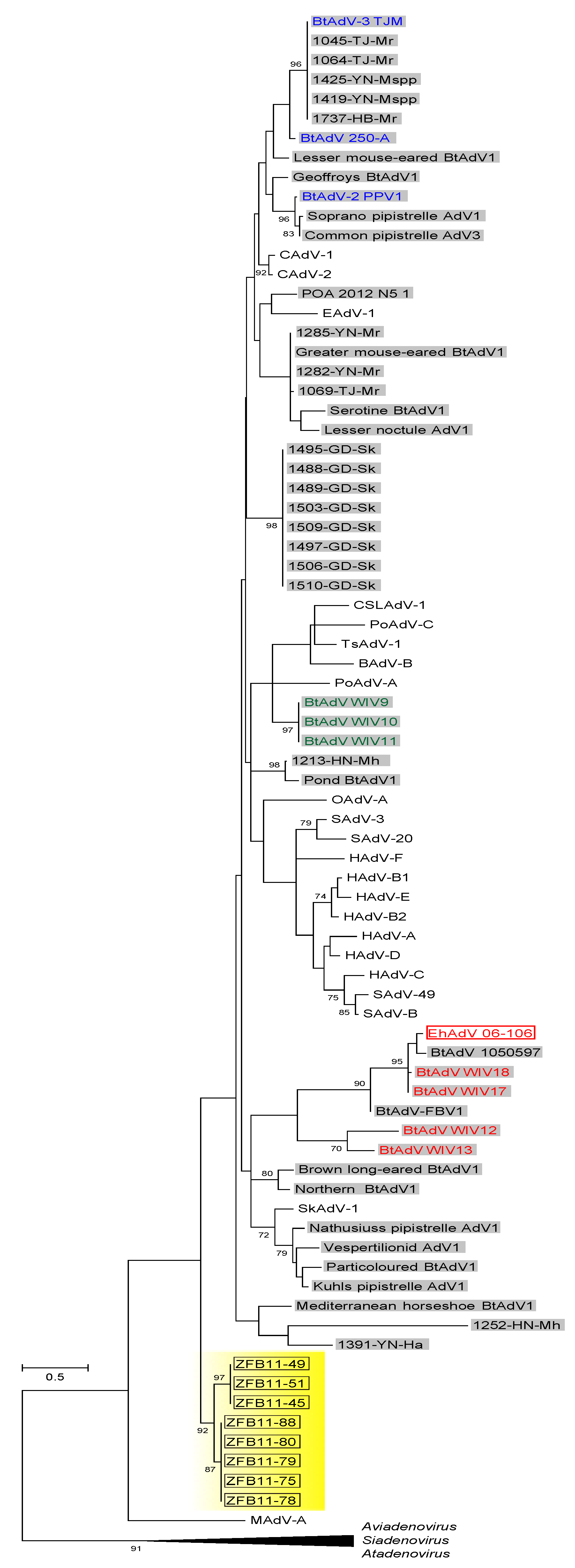
3.3. Phylogenetic Analyses of the Pol and Hexon Proteins
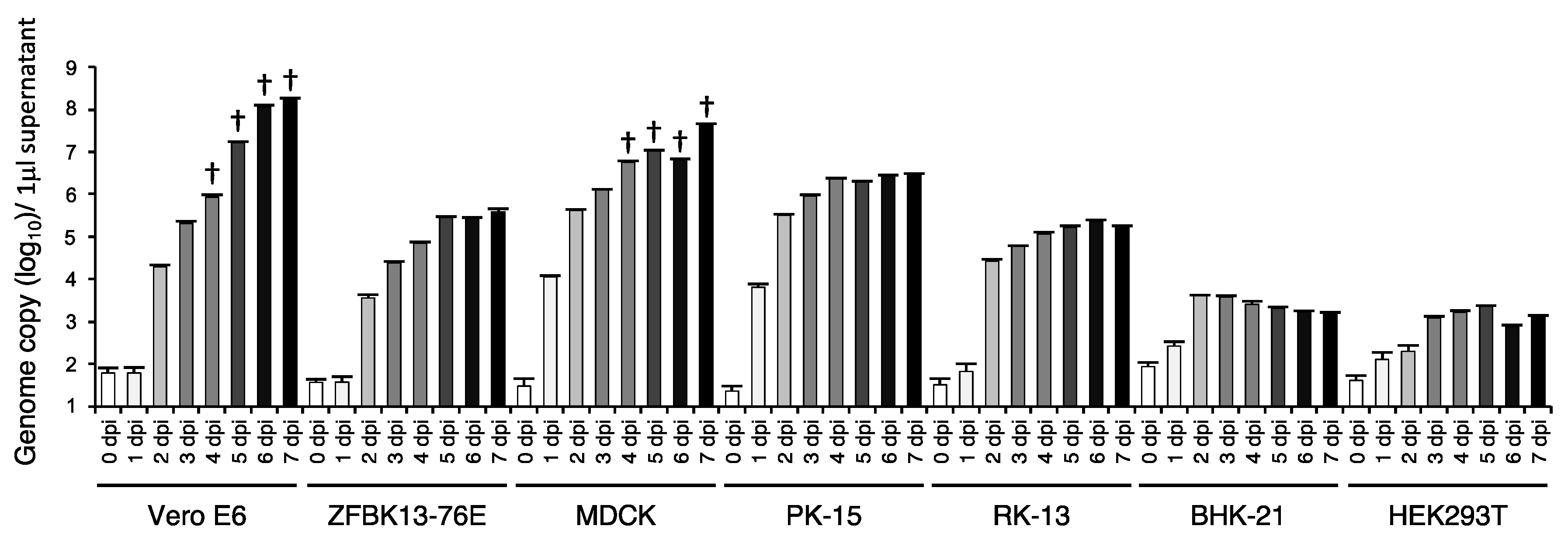
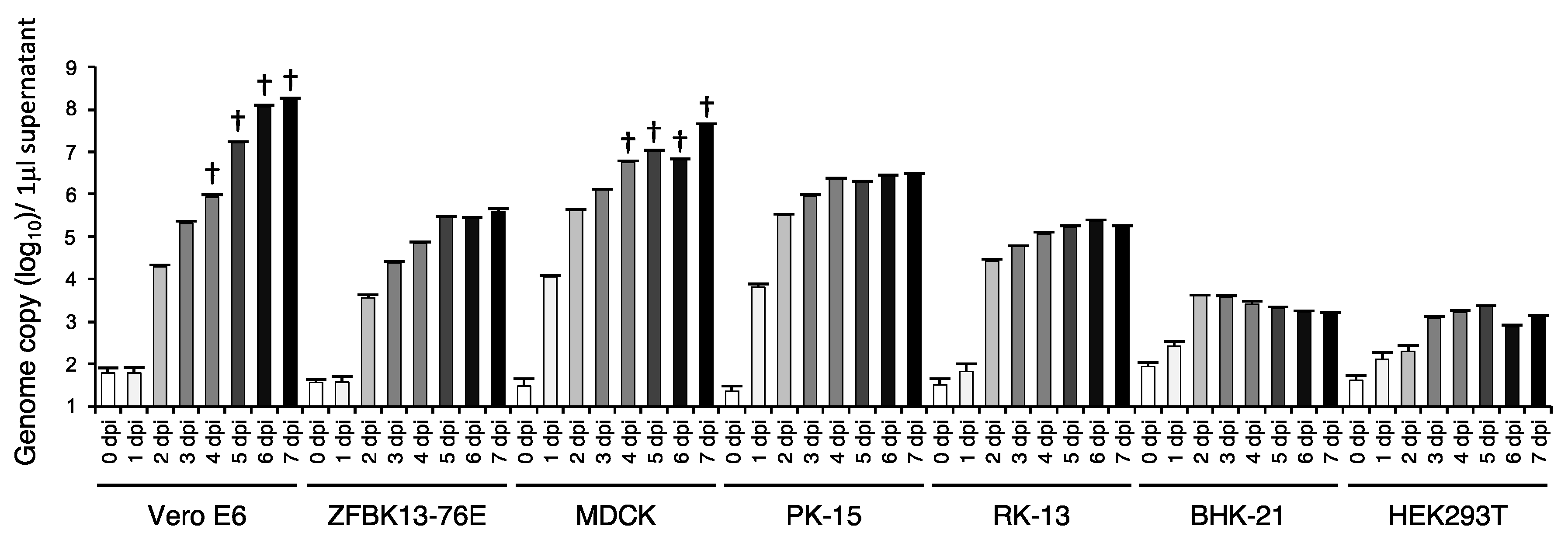
3.4. In Vitro Cell Tropism of EhAdV 06-106
3.5. Prevalence of Bat Adenoviruses in Zambia
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Jong, C.; Field, H.; Newman, S.H.; Epstein, J.H. Emerging infectious dieses. In Investigating the Role of Bats in Emerging Zoonoses: Balancing Ecology, Conservation and Public Health Interests; Newman, S.H., Field, H.E., de Jong, C.E., Epstein, J.H., Eds.; FAO: Rome, Italy, 2011; pp. 1–13. ISBN 978-92-5-107028-4. [Google Scholar]
- Calisher, C.H.; Childs, J.E.; Field, H.E.; Holmes, K.V.; Schountz, T. Bats: Important reservoir hosts of emerging viruses. Clin. Microbiol. Rev. 2006, 19, 531–545. [Google Scholar] [CrossRef] [PubMed]
- Muleya, W.; Sasaki, M.; Orba, Y.; Ishii, A.; Thomas, Y.; Nakagawa, E.; Ogawa, H.; Hang’ombe, B.; Namangala, B.; Mweene, A.; et al. Molecular epidemiology of paramyxoviruses in frugivorous Eidolon helvum bats in Zambia. J. Vet. Med. Sci. 2014, 76, 611–614. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, H.; Koizumi, N.; Ohnuma, A.; Mutemwa, A.; Hang’ombe, B.M.; Mweene, A.S.; Takada, A.; Sugimoto, C.; Suzuki, Y.; Kida, H.; et al. Molecular epidemiology of pathogenic Leptospira spp. in the straw-colored fruit bat (Eidolon helvum) migrating to Zambia from the Democratic Republic of Congo. Infect. Genet. Evol. 2015, 32, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, H.; Miyamoto, H.; Nakayama, E.; Yoshida, R.; Nakamura, I.; Sawa, H.; Ishii, A.; Thomas, Y.; Nakagawa, E.; Matsuno, K.; et al. Seroepidemiological Prevalence of Multiple Species of Filoviruses in Fruit Bats (Eidolon helvum) Migrating in Africa. J. Infect. Dis. 2015, 212, S101–S108. [Google Scholar] [CrossRef] [PubMed]
- Harrach, B.; Benkö, M.; Both, G.; Brown, M.; Davison, A.; Echavarría, M.; Hess, M.; Jones, M.; Kajon, A.; Lehmkuhl, H.; et al. Family Adenoviridae. In Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses; King, A., Adams, M., Carstens, E., Lefkowitz, E., Eds.; Elsevier: San Diego, CA, USA, 2011; pp. 125–141. ISBN 978-0-12-384684-6. [Google Scholar]
- Berk, A.J. Adenoviridae . In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 2, pp. 1704–1731. ISBN 978-1451105636. [Google Scholar]
- Benkö, M.; Harrach, B.; Kremer, E.J. Do nonhuman primate or bat adenoviruses pose a risk for human health? Future Microbiol. 2014, 9, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Buonavoglia, C.; Martella, V. Canine respiratory viruses. Vet. Res. 2007, 38, 355–373. [Google Scholar] [CrossRef] [PubMed]
- International Committee on Taxonomy of Viruses (ICTV) Virus Taxonomy: 2016 Release. Available online: http://talk.ictvonline.org/taxonomy/ (accessed on 20 September 2017).
- Maeda, K.; Hondo, E.; Terakawa, J.; Kiso, Y.; Nakaichi, N.; Endoh, D.; Sakai, K.; Morikawa, S.; Mizutani, T. Isolation of novel adenovirus from fruit bat (Pteropus dasymallus yayeyamae). Emerg. Infect. Dis. 2008, 14, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Vidovszky, M.; Kohl, C.; Boldogh, S.; Gőrfől, T.; Wibbelt, G.; Kurth, A.; Harrach, B. Random sampling of the Central European bat fauna reveals the existence of numerous hitherto unknown adenoviruses. Acta Vet. Hung. 2015, 63, 508–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jánoska, M.; Vidovszky, M.; Molnár, V.; Liptovszky, M.; Harrach, B.; Benkő, M. Novel adenoviruses and herpesviruses detected in bats. Vet. J. 2011, 189, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ge, X.; Zhang, H.; Zhou, P.; Zhu, Y.; Zhang, Y.; Yuan, J.; Wang, L.F.; Shi, Z. Host range, prevalence, and genetic diversity of adenoviruses in bats. J. Virol. 2010, 84, 3889–3897. [Google Scholar] [CrossRef] [PubMed]
- Raut, C.G.; Yadav, P.D.; Towner, J.S.; Amman, B.R.; Erickson, B.R.; Cannon, D.L.; Sivaram, A.; Basu, A.; Nichol, S.T.; Mishra, A.C.; et al. Isolation of a novel adenovirus from Rousettus leschenaultii bats from India. Intervirology 2012, 55, 488–490. [Google Scholar] [CrossRef] [PubMed]
- Sonntag, M.; Mühldorfer, K.; Speck, S.; Wibbelt, G.; Kurth, A. New adenovirus in bats, Germany. Emerg. Infect. Dis. 2009, 15, 2052–2055. [Google Scholar] [CrossRef] [PubMed]
- International Committee on Taxonomy of Viruses (ICTV) Virus Taxonomy History: Bat Mastadenovirus A. Available online: http://talk.ictvonline.org/taxonomy/p/taxonomy-history?taxnode_id=20161725 (accessed on 8 September 2017).
- Kohl, C.; Vidovszky, M.Z.; Mühldorfer, K.; Dabrowski, P.W.; Radonic, A.; Nitsche, A.; Wibbelt, G.; Kurth, A.; Harrach, B. Genome analysis of bat adenovirus 2: Indications of interspecies transmission. J. Virol. 2012, 86, 1888–1892. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.; Yang, X.L.; Ge, X.Y.; Peng, C.; Zhang, Y.Z.; Zhang, L.B.; Shi, Z.L. Novel bat adenoviruses with an extremely large E3 gene. J. Gen. Virol. 2016, 97, 1625–1635. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.; Yang, X.L.; Ge, X.Y.; Peng, C.; Liu, H.Z.; Zhang, Y.Z.; Zhang, L.B.; Shi, Z.L. Novel bat adenoviruses with low G + C content shed new light on the evolution of adenoviruses. J. Gen. Virol. 2017, 98, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Hinojosa, G.; Gulland, F.M.; Goldstein, T.; Venn-Watson, S.; Rivera, R.; Waltzek, T.B.; Salemi, M.; Wellehan, J.F., Jr. Phylogenomic characterization of California sea lion adenovirus-1. Infect. Genet. Evol. 2015, 31, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Nao, N.; Yamagishi, J.; Miyamoto, H.; Igarashi, M.; Manzoor, R.; Ohnuma, A.; Tsuda, Y.; Furuyama, W.; Shigeno, A.; Kajihara, M.; et al. Genetic predisposition to acquire a polybasic cleavage site for highly pathogenic avian influenza virus hemagglutinin. MBio 2017, 8, e02298-16. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved splice site detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, J.; Miyamoto, H.; Kajihara, M.; Ogawa, H.; Maeda, K.; Sakoda, Y.; Yoshida, R.; Takada, A. Characterization of the envelope glycoprotein of a novel filovirus, lloviu virus. J. Virol. 2014, 88, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Chroboczek, J.; Bieber, F.; Jacrot, B. The sequence of the genome of adenovirus type 5 and its comparison with the genome of adenovirus type 2. Virology 1992, 186, 280–285. [Google Scholar] [CrossRef]
- Hackenbrack, N.; Rogers, M.B.; Ashley, R.E.; Keel, M.K.; Kubiski, S.V.; Bryan, J.A.; Ghedin, E.; Holmes, E.C.; Hafenstein, S.L.; Allison, A.B. Evolution and cryo-electron microscopy capsid structure of a north American bat adenovirus and its relationship to other Mastadenoviruses. J. Virol. 2017, 91, e01504-16. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.J.; Benko, M.; Harrach, B. Genetic content and evolution of adenoviruses. J. Gen. Virol. 2003, 84, 2895–2908. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.S.; Leggett, R.M.; Bexfield, N.H.; Alston, M.; Daly, G.; Todd, S.; Tachedjian, M.; Holmes, C.E.; Crameri, S.; Wang, L.F.; et al. Metagenomic study of the viruses of African straw-coloured fruit bats: Detection of a chiropteran poxvirus and isolation of a novel adenovirus. Virology 2013, 441, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.S.; Idamakanti, N.; Zakhartchouk, A.N.; Baxi, M.K.; Lee, J.B.; Pyne, C.; Babiuk, L.A.; Tikoo, S.K. Nucleotide sequence, genome organization, and transcription map of bovine adenovirus type 3. J. Virol. 1998, 72, 1394–1402. [Google Scholar] [PubMed]
- DeFrees, S.L.; Wilson, D.E. Eidolon helvum. Mamm. Species 1988, 312, 1–5. [Google Scholar] [CrossRef]
- Richter, H.V.; Cumming, G.S. First application of satellite telemetry to track African straw-coloured fruit bat migration. J. Zool. 2008, 275, 172–176. [Google Scholar] [CrossRef]
- Benkö, M.; Harrach, B. A proposal for a new (third) genus within the family Adenoviridae. Arch. Virol. 1998, 143, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Benkö, M.; Harrach, B. Molecular evolution of adenoviruses. Curr. Top. Microbiol. Immunol. 2003, 272, 3–35. [Google Scholar] [CrossRef] [PubMed]
- Wellehan, J.F.; Johnson, A.J.; Harrach, B.; Benko, M.; Pessier, A.P.; Johnson, C.M.; Garner, M.M.; Childress, A.; Jacobson, E.R. Detection and analysis of six lizard adenoviruses by consensus primer PCR provides further evidence of a reptilian origin for the atadenoviruses. J. Virol. 2004, 78, 13366–13369. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, Y.; Takeuchi, O.; Akira, S. TLR9 as a key receptor for the recognition of DNA. Adv. Drug. Deliv. Rev. 2008, 60, 795–804. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Strain | ICTV Species * | Host | Country | Full-Genome (bp) | ITR (bp) | E3 Region | G + C Content (%) | Reference | |
|---|---|---|---|---|---|---|---|---|---|---|
| Product | Total Length (bp) † | |||||||||
| Group 1 | PPV1 | Bat mastadenovirus B | Pipistrellus pipistrellus | Germany | 31,616 | 137 | 12.5K, ORF1 | 1524 | 53.5 | [18] |
| TJM | Bat mastadenovirus A | Myotis ricketti | China | 31,681 | 128 | ORFA | 1149 | 56.9 | [14] | |
| 250-A | (Bat mastadenovirus D) | Corynorhinus rafinesquii | United States | 31,484 | 239 | 12.5K, ORFA | 1515 | 49.8 | [28] | |
| Group 2 | WIV9 | (Bat mastadenovirus C) | Rhinolophus sinicus | China | 37,545 | 35 | 12.5K, E3L, E3s | 5145 | 55.5 | [19] |
| WIV10 | (Bat mastadenovirus C) | Rhinolophus sinicus | China | 37,556 | 51 | 12.5K, E3L, E3s | 5133 | 55.2 | [19] | |
| WIV11 | (Bat mastadenovirus C) | Rhinolophus sinicus | China | 38,073 | 51 | 12.5K, E3L, E3s | 5631 | 55.0 | [19] | |
| Group 3 | WIV12 | (Bat mastadenovirus E) | Miniopterus schreibersii | China | 29,581 | 73 | 14.7K | 390 | 34.2 | [20] |
| WIV13 | (Bat mastadenovirus F) | Miniopterus schreibersii | China | 29,162 | 61 | 14.7K | 363 | 31.3 | [20] | |
| WIV17 | (Bat mastadenovirus G) | Rousettus Ieschenaultii | China | 29,923 | 178 | 14.7K | 396 | 34.3 | [20] | |
| WIV18 | (Bat mastadenovirus G) | Rousettus Ieschenaultii | China | 29,812 | 177 | 14.7K | 396 | 34.2 | [20] | |
| EhAdV 06-106 | (Bat mastadenovirus H) | Eidolon helvum | Zambia | 30,134 | 102 | 14.7K | 378 | 35.2 | This study | |
| EhAdV 06-106 | Bat Adenovirus WIV17 | Bat Adenovirus WIV18 | |||||
|---|---|---|---|---|---|---|---|
| Gene Product | Genomic Position * | Gene (nt) | Protein (AA) | Protein (AA) | Identity (%) | Protein (AA) | Identity (%) |
| ITR | 1–102 | 102 | N.A. † | N.A. | N.A. | N.A. | N.A. |
| E1A | 487–1090, 1230–1294 | 669 | 222 | 195 | 46.1 | 195 | 44.1 |
| E1B small | 1529–2014 | 486 | 161 | 168 | 53.4 | 168 | 44.7 |
| E1B large | 1909–3180 | 1272 | 423 | 425 | 59.5 | 425 | 60.2 |
| IX | 3240–3506 | 267 | 88 | 83 | 46.9 | 82 | 51.2 |
| Iva2 | 3550–4874c, 5153–5165c | 1338 | 445 | 422 | 82.7 | 422 | 82.7 |
| Pol | 4638–7895c | 3258 | 1085 | 1086 | 80.4 | 1086 | 81.1 |
| pTP | 7892–9682c, 12,334–12,342c | 1780 | 599 | 556 | 78.8 | 556 | 80.2 |
| 52K | 9697–10821 | 1125 | 374 | 373 | 80.4 | 373 | 79.3 |
| pIIIa | 10,742–12,319 | 1578 | 525 | 521 | 82.8 | 521 | 82.4 |
| Penton | 12,377–13,768 | 1392 | 463 | 464 | 82.9 | 464 | 83.8 |
| pVII | 13,771–14,115 | 345 | 114 | 118 | 83.9 | 118 | 83.9 |
| V | 14,159–16,048 | 1890 | 629 | 582 | 63.6 | 571 | 65.9 |
| pX | 16,074–16,280 | 207 | 68 | 66 | 83.3 | 66 | 84.8 |
| pVI | 16,341–16,970 | 630 | 209 | 218 | 81.8 | 218 | 82.8 |
| Hexon | 17,053–19,785 | 2733 | 910 | 910 | 82.1 | 910 | 82.5 |
| Protease | 19,786–20,394 | 609 | 202 | 200 | 77.0 | 200 | 76.0 |
| DBP | 20,457–21,695c | 1239 | 412 | 413 | 72.2 | 413 | 72.0 |
| 100K | 21,710–23,716 | 2007 | 668 | 650 | 77.9 | 650 | 78.5 |
| 33K | 23,595–23,736, 23,897–24,153 | 399 | 132 | 142 | 77.2 | 140 | 75.7 |
| 22K | 23,595–23,921 | 327 | 106 | 118 | 75.9 | 116 | 74.0 |
| pVIII | 24,153–24,794 | 642 | 213 | 213 | 85.9 | 213 | 84.9 |
| E3 14.7K | 24,797–25,174 | 378 | 125 | 131 | 52.7 | 131 | 48.0 |
| U exon | 25,191–25,361c | 171 | 56 | 55 | 70.9 | 55 | 69.0 |
| Fiber | 25,360–27,264 | 1905 | 634 | 593 | 37.6 | 579 | 38.5 |
| E4 ORF6/7 | 27,287–27,514c, 28,217–28,255c | 267 | 88 | 88 | 52.2 | 88 | 52.2 |
| E4 34K | 27,515–28,255 | 741 | 246 | 245 | 62.4 | 245 | 63.2 |
| E4 ORF3 | 28,258–28,662c | 405 | 134 | 141 | 41.0 | 141 | 41.0 |
| E4 ORF2 | 28,674–29,081c | 408 | 135 | 135 | 35.0 | 135 | 35.0 |
| E4 unknown2 | 29,164–29,292c | 129 | 42 | N.A. | N.A. | N.A. | N.A. |
| E4 unknown1 | 29,329–29,556c | 228 | 75 | N.A. | N.A. | N.A. | N.A. |
| ITR | 30,033–30,134 | 102 | N.A. | N.A. | N.A. | N.A. | N.A. |
| Year | Sample ID | Location | No. of Samples | ||
|---|---|---|---|---|---|
| Total | Positive | Positive Rate (%) | |||
| 2010 | ZFB10-01–ZFB10-47 | Kasanka National Park | 47 | 0 | 0 |
| ZFB10-48–ZFB10-52 | Ndola | 4 | 0 | 0 | |
| 2011 | ZFB11-01–ZFB11-38 | Ndola | 38 | 0 | 0 |
| ZFB11-39–ZFB11-95 | Kasanka National Park | 57 | 8 | 14.04 | |
| 2012 | ZFB12-01–ZFB12-60 | Ndola | 60 | 0 | 0 |
| ZFB12-61–ZFB12-110 * | Kasanka National Park | 49 | 0 | 0 | |
| 2013 | ZFB13-01–ZFB13-76 | Ndola | 76 | 0 | 0 |
| ZFB13-77–ZFB13-111 † | Kasanka National Park | 34 | 0 | 0 | |
| Total | 365 | 8 | 2.19 | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogawa, H.; Kajihara, M.; Nao, N.; Shigeno, A.; Fujikura, D.; Hang’ombe, B.M.; Mweene, A.S.; Mutemwa, A.; Squarre, D.; Yamada, M.; et al. Characterization of a Novel Bat Adenovirus Isolated from Straw-Colored Fruit Bat (Eidolon helvum). Viruses 2017, 9, 371. https://doi.org/10.3390/v9120371
Ogawa H, Kajihara M, Nao N, Shigeno A, Fujikura D, Hang’ombe BM, Mweene AS, Mutemwa A, Squarre D, Yamada M, et al. Characterization of a Novel Bat Adenovirus Isolated from Straw-Colored Fruit Bat (Eidolon helvum). Viruses. 2017; 9(12):371. https://doi.org/10.3390/v9120371
Chicago/Turabian StyleOgawa, Hirohito, Masahiro Kajihara, Naganori Nao, Asako Shigeno, Daisuke Fujikura, Bernard M. Hang’ombe, Aaron S. Mweene, Alisheke Mutemwa, David Squarre, Masao Yamada, and et al. 2017. "Characterization of a Novel Bat Adenovirus Isolated from Straw-Colored Fruit Bat (Eidolon helvum)" Viruses 9, no. 12: 371. https://doi.org/10.3390/v9120371