1. Introduction
To maintain their integrity, cells engage various processes including autophagy, a lysosomal-dependent catabolic process, which allows the degradation of deleterious cytoplasmic components [
1]. Macroautophagy, thereafter referred to as autophagy, is particularly efficient in this function as this form of autophagy is the only one that permits the recycling of very large portions of the cytoplasm after their sequestration within de novo formed autophagosomal vesicles. Thus, among intracellular substrates, autophagosomes can surround invading intracellular pathogens to target them to the lysosomal pathway; the degradation of pathogens through the autophagy pathway is known as xenophagy [
2]. However, most infectious pathogens have evolved strategies to escape autophagy or even to use some properties of this cellular mechanism to optimize their intracellular life cycle; measles virus (MeV) is a striking example of such an optimization [
3,
4].
MeV, which is responsible for measles, is among the most contagious human pathogens [
5]. This virus first affects the respiratory tract, before disseminating within the whole body. Among measles, the clinical symptoms are a fever, cough, and generalized maculopapular rash. Moreover, one to two weeks after MeV infection, a profound immunosuppression state is established which, although transient, can lead to the establishment of secondary opportunistic infections responsible for most of the MeV infection-induced complications [
6]. Despite the existence of an efficient vaccine, MeV is still responsible for a significant proportion of mortality worldwide, especially in developing countries, and recent outbreaks have highlighted the importance of better understanding how the virus deals with the human host cell defenses to establish a productive infection [
5].
MeV is an enveloped virus with a negative-stranded RNA genome [
7]. After entering a cell, eight viral proteins are synthetized: six structural factors, which ensure viral genome replication and new particles formation (MeV-N, MeV-P, MeV-L, MeV-M and MeV-H, and MeV-F); and two non-structural proteins, which counteract, or hijack, cellular pathways to optimize intracellular replication (MeV-V ad MeV-C). Replication and virus assembly take place within the cytosol, and newly formed infectious particles bud from the plasma membrane before secondary infections. Finally, infected cells can fuse with uninfected cells to form syncytia, allowing the virus to spread from one cell to another without virus exposition outside of the infected cells.
Our group reported that upon infection, MeV can induce autophagy through three independent pathways [
3,
4,
8,
9,
10,
11]. First, the engagement of CD46, one of the MeV cell surface receptors, induces autophagy upon virus entry: this pathway only concerns attenuated/vaccinal strains of MeV, which use CD46 to infect cells [
4,
10,
11,
12]. A few hours post infection, a second signaling pathway leads to autophagy induction following the expression of MeV-C and its interaction with the autophagy-regulating protein IRGM (Immunity-Related GTPase family M protein) [
3,
8,
9]. Finally, cell-cell fusion can also trigger autophagy, which contributes to sustaining both infected-syncytia viability and MeV replication [
4]. Thus, MeV displays a very intricate relationship with autophagy and benefits from this process, only if completed, in order to efficiently produce new infectious particles. However, it remains to be understood how MeV escapes from autophagy degradation, especially in regards to its putative detection by autophagy receptors, whose function is to transfer pathogens to the autophagy machinery for degradation.
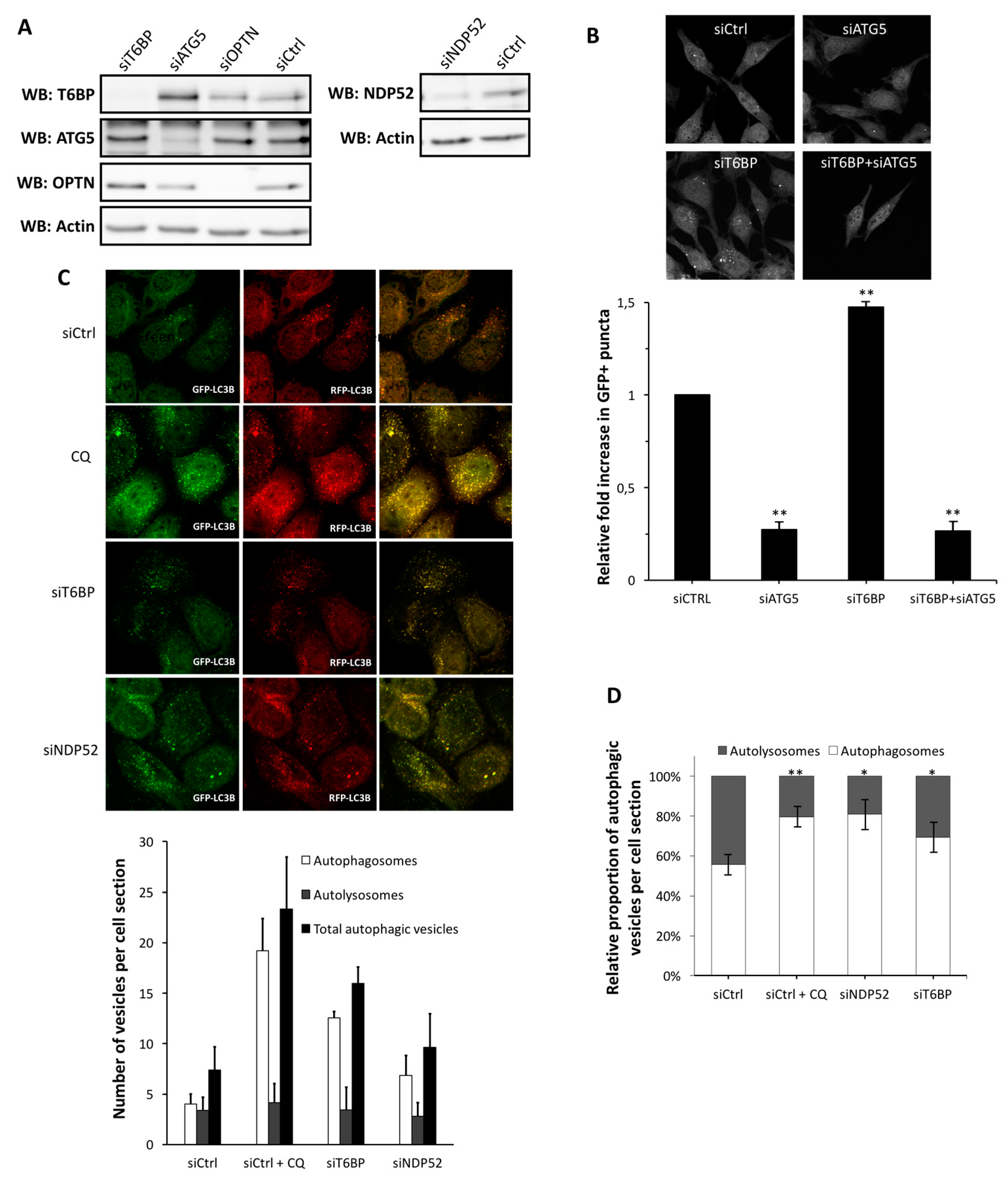
Autophagy receptors have the ability to bind intracellular pathogens or components of these pathogens and to target them toward growing autophagosomes. To this end, autophagy receptors contain LC3 interacting regions (LIR) in their primary sequence that are able to bind-members of the
autophagy-related protein 8 (ATG8) family (LC3 and GABARAP (gamma-aminobutyric acid receptor-associated protein family) members in mammals), and which are essential factors anchored in the membrane of phagophores in order to drive autophagosome formation [
13]. Among autophagy receptors, NDP52, optineurin (OPTN), and T6BP were concomitantly associated with the biogenesis of phagophores [
14]. Independently of this role, we recently reported that NDP52 and OPTN can ensure the maturation of autophagosomes by themselves, resulting in the fusion of autophagosomes with lysosomes [
15,
16]. Thus, during xenophagy, NDP52 and OPTN can play a dual function: (i) they function as autophagy receptors that target pathogens to autophagy; and (ii) they also behave as autophagy adaptors that regulate autophagosome-lysosome fusion in order to degrade the entrapped pathogens. T6BP is another autophagy receptor which might have such a dual function, as recently reported in the context of a bacterial infection [
17].
The role and regulation of autophagosome maturation during infection remain poorly understood. Moreover, although the role of autophagy receptors has been widely studied in the context of bacterial infection, little is known in relation to their functions upon viral infections. Since a complete autophagy flux is necessary for efficient MeV replication, we investigated the question of the requirement of autophagy receptors in autophagosome maturation and MeV replication.
3. Discussion
In the course of infection, viruses have to face cellular immune protection mechanisms [
24]. Among them, viral components can be detected by autophagy receptors and degraded through the lysosomal pathway to fight viral infection [
25,
26,
27,
28,
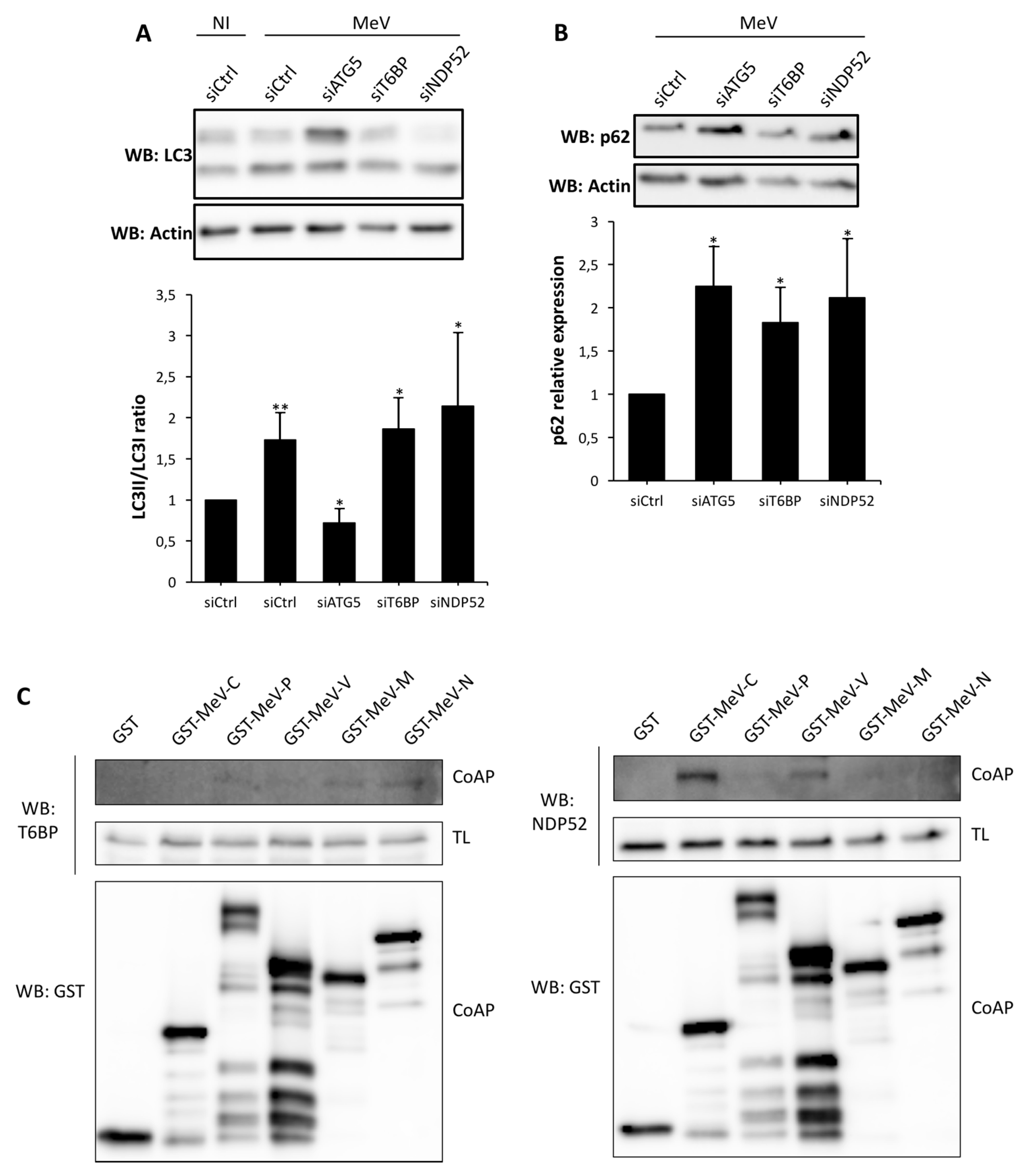
29]. However, MeV infection, although inducing a complete and productive autophagy flux, meaning from the formation of an isolated phagophore to the degradation and recycling of autophagy substrates by autolysosomes, seems insensitive to autophagy, but instead, uses this process for an optimal replication [
3,
4]. Here, we report that autophagy receptors, which also play an important role in the maturation process of autophagosome-lysosome fusion, are not used equivalently by MeV during cell infection.
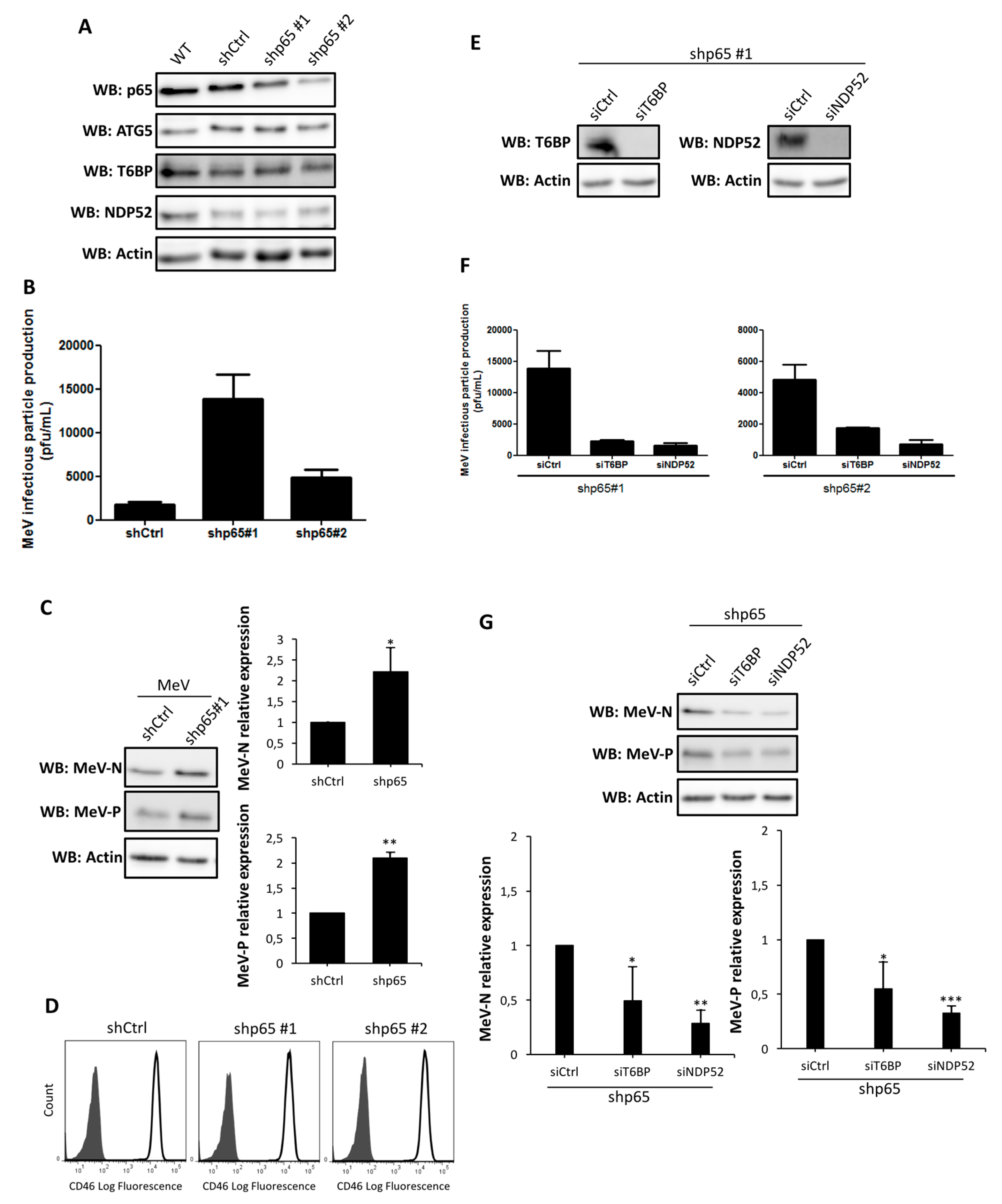
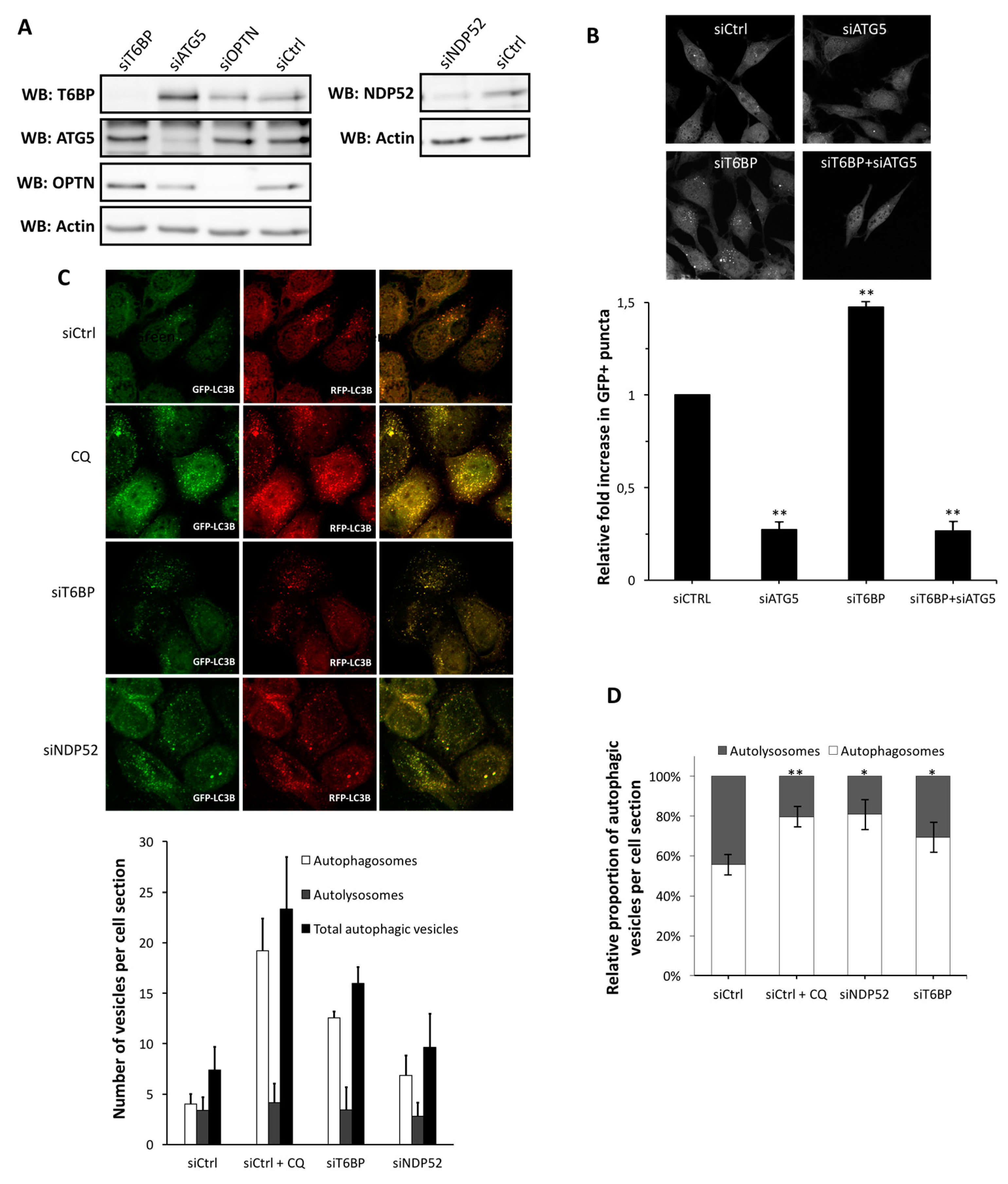
Autophagy receptors ensure the recognition of cytosolic substrates to target them to the autophagy machinery [
30]. We recently reported that the two autophagy receptors NDP52 and OPTN also regulate the fusion between autophagosomes and lysosomes, and therefore, play the dual role of autophagy receptors and autophagy adaptors, for an efficient degradation by autophagy [
15,
16]. Here, we extend this discovery to T6BP, which also regulates the maturation of autophagosomes, since the reduced expression of this protein led to an accumulation of autophagosomes. T6BP (also called TAXIBP1 or CALCOCO
3) has several homologies with NDP52 (also called CALCOCO
2), which might explain how this protein contributes to autophagy maturation. NDP52 contains a MYOSIN VI binding domain and a LIR motif, which interact with MYOSIN VI and LC3B, respectively [
15,
31]. These two binding sites were shown to be essential for NDP52-mediated autophagosome maturation [
15]. Indeed, MYOSIN VI interacts with the endosomal protein TOM-1 [
14], and LC3B is anchored in the autophagosomal membrane. By interacting with MYOSIN VI and LC3B, NDP52 connects the autophagosome with the endosomal pathway. Similarly to NDP52, T6BP was also reported for its potency to physically bind MYOSIN VI via two essential residues, C688 and C715 [
30]. T6BP also contains a LIR domain, allowing its co-localisation and interaction with LC3B [
17,
32]. Thus, through the concomitant interaction with MYOSIN VI and LC3B, T6BP could govern the maturation of autophagosomes, similarly to NDP52. Indeed, a recent work described the essential role for both T6BP and MYOSIN VI in the late phase of autophagy for an efficient clearing of intracellular infection by
Salmonella typhimurium [
17]. Both T6BP and NDP52, but not OPTN, contain a so-called SKICH domain whose function is undetermined. Whether this domain plays a role in the differential impact of these receptors on MeV replication remains to be studied.
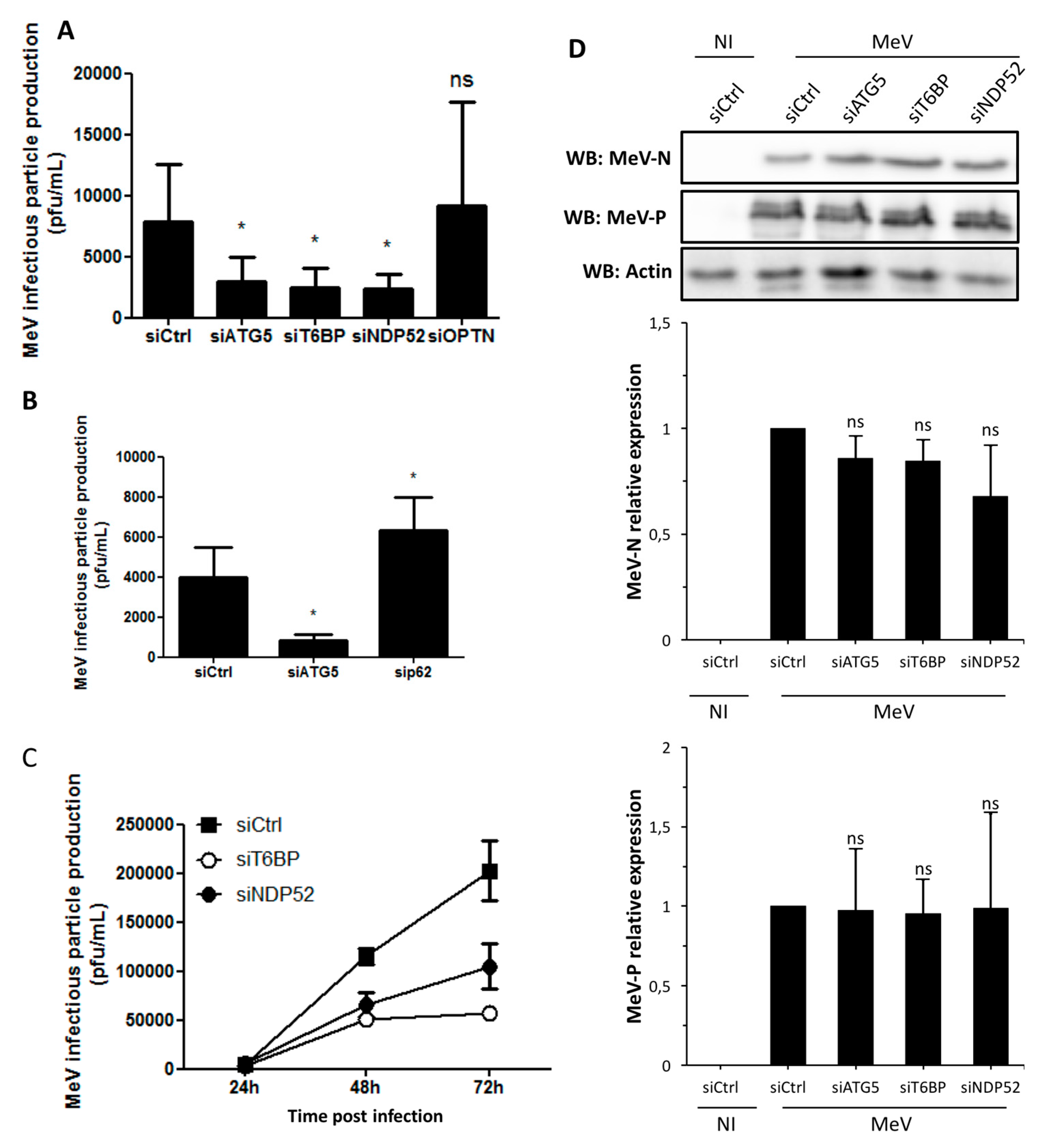
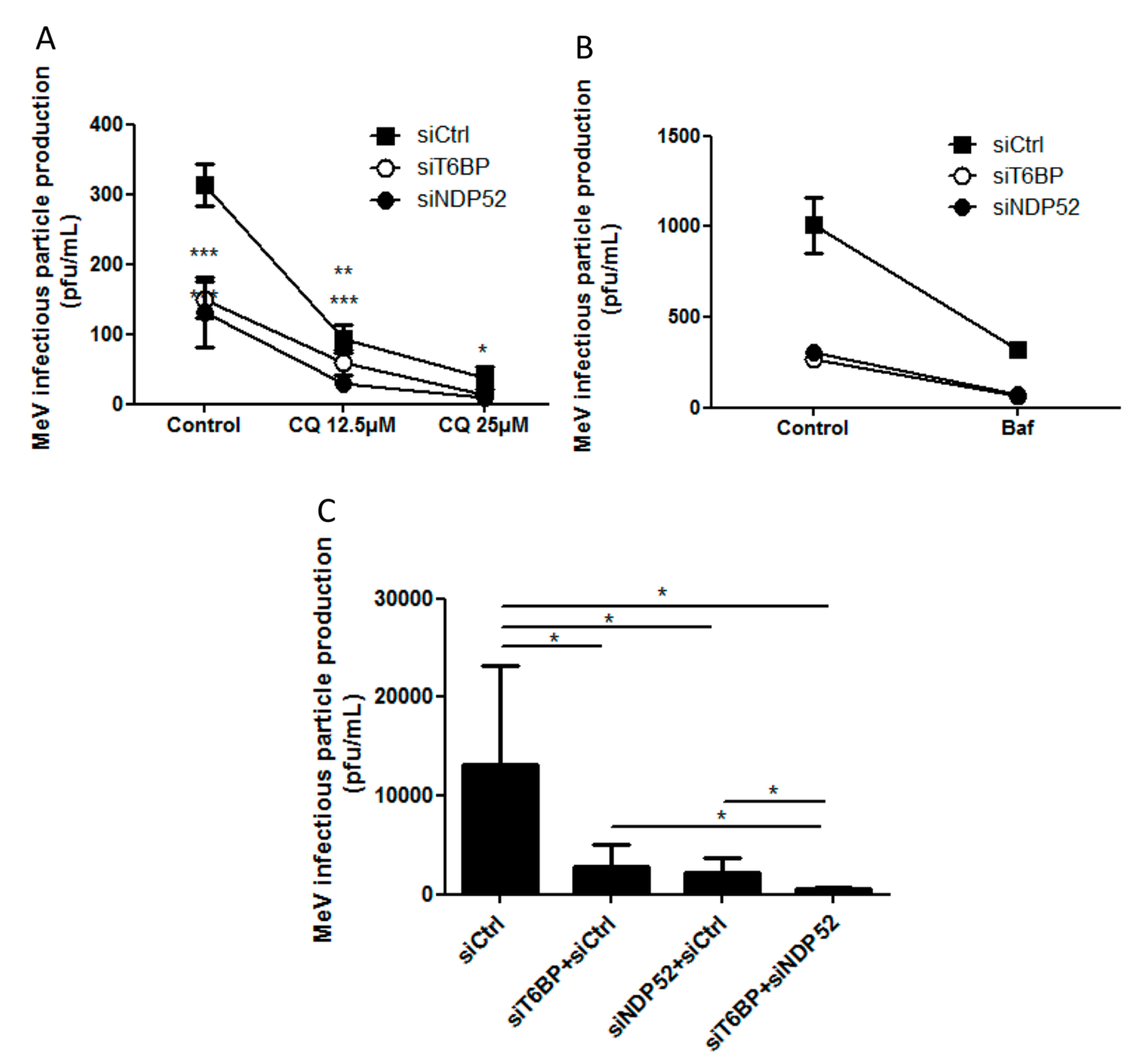
T6BP and NDP52 are both involved in a negative regulation of the canonical NF-κB signaling pathway [
19,
20]. Since viral infections can be regulated by the NF-κB pathway [
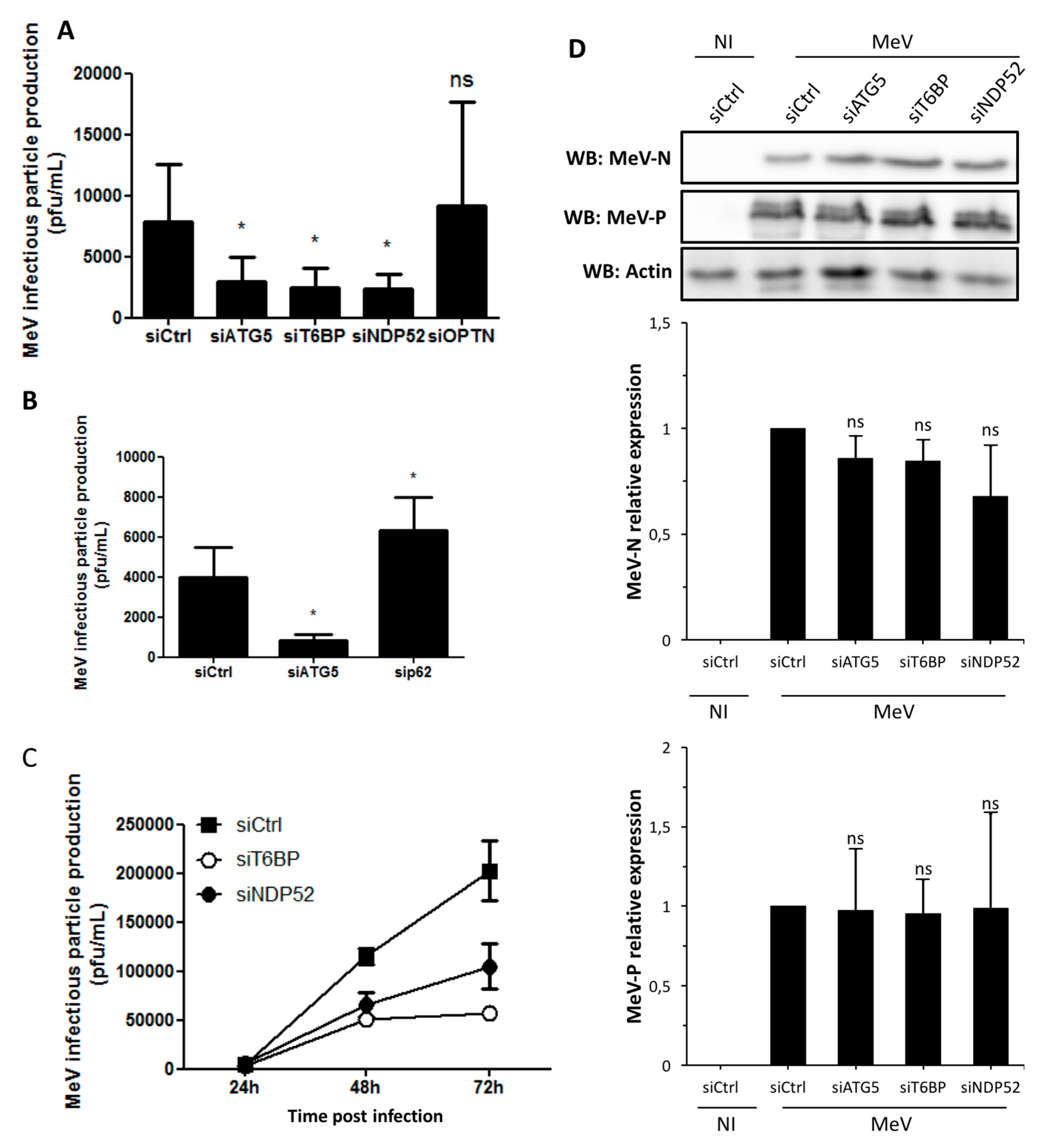
21], the impact of the reduced expression of T6BP or NDP52 on MeV replication could have been due to its role in an autophagy-independent mechanism. However, the use of NF-κB defective cells allowed us to demonstrate that the absence of T6BP or NDP52 impacted MeV replication, independently of the activation of this transcription factor. Thus, although we cannot exclude that a T6BP/NDP52-dependent regulation of NF-κB could contribute to the partial control of MeV replication, the role of these autophagy receptors in the maturation of autophagosomes appears to be predominantly required for an efficient MeV replication. OPTN has also been reported to either positively or negatively regulate the NF-κB signaling pathway [
33,
34]. The fact that siOPTN did not affect MeV replication also suggests that the potential role of T6BP and NDP52 in NF-κB signaling has no significant role in the course of MeV infection in HeLa cells.
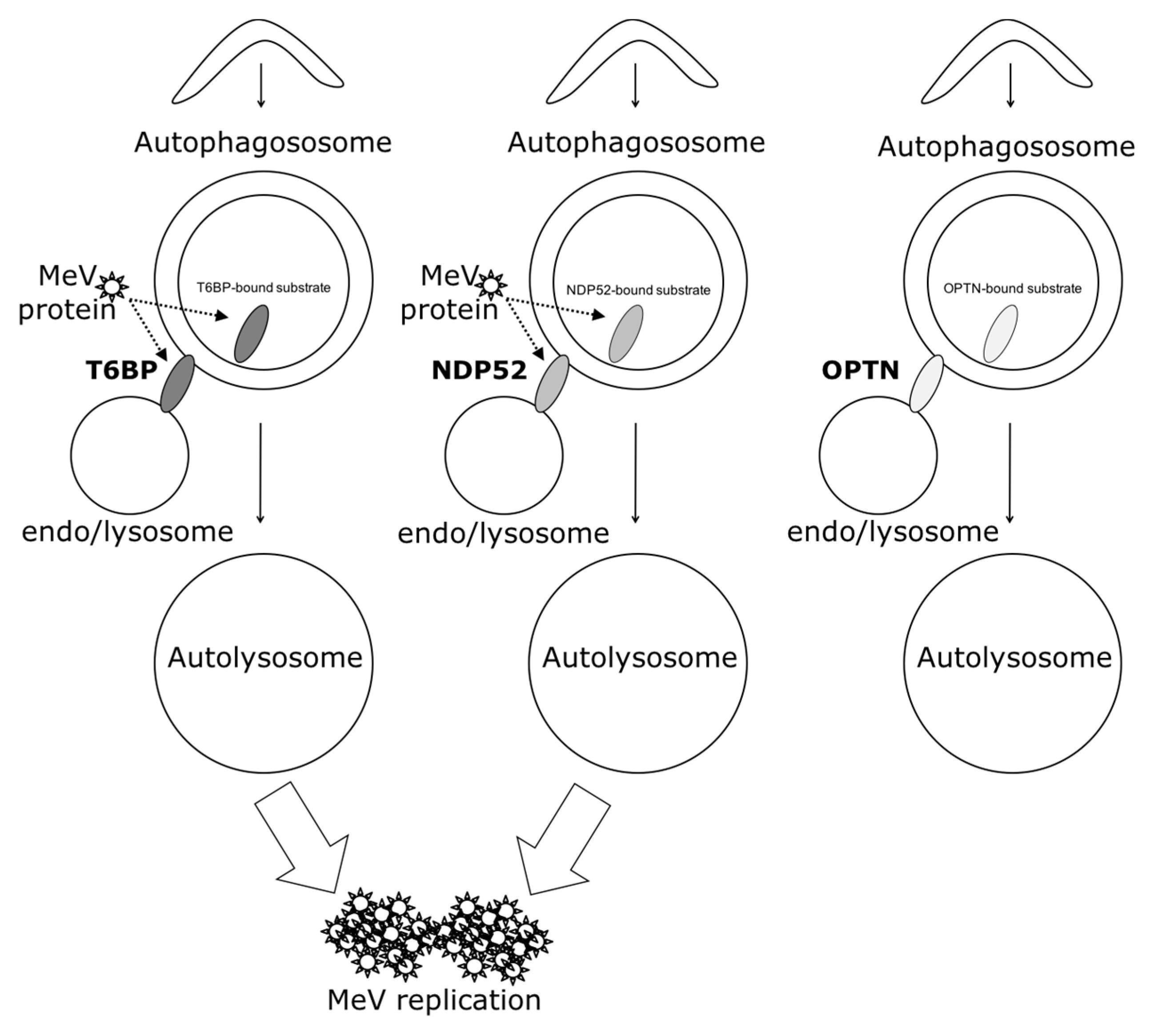
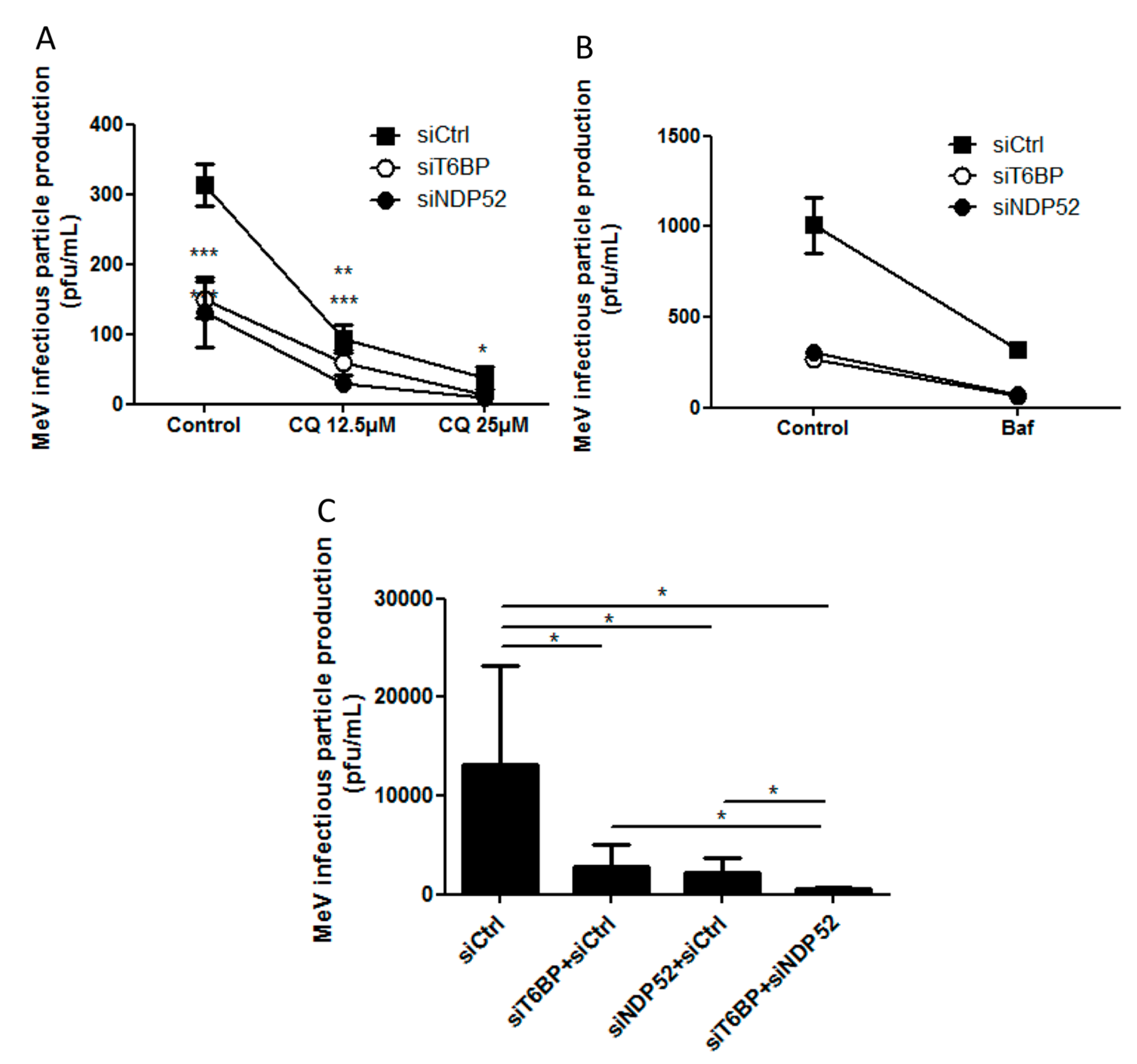
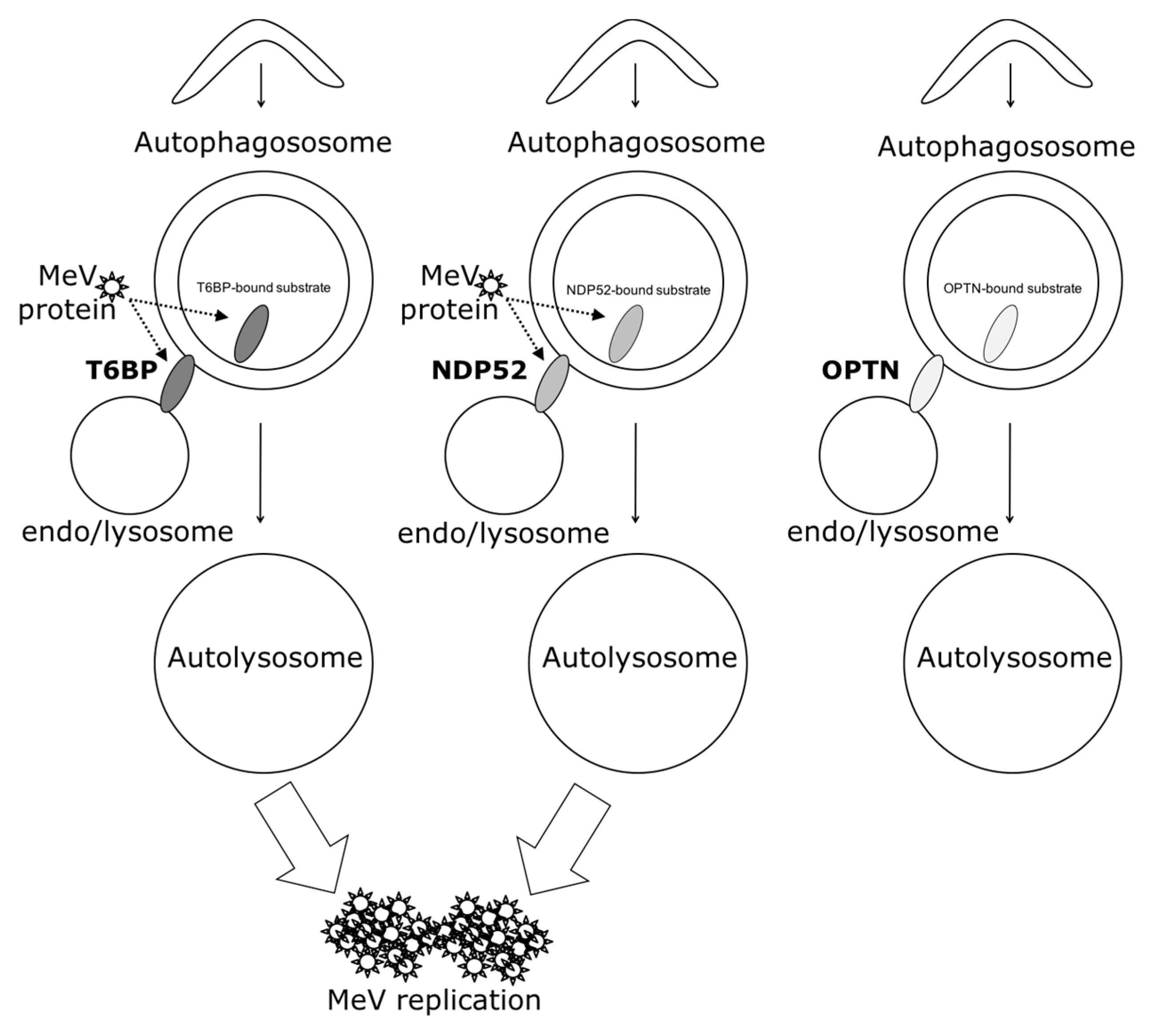
In our work, several lines of evidence suggest that autophagosome maturation could be regulated independently by each autophagy receptor/adaptor and that they could be exploited individually by MeV to replicate. As described, beyond their function as autophagy receptors, NDP52, T6BP, and OPTN also have a role in the maturation of autophagosomes. Strikingly, the reduced expression of OPTN did not impact the production of infectious MeV particles, contrary to the reduced expression of T6BP or NDP52. Thus, not all maturated autophagosomes seem to be involved in MeV replication (e.g., the ones regulated by OPTN), but only some of them, such as those regulated by NDP52 or T6BP. How MeV makes the distinction between individual autophagosomes remains to be fully depicted. This could occur through the interaction of viral proteins with either NDP52 or T6BP, which could potentiate the maturation of autophagosomes regulated by these two proteins. Indeed, we found that distinct MeV proteins have the ability to interact with either NDP52 or T6BP, but whether such interactions take place in the course of infection and drive autophagosome maturation remains to be fully investigated. Alternatively, T6BP and NDP52, but not OPTN, could target selective substrates to autophagy whose degradation is required for MeV replication (
Figure 6). Since MeV-induced autophagy contributes to delay the death of infected cells [
4], such selective substrates could be infection-induced apoptotic factors, but this also needs to be analyzed.
Another piece of evidence suggesting a distinct usage of individual NDP52-mediated and T6BP-mediated autophagosome maturation for MeV replication is the fact that the co-reduced expression of NDP52 and T6BP impacted MeV replication more efficiently than their individual reduction. If NDP52 or T6BP were both co-engaged in the maturation process of all autophagosomes, their single reduction would have impacted the function of both receptors on MeV replication. Although used at saturating concentrations, we cannot exclude that siRNA treatments are not completely efficient at reducing the expression of individual proteins. Thus, siNDP52 (or siT6BP) could permit some NDP52/T6BP-dependent autophagosome maturation, which would not have been impacted due to residual endogenous NDP52 upon siNDP52 treatment (or T6BP upon siT6BP treatment). However, if all autophagy receptors were engaged in the regulation of each individual autophagosome, we would have expected that the reduced expression of OPTN, which also impacts autophagosome maturation, would have decreased MeV replication. Thus, although we cannot exclude that a unique molecular machinery involving NDP52, T6BP, and OPTN is required for the maturation of autophagosomes, our results suggest a very fine tuned molecular regulation of autophagosome maturation, which could be exploited by MeV to replicate (
Figure 6).
In the course of infection, the interplay of MeV with the autophagy process is very intricate, as we already reported that several signaling pathways are involved. The deeper study of this specific host-pathogen interaction allowed us to reveal here a potential individual regulation of autophagosome maturation by individual autophagy receptors. Our study offers interesting perspectives in regards to both the understanding of autophagy molecular regulation, a cellular process whose deregulation is associated with several human pathologies, and the potential development of strategies to fight MeV infection, one of the most contagious human diseases, possibly by targeting individual molecules involved in the specific maturation of autophagosomes, without altering the complete autophagy process.
4. Experimental Procedures
4.1. Antibodies and Reagents
Antibodies used were: anti-T6BP (HPA024432) anti-NDP52 (HPA023195), anti-LC3B (L7543), anti-actin (A2066), and anti-ATG5 (A0856), all from Sigma-Aldrich, anti-OPTN (Abcam, ab23666 Paris, France), anti-SQSTM1/p62 (Santa Cruz Biotechnology, sc-28359, Heidelberg, Germany), and anti-p65/RelA (Millipore #06-418, Molsheim, France). Anti-MeV-N (mouse monoclonal, clone 120) and anti-MeV-P (rabbit polyclonal, clone J37171) were used. Anti-CD46 conjugated to the PE antibody (8E2 clone) was from ThermoFisher Scientific (12-0469-42, Courtaboeuf, France). Secondary antibodies used were: anti-Mouse conjugated to Peroxydase (A2304) from Sigma-Aldrich and anti-Rabbit conjugated to HRP (NA9340). Pharmacological agents used were Bafilomycin A1 (25 nM) (InvivoGen #tlrl-baf1, Toulouse, France) and Chloroquine (25 µM or 12.5 µM) (C6628, Sigma-Aldrich, St. Quentin Fallavier, France).
4.2. Cell Culture
HeLa, GFP-LC3-HeLa, mRFP-GFP-LC3-HeLa, shp65-HeLa, and Vero cells were maintained in DMEM medium, supplemented with 10%FBS, 0.1% Gentamicin. An additional 500 µg/mL of Geneticin/G418 was added for GFP-LC3-HeLa, mRFP-GFP-LC3-HeLa, and shp65-HeLa cell cultures. The shCtrl, shp65#1, and shp65#2 HeLa cell lines used in this study were the HeLa-cont#1, HeLa-p65 KD#1, and HeLa-p65 KD#2 cell lines used in [
21], respectively.
4.3. siRNA Transfection
The day before transfection with siRNA, the cells were seeded in six-well plates with 1 × 105 cells per well in OPTIMEM complemented with 10% fetal bovine serum (FBS), 2 mM of l-glutamine, 50 mg/mL of Gentamycin, 0.1 mM non-essential amino acid, 0.1 mM pyruvate sodium, and 0.1 g/L bicarbonate sodium. The cells were transfected with 100 pmol of total siRNA using Lipofectamine RNAiMAX from Invitrogen (13778-150, Courtaboeuf, France), according to the manufacturer’s instructions. Protein expression level was assessed by Western blotting four days post transfection (lysis buffer: PBS 1X, 0.5% Nonidet P40, and protease inhibitor) (Complete Mini EDTA free, Roche Applied Science #04693159001, Meylan, France).
For titration experiments, the cells were transferred 48h after siRNA transfection to a 24-well plate at 2 × 104 cells per well. Five hours after the transfer, cells were infected with MeV.
4.4. MeV Strains and Titration by Plaque Assay
Measles Virus Edmonston strain (MeV) was obtained from ATCC. HeLa cells were infected with MeV at the indicated MOI. After the indicated period of infection, cells were submitted to five freeze (−80 °C)-thaw cycles (ambient temperature) and infectious viral particles were quantified by limiting dilution on confluent Vero cells. Briefly, supernatants were diluted in DMEM culture medium with 2% FBS. The dilutions covered the range from 1/2 to 1/810 and each dilution was tested in duplicate. A total of 0.45 mL of each dilution was loaded onto the Vero cell monolayer. After 1.5 h of adsorption at 37 °C, 800 µL of DMEM culture medium with 2% FBS was added and cells were further incubated at 37 °C, 5% CO2 for 48 h. Then, Vero cells were fixed and stained with Methylene Blue. Plaque-forming units (pfu) were numerated after the cell layers had been washed and left to dry. Only dilutions which displayed at least 10 pfu were taken into account. At least three dilutions were considered when calculating the viral titers for each duplicate in each experiment and the mean of the duplicates was calculated. Results are represented as a fold increase normalized to the control condition relevant to a given experiment.
4.5. Molecular Cloning
For mammalian cell expression, viral proteins were engineered into a pDEST27 plasmid, allowing the expression of Glutathione S-transferase (GST) tagged proteins for co-affinity purification experiments.
4.6. GST Co-Affinity Purification Assays
HeLa cells were seeded at 2.5 × 105 cells per well in six-well plates. Twenty-four hours later, cells were transfected with 2 μg/well of plasmid encoding the GST-tagged genes. The cells were harvested 48 h late and lysed in PBS 1×, containing Calcium and Magnesium with 0.5% of Nonidet P40 and protease inhibitor cocktail (Complete Mini EDTA free, Roche Applied Science #04693159001, Meylan, France). The purified lysate was incubated overnight at 4 °C on Glutathione Sepharose beads (GE Healthcare #17-0746-01, Courtboeuf, France). Elution and Western blotting were performed the next day.
4.7. Confocal Microscopy
All images were taken on a confocal Zeiss LSM 710 (Marly le Roi, France) with a plan apochromat 40× objective. The quantification of fluorescent vesicles was carried out using ImageJ. The cells were cultured in 24 well-plates with a sterile coverslip in each well. The cells were fixed in ice cold acetone. At least 100 cells per individual experiment were numerated.
4.8. Immunofluorescence-coupled Flow Cytometry
For CD46 staining, 0.25–0.5 × 106 cells were incubated in microtiter U-bottom plates with saturating concentrations of labeled monoclonal antibody (mAb) in 20 μL PBS 2% FCS/0.1% NaN3for 30 min on ice. Cells were washed twice and analyzed immediately, without fixation. The anti-human CD46 mAb used was the phycoerythrin (PE)-conjugated 8E2 clone (mouse IgG1κ) from eBioscience. A LSRII flow cytometer (Becton Dickinson, Pont-de-Claix, France) and the FlowJo software (Tristar, Ashland OR, USA) were used to collect and analyze the data. Nonviable cells were excluded using forward and side scatter electronic gating. In some experiments, results were confirmed by using the fluorescein isothiocyanate (FITC)-conjugated anti-human CD46 E4.3 mAb (mouse IgG2aκ) from BD Biosciences (Pont-de-Claix, France).
4.9. Statistical Analysis
All
p-values were calculated using a one-tailed Welch’s
t-test (Student’s
t-test assuming non-equal variances of the samples), except for the result of
Figure 5A,B for which an Anova2 Bonferroni
post hoc test was applied; *
p < 0.05, **
p < 0.01, ***
p < 0.001.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}