Mutations in HPV18 E1^E4 Impact Virus Capsid Assembly, Infectivity Competence, and Maturation

Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
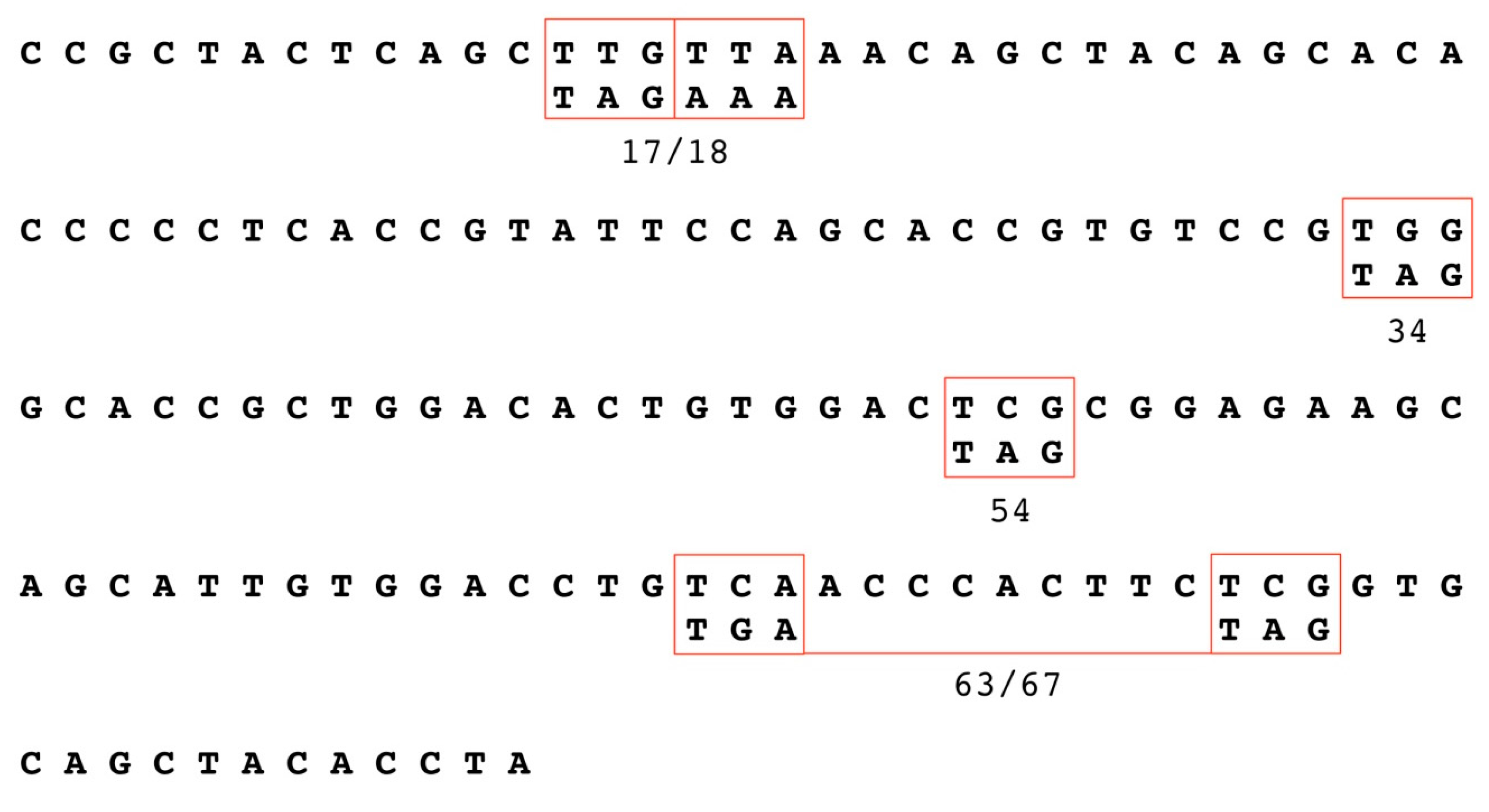
2.2. Generation of HPV18 E1^E4 Mutants
2.3. Keratinocyte Cultures and Electroporation
2.4. Organotypic Raft Culture Derived Native Virion Production
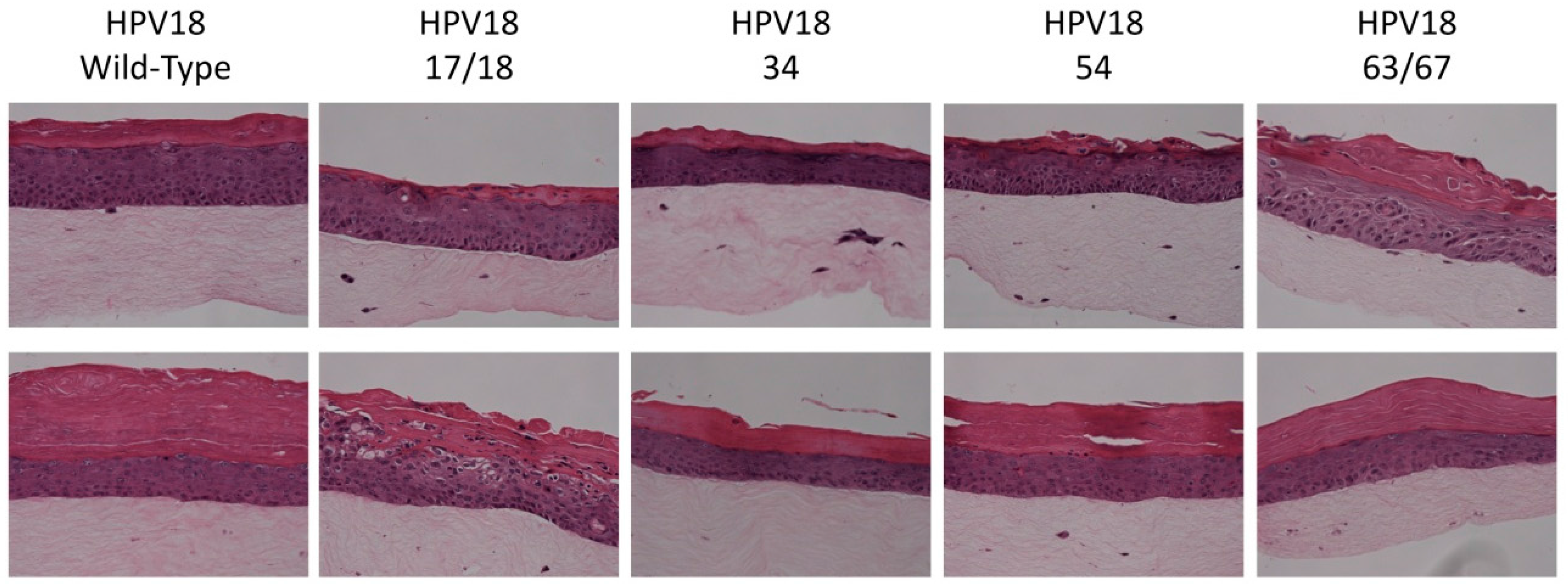
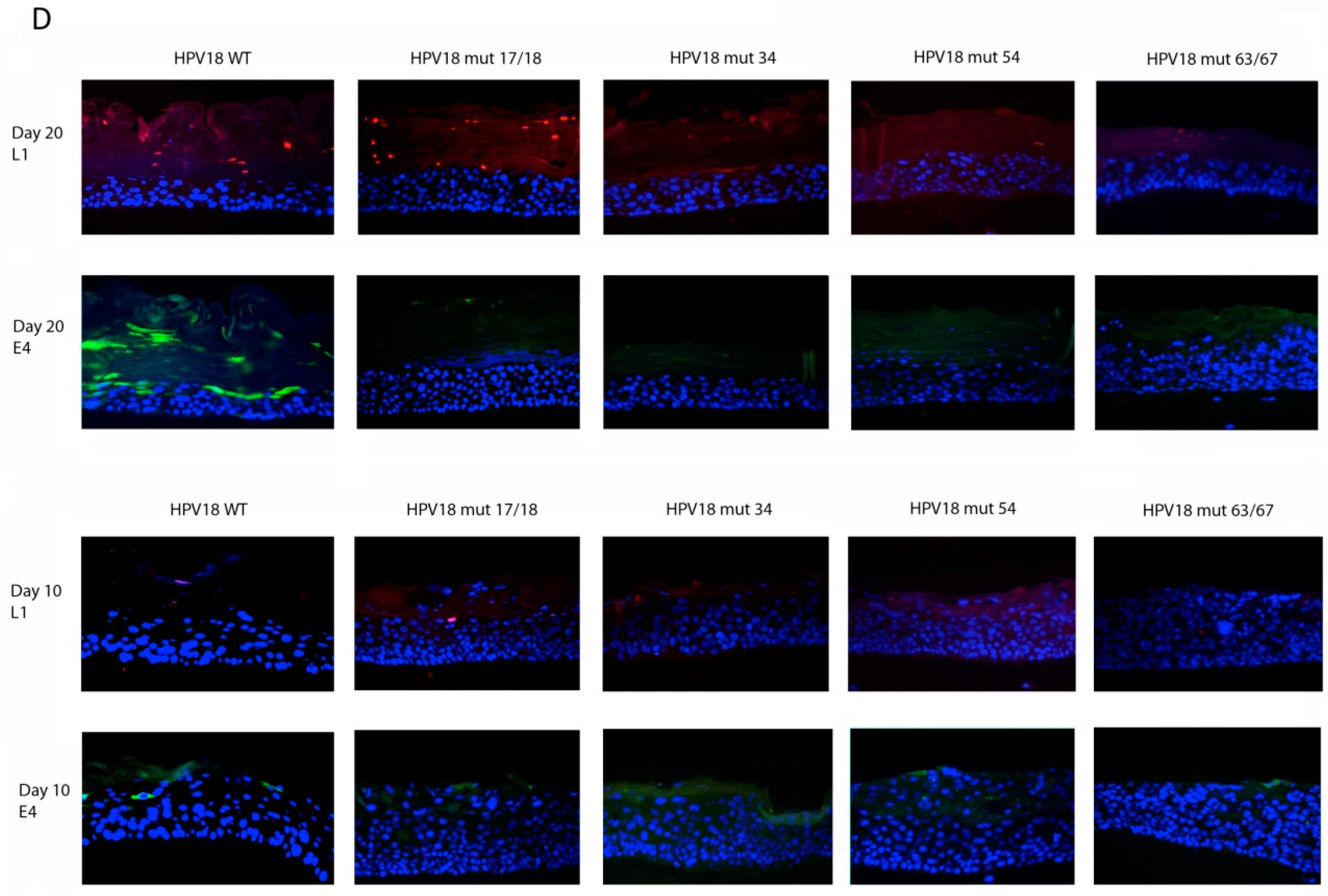
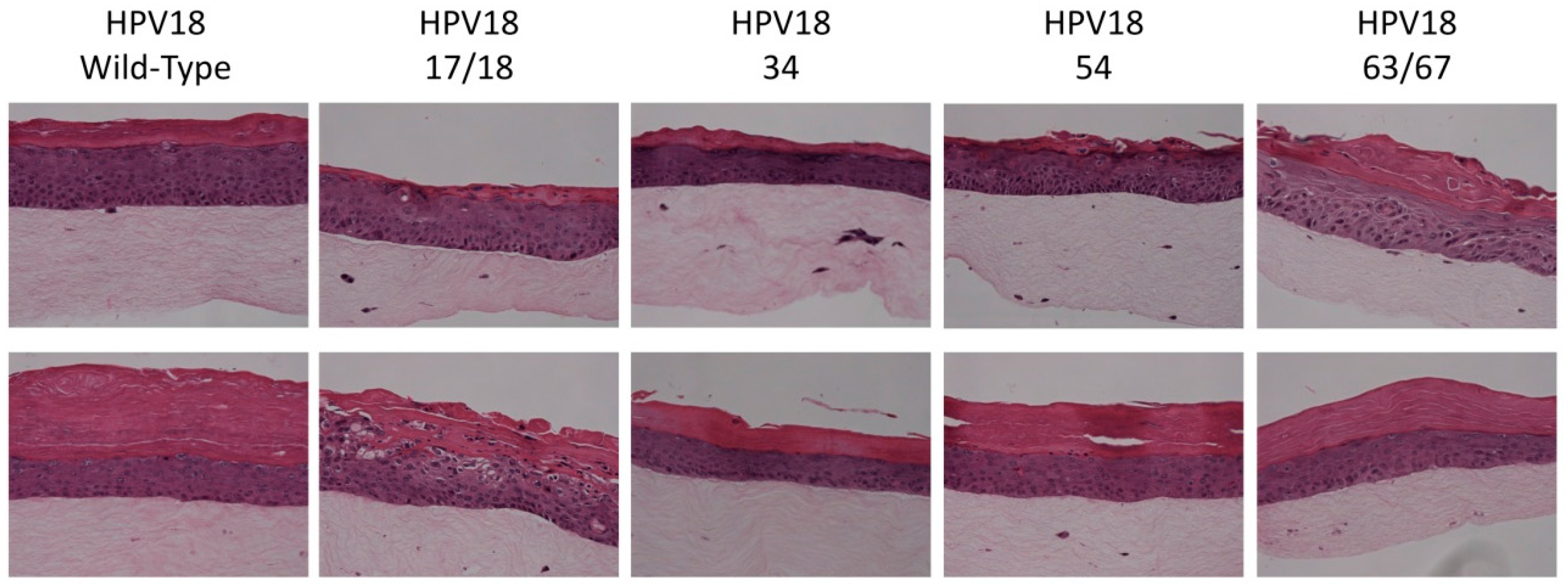
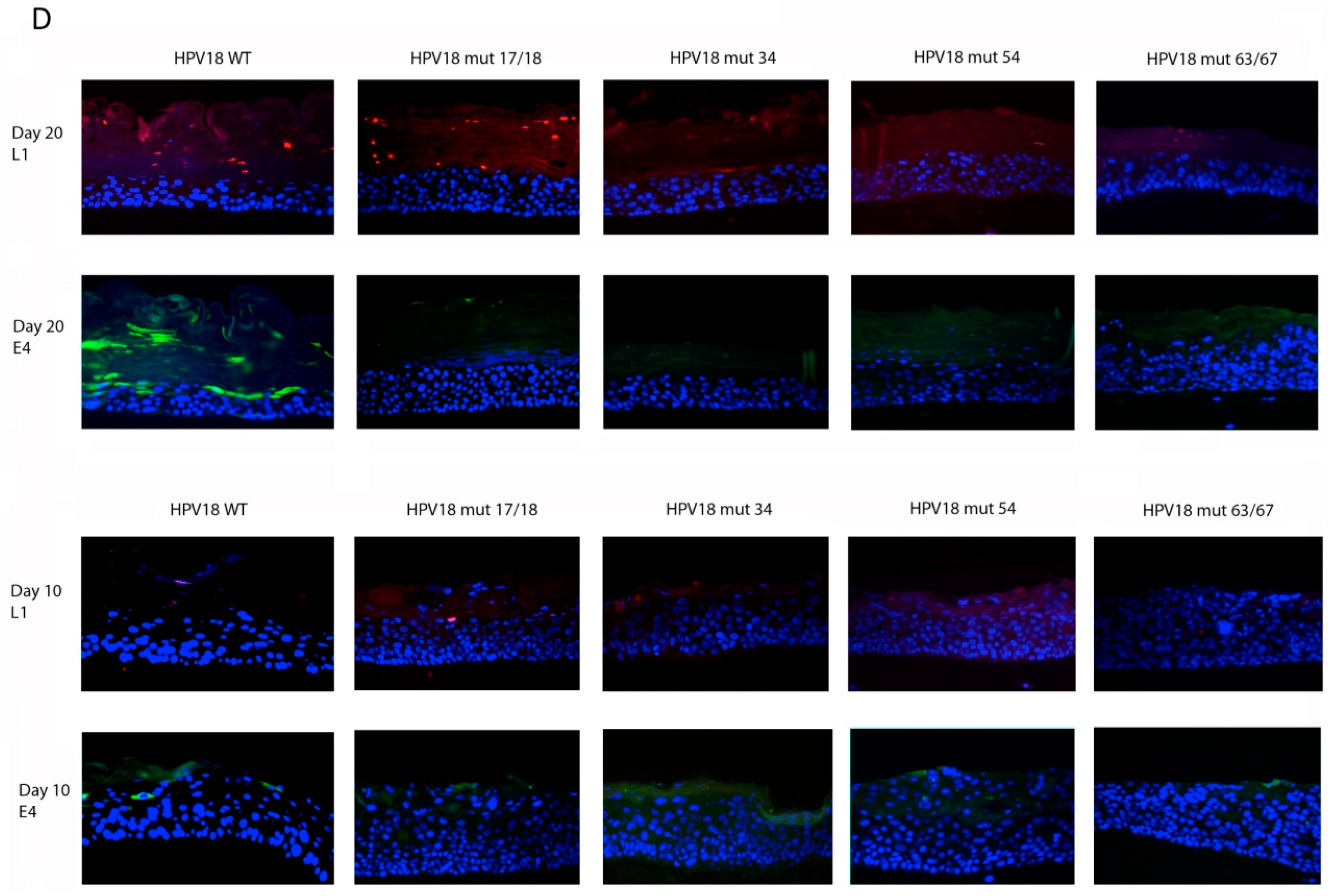
2.5. Histology and Immunofluorescent (IF) Staining of Tissue Sections
2.6. Virus Harvest and Isolation
2.7. OptiPrep Purification of Virions
2.8. Viral Titers
2.9. Infectivity Assays
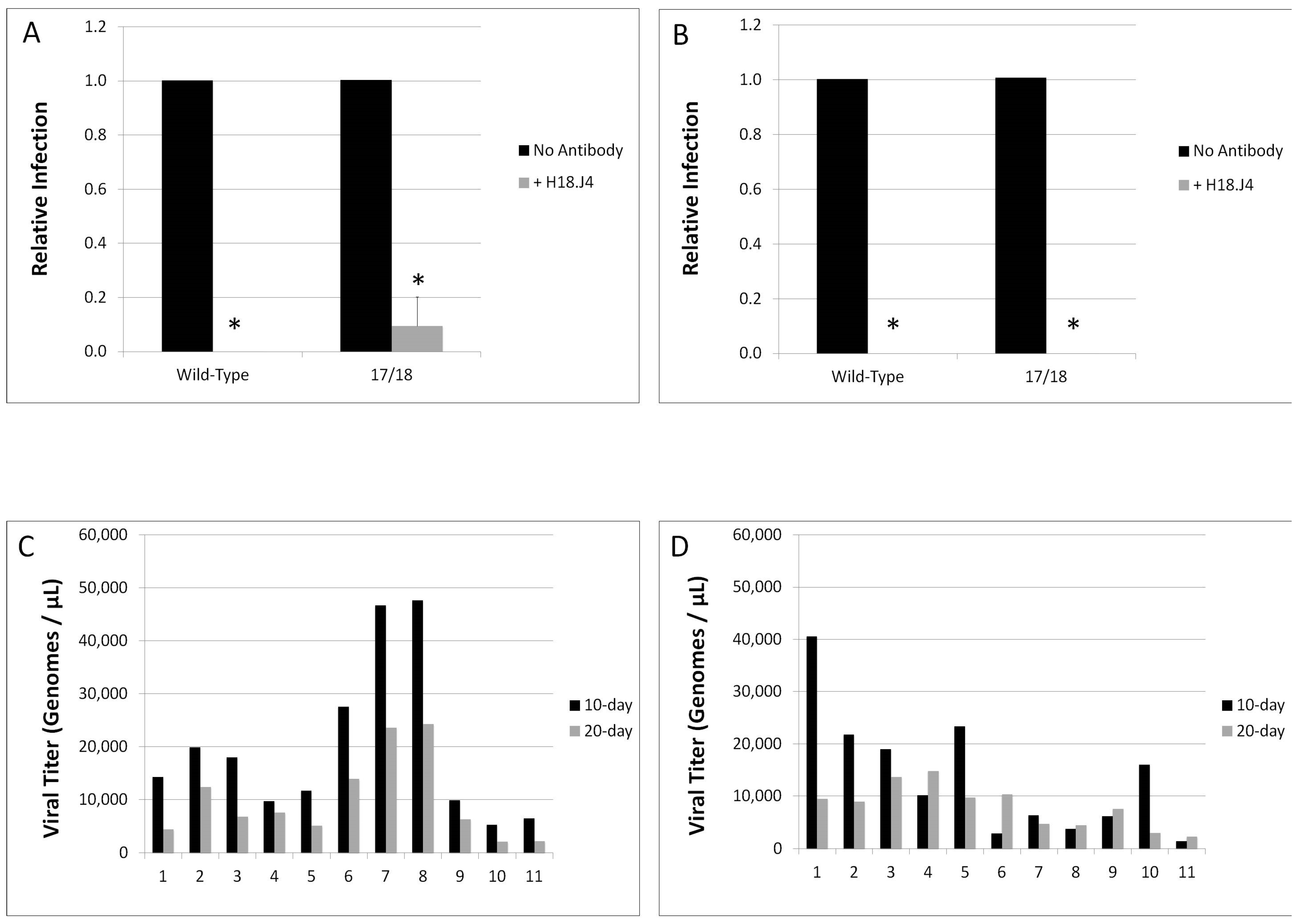
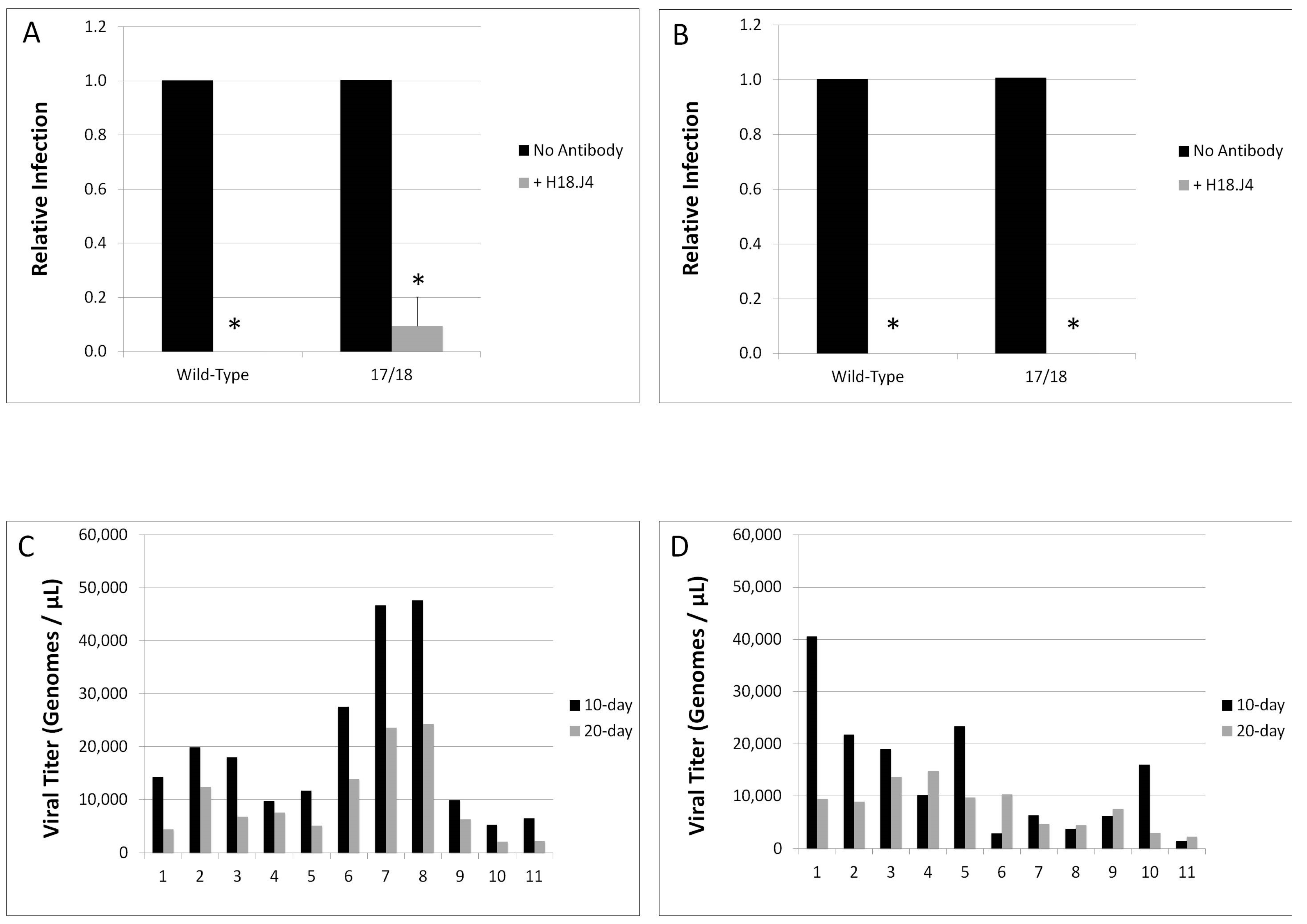
2.10. Neutralization Assays
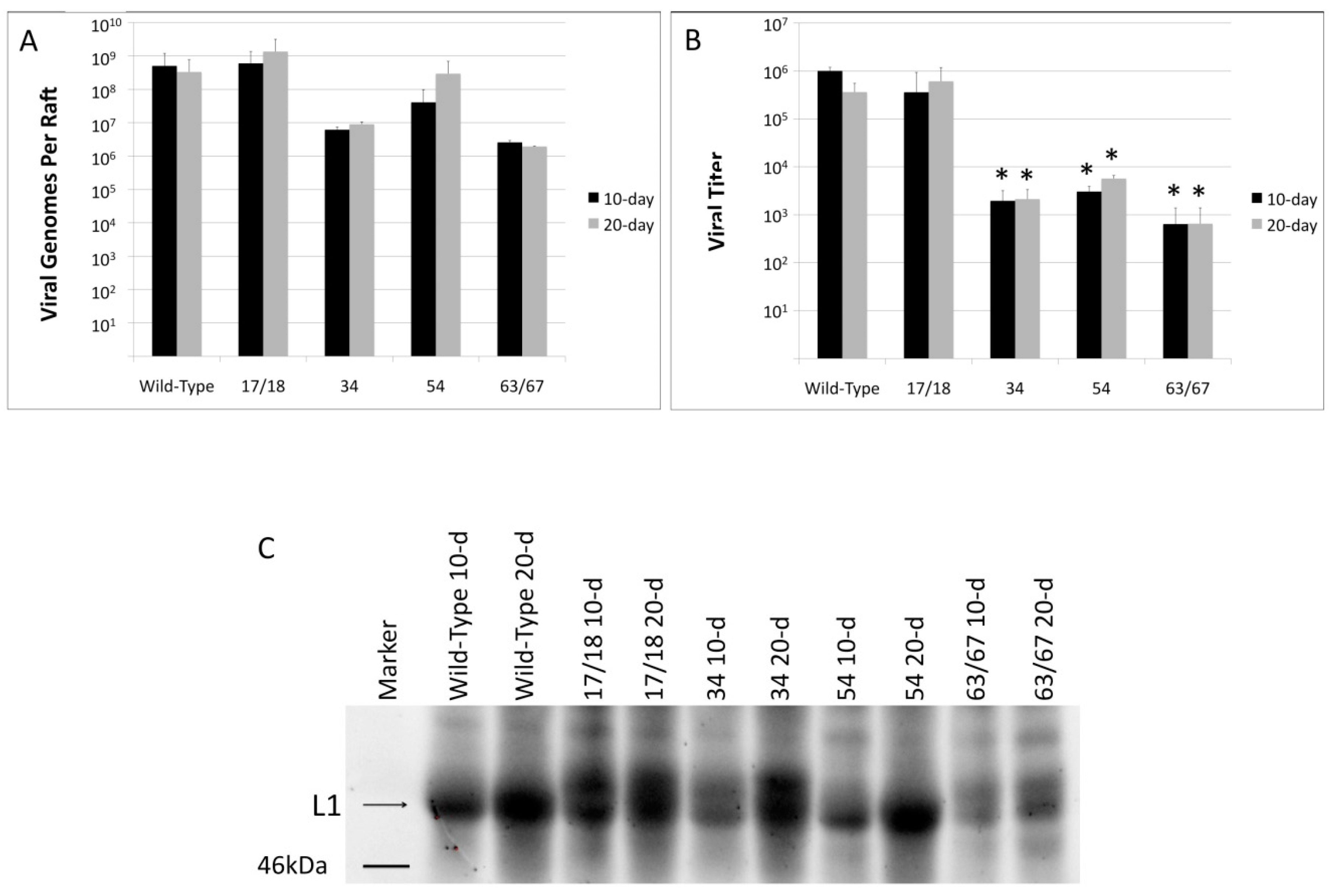
2.11. SDS-PAGE and L1 Western Blot
3. Results
3.1. Establishment of HPV18 17/18, 34, 54, and 63/67 Cell Lines
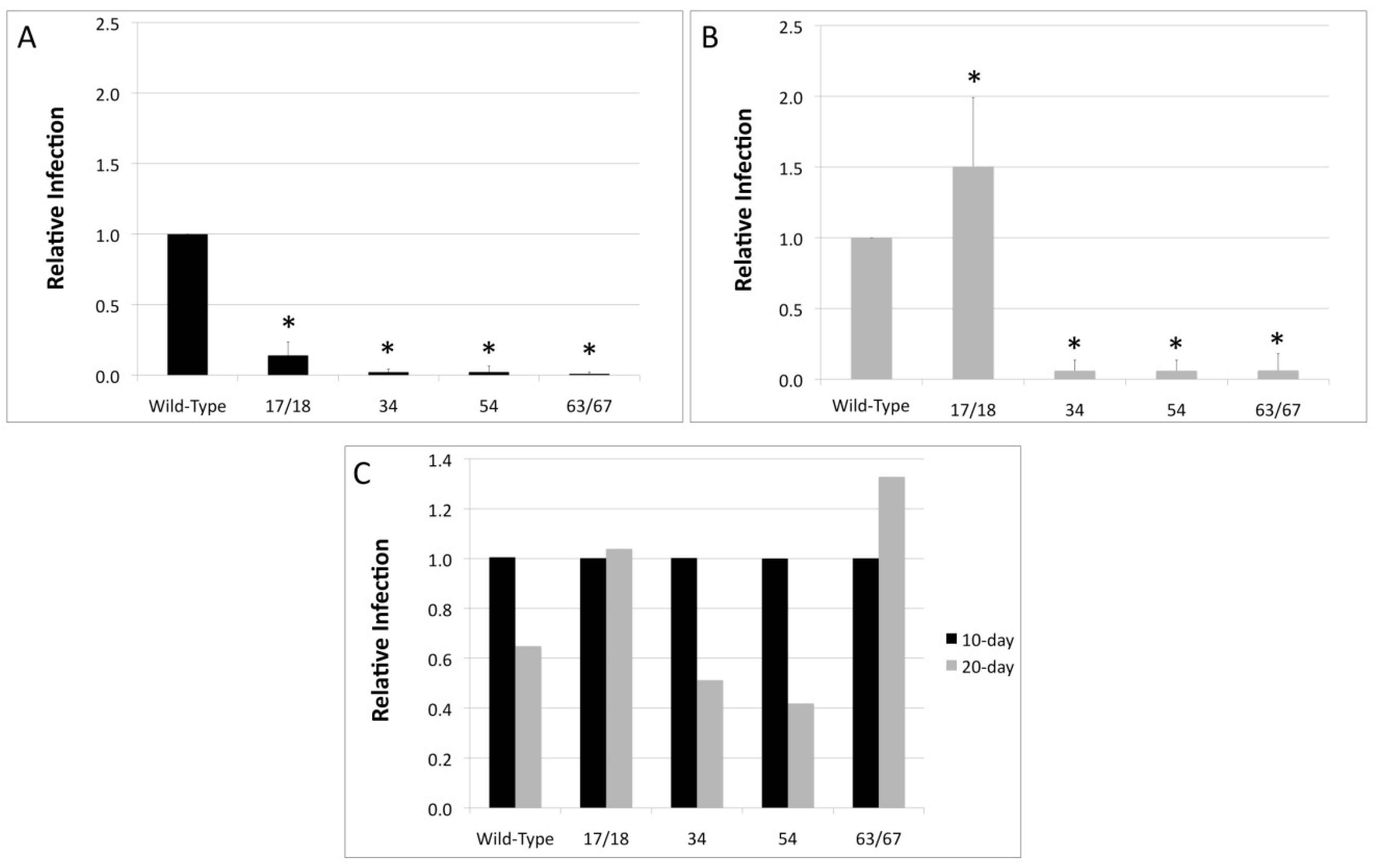
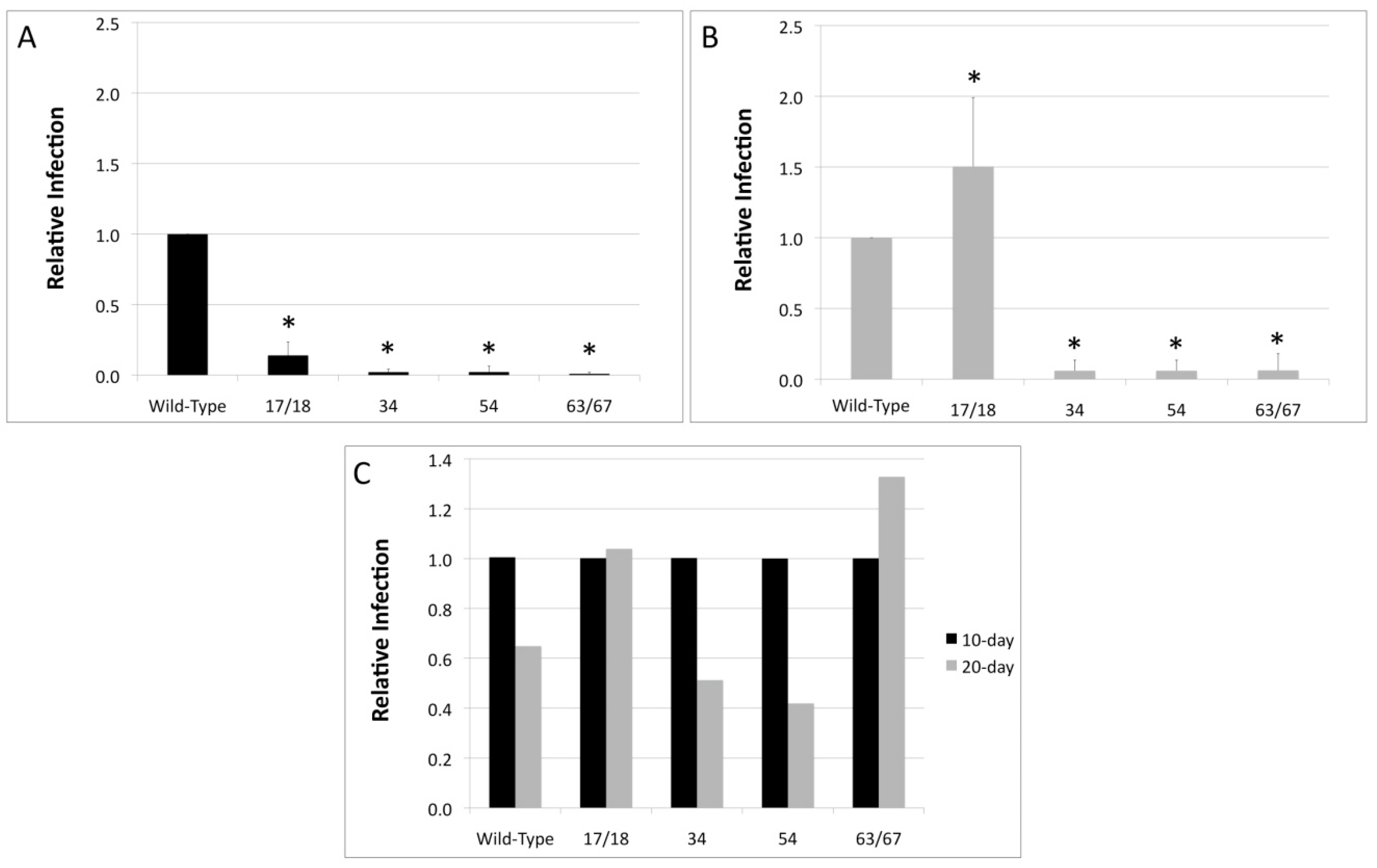
3.2. Viral Titers Are Reduced in HPV18 Truncation Mutants
3.3. Relative Infectivity Is Negatively Impacted by Mutations in E1^E4
3.4. Mutations in E1^E4 Effect Stability but Not Capsid Conformation
4. Discussion
Author Contributions
Conflicts of Interest
References
- Zur Hausen, H. Papillomaviruses in the causation of human cancers—A brief historical account. Virology 2009, 384, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Munoz, N.; Castellsague, X.; de Gonzalez, A.B.; Gissmann, L. HPV in the etiology of human cancer. Vaccine 2006, 24 (Suppl. 3), S1–S10. [Google Scholar] [CrossRef] [PubMed]
- Syrjanen, S.; Lodi, G.; von Bultzingslowen, I.; Aliko, A.; Arduino, P.; Campisi, G.; Challacombe, S.; Ficarra, G.; Flaitz, C.; Zhou, H.M.; et al. Human papillomaviruses in oral carcinoma and oral potentially malignant disorders: A systematic review. Oral Dis. 2011, 17 (Suppl. 1), 58–72. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, G.; Kreimer, A.R.; Viscidi, R.; Pawlita, M.; Fakhry, C.; Koch, W.M.; Westra, W.H.; Gillison, M.L. Case-control study of human papillomavirus and oropharyngeal cancer. N. Engl. J. Med. 2007, 356, 1944–1956. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Kjaer, S.K. Natural history of anogenital human papillomavirus infection and neoplasia. J. Natl. Cancer Inst. Monogr. 2003, 31, 14–19, Chapter 2. [Google Scholar] [CrossRef]
- De Villiers, E.M.; Fauquet, C.; Broker, T.R.; Bernard, H.U.; zur Hausen, H. Classification of papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Lowy, D.R.; Schiller, J.T. Reducing HPV-associated cancer globally. Cancer Prev. Res. Phila. 2012, 5, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Winer, R.L.; Hughes, J.P.; Feng, Q.; O’Reilly, S.; Kiviat, N.B.; Holmes, K.K.; Koutsky, L.A. Condom use and the risk of genital human papillomavirus infection in young women. N. Engl. J. Med. 2006, 354, 2645–2654. [Google Scholar] [CrossRef] [PubMed]
- Munoz, N.; Bosch, F.X.; de Sanjose, S.; Herrero, R.; Castellsague, X.; Shah, K.V.; Snijders, P.J.; Meijer, C.J. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 2003, 348, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.M.; Baker, C.C. Papillomavirus genome structure, expression, and post-transcriptional regulation. Front. Biosci. 2006, 11, 2286–2302. [Google Scholar] [CrossRef] [PubMed]
- Favre, M.; Orth, G.; Croissant, O.; Yaniv, M. Human papillomavirus DNA: Physical map. Proc. Natl. Acad. Sci. USA 1975, 72, 4810–4814. [Google Scholar] [CrossRef] [PubMed]
- Hummel, M.; Hudson, J.B.; Laimins, L.A. Differentiation-induced and constitutive transcription of human papillomavirus type 31B in cell lines containing viral episomes. J. Virol. 1992, 66, 6070–6080. [Google Scholar] [PubMed]
- Doorbar, J.; Campbell, D.; Grand, R.J.; Gallimore, P.H. Identification of the human papilloma virus-1A E4 gene products. Embo J. 1986, 5, 355–362. [Google Scholar] [PubMed]
- Conger, K.L.; Liu, J.S.; Kuo, S.R.; Chow, L.T.; Wang, T.S. Human papillomavirus DNA replication. Interactions between the viral E1 protein and two subunits of human DNA polymerase α/primase. J. Biol. Chem. 1999, 274, 2696–2705. [Google Scholar] [CrossRef] [PubMed]
- Cripe, T.P.; Haugen, T.H.; Turk, J.P.; Tabatabai, F.; Schmid, P.G.; Durst, M.; Gissmann, L.; Roman, A.; Turek, L.P. Transcriptional regulation of the human papillomavirus-16 E6–E7 promoter by a keratinocyte-dependent enhancer, and by viral E2 trans-activator and repressor gene products: Implications for cervical carcinogenesis. EMBO J. 1987, 6, 3745–3753. [Google Scholar] [PubMed]
- Gloss, B.; Bernard, H.U. The E6/E7 promoter of human papillomavirus type 16 is activated in the absence of E2 proteins by a sequence-aberrant SP1 distal element. J. Virol. 1990, 64, 5577–5584. [Google Scholar] [PubMed]
- Mohr, I.J.; Clark, R.; Sun, S.; Androphy, E.J.; MacPherson, P.; Botchan, M.R. Targeting the E1 replication protein to the papillomavirus origin of replication by complex formation with the E2 transactivator. Science 1990, 250, 1694–1699. [Google Scholar] [CrossRef] [PubMed]
- Bastien, N.; McBride, A.A. Interaction of the papillomavirus E2 protein with mitotic chromosomes. Virology 2000, 270, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Androphy, E.J.; Hubbert, N.L.; Schiller, J.T.; Lowy, D.R. Identification of the HPV-16 E6 protein from transformed mouse cells and human cervical carcinoma cell lines. EMBO J. 1987, 6, 989–992. [Google Scholar] [PubMed]
- Neary, K.; DiMaio, D. Open reading frames E6 and E7 of bovine papillomavirus type 1 are both required for full transformation of mouse C127 cells. J. Virol. 1989, 63, 259–266. [Google Scholar] [PubMed]
- Phelps, W.C.; Yee, C.L.; Munger, K.; Howley, P.M. The human papillomavirus type 16 E7 gene encodes transactivation and transformation functions similar to those of adenovirus E1A. Cell 1988, 53, 539–547. [Google Scholar] [CrossRef]
- Dyson, N.; Howley, P.M.; Munger, K.; Harlow, E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 1989, 243, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Longworth, M.S.; Laimins, L.A. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol. Mol. Biol. Rev. 2004, 68, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Meyers, C.; Frattini, M.G.; Hudson, J.B.; Laimins, L.A. Biosynthesis of human papillomavirus from a continuous cell line upon epithelial differentiation. Science 1992, 257, 971–973. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Gallimore, P.H. Identification of proteins encoded by the L1 and L2 open reading frames of human papillomavirus 1A. J. Virol. 1987, 61, 2793–2799. [Google Scholar] [PubMed]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life cycle of human papillomaviruses. Vaccine 2012, 30 (Suppl. 5), F55–F70. [Google Scholar] [CrossRef] [PubMed]
- Bryan, J.T.; Brown, D.R. Transmission of human papillomavirus type 11 infection by desquamated cornified cells. Virology 2001, 281, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Bryan, J.T.; Brown, D.R. Association of the human papillomavirus type 11 E1^E4 protein with cornified cell envelopes derived from infected genital epithelium. Virology 2000, 277, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Ozbun, M.A.; Meyers, C. Characterization of late gene transcripts expressed during vegetative replication of human papillomavirus type 31B. J. Virol. 1997, 71, 5161–5172. [Google Scholar] [PubMed]
- Wang, X.; Meyers, C.; Wang, H.K.; Chow, L.T.; Zheng, Z.M. Construction of a full transcription map of human papillomavirus type 18 during productive viral infection. J. Virol. 2011, 85, 8080–8092. [Google Scholar] [CrossRef] [PubMed]
- Milligan, S.G.; Veerapraditsin, T.; Ahamet, B.; Mole, S.; Graham, S.V. Analysis of novel human papillomavirus type 16 late mRNAs in differentiated W12 cervical epithelial cells. Virology 2007, 360, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.T.; Reilly, S.S.; Broker, T.R.; Taichman, L.B. Identification and mapping of human papillomavirus type 1 RNA transcripts recovered from plantar warts and infected epithelial cell cultures. J. Virol. 1987, 61, 1913–1918. [Google Scholar] [PubMed]
- Chow, L.T.; Nasseri, M.; Wolinsky, S.M.; Broker, T.R. Human papillomavirus types 6 and 11 mRNAs from genital condylomata acuminata. J. Virol. 1987, 61, 2581–2588. [Google Scholar] [PubMed]
- Doorbar, J.; Parton, A.; Hartley, K.; Banks, L.; Crook, T.; Stanley, M.; Crawford, L. Detection of novel splicing patterns in a HPV16-containing keratinocyte cell line. Virology 1990, 178, 254–262. [Google Scholar] [CrossRef]
- Peh, W.L.; Brandsma, J.L.; Christensen, N.D.; Cladel, N.M.; Wu, X.; Doorbar, J. The viral E4 protein is required for the completion of the cottontail rabbit papillomavirus productive cycle in vivo. J. Virol. 2004, 78, 2142–2151. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Foo, C.; Coleman, N.; Medcalf, L.; Hartley, O.; Prospero, T.; Napthine, S.; Sterling, J.; Winter, G.; Griffin, H. Characterization of events during the late stages of HPV16 infection in vivo using high-affinity synthetic fabs to E4. Virology 1997, 238, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Middleton, K.; Peh, W.; Southern, S.; Griffin, H.; Sotlar, K.; Nakahara, T.; El-Sherif, A.; Morris, L.; Seth, R.; Hibma, M.; et al. Organization of human papillomavirus productive cycle during neoplastic progression provides a basis for selection of diagnostic markers. J. Virol. 2003, 77, 10186–10201. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Ely, S.; Sterling, J.; McLean, C.; Crawford, L. Specific interaction between HPV-16 E1-E4 and cytokeratins results in collapse of the epithelial cell intermediate filament network. Nature 1991, 352, 824–827. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.; Ashmole, I.; Johnson, G.D.; Kreider, J.W.; Gallimore, P.H. Cutaneous and mucosal human papillomavirus E4 proteins form intermediate filament-like structures in epithelial cells. Virology 1993, 197, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Raj, K.; Berguerand, S.; Southern, S.; Doorbar, J.; Beard, P. E1 empty set E4 protein of human papillomavirus type 16 associates with mitochondria. J. Virol. 2004, 78, 7199–7207. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Elston, R.C.; Napthine, S.; Raj, K.; Medcalf, E.; Jackson, D.; Coleman, N.; Griffin, H.M.; Masterson, P.; Stacey, S.; et al. The E1^E4 protein of human papillomavirus type 16 associates with a putative RNA helicase through sequences in its c terminus. J. Virol. 2000, 74, 10081–10095. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.; Hillman, M.L.; Knight, G.L.; Gallimore, P.H. The ND10 component promyelocytic leukemia protein relocates to human papillomavirus type 1 E4 intranuclear inclusion bodies in cultured keratinocytes and in warts. J. Virol. 2003, 77, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Nishimura, A.; Tanaka, M.; Ueno, T.; Ishimoto, A.; Sakai, H. Modulation of the cell division cycle by human papillomavirus type 18 E4. J. Virol. 2002, 76, 10914–10920. [Google Scholar] [CrossRef] [PubMed]
- Davy, C.E.; Jackson, D.J.; Raj, K.; Peh, W.L.; Southern, S.A.; Das, P.; Sorathia, R.; Laskey, P.; Middleton, K.; Nakahara, T.; et al. Human papillomavirus type 16 E1 E4-induced G2 arrest is associated with cytoplasmic retention of active Cdk1/cyclin B1 complexes. J. Virol. 2005, 79, 3998–4011. [Google Scholar] [CrossRef] [PubMed]
- Knight, G.L.; Grainger, J.R.; Gallimore, P.H.; Roberts, S. Cooperation between different forms of the human papillomavirus type 1 E4 protein to block cell cycle progression and cellular DNA synthesis. J. Virol. 2004, 78, 13920–13933. [Google Scholar] [CrossRef] [PubMed]
- Davy, C.E.; Jackson, D.J.; Wang, Q.; Raj, K.; Masterson, P.J.; Fenner, N.F.; Southern, S.; Cuthill, S.; Millar, J.B.; Doorbar, J. Identification of a G2 arrest domain in the E1 wedge E4 protein of human papillomavirus type 16. J. Virol. 2002, 76, 9806–9818. [Google Scholar] [CrossRef] [PubMed]
- Egawa, N.; Wang, Q.; Griffin, H.M.; Murakami, I.; Jackson, D.; Mahmood, R.; Doorbar, J. HPV16 and 18 genome amplification show different E4-dependence, with 16E4 enhancing E1 nuclear accumulation and replicative efficiency via its cell cycle arrest and kinase activation functions. PLoS Pathog. 2017, 13, e1006282. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Wilson, S.; Mullikin, B.; Suzich, J.; Meyers, C. Human papillomavirus type 45 propagation, infection, and neutralization. Virology 2003, 312, 1–7. [Google Scholar] [CrossRef]
- Meyers, C.; Mayer, T.J.; Ozbun, M.A. Synthesis of infectious human papillomavirus type 18 in differentiating epithelium transfected with viral DNA. J. Virol. 1997, 71, 7381–7386. [Google Scholar] [PubMed]
- McLaughlin-Drubin, M.E.; Christensen, N.D.; Meyers, C. Propagation, infection, and neutralization of authentic HPV16 virus. Virology 2004, 322, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Conway, M.J.; Alam, S.; Ryndock, E.J.; Cruz, L.; Christensen, N.D.; Roden, R.B.; Meyers, C. Tissue-spanning redox gradient-dependent assembly of native human papillomavirus type 16 virions. J. Virol. 2009, 83, 10515–10526. [Google Scholar] [CrossRef] [PubMed]
- Conway, M.J.; Cruz, L.; Alam, S.; Christensen, N.D.; Meyers, C. Differentiation-dependent interpentameric disulfide bond stabilizes native human papillomavirus type 16. PLoS ONE 2011, 6, e22427. [Google Scholar] [CrossRef] [PubMed]
- Griffin, H.; Wu, Z.; Marnane, R.; Dewar, V.; Molijn, A.; Quint, W.; Van Hoof, C.; Struyf, F.; Colau, B.; Jenkins, D.; et al. E4 antibodies facilitate detection and type-assignment of active HPV infection in cervical disease. PLoS ONE 2012, 7, e49974. [Google Scholar] [CrossRef] [PubMed]
- Griffin, H.; Soneji, Y.; Van Baars, R.; Arora, R.; Jenkins, D.; van de Sandt, M.; Wu, Z.; Quint, W.; Jach, R.; Okon, K.; et al. Stratification of HPV-induced cervical pathology using the virally encoded molecular marker E4 in combination with p16 or MCM. Mod. Pathol. 2015, 28, 977–993. [Google Scholar] [CrossRef] [PubMed]
- McLean, C.S.; Churcher, M.J.; Meinke, J.; Smith, G.L.; Higgins, G.; Stanley, M.; Minson, A.C. Production and characterisation of a monoclonal antibody to human papillomavirus type 16 using recombinant vaccinia virus. J. Clin. Pathol. 1990, 43, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Griffin, H.; Doorbar, J. Detection of papillomavirus gene expression patterns in tissue sections. Curr. Protoc. Microbiol. 2016, 41, 14b.17.11–14b.17.20. [Google Scholar] [PubMed]
- Conway, M.J.; Cruz, L.; Alam, S.; Christensen, N.D.; Meyers, C. Cross-neutralization potential of native human papillomavirus N-terminal L2 epitopes. PLoS ONE 2011, 6, e16405. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Peh, W.L.; Doorbar, J.; Lee, D.; Lambert, P.F. Human papillomavirus type 16 E1circumflexE4 contributes to multiple facets of the papillomavirus life cycle. J. Virol. 2005, 79, 13150–13165. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J. The E4 protein; structure, function and patterns of expression. Virology 2013, 445, 80–98. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.; Fehrmann, F.; Laimins, L.A. Role of the E1^E4 protein in the differentiation-dependent life cycle of human papillomavirus type 31. J. Virol. 2005, 79, 6732–6740. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.; Ryan, G.B.; Knight, G.L.; Laimins, L.A.; Roberts, S. The full-length E1E4 protein of human papillomavirus type 18 modulates differentiation-dependent viral DNA amplification and late gene expression. Virology 2007, 362, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.S.; Conway, M.J.; Christensen, N.D.; Alam, S.; Meyers, C. Papillomavirus capsid proteins mutually impact structure. Virology 2011, 412, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Ryndock, E.J.; Conway, M.J.; Alam, S.; Gul, S.; Murad, S.; Christensen, N.D.; Meyers, C. Roles for human papillomavirus type 16 L1 cysteine residues 161, 229, and 379 in genome encapsidation and capsid stability. PLoS ONE 2014, 9, e99488. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Pastrana, D.V.; Lowy, D.R.; Schiller, J.T. Efficient intracellular assembly of papillomaviral vectors. J. Virol. 2004, 78, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Budgeon, L.R.; Doorbar, J.; Briggs, E.R.; Howett, M.K. The human papillomavirus type 11 E1^E4 protein is not essential for viral genome amplification. Virology 2006, 351, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Fan, L.; Jones, J.; Bryan, J. Colocalization of human papillomavirus type 11 E1^E4 and L1 proteins in human foreskin implants grown in athymic mice. Virology 1994, 201, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Perez-Berna, A.J.; Ortega-Esteban, A.; Menendez-Conejero, R.; Winkler, D.C.; Menendez, M.; Steven, A.C.; Flint, S.J.; de Pablo, P.J.; San Martin, C. The role of capsid maturation on adenovirus priming for sequential uncoating. J. Biol. Chem. 2012, 287, 31582–31595. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.; Nagashima, K.; Freed, E.O. Mutation of dileucine-like motifs in the human immunodeficiency virus type 1 capsid disrupts virus assembly, GAG-GAG interactions, GAG-membrane binding, and virion maturation. J. Virol. 2006, 80, 7939–7951. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus Type | Stability |
|---|---|
| HPV18 Wild-Type 10-day | 0.470 |
| HPV18 Wild-Type 20-day | 0.457 |
| HPV18 E1^E4 17/18 10-day | 4.76 |
| HPV18 E1^E4 17/18 20-day | 1.73 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biryukov, J.; Myers, J.C.; McLaughlin-Drubin, M.E.; Griffin, H.M.; Milici, J.; Doorbar, J.; Meyers, C. Mutations in HPV18 E1^E4 Impact Virus Capsid Assembly, Infectivity Competence, and Maturation. Viruses 2017, 9, 385. https://doi.org/10.3390/v9120385
Biryukov J, Myers JC, McLaughlin-Drubin ME, Griffin HM, Milici J, Doorbar J, Meyers C. Mutations in HPV18 E1^E4 Impact Virus Capsid Assembly, Infectivity Competence, and Maturation. Viruses. 2017; 9(12):385. https://doi.org/10.3390/v9120385
Chicago/Turabian StyleBiryukov, Jennifer, Jocelyn C. Myers, Margaret E. McLaughlin-Drubin, Heather M. Griffin, Janice Milici, John Doorbar, and Craig Meyers. 2017. "Mutations in HPV18 E1^E4 Impact Virus Capsid Assembly, Infectivity Competence, and Maturation" Viruses 9, no. 12: 385. https://doi.org/10.3390/v9120385