
Interplay between Autophagy, Exosomes and HIV-1 Associated Neurological Disorders: New Insights for Diagnosis and Therapeutic Applications

 , ,
, , {kind=link}
Abstract
:1. Introduction
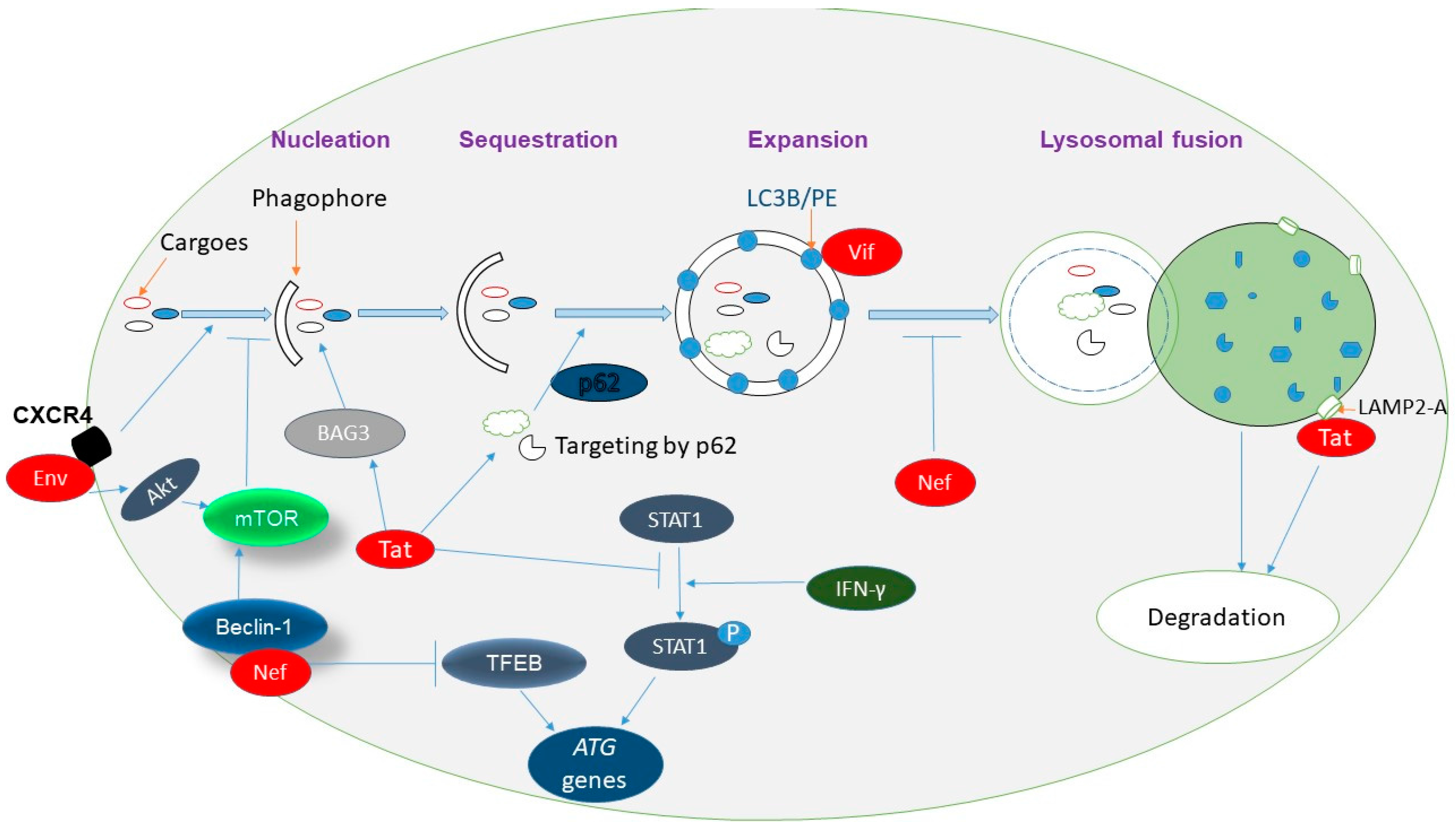
2. Autophagy and Its Regulation
3. Autophagy and Exosomes
4. Viral Regulation of Autophagy
5. Complex Interaction between HIV-1 and Autophagy
5.1. HIV-1 Induces Autophagy
5.2. Autophagy Restricts HIV-1 Infection and Disease Progression
5.3. HIV-1 Has Evolved Mechanisms to Defend against Autophagy
6. HIV-1, Exosomes and Autophagy
7. Autophagy Modulation by HIV-1 in Central Nervous System Cells
7.1. Neurons
7.2. Microglia
7.3. Astrocytes
8. Therapeutic Approaches for HIV-1 Based on Autophagy
8.1. Induction of Autophagy
8.1.1. mTOR Inhibitors
8.1.2. Tat-Beclin-1 Fusion Peptide
8.1.3. Vitamin D, Trehalose and Nitric Oxide Inhibitors
8.1.4. Histone Deacetylase Inhibitors
8.2. Inhibition of Autophagy
8.3. Silencing of Beclin-1
8.4. Downregulation of ATG Genes
8.5. Upregulation of SQSTM1/p62
8.6. Targeting Viral Proteins That Directly Affect Autophagy
9. Nano-Formulation of Targeted Drugs
10. Conclusions and Perspective
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Davis, L.E.; Hjelle, B.L.; Miller, V.E.; Palmer, D.L.; Llewellyn, A.L.; Merlin, T.L.; Young, S.A.; Mills, R.G.; Wachsman, W.; Wiley, C.A. Early viral brain invasion in iatrogenic human immunodeficiency virus infection. Neurology 1992, 42, 1736–1739. [Google Scholar] [CrossRef] [PubMed]
- Heaton, R.K.; Franklin, D.R.; Ellis, R.J.; McCutchan, J.A.; Letendre, S.L.; Leblanc, S.; Corkran, S.H.; Duarte, N.A.; Clifford, D.B.; Woods, S.P.; et al. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: Differences in rates, nature, and predictors. J. Neurovirol. 2011, 17, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Pinkevych, M.; Cromer, D.; Tolstrup, M.; Grimm, A.J.; Cooper, D.A.; Lewin, S.R.; Søgaard, O.S.; Rasmussen, T.A.; Kent, S.J.; Kelleher, A.D.; et al. HIV reactivation from latency after treatment interruption occurs on average every 5-8 days—Implications for HIV remission. PLoS Pathog. 2015, 11, e1005000. [Google Scholar] [CrossRef] [PubMed]
- Churchill, M.J.; Gorry, P.R.; Cowley, D.; Lal, L.; Sonza, S.; Purcell, D.F.; Thompson, K.A.; Gabuzda, D.; McArthur, J.C.; Pardo, C.A.; et al. Use of laser capture microdissection to detect integrated HIV-1 DNA in macrophages and astrocytes from autopsy brain tissues. J. Neurovirol. 2006, 12, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.A.; Cherry, C.L.; Bell, J.E.; McLean, C.A. Brain cell reservoirs of latent virus in presymptomatic HIV-infected individuals. Am. J. Pathol. 2011, 179, 1623–1629. [Google Scholar] [CrossRef] [PubMed]
- Saylor, D.; Dickens, A.M.; Sacktor, N.; Haughey, N.; Slusher, B.; Pletnikov, M.; Mankowski, J.L.; Brown, A.; Volsky, D.J.; McArthur, J.C. HIV-associated neurocognitive disorder—Pathogenesis and prospects for treatment. Nat. Rev. Neurol. 2016, 12, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Maschke, M.; Kastrup, O.; Esser, S.; Ross, B.; Hengge, U.; Hufnagel, A. Incidence and prevalence of neurological disorders associated with HIV since the introduction of highly active antiretroviral therapy (HAART). J. Neurol. Neurosurg. Psychiatry 2000, 69, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Heaton, R.K.; Clifford, D.B.; Franklin, D.R.; Woods, S.P.; Ake, C.; Vaida, F.; Ellis, R.J.; Letendre, S.L.; Marcotte, T.D.; Atkinson, J.H.; et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology 2010, 75, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Samji, H.; Cescon, A.; Hogg, R.S.; Modur, S.P.; Althoff, K.N.; Buchacz, K.; Burchell, A.N.; Cohen, M.; Gebo, K.A.; Gill, M.J.; et al. closing the gap: Increases in life expectancy among treated HIV-positive individuals in the united states and canada. PLoS ONE 2013, 8, e81355. [Google Scholar] [CrossRef] [PubMed]
- Tavazzi, E.; Morrison, D.; Sullivan, P.; Morgello, S.; Fischer, T. Brain inflammation is a common feature of HIV-infected patients without HIV encephalitis or productive brain infection. Curr. HIV Res. 2014, 12, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Decloedt, E.H.; Rosenkranz, B.; Maartens, G.; Joska, J. Central nervous system penetration of antiretroviral drugs: Pharmacokinetic, pharmacodynamic and pharmacogenomic considerations. Clin. Pharmacokinet. 2015, 54, 581–598. [Google Scholar] [CrossRef] [PubMed]
- Ene, L.; Duiculescu, D.; Ruta, S.M. How much do antiretroviral drugs penetrate into the central nervous system? J. Med. Life 2011, 4, 432–439. [Google Scholar] [PubMed]
- Shah, A.; Gangwani, M.R.; Chaudhari, N.S.; Glazyrin, A.; Bhat, H.K.; Kumar, A. Neurotoxicity in the post-HAART era: Caution for the antiretroviral therapeutics. Neurotox. Res. 2016, 30, 677–697. [Google Scholar] [CrossRef] [PubMed]
- Kranick, S.M.; Nath, A. Neurologic complications of HIV-1 infection and its treatment in the era of antiretroviral therapy. Continuum 2012, 18, 1319–1337. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Zhang, T.; Qu, X.; Zhang, Y.; Putatunda, R.; Xiao, X.; Li, F.; Xiao, W.; Zhao, H.; Dai, S.; et al. In vivo excision of HIV-1 provirus by saCas9 and multiplex single-guide RNAs in animal models. Mol. Ther. 2017, 25, 1168–1186. [Google Scholar] [CrossRef] [PubMed]
- Bobbin, M.L.; Burnett, J.C.; Rossi, J.J.; Mitsuya, H.; Weinhold, K.; Furman, P.; Clair, M.S.; Lehrman, S.; Gallo, R.; Burnett, J.; et al. RNA interference approaches for treatment of HIV-1 infection. Genome Med. 2015, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Khalili, K.; White, M.K.; Jacobson, J.M. Novel AIDS therapies based on gene editing. Cell. Mol. Life Sci. 2017, 74, 2439–2450. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Masliah, E.; Spector, S.A. Autophagy is increased in postmortem brains of persons with HIV-1-associated encephalitis. J. Infect. Dis. 2011, 203, 1647–1657. [Google Scholar] [CrossRef] [PubMed]
- Espert, L.; Varbanov, M.; Robert-Hebmann, V.; Sagnier, S.; Robbins, I.; Sanchez, F.; Lafont, V.; Biard-Piechaczyk, M. Differential role of autophagy in CD4 T cells and macrophages during X4 and R5 HIV-1 infection. PLoS ONE 2009, 4, e5787. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Dever, S.M.; Rodriguez, M.; Lapierre, J.; Costin, B.N.; El-Hage, N. Differing roles of autophagy in HIV-associated neurocognitive impairment and encephalitis with implications for morphine co-exposure. Front. Microbiol. 2015, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chiramel, A.I.; Brady, N.R.; Bartenschlager, R. Divergent roles of autophagy in virus infection. Cells 2013, 2, 83–104. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Son, J.H.; Shim, J.H.; Kim, K.-H.; Ha, J.-Y.; Han, J.Y. Neuronal autophagy and neurodegenerative diseases. Exp. Mol. Med. 2012, 44, 89. [Google Scholar] [CrossRef] [PubMed]
- Deter, R.L.; De Duve, C. Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J. Cell. Biol. 1967, 33, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, S.; Kuma, A.; Murakami, M.; Kishi, C.; Yamamoto, A.; Mizushima, N. Autophagy is essential for preimplantation development of mouse embryos. Science 2008, 321, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Jagannath, C.; Liu, X.-D.; Sharafkhaneh, A.; Kolodziejska, K.E.; Eissa, N.T. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 2007, 27, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, S.T.; Saitoh, T.; Nowag, H.; Münz, C.; Yoshimori, T. Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 2015, 16, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Alva, A.; Su, H.; Dutt, P.; Freundt, E.; Welsh, S.; Baehrecke, E.H.; Lenardo, M.J. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 2004, 304, 1500–1502. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y.; Shimizu, S. Another way to die: Autophagic programmed cell death. Cell Death Differ. 2005, 12, 1528–1534. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, T.; Klionsky, D.J. Autophagy: Molecular machinery for self-eating. Cell Death Differ. 2005, 12, 1542–1552. [Google Scholar] [CrossRef] [PubMed]
- Badadani, M.; Badadani, M. Autophagy mechanism, regulation, functions, and disorders. ISRN Cell Biol. 2012, 2012, 927064. [Google Scholar] [CrossRef]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, Y.C.; Fang, C.; Russell, R.C.; Kim, J.H.; Fan, W.; Liu, R.; Zhong, Q.; Guan, K.-L. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 2013, 152, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Zalckvar, E.; Berissi, H.; Mizrachy, L.; Idelchuk, Y.; Koren, I.; Eisenstein, M.; Sabanay, H.; Pinkas-Kramarski, R.; Kimchi, A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009, 10, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.C.; Wei, Y.; An, Z.; Zou, Z.; Xiao, G.; Bhagat, G.; White, M.; Reichelt, J.; Levine, B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 2012, 338, 956–959. [Google Scholar] [CrossRef] [PubMed]
- Cherra, S.J.; Kulich, S.M.; Uechi, G.; Balasubramani, M.; Mountzouris, J.; Day, B.W.; Chu, C.T. Regulation of the autophagy protein LC3 by phosphorylation. J. Cell Biol. 2010, 190, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, G.; Wada, K.; Okuno, M.; Kurosawa, M.; Nukina, N. Serine 403 Phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol. Cell 2011, 44, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.-J.R.; Kim, H.; Kim, K.I.L.; Baek, S.H. Epigenetic and transcriptional regulation of autophagy. Autophagy 2016, 12, 2248–2249. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.C.; Vacca, F.; Gruenberg, J. Endosome maturation, transport and functions. Semin. Cell Dev. Biol. 2014, 31, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Février, B.; Raposo, G. Exosomes: Endosomal-derived vesicles shipping extracellular messages. Curr. Opin. Cell Biol. 2004, 16, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569. [Google Scholar] [PubMed]
- Vlassov, A.V.; Magdaleno, S.; Setterquist, R.; Conrad, R. Exosomes: Current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Fader, C.M.; Sánchez, D.; Furlán, M.; Colombo, M.I. Induction of Autophagy Promotes Fusion of Multivesicular Bodies with Autophagic Vacuoles in K562 Cells. Traffic 2007, 9, 230–250. [Google Scholar] [CrossRef] [PubMed]
- Fader, C.M.; Colombo, M.I. Multivesicular bodies and autophagy in erythrocyte maturation. Autophagy 2006, 2, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.T. Viruses and the autophagy pathway. Virology 2015, 479, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Lussignol, M.; Queval, C.; Bernet-Camard, M.-F.; Cotte-Laffitte, J.; Beau, I.; Codogno, P.; Esclatine, A. The herpes simplex virus 1 Us11 protein inhibits autophagy through its interaction with the protein kinase PKR. J. Virol. 2013, 87, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; Alexander, D.; Tallóczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 Confers Neurovirulence by Targeting the Beclin 1 Autophagy Protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Ku, B.; Woo, J.-S.; Liang, C.; Lee, K.-H.; Hong, H.-S.; E, X.; Kim, K.-S.; Jung, J.U.; Oh, B.H. Structural and biochemical bases for the inhibition of autophagy and apoptosis by viral BCL-2 of murine γ-Herpesvirus 68. PLoS Pathog. 2008, 4, e25. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Mei, Y.; Sanishvili, R.; Levine, B.; Colbert, C.L.; Sinha, S. Targeting γ-herpesvirus 68 Bcl-2-mediated down-regulation of autophagy. J. Biol. Chem. 2014, 289, 8029–8040. [Google Scholar] [CrossRef] [PubMed]
- Gannagé, M.; Dormann, D.; Albrecht, R.; Dengjel, J.; Torossi, T.; Rämer, P.C.; Lee, M.; Strowig, T.; Arrey, F.; Conenello, G.; et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 2009, 6, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Beale, R.; Wise, H.; Stuart, A.; Ravenhill, B.J.; Digard, P.; Randow, F.; Schwartzman, L.M.; Kash, J.C.; Fodor, E.; Firth, A.E.; et al. A LC3-interacting motif in the influenza a virus M2 protein is required to subvert autophagy and maintain virion stability. Cell Host Microbe 2014, 15, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Zhang, G.; Yang, X.; Zhang, S.; Chen, L.; Yan, Q.; Xu, M.; Banerjee, A.K.; Chen, M. Phosphoprotein of human parainfluenza virus type 3 blocks autophagosome-lysosome fusion to increase virus production. Cell Host Microbe 2014, 15, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Suhy, D.A.; Giddings, T.H.; Kirkegaard, K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: An autophagy-like origin for virus-induced vesicles. J. Virol. 2000, 74, 8953–8965. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.P.; Kirkegaard, K. Modification of cellular autophagy protein LC3 by poliovirus. J. Virol. 2007, 81, 12543–12553. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.H.; Rangarajan, A. Simian virus 40 small T antigen activates AMPK and triggers autophagy to protect cancer cells from nutrient deprivation. J. Virol. 2009, 83, 8565–8574. [Google Scholar] [CrossRef] [PubMed]
- Guévin, C.; Manna, D.; Bélanger, C.; Konan, K.V.; Mak, P.; Labonté, P. Autophagy protein ATG5 interacts transiently with the hepatitis C virus RNA polymerase (NS5B) early during infection. Virology 2010, 405, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.-H.; Chen, L.-W.; Wang, W.-H.; Chang, P.-J.; Chiu, Y.-F.; Hung, C.-C.; Lin, Y.-J.; Liou, J.-Y.; Tsai, W.-J.; Hung, C.-L.; et al. Regulation of autophagic activation by Rta of Epstein-Barr virus via the extracellular signal-regulated kinase pathway. J. Virol. 2014, 88, 12133–12145. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Fang, M.; Hu, Y.; Huang, B.; Li, N.; Chang, C.; Huang, R.; Xu, X.; Yang, Z.; Chen, Z.; et al. Hepatitis B virus X protein inhibits autophagic degradation by impairing lysosomal maturation. Autophagy 2014, 10, 416–430. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Da, L.; Tang, H.; Yang, J.; Lei, Y.; Tiollais, P.; Li, T.; Zhao, M. Hepatitis B virus X protein reduces starvation-induced cell death through activation of autophagy and inhibition of mitochondrial apoptotic pathway. Biochem. Biophys. Res. Commun. 2011, 415, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Bhanja Chowdhury, J.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus upregulates Beclin1 for induction of autophagy and activates mTOR signaling. J. Virol. 2012, 86, 8705–8712. [Google Scholar] [CrossRef] [PubMed]
- Su, W.-C.; Chao, T.-C.; Huang, Y.-L.; Weng, S.-C.; Jeng, K.-S.; Lai, M.M.C. Rab5 and Class III phosphoinositide 3-kinase Vps34 are involved in Hepatitis C virus NS4B-induced autophagy. J. Virol. 2011, 85, 10561–10571. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Da, L.; Mao, Y.; Li, Y.; Li, D.; Xu, Z.; Li, F.; Wang, Y.; Tiollais, P.; Li, T.; et al. Hepatitis B virus X protein sensitizes cells to starvation-induced autophagy via up-regulation of beclin 1 expression. Hepatology 2009, 49, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-T.; Chen, G.G.; Hu, B.-G.; Zhang, Z.-Y.; Yun, J.-P.; He, M.-L.; Lai, P.B.S. Hepatitis B virus x protein induces autophagy via activating death-associated protein kinase. J. Viral. Hepat. 2014, 21, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Krejbich-Trotot, P.; Gay, B.; Li-Pat-Yuen, G.; Hoarau, J.-J.; Jaffar-Bandjee, M.-C.; Briant, L.; Gasque, P.; Denizot, M. Chikungunya triggers an autophagic process which promotes viral replication. Virol. J. 2011, 8, 432. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, Z.; Ge, X.; Guo, X.; Yang, H. Autophagy promotes the replication of encephalomyocarditis virus in host cells. Autophagy 2011, 7, 613–628. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Santarelli, R.; Farina, A.; Gonnella, R.; Lotti, L.V.; Faggioni, A.; Cirone, M. Epstein-Barr virus blocks the autophagic flux and appropriates the autophagic machinery to enhance viral replication. J. Virol. 2014, 88, 12715–12726. [Google Scholar] [CrossRef] [PubMed]
- El-Hage, N.; Rodriguez, M.; Dever, S.M.; Masvekar, R.R.; Gewirtz, D.A.; Shacka, J.J. HIV-1 and morphine regulation of autophagy in microglia: Limited interactions in the context of HIV-1 infection and opioid abuse. J. Virol. 2015, 89, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Van Grol, J.; Subauste, C.; Andrade, R.M.; Fujinaga, K.; Nelson, J.; Subauste, C.S. HIV-1 inhibits autophagy in bystander macrophage/monocytic cells through Src-Akt and STAT3. PLoS ONE 2010, 5, e11733. [Google Scholar] [CrossRef] [PubMed]
- Sardo, L.; Iordanskiy, S.; Klase, Z.; Kashanchi, F. HIV-1 Nef blocks autophagy in human astrocytes. Cell Cycle 2015, 14, 3781–3782. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gao, Y.; Tan, J.; Devadas, K.; Ragupathy, V.; Takeda, K.; Zhao, J.; Hewlett, I. HIV-1 and HIV-2 infections induce autophagy in Jurkat and CD4+ T cells. Cell Signal. 2012, 24, 1414–1419. [Google Scholar] [CrossRef] [PubMed]
- Espert, L.; Denizot, M.; Grimaldi, M.; Robert-Hebmann, V.; Gay, B.; Varbanov, M.; Codogno, P.; Biard-Piechaczyk, M.; Maldarelli, F.; Sato, H.; et al. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J. Clin. Investig. 2006, 116, 2161–2172. [Google Scholar] [CrossRef] [PubMed]
- Varbanov, M.; Blanco, J.; García, E.; Curriu, M.; Sagnier, S.; Espert, L.; Denizot, M.; Robert, V.; Biard-Piechaczyk, M.; Mamoun, R.Z. HIV-1 gp41 fusogenic function triggers autophagy in uninfected cells. Autophagy 2008, 4, 998–1008. [Google Scholar]
- Bruno, A.P.; De Simone, F.I.; Iorio, V.; De Marco, M.; Khalili, K.; Sariyer, I.K.; Capunzo, M.; Nori, S.L.; Rosati, A. HIV-1 Tat protein induces glial cell autophagy through enhancement of BAG3 protein levels. Cell Cycle 2014, 13, 3640–3644. [Google Scholar] [CrossRef] [PubMed]
- Fields, J.; Dumaop, W.; Elueteri, S.; Campos, S.; Serger, E.; Trejo, M.; Kosberg, K.; Adame, A.; Spencer, B.; Rockenstein, E.; et al. HIV-1 Tat alters neuronal autophagy by modulating autophagosome fusion to the lysosome: Implications for HIV-associated neurocognitive disorders. J. Neurosci. 2015, 35, 1921–1938. [Google Scholar] [CrossRef] [PubMed]
- Sagnier, S.; Daussy, C.F.; Borel, S.; Robert-Hebmann, V.; Faure, M.; Blanchet, F.P.; Beaumelle, B.; Biard-Piechaczyk, M.; Espert, L. Autophagy restricts HIV-1 infection by selectively degrading Tat in CD4+ T lymphocytes. J. Virol. 2015, 89, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Nardacci, R.; Amendola, A.; Ciccosanti, F.; Corazzari, M.; Esposito, V.; Vlassi, C.; Taibi, C.; Fimia, G.M.; Del Nonno, F.; Ippolito, G.; et al. Autophagy plays an important role in the containment of HIV-1 in nonprogressor-infected patients. Autophagy 2014, 10, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Borel, S.; Robert-Hebmann, V.; Alfaisal, J.; Jain, A.; Faure, M.; Espert, L.; Chaloin, L.; Paillart, J.-C.; Johansen, T.; Biard-Piechaczyk, M. HIV-1 viral infectivity factor interacts with light chain 3 and inhibits autophagy. AIDS 2014, 29, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Rawat, P.; Bruckman, R.S.; Spector, S.A. Human immunodeficiency virus type 1 Nef inhibits autophagy through transcription factor EB sequestration. PLoS Pathog. 2015, 11, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Li, J.C.; Au, K.; Fang, J.; Yim, H.C.; Chow, K.; Ho, P.; Lau, A.S. HIV-1 trans-activator protein dysregulates IFN-γ signaling and contributes to the suppression of autophagy induction. AIDS 2011, 25, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Daussy, C.F.; Beaumelle, B.; Espert, L. Autophagy restricts HIV-1 infection. Oncotarget 2015, 6, 20752–20753. [Google Scholar] [CrossRef] [PubMed]
- Saribas, A.S.; Khalili, K.; Sariyer, I.K. Dysregulation of autophagy by HIV-1 Nef in human astrocytes. Cell Cycle 2015, 14, 2899–2904. [Google Scholar] [CrossRef] [PubMed]
- Dinkins, C.; Pilli, M.; Kehrl, J.H. Roles of autophagy in HIV infection. Immunol. Cell Biol. 2015, 93, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo-Useros, N.; Naranjo-Gómez, M.; Archer, J.; Hatch, S.C.; Erkizia, I.; Blanco, J.; Borràs, F.E.; Puertas, M.C.; Connor, J.H.; Fernández-Figueras, M.T.; et al. Capture and transfer of HIV-1 particles by mature dendritic cells converges with the exosome-dissemination pathway. Blood 2009, 113, 2732–2741. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Wu, N.; Gan, X.; Yan, W.; Morrell, J.C.; Gould, S.J. Higher-order oligomerization targets plasma membrane proteins and HIV Gag to exosomes. PLoS Biol. 2007, 5. [Google Scholar] [CrossRef] [PubMed]
- Garrus, J.E.; von Schwedler, U.K.; Pornillos, O.W.; Morham, S.G.; Zavitz, K.H.; Wang, H.E.; Wettstein, D.A.; Stray, K.M.; Côté, M.; Rich, R.L.; et al. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 2001, 107, 55–65. [Google Scholar] [CrossRef]
- Martin-Serrano, J.; Zang, T.; Bieniasz, P.D. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 2001, 7, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liu, X.; Li, Z.; Zhan, P.; De Clercq, E. TSG101: A novel anti-HIV-1 drug target. Curr. Med. Chem. 2010, 17, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Raymond, A.D.; Campbell-Sims, T.C.; Khan, M.; Lang, M.; Huang, M.B.; Bond, V.C.; Powell, M.D. HIV type 1 Nef is released from infected cells in CD45+ microvesicles and is present in the plasma of HIV-infected individuals. AIDS Res. Hum. Retrovir. 2011, 27, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Columba Cabezas, S.; Federico, M. Sequences within RNA coding for HIV-1 Gag p17 are efficiently targeted to exosomes. Cell Microbiol. 2013, 15, 412–429. [Google Scholar] [CrossRef] [PubMed]
- Bernard, M.A.; Zhao, H.; Yue, S.C.; Anandaiah, A.; Koziel, H.; Tachado, S.D. Novel HIV-1 miRNAs Stimulate TNFα Release in Human Macrophages via TLR8 Signaling Pathway. PLoS ONE 2014, 9, e106006. [Google Scholar] [CrossRef] [PubMed]
- Campbell, T.D.; Khan, M.; Huang, M.-B.; Bond, V.C.; Powell, M.D. HIV-1 Nef protein is secreted into vesicles that can fuse with target cells and virions. Ethn. Dis. 2008, 18 (Suppl. 2), 14–19. [Google Scholar]
- Narayanan, A.; Iordanskiy, S.; Das, R.; Van Duyne, R.; Santos, S.; Jaworski, E.; Guendel, I.; Sampey, G.; Dalby, E.; Iglesias-Ussel, M.; et al. Exosomes derived from HIV-1-infected cells contain trans-activation response element RNA. J. Biol. Chem. 2013, 288, 20014–20033. [Google Scholar] [CrossRef] [PubMed]
- Lenassi, M.; Cagney, G.; Liao, M.; Vaupotič, T.; Bartholomeeusen, K.; Cheng, Y.; Krogan, N.J.; Plemenitaš, A.; Peterlin, B.M. HIV Nef is Secreted in Exosomes and Triggers Apoptosis in Bystander CD4+ T Cells. Traffic 2010, 11, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Sampey, G.C.; Saifuddin, M.; Schwab, A.; Barclay, R.; Punya, S.; Chung, M.-C.; Hakami, R.M.; Zadeh, M.A.; Lepene, B.; Klase, Z.A.; et al. Exosomes from HIV-1-infected cells stimulate production of pro-inflammatory cytokines through trans-activating response (TAR) RNA. J. Biol. Chem. 2016, 291, 1251–1266. [Google Scholar] [CrossRef] [PubMed]
- Baixauli, F.; López-Otín, C.; Mittelbrunn, M. Exosomes and autophagy: Coordinated mechanisms for the maintenance of cellular fitness. Front. Immunol. 2014, 5, 403. [Google Scholar] [CrossRef] [PubMed]
- Toborek, M.; Lee, Y.W.; Pu, H.; Malecki, A.; Flora, G.; Garrido, R.; Hennig, B.; Bauer, H.-C.; Nath, A. HIV-Tat protein induces oxidative and inflammatory pathways in brain endothelium. J. Neurochem. 2003, 84, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Alirezaei, M.; Kiosses, W.B.; Fox, H.S. Decreased neuronal autophagy in HIV dementia: A mechanism of indirect neurotoxicity. Autophagy 2008, 4, 963–966. [Google Scholar] [CrossRef] [PubMed]
- Alirezaei, M.; Kiosses, W.B.; Flynn, C.T.; Brady, N.R.; Fox, H.S.; Komatsu, M.; Ueno, T.; Waguri, S.; Uchiyama, Y.; Kominami, E.; et al. Disruption of neuronal autophagy by infected microglia results in neurodegeneration. PLoS ONE 2008, 3, e2906. [Google Scholar] [CrossRef] [PubMed]
- Su, P.; Zhang, J.; Wang, D.; Zhao, F.; Cao, Z.; Aschner, M.; Luo, W. The role of autophagy in modulation of neuroinflammation in microglia. Neuroscience 2016, 319, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Di Malta, C.; Fryer, J.D.; Settembre, C.; Ballabio, A. Autophagy in astrocytes: A novel culprit in lysosomal storage disorders. Autophagy 2012, 8, 1871–1872. [Google Scholar] [CrossRef] [PubMed]
- Kramer-Hämmerle, S.; Rothenaigner, I.; Wolff, H.; Bell, J.E.; Brack-Werner, R. Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. Virus Res. 2005, 111, 194–213. [Google Scholar] [CrossRef] [PubMed]
- Gorry, P.R.; Ong, C.; Thorpe, J.; Bannwarth, S.; Thompson, K.A.; Gatignol, A.; Vesselingh, S.L.; Purcell, D.F. J. Astrocyte infection by HIV-1: Mechanisms of restricted virus replication, and role in the pathogenesis of HIV-1-associated dementia. Curr. HIV Res. 2003, 1, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Mehla, R.; Chauhan, A. HIV-1 differentially modulates autophagy in neurons and astrocytes. J. Neuroimmunol. 2015, 285, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Fu, M.; Kumar, S.; Kumar, A. Methamphetamine potentiates HIV-1 gp120-mediated autophagy via Beclin-1 and Atg5/7 as a pro-survival response in astrocytes. Cell Death Dis. 2016, 7, e2425. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Lapierre, J.; Ojha, C.R.; Estrada-Bueno, H.; Dever, S.M.; Gewirtz, D.A.; Kashanchi, F.; El-Hage, N. Importance of autophagy in mediating HIV and morphine-induced metabolic dysfunction and inflammation in human astrocytes. Viruses 2017. under review. [Google Scholar]
- Tang, Y.; Hamed, H.A.; Cruickshanks, N.; Fisher, P.B.; Grant, S.; Dent, P. Obatoclax and lapatinib interact to induce toxic autophagy through NOXA. Mol. Pharmacol. 2012, 81, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zou, Z.; Becker, N.; Anderson, M.; Sumpter, R.; Xiao, G.; Kinch, L.; Koduru, P.; Christudass, C.S.; Veltri, R.W.; et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 2013, 154, 1269–1284. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Kepp, O.; Michaud, M.; Martins, I.; Minoux, H.; Mativier, D.; Maiuri, M.C.; Kroemer, R.T.; Kroemer, G. Association and dissociation of autophagy, apoptosis and necrosis by systematic chemical study. Oncogene 2011, 30, 4544–4556. [Google Scholar] [CrossRef] [PubMed]
- Scharl, M.; Rogler, G. Inflammatory bowel disease: Dysfunction of autophagy? Dig. Dis. 2012, 30, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.-F.; Guo, B.-S.; Liu, Y.-C.; Wu, C.-C.; Yang, C.-H.; Tsai, K.-J.; Shen, C.-K.J. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc. Natl. Acad. Sci. USA 2012, 109, 15024–15029. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.D.O.; Ladha, S.; Ehrnhoefer, D.E.; Hayden, M.R. Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 2015, 38, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Spector, S.A. Autophagy induction by vitamin D inhibits both Mycobacterium tuberculosis and human immunodeficiency virus type 1. Autophagy 2012, 8, 1523–1525. [Google Scholar] [CrossRef] [PubMed]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013, 494, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Rai, P.; Plagov, A.; Kumar, D.; Pathak, S.; Ayasolla, K.R.; Chawla, A.K.; Mathieson, P.W.; Saleem, M.A.; Husain, M.; Malhotra, A.; et al. Rapamycin-induced modulation of HIV gene transcription attenuates progression of HIVAN. Exp. Mol. Pathol. 2013, 94, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Heredia, A.; Latinovic, O.; Gallo, R.C.; Melikyan, G.; Reitz, M.; Le, N.; Redfield, R.R. Reduction of CCR5 with low-dose rapamycin enhances the antiviral activity of vicriviroc against both sensitive and drug-resistant HIV-1. Proc. Natl. Acad. Sci. USA 2008, 105, 20476–20481. [Google Scholar] [CrossRef] [PubMed]
- Heredia, A.; Le, N.; Gartenhaus, R.B.; Sausville, E.; Medina-Moreno, S.; Zapata, J.C.; Davis, C.; Gallo, R.C.; Redfield, R.R. Targeting of mTOR catalytic site inhibits multiple steps of the HIV-1 lifecycle and suppresses HIV-1 viremia in humanized mice. Proc. Natl. Acad. Sci. USA 2015, 112, 9412–9417. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. FDA approves Rapamune to treat LAM, a Very Rare Lung Disease. 2015. Available online: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm448523.htm (accessed on 23-04-2017).
- Salmon, A.B. About-face on the metabolic side effects of rapamycin. Oncotarget 2015, 6, 2585–2586. [Google Scholar] [CrossRef] [PubMed]
- Feldman, M.E.; Apsel, B.; Uotila, A.; Loewith, R.; Knight, Z.A.; Ruggero, D.; Shokat, K.M. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009, 7, e38. [Google Scholar] [CrossRef] [PubMed]
- García-Martínez, J.M.; Moran, J.; Clarke, R.G.; Gray, A.; Cosulich, S.C.; Chresta, C.M.; Alessi, D.R. Ku-0063794 is a specific inhibitor of the mammalian target of rapamycin (mTOR). Biochem. J. 2009, 421, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Kwiatkowski, D.J. Equivalent benefit of rapamycin and a potent mTOR ATP-competitive inhibitor, MLN0128 (INK128), in a mouse model of tuberous sclerosis. Mol. Cancer Res. 2013, 11, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [PubMed]
- Buzzai, M.; Jones, R.G.; Amaravadi, R.K.; Lum, J.J.; DeBerardinis, R.J.; Zhao, F.; Viollet, B.; Thompson, C.B. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007, 67, 6745–6752. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Spector, S.A. Vitamin D inhibits human immunodeficiency virus type 1 and Mycobacterium tuberculosis infection in macrophages through the induction of autophagy. PLoS Pathog. 2012, 8, e1002689. [Google Scholar] [CrossRef] [PubMed]
- Dao, C.N.; Patel, P.; Overton, E.T.; Rhame, F.; Pals, S.L.; Johnson, C.; Bush, T.; Brooks, J.T. Low vitamin D among HIV-infected adults: Prevalence of and risk factors for low vitamin D Levels in a cohort of HIV-infected adults and comparison to prevalence among adults in the US general population. Clin. Infect. Dis. 2011, 52, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Spector, S.A. Hormonally active vitamin D3 (1alpha,25-dihydroxycholecalciferol) triggers autophagy in human macrophages that inhibits HIV-1 infection. J. Biol. Chem. 2011, 286, 18890–18902. [Google Scholar] [CrossRef] [PubMed]
- Pauza, C.D.; Kornbluth, R.; Emau, P.; Richman, D.D.; Deftos, L.J. Vitamin D3 compounds regulate human immunodeficiency virus type 1 replication in U937 monoblastoid cells and in monocyte-derived macrophages. J. Leukoc. Biol. 1993, 53, 157–164. [Google Scholar] [PubMed]
- Yuk, J.-M.; Shin, D.-M.; Lee, H.-M.; Yang, C.-S.; Jin, H.S.; Kim, K.-K.; Lee, Z.-W.; Lee, S.-H.; Kim, J.-M.; Jo, E.-K. Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe 2009, 6, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Spector, S.A. Vitamin D and HIV: Letting the sun shine in. Top. Antivir. Med. 2011, 19, 6–10. [Google Scholar] [PubMed]
- McComsey, G.A.; Kendall, M.A.; Tebas, P.; Swindells, S.; Hogg, E.; Alston-Smith, B.; Suckow, C.; Gopalakrishnan, G.; Benson, C.; Wohl, D.A. Alendronate with calcium and vitamin D supplementation is safe and effective for the treatment of decreased bone mineral density in HIV. AIDS 2007, 21, 2473–2482. [Google Scholar] [CrossRef] [PubMed]
- Mardones, P.; Rubinsztein, D.C.; Hetz, C. Mystery solved: Trehalose kickstarts autophagy by blocking glucose transport. Sci. Signal. 2016, 9, fs2. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem. 2007, 282, 5641–5652. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Korolchuk, V.I.; Renna, M.; Imarisio, S.; Fleming, A.; Williams, A.; Garcia-Arencibia, M.; Rose, C.; Luo, S.; Underwood, B.R.; et al. Complex inhibitory effects of nitric oxide on autophagy. Mol. Cell. 2011, 43, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Torre, D.; Pugliese, A.; Speranza, F. Role of nitric oxide in HIV-1 infection: Friend or foe? Lancet Infect. Dis. 2002, 2, 273–280. [Google Scholar] [CrossRef]
- Chen, F.; Lu, Y.; Castranova, V.; Rojanasakul, Y.; Miyahara, K.; Shizuta, Y.; Vallyathan, V.; Shi, X.; Demers, L.M. Nitric oxide inhibits HIV tat-induced NF-kappaB activation. Am. J. Pathol. 1999, 155, 275–284. [Google Scholar] [CrossRef]
- Shirakawa, K.; Chavez, L.; Hakre, S.; Calvanese, V.; Verdin, E. Reactivation of latent HIV by histone deacetylase inhibitors. Trends Microbiol. 2013, 21, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Bruckman, R.S.; Chu, Y.-L.; Spector, S.A. Autophagy induction by histone deacetylase inhibitors inhibits HIV type 1. J. Biol. Chem. 2015, 290, 5028–5040. [Google Scholar] [CrossRef] [PubMed]
- Jeng, M.Y.; Ali, I.; Ott, M. Manipulation of the host protein acetylation network by human immunodeficiency virus type 1. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 314–325. [Google Scholar] [PubMed]
- Margolis, D.M. Histone deacetylase inhibitors and HIV latency. Curr. Opin. HIV AIDS 2011, 6, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Marban, C.; Forouzanfar, F.; Ait-Ammar, A.; Fahmi, F.; El Mekdad, H.; Daouad, F.; Rohr, O.; Schwartz, C. Targeting the brain reservoirs: Toward an HIV cure. Front. Immunol. 2016, 7, 397. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell. Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Spector, S.A. Inhibition of human immunodeficiency virus type-1 through autophagy. Curr. Opin. Microbiol. 2013, 16, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Harhaji-Trajkovic, L.; Arsikin, K.; Kravic-Stevovic, T.; Petricevic, S.; Tovilovic, G.; Pantovic, A.; et al. Chloroquine-mediated lysosomal dysfunction enhances the anticancer effect of nutrient deprivation. Pharm. Res. 2012, 29, 2249–2263. [Google Scholar] [CrossRef] [PubMed]
- Savarino, A.; Boelaert, J.R.; Cassone, A.; Majori, G.; Cauda, R. Effects of chloroquine on viral infections: An old drug against today’s diseases. Lancet Infect. Dis. 2003, 3, 722–727. [Google Scholar] [CrossRef]
- Neely, M.; Kalyesubula, I.; Bagenda, D.; Myers, C.; Olness, K. Effect of chloroquine on human immunodeficiency virus (HIV) vertical transmission. Afr. Health Sci. 2003, 3, 61–67. [Google Scholar] [PubMed]
- Boelaert, J.R.; Sperber, K.; Piette, J. The additive in vitro anti-HIV-1 effect of chloroquine, when combined with zidovudine and hydroxyurea. Biochem. Pharmacol. 2001, 61, 1531–1535. [Google Scholar] [CrossRef]
- Yang, Y.; Hu, L.; Zheng, H.; Mao, C.; Hu, W.; Xiong, K.; Wang, F.; Liu, C. Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol. Sin. 2013, 34, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Yuan, N.; Song, L.; Zhang, S.; Lin, W.; Cao, Y.; Xu, F.; Fang, Y.; Wang, Z.; Zhang, H.; Li, X.; et al. Bafilomycin A1 targets both autophagy and apoptosis pathways in pediatric B-cell acute lymphoblastic leukemia. Haematologica 2015, 100, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Kyei, G.B.; Dinkins, C.; Davis, A.S.; Roberts, E.; Singh, S.B.; Dong, C.; Wu, L.; Kominami, E.; Ueno, T.; Yamamoto, A.; et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009, 186, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Münz, C. Beclin-1 targeting for viral immune escape. Viruses 2011, 3, 1166–1178. [Google Scholar] [CrossRef] [PubMed]
- Raben, N.; Shea, L.; Hill, V.; Plotz, P. Monitoring autophagy in lysosomal storage disorders. Methods Enzymol. 2009, 453, 417–449. [Google Scholar] [PubMed]
- Eekels, J.J.; Sagnier, S.; Geerts, D.; Jeeninga, R.E.; Biard-Piechaczyk, M.; Berkhout, B.; Mizushima, N.; Klionsky, D.; Qu, X.; Yu, J.; et al. Inhibition of HIV-1 replication with stable RNAi-mediated knockdown of autophagy factors. Virol. J. 2012, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Ireland, J.M.; Unanue, E.R. Autophagy in antigen-presenting cells results in presentation of citrullinated peptides to CD4 T cells. J. Exp. Med. 2011, 208, 2625–2632. [Google Scholar] [CrossRef] [PubMed]
- Andersen, A.N.; Landsverk, O.J.; Simonsen, A.; Bogen, B.; Corthay, A.; Øynebråten, I. Coupling of HIV-1 antigen to the selective autophagy receptor SQSTM1/p62 promotes T-Cell-mediated immunity. Front. Immunol. 2016, 7, 167. [Google Scholar] [CrossRef] [PubMed]
- Mbita, Z.; Hull, R.; Dlamini, Z. Human immunodeficiency virus-1 (HIV-1)-mediated apoptosis: New therapeutic targets. Viruses 2014, 6, 3181–3227. [Google Scholar] [CrossRef] [PubMed]
- Mousseau, G.; Kessing, C.F.; Fromentin, R.; Trautmann, L.; Chomont, N.; Valente, S.T. The Tat inhibitor didehydro-cortistatin a prevents HIV-1 reactivation from latency. MBio 2015, 6, e00465. [Google Scholar] [CrossRef] [PubMed]
- Smithgall, T.E.; Thomas, G. Small molecule inhibitors of the HIV-1 virulence factor, Nef. Drug Discov. Today Technol. 2013, 10, e523–e529. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.K.; Griffiths, C.; Lea, S.M.; James, W. Structural characterization of an anti-gp120 RNA aptamer that neutralizes R5 strains of HIV-1. RNA 2005, 11, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Neff, C.P.; Zhou, J.; Remling, L.; Kuruvilla, J.; Zhang, J.; Li, H.; Smith, D.D.; Swiderski, P.; Rossi, J.J.; Akkina, R. An aptamer-siRNA chimera suppresses HIV-1 viral loads and protects from helper CD4+ T Cell decline in humanized mice. Sci Transl. Med. 2011, 3, 66ra6. [Google Scholar] [CrossRef] [PubMed]
- Giardiello, M.; Liptrott, N.J.; McDonald, T.O.; Moss, D.; Siccardi, M.; Martin, P.; Smith, D.; Gurjar, R.; Rannard, S.P.; Owen, A.; et al. Accelerated oral nanomedicine discovery from miniaturized screening to clinical production exemplified by paediatric HIV nanotherapies. Nat. Commun. 2016, 7, 13184. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, A.; Jayant, R.D.; Nair, M. Advancements in nano-enabled therapeutics for neuroHIV management. Int. J. Nanomed. 2016, 11, 4317–4325. [Google Scholar] [CrossRef] [PubMed]
- Nair, M. Personalized NanoMedicine: Towards new Theranostic Approach. J. Pers. Nanomed. 2015, 1, 1–2. [Google Scholar] [PubMed]
- Rodriguez, M.; Kaushik, A.; Lapierre, J.; Dever, S.M.; El-Hage, N.; Nair, M. electro-magnetic nano-particle bound Beclin1 siRNA crosses the blood–brain barrier to attenuate the inflammatory effects of HIV-1 infection in vitro. J. Neuroimmune Pharmacol. 2017, 12, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Lapierre, J.; Ojha, C.R.; Kaushik, A.; Batrakova, E.; Kashanchi, F.; Dever, S.M.; Nair, M.; El-Hage, N. Intranasal drug delivery of small interfering RNA targeting Beclin1 encapsulated with polyethylenimine (PEI) in mouse brain to achieve HIV attenuation. Sci. Rep. 2017, 7, 1862. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Guo, D.; Dash, P.K.; Araínga, M.; Wiederin, J.L.; Haverland, N.A.; Knibbe-Hollinger, J.; Martinez-Skinner, A.; Ciborowski, P.; Goodfellow, V.S.; et al. The mixed lineage kinase-3 inhibitor URMC-099 improves therapeutic outcomes for long-acting antiretroviral therapy. Nanomed. Nanotechnol. 2016, 12, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Marker, D.F.; Tremblay, M.-E.; Puccini, J.M.; Barbieri, J.; Gantz Marker, M.A.; Loweth, C.J.; Muly, E.C.; Lu, S.-M.; Goodfellow, V.S.; Dewhurst, S.; et al. The new small-molecule mixed-lineage kinase 3 inhibitor URMC-099 is neuroprotective and anti-inflammatory in models of human immunodeficiency virus-associated neurocognitive disorders. J. Neurosci. 2013, 33, 9998–10010. [Google Scholar] [CrossRef] [PubMed]
- Gnanadhas, D.P.; Dash, P.K.; Sillman, B.; Bade, A.N.; Lin, Z.; Palandri, D.L.; Gautam, N.; Alnouti, Y.; Gelbard, H.A.; Mcmillan, J.; et al. Autophagy facilitates macrophage depots of sustained-release nanoformulated antiretroviral drugs. J. Clin. Investig. 2017, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Richman, D.D. Introduction: Challenges to finding a cure for HIV infection. Curr. Opin. HIV AIDS 2011, 6, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Churchill, M.J.; Deeks, S.G.; Margolis, D.M.; Siliciano, R.F.; Swanstrom, R. HIV reservoirs: What, where and how to target them. Nat. Rev. Microbiol. 2015, 14, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Coley, W.; Kehn-Hall, K.; Van Duyne, R.; Kashanchi, F. Novel HIV-1 therapeutics through targeting altered host cell pathways. Expert Opin. Biol. Ther. 2009, 9, 1369–1382. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Spector, S.A. Human immunodeficiency virus type-1 infection inhibits autophagy. AIDS 2008, 22, 695–699. [Google Scholar] [CrossRef] [PubMed]
- LeBrasseur, N. HIV uses autophagy for its own means. J. Cell Biol. 2009, 186, 165. [Google Scholar] [CrossRef]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ojha, C.R.; Lapierre, J.; Rodriguez, M.; Dever, S.M.; Zadeh, M.A.; DeMarino, C.; Pleet, M.L.; Kashanchi, F.; El-Hage, N. Interplay between Autophagy, Exosomes and HIV-1 Associated Neurological Disorders: New Insights for Diagnosis and Therapeutic Applications. Viruses 2017, 9, 176. https://doi.org/10.3390/v9070176
Ojha CR, Lapierre J, Rodriguez M, Dever SM, Zadeh MA, DeMarino C, Pleet ML, Kashanchi F, El-Hage N. Interplay between Autophagy, Exosomes and HIV-1 Associated Neurological Disorders: New Insights for Diagnosis and Therapeutic Applications. Viruses. 2017; 9(7):176. https://doi.org/10.3390/v9070176
Chicago/Turabian StyleOjha, Chet Raj, Jessica Lapierre, Myosotys Rodriguez, Seth M. Dever, Mohammad Asad Zadeh, Catherine DeMarino, Michelle L. Pleet, Fatah Kashanchi, and Nazira El-Hage. 2017. "Interplay between Autophagy, Exosomes and HIV-1 Associated Neurological Disorders: New Insights for Diagnosis and Therapeutic Applications" Viruses 9, no. 7: 176. https://doi.org/10.3390/v9070176