1. Introduction
Citrus sudden death (CSD) is a disease that was first detected in 1999 in citrus groves located in the municipality of Comendador Gomes (southwestern Minas Gerais State), Brazil [
1]. At that time, CSD was found to affect only plants of sweet orange (
Citrus sinensis L. Osbeck) grafted on Rangpur lime rootstock (
Citrus limonia L. Osb.), a very important drought-resistant rootstock used in Brazil [
1]. However, CSD quickly spread into the northern part of São Paulo State and, since then, has caused the death of four million orange trees [
2,
3]. The symptoms of CSD are characterized by a general decline, including pale green coloration of the leaves, overall defoliation, death of the roots and presence of a characteristic yellow stain in the rootstock bark [
4]. Later, CSD-symptoms were also detected in sweet oranges grafted on the other rootstocks, such as
Citrus volkameriana [
3],
Citrus jambiri and
Citrus pennivisiculata Lush [
5].
The main challenge in studying CSD is that the etiology has not been definitively determined, even after seventeen years from its first detection. Similarities with quick-decline form of citrus tristeza disease, mainly on the symptoms and distribution of the CSD-affected plants, have led previous works to hypothesize that a new variant of Citrus tristeza virus (CTV), a member of the family
Closteroviridae and one of the most economically important citrus viruses, might be associated in developing CSD symptoms [
2,
3,
6,
7]. However, several attempts in trying to identify an isolate or a new variant of CTV associated with CSD, have failed [
2,
6,
8,
9]. Conventional shotgun sequencing (which did not make use of the next-generation sequencing (NGS) technology) of complementary DNA (cDNA) derived from double-stranded RNA (dsRNA) isolated from plants showing CSD symptoms was able to identify a novel virus from the family
Tymoviridae [
9]. It was suggested that this new virus was likely to be associated with CSD and was named Citrus sudden death-associated virus (CSDaV). However, the role of this virus in CSD-affected plants is still not completely clear. The conventional shotgun sequencing approach, after the low-quality reads were removed, generated a low number of valid reads [
9], which made it difficult to study the frequency of detected viruses, to differentiate virus isolates and to discover novel viruses that might be involved with CSD disease. In addition, conventional approaches for virus detection require prior knowledge of genome sequences [
10], thereby allowing for identification only of specific known viruses, and thus is not suitable to study the virome within plants [
10,
11].
NGS has been widely and successfully used for improved detection and characterization of known and novel viruses in infected plant hosts [
11,
12]. Deep sequencing of the transcriptome (RNA-seq) and small RNAs (sRNAs) have been shown to be a promising and powerful approach in detecting both RNA and DNA viruses [
10,
13]. Consequently, this approach can be used to better understand plant diseases, especially when the viral etiology is unknown, as well as to explore plant virus–host interactions [
12,
14].
In order to identify putative viruses associated with citrus plants affected by CSD, and compare the frequency and diversity of viruses between CSD-symptomatic and -asymptomatic plants, we have performed a high-throughput sequencing analysis of the transcriptomes and sRNAs from CSD-symptomatic and -asymptomatic citrus plants, all grown in a CSD-affected region. Our work was able to effectively identify both CSDaV and CTV in multiple virus infections and to differentiate two CTV and CSDaV genotypes.
4. Discussion
Our study demonstrated a putative association of CSD-symptomatic plants with a specific CSDaV genotype and with Citrus endogenous pararetrovirus (CitPRV) as well. We were also able to identify two putative novel viruses that were shown to be more associated with CSD-asymptomatic plants. This work provides new insights into the role of the identified viruses in citrus plants affected by CSD, which will be able to contribute to further studies.
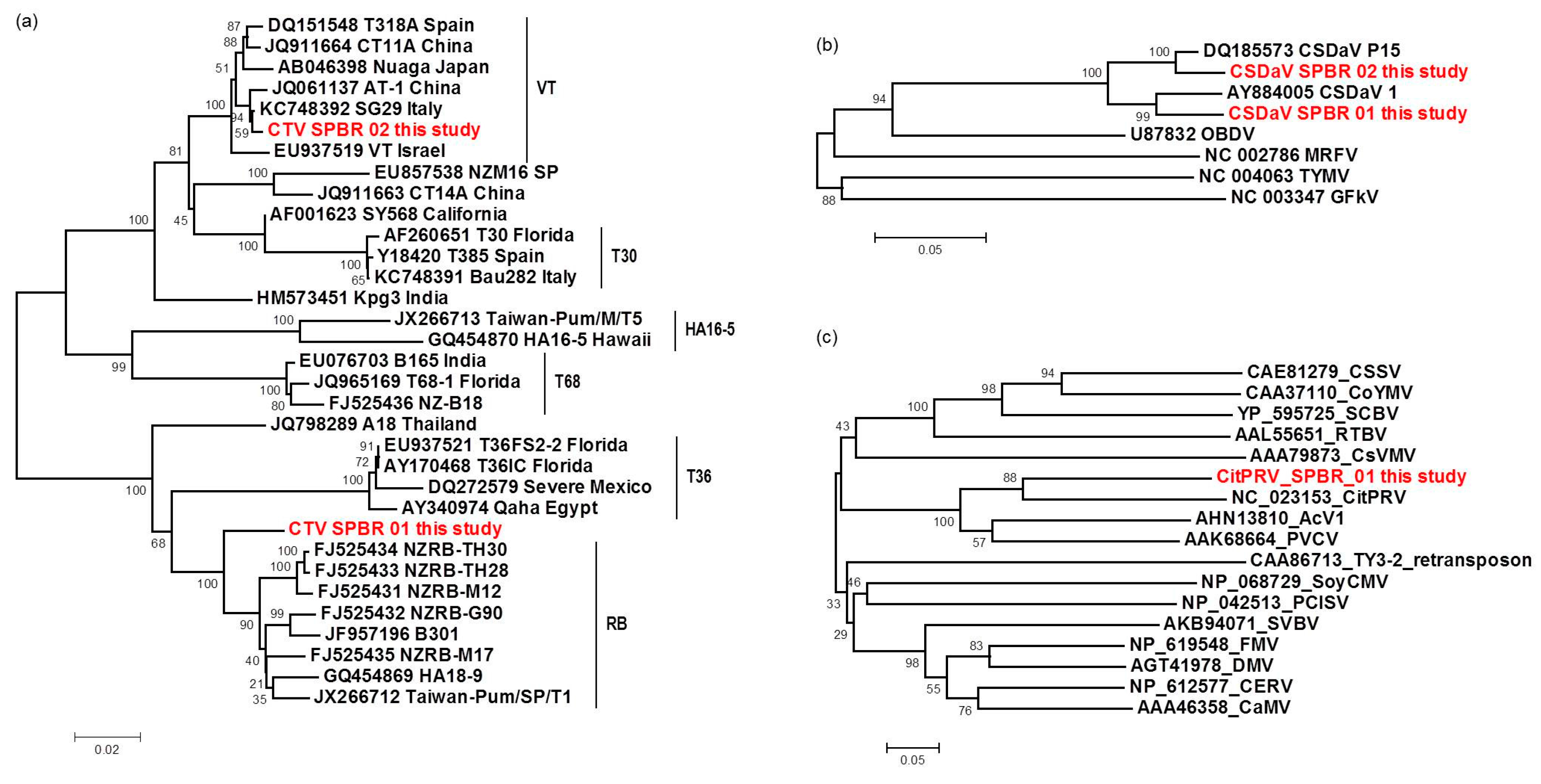
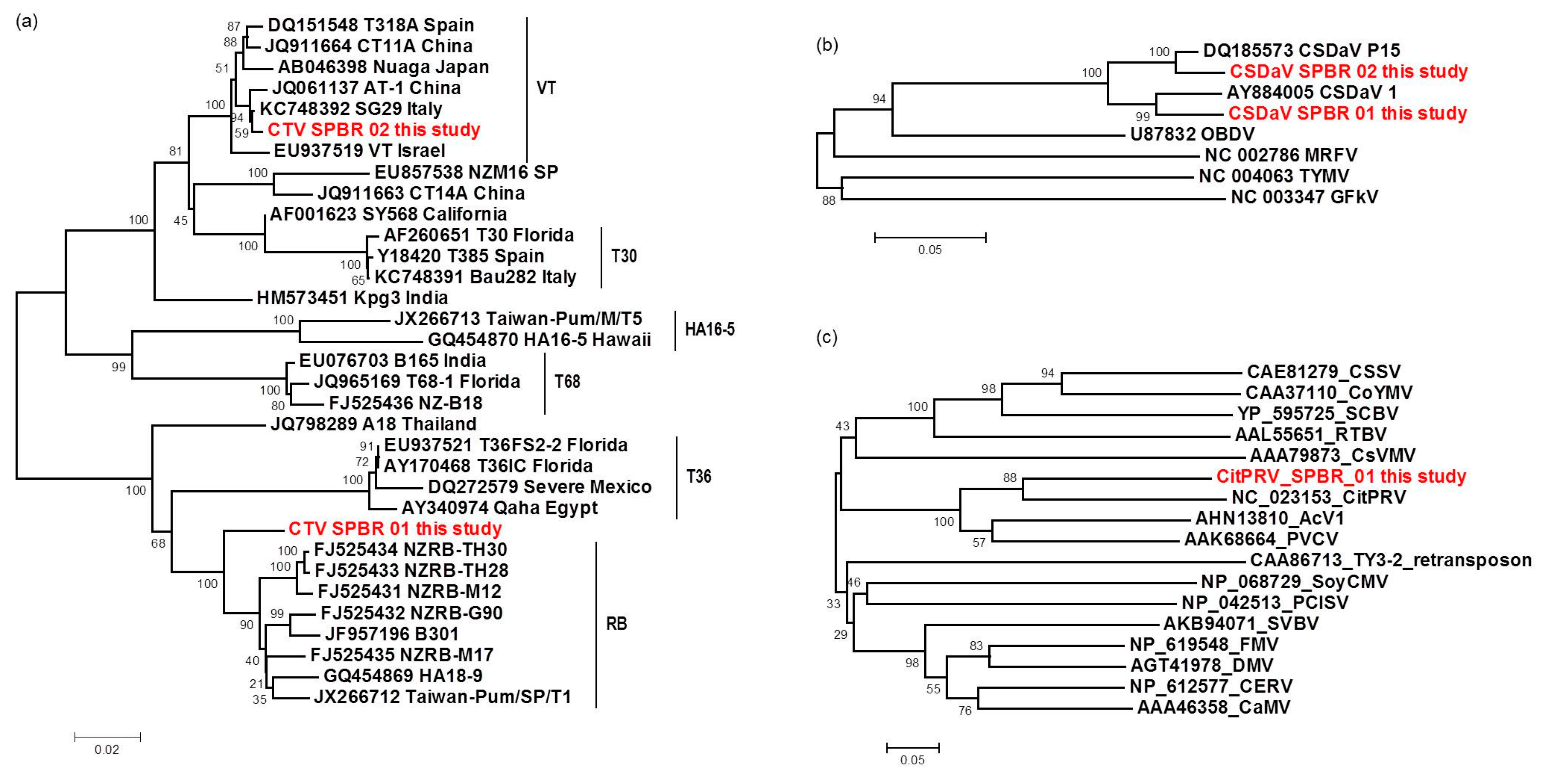
In this work, Illumina high-throughput sequencing of the transcriptome and sRNAs from citrus plants grown in a region affected by citrus sudden death disease has allowed us to identify and compare viral sequences presenting in these plants. The deep sequencing analyses were sensitive and sufficient to identify the predominant viruses, to obtain information about their genetic diversity, and to demonstrate the presence of putative novel and low-titer viruses. CTV was the most predominant virus identified here, represented by 97.4% of total reads, followed by CSDaV, which corresponded to 1.94% of the reads, CitPRV with 0.53% of the reads, and other viruses represented by 0.13% of the reads. The presence of the two first mentioned viruses was not a surprising finding because in attempts to discover the causal agent of CSD, both of these viruses were detected and associated with CSD-affected plants. Maccheroni et al. [
9] reported a high correlation at 99.7% of the assessed plants between CSD symptoms and the presence of CSDaV, but the role of this virus in CSD is not yet clear. CTV is an endemic virus in Brazil, mainly due to the cross-protection program and the presence of aphid transmitters [
26]. Previous published works have used different approaches to identify an isolate or new variant of CTV associated with CSD, but all attempts have failed so far [
2,
6,
7,
8]. Although previous works have shown that citrus plants affected by CSD are infected by a mixed population of divergent CTV variants [
2,
7,
8,
9], it is still unknown which specific CTV genotypes are present in those plants. To our knowledge, this is the first high-throughput sequencing-based study of the viral sequences present in citrus plants affected by the CSD disease. Our work reveals mixed viral infections in both CSD-symptomatic and -asymptomatic plants, including CTV, CSDaV, CitPRV and two putative novel viruses tentatively named in this study as CJLV and CVLV.
For the first time, the full consensus sequences of the two predominant Brazilian CTV genotypes present in those CSD-affected plants were obtained and identified here as CTV_SPBR-01 and CTV_SPBR_02. Phylogenetic analysis clustered CTV_SPBR_01 within RB-like CTV isolates, whereas CTV_SPBR_02 was clustered within VT-like CTV isolates. Both of the resistance breaking (RB) and VT strains have been characterized as severe or aggressive strains, which are associated with decline symptoms of citrus trees propagated on sour orange rootstock (
Citrus aurantium L.) or stem pitting (SP) of the scion regardless of the rootstocks [
27,
28,
29,
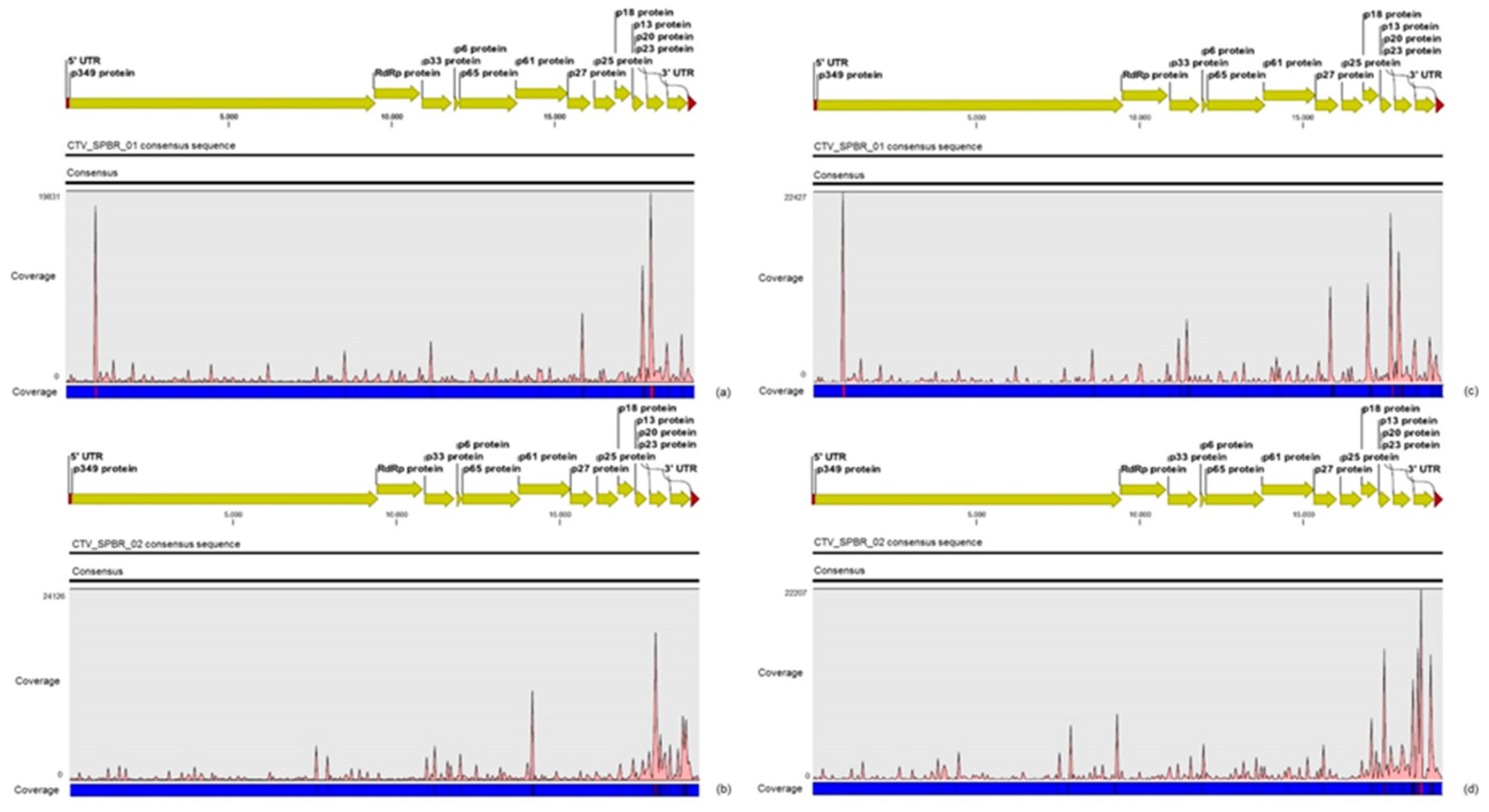
30]. Although re-assembly analysis comparing CTV mapped reads between libraries from asymptomatic and symptomatic plants did not show significant differences regarding average coverage values, differences on the read distribution and hotspot regions between these two libraries were noticed. Similar to the results obtained here, previous works reported that CTV infection induces accumulation of sRNAs mapping preferentially at the 3′-terminal region of the viral genome [
21,
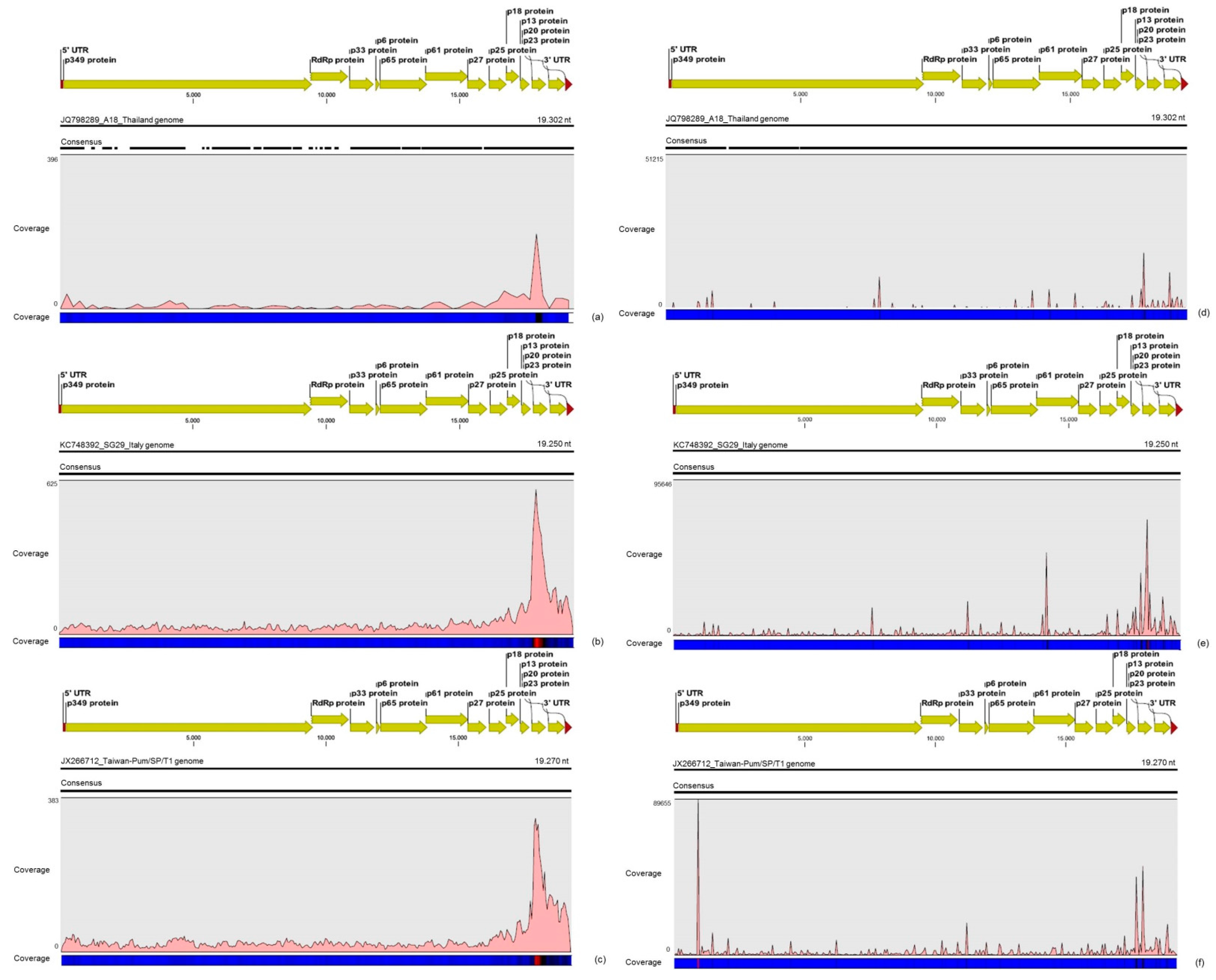
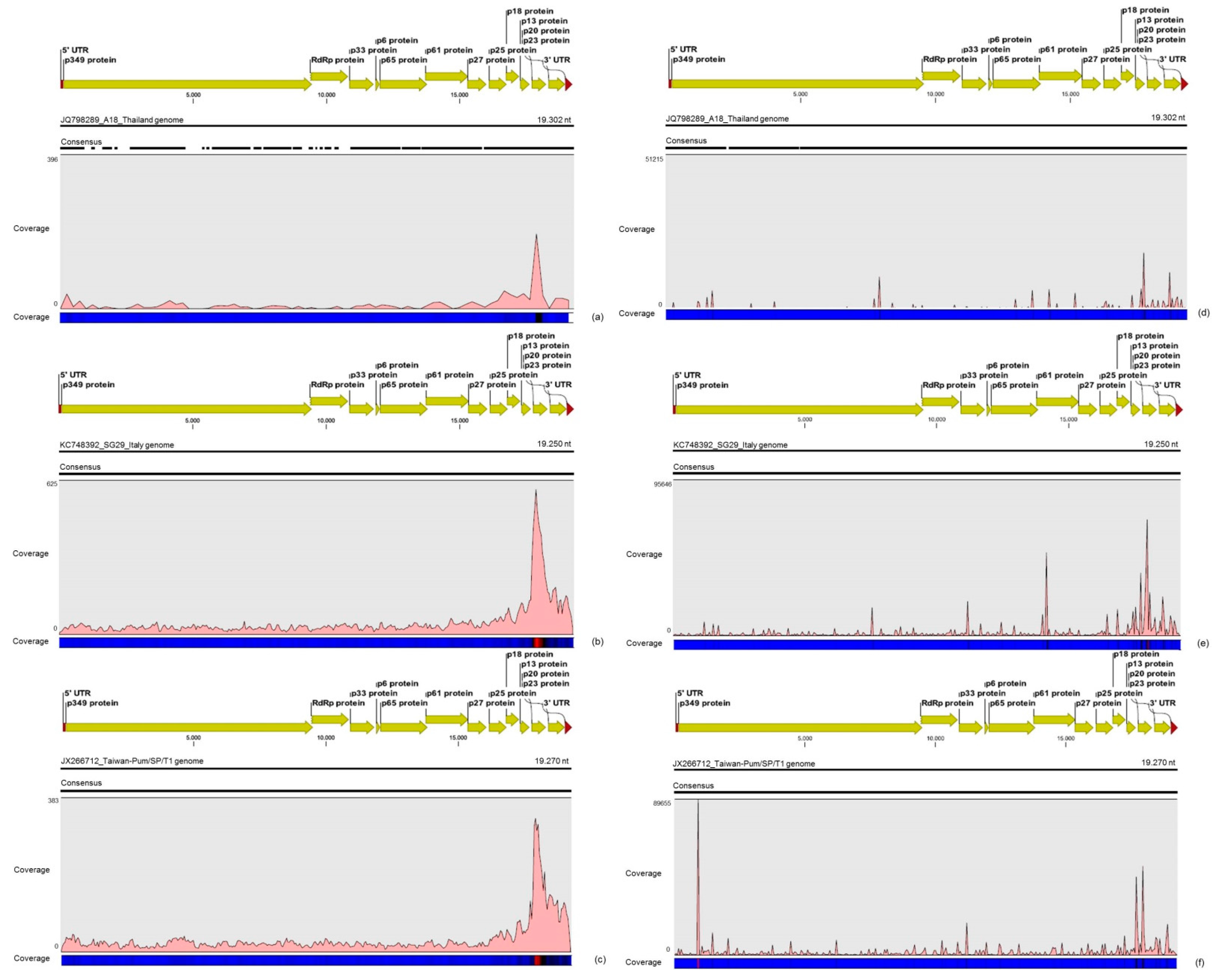
31]. Interestingly, mapping reads from asymptomatic libraries for both CTV genotypes identified here has shown a hotspot over the silencing suppressor gene p20, besides other lower hotspots as well, whereas mapping reads from symptomatic libraries showed an increased read coverage over the host range associated genes: the p13, p18 and p33 when CTV_SPBR_01 consensus sequence was used as reference, and the
p13, when CTV_SPBR_02 was used as reference. CTV_SPBR_02 also showed a hotspot over the p23 gene, which is a multifunctional gene and is also associated with silencing suppressor activity [
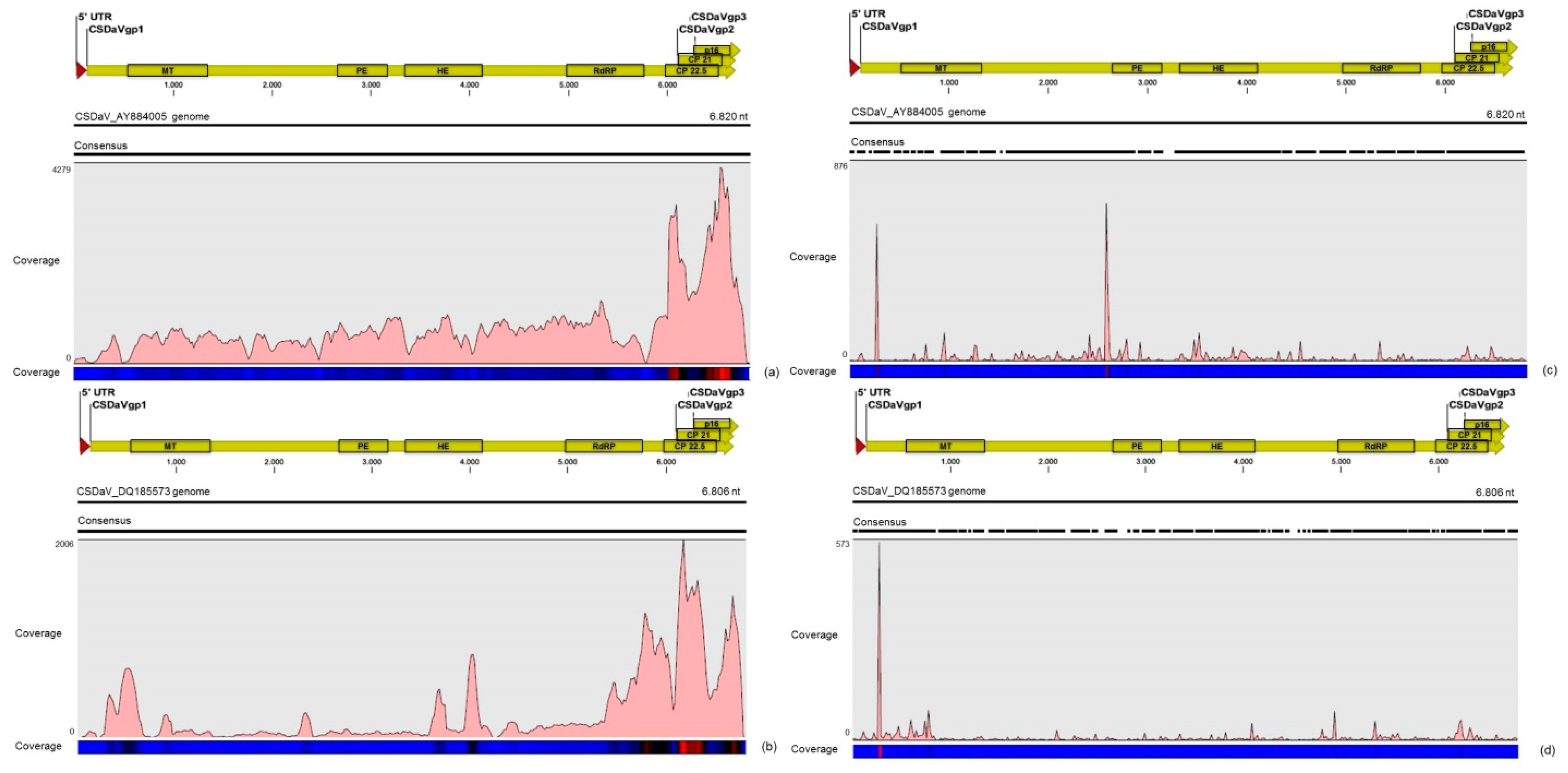
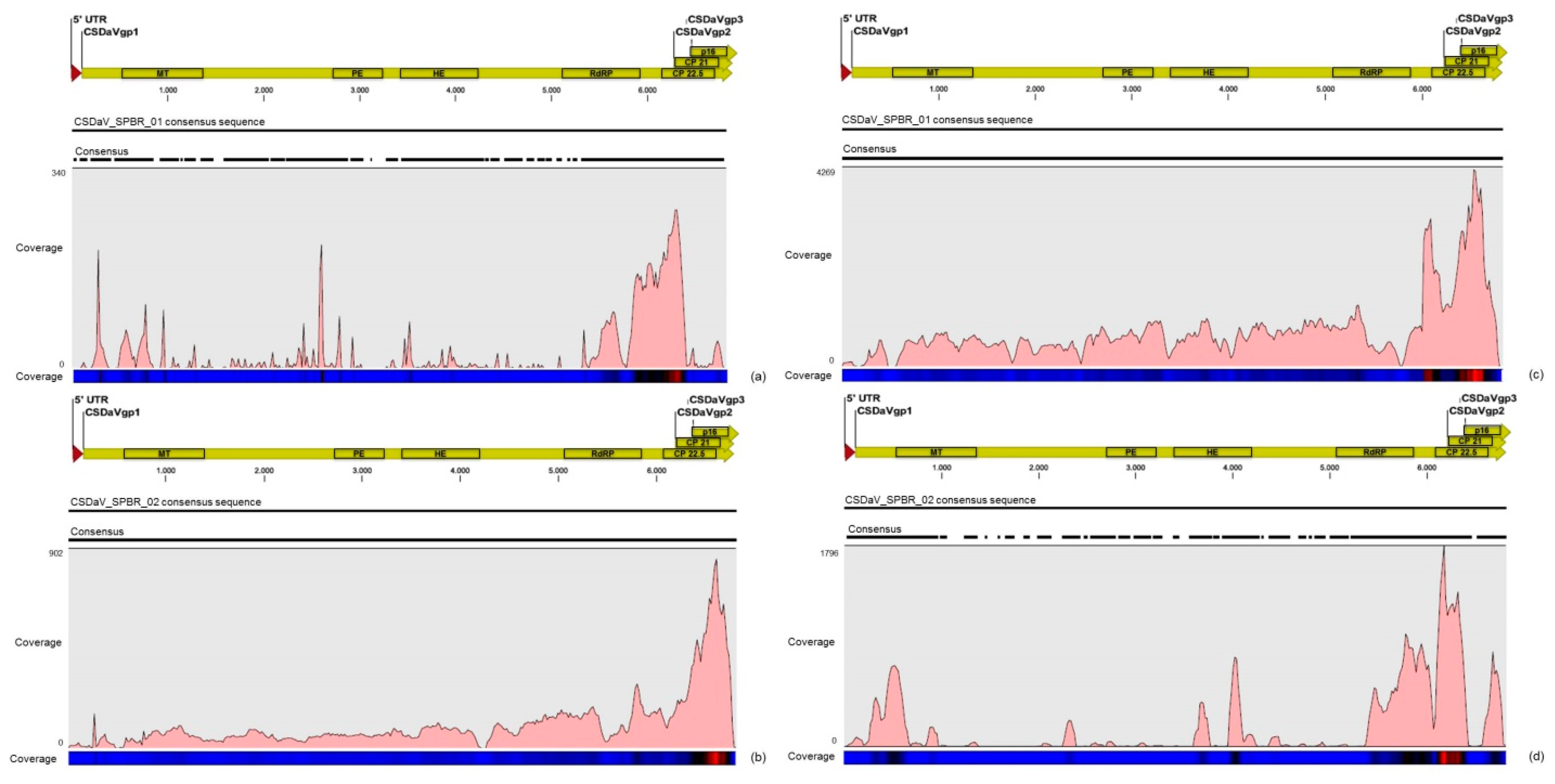
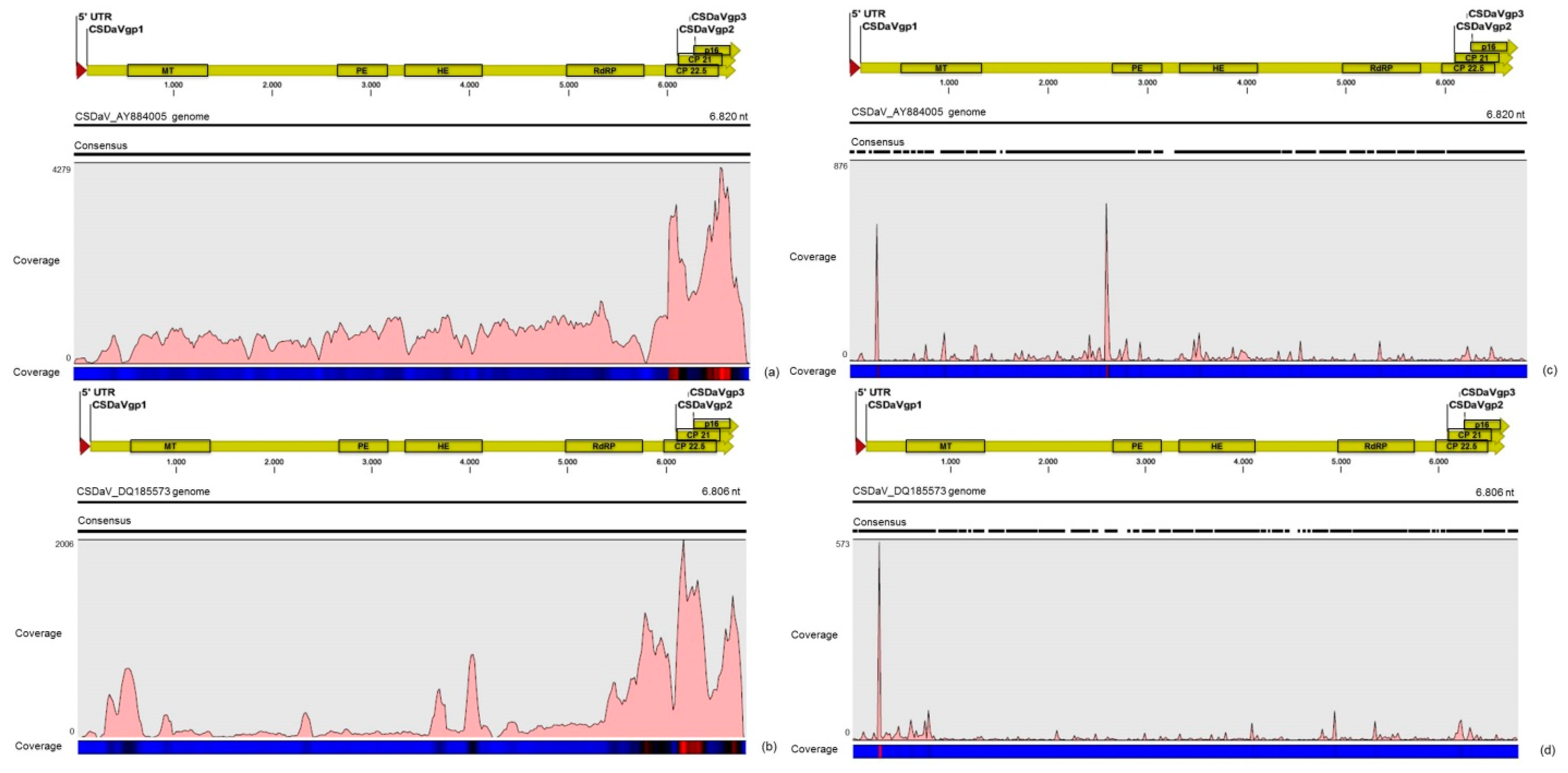
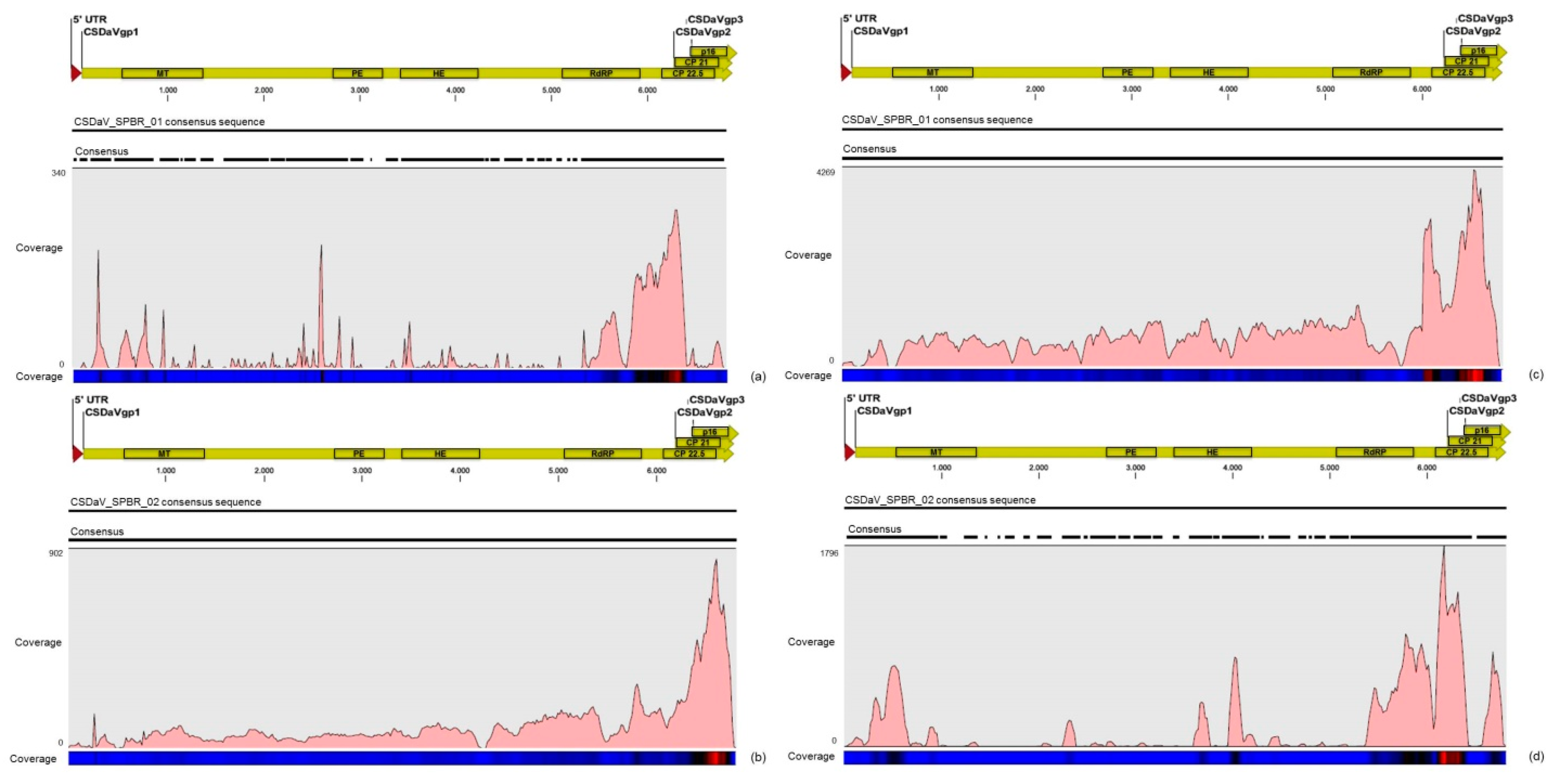
32]. Based on these results, the association of these two predominant, severe-like CTV isolates with CSD-symptomatic plants is not clear, however, the results led us to think about a new question concerning these CTV isolates: Could these isolates be the helpers in mixed virus infections by using their silencing suppressor and host range genes/proteins to facilitate the systemic infection of the other virus(es)? CSDaV could be this other virus and involved with CSD. Interestingly, the CSDaV consensus sequence obtained from libraries constructed from the symptomatic plants (CSDaV_SPBR_01) showed to be phylogenetically distant from the CSDaV consensus sequence extracted from the asymptomatic libraries (CSDaV_SPBR_02), showing at about 13% nucleotide diversity between them, which is consistent with our previous work on the CSDaV genetic diversity [
33]. Furthermore, a remarkable 29 times higher average coverage was found in mapping reads from symptomatic libraries on the CSDaV_SPBR_01 consensus sequence, compared to mapping reads from asymptomatic libraries. The average coverage of the CSDaV_SPBR_02 genotype using reads from the symptomatic plants was only 1.6 times higher than mapping reads from asymptomatic libraries on the same genotype. These results strongly support an association of CSDaV with CSD symptoms and suggest that there is a specific CSDaV genotype that could be more associated with this disease.
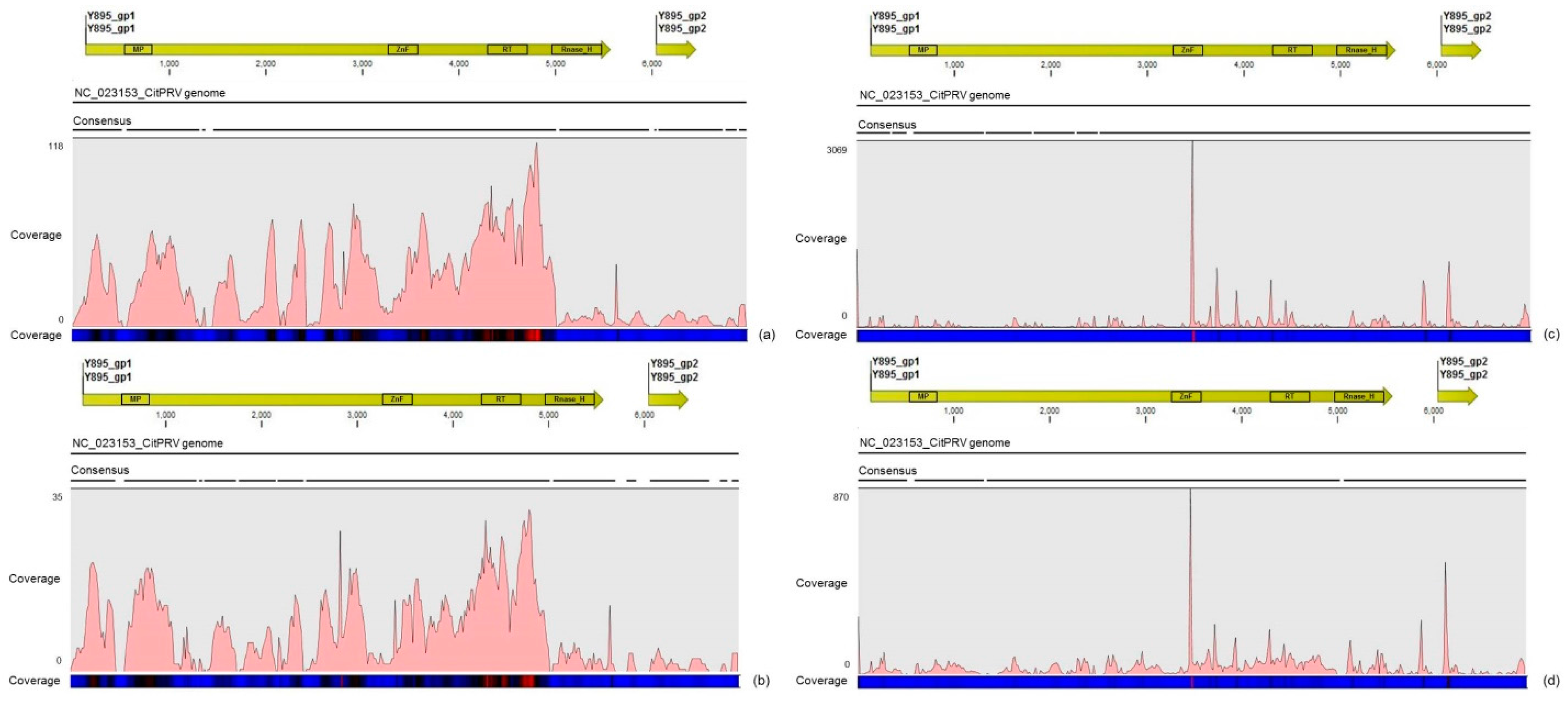
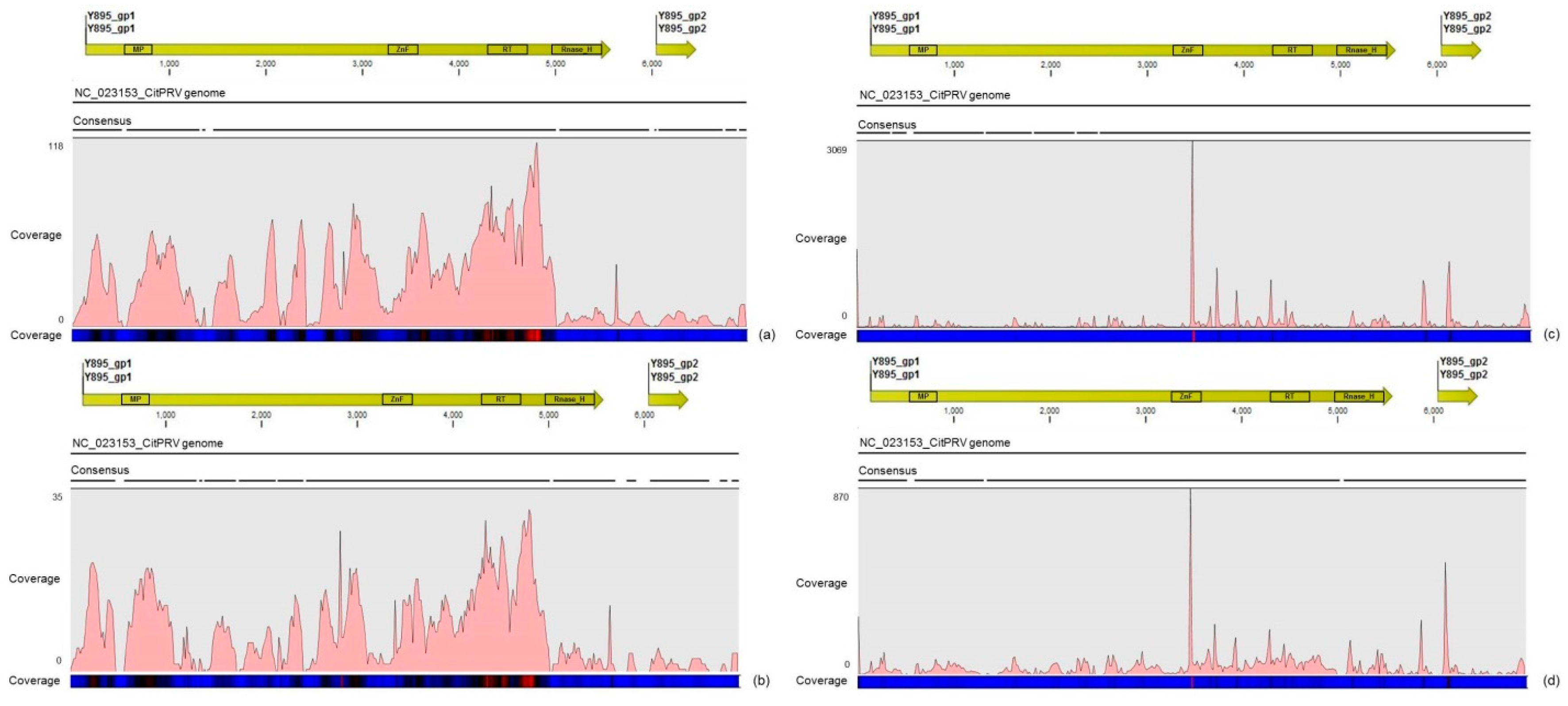
Another interesting result came from the comparative analysis between mapped reads from the asymptomatic and symptomatic libraries on the endogenous CitPRV genome. Besides the higher average coverage of this virus in symptomatic libraries (at about five times), it was also noticed that symptomatic libraries have higher number of small RNAs, compared to the asymptomatic libraries. It has been shown that other plant pararetroviruses, such as endogenous Petunia vein clearing virus (PVCV) and Tobacco vein clearing virus (TVCV), can be in some way induced, culminating to the development of viral symptoms and sRNA accumulation [
34,
35,
36]. Although the difference regarding the average coverage of the CitPRV between symptomatic and asymptomatic libraries was lower than that we obtained for CSDaV, this result cannot be ignored. As far as we know, this is the first time that CitPRV was identified in citrus plants in Brazil, and it represents the initial step in studying the possible role of CitPRV in CSD symptoms in these plants.
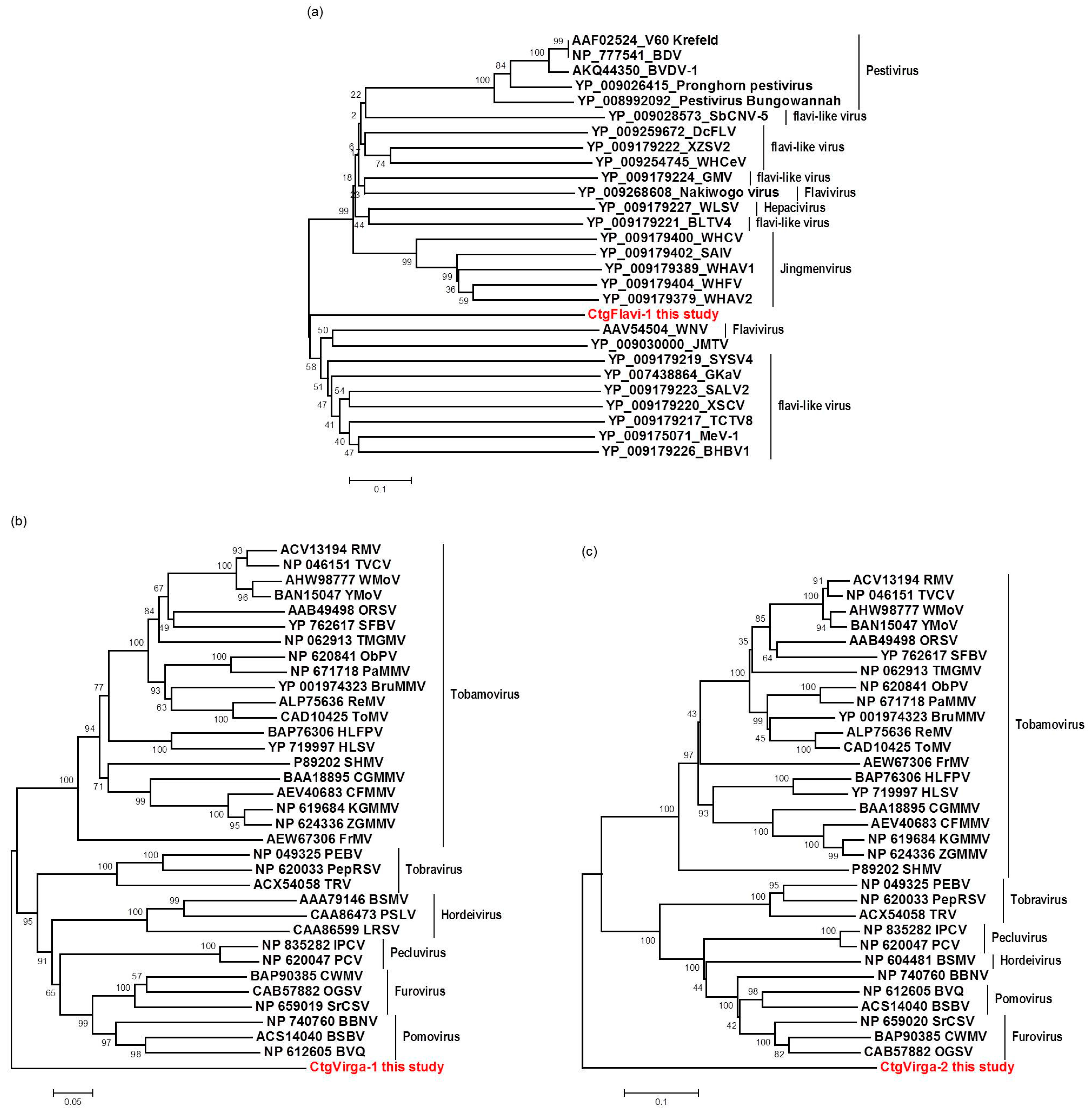
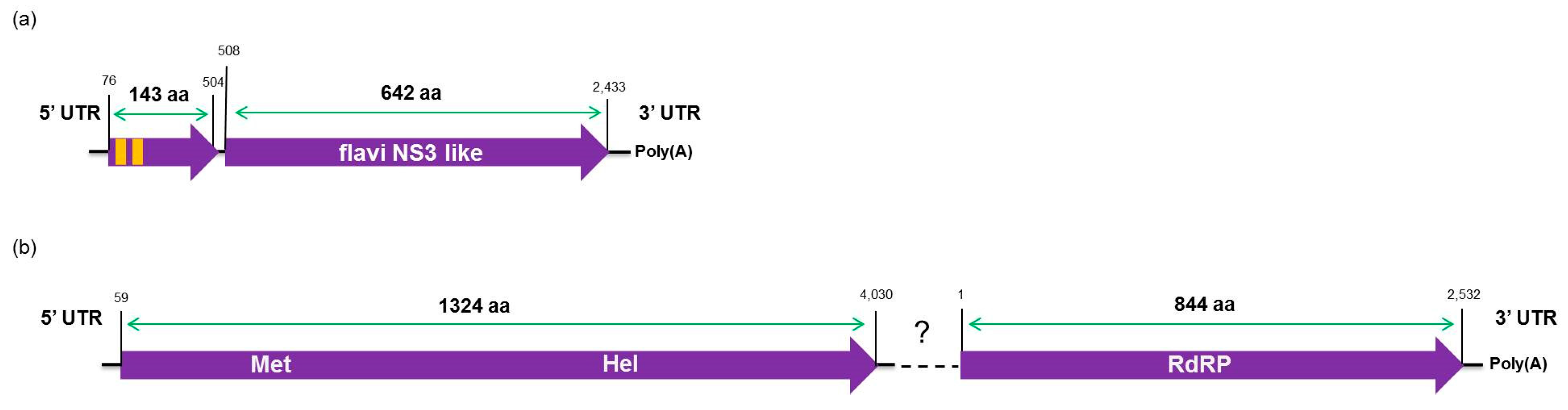
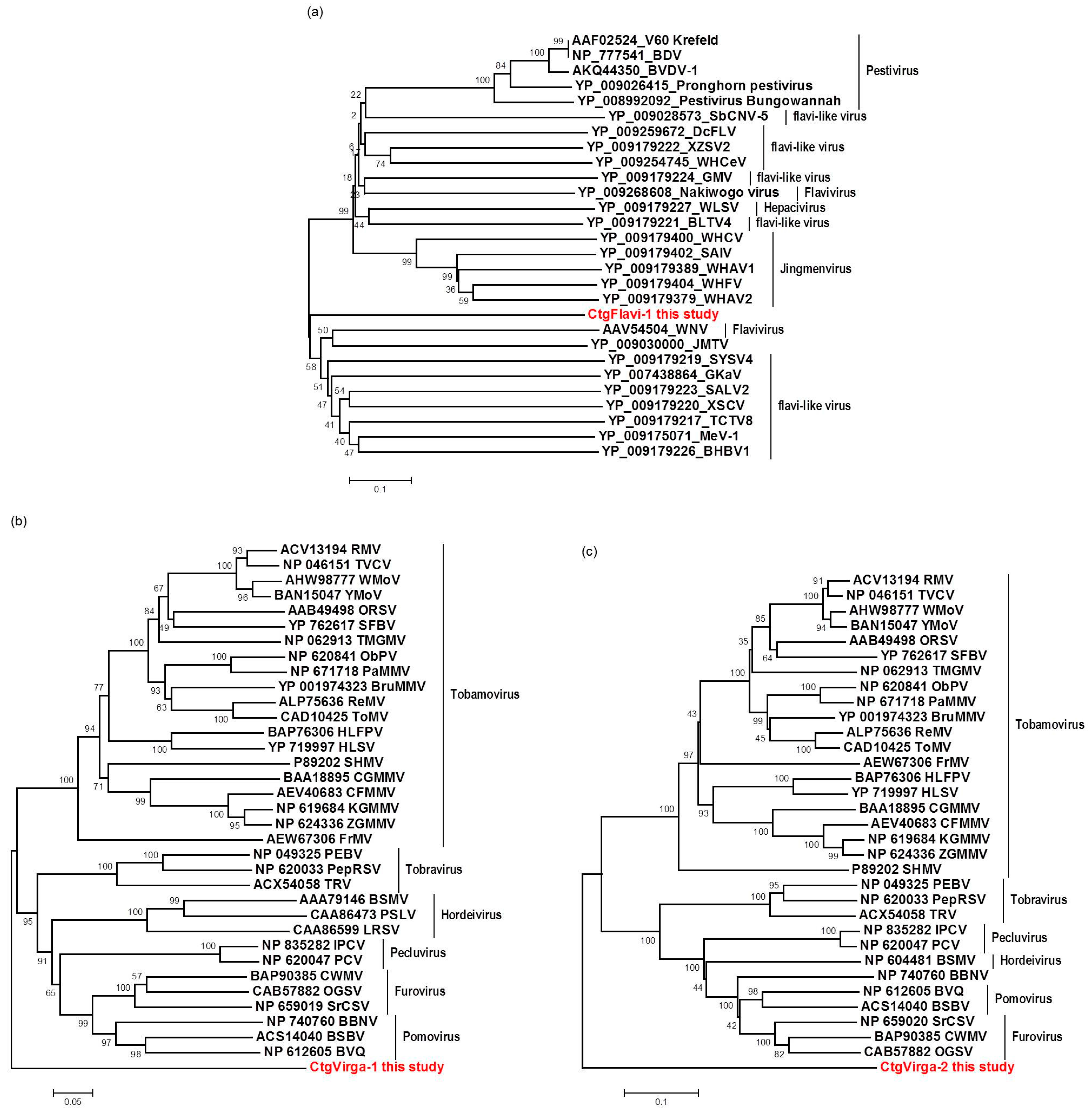
The high-throughput sequencing approach also allowed the identification of two putative novel viruses infecting the plants studied here, which shows low amino acid identity to viruses from the families Flaviviridae and Virgaviridae. Interestingly, although the genomes of both viruses are not completed using our data here, results obtained from the re-assembly analysis on the contigs from the CJLV and CVLV demonstrated a remarkable higher average coverage for both viruses in asymptomatic libraries. Besides that, it was also observed a higher diversity of viral sequences in these libraries. Of 27 viral species identified in the BLASTx analysis using assembled contigs obtained in this study as queries, 21 of them were found only in asymptomatic libraries. The lower viral diversity in libraries constructed from symptomatic plants is might be attributed to a strong competition among different viruses within the host for adequate replication conditions. Our results might suggest two things: (1) in the CSD-affected plants, viruses that are associated to developing CSD symptoms (i.e., CTV, CSDaV and/or CitPRV) are the fittest viruses, eliminating or suppressing other viruses from the within-host competition; and (2) in plants not affected by CSD, other viruses (i.e., CJLV and/or CVLV) could play a role in suppressing infections by virus(es) putatively associated in developing CSD symptoms.
In summary, this work has shown that high throughput sequencing analysis, combining data from RNA-seq and sRNA libraries, provided a wide range of information and it was a valid approach to identify and compare viral sequences in citrus plants grown in regions affected by CSD. The correlation of the viruses with the CSD disease indicated a higher association of the CSD-symptomatic plants with a specific CSDaV isolate/genotype and a likely association with CitPRV. We have identified two putative novel viruses that, interestingly, showed to be more associated with the CSD-asymptomatic plants. This study also contributed to describing, for the first time, the specific predominant CTV isolates/genotypes infecting citrus plants grown in the CSD-affected region. All data obtained in this work could together provide new insights into the role of the identified viruses in citrus plants affected by CSD and contribute to further epidemiological studies.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}