
Analysis of Class I Major Histocompatibility Complex Gene Transcription in Human Tumors Caused by Human Papillomavirus Infection

 , and
, and 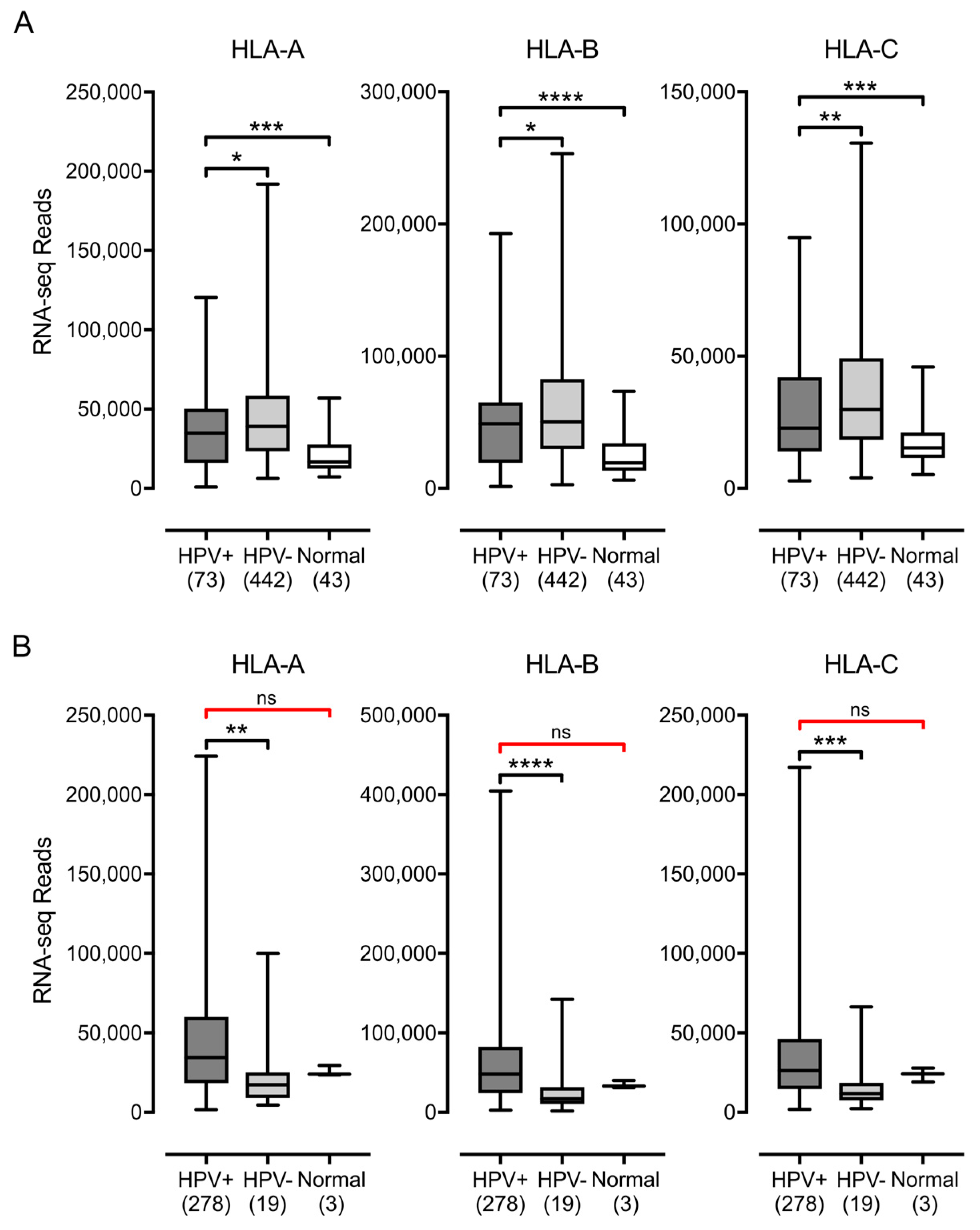{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. RNA Expression Comparisons and Statistical Analysis
2.2. Somatic Mutation Frequency Comparisons and Statistical Analysis
3. Results
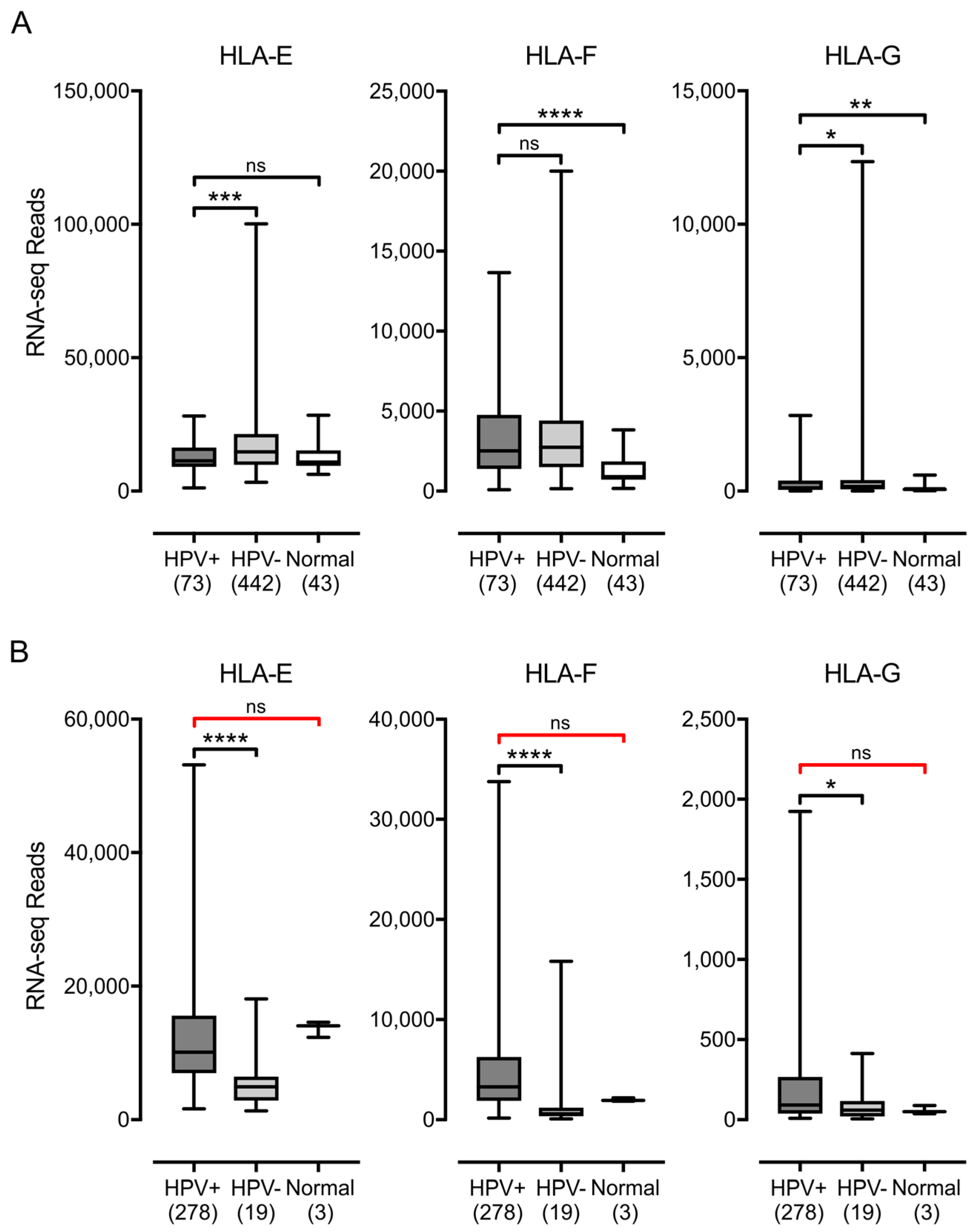
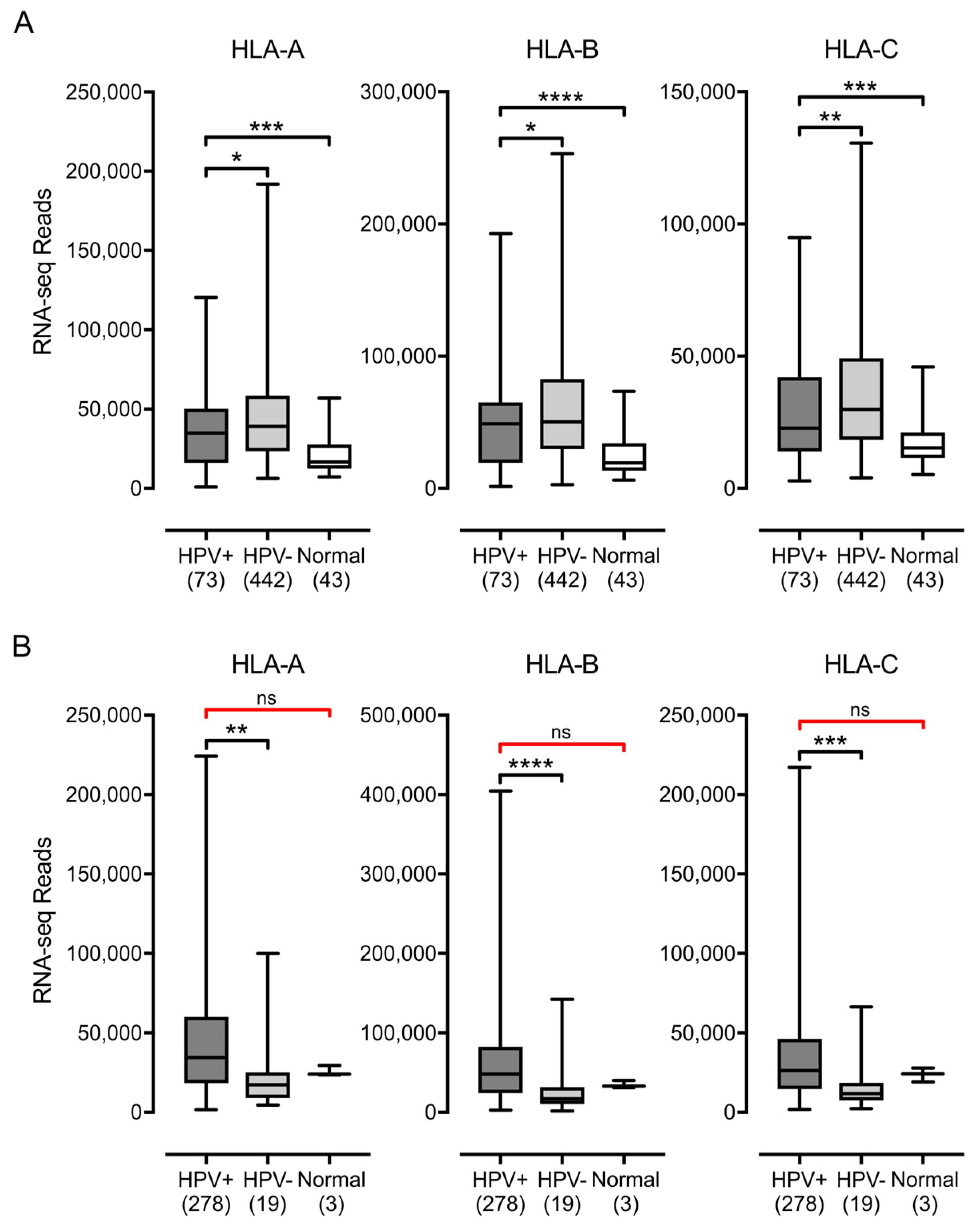
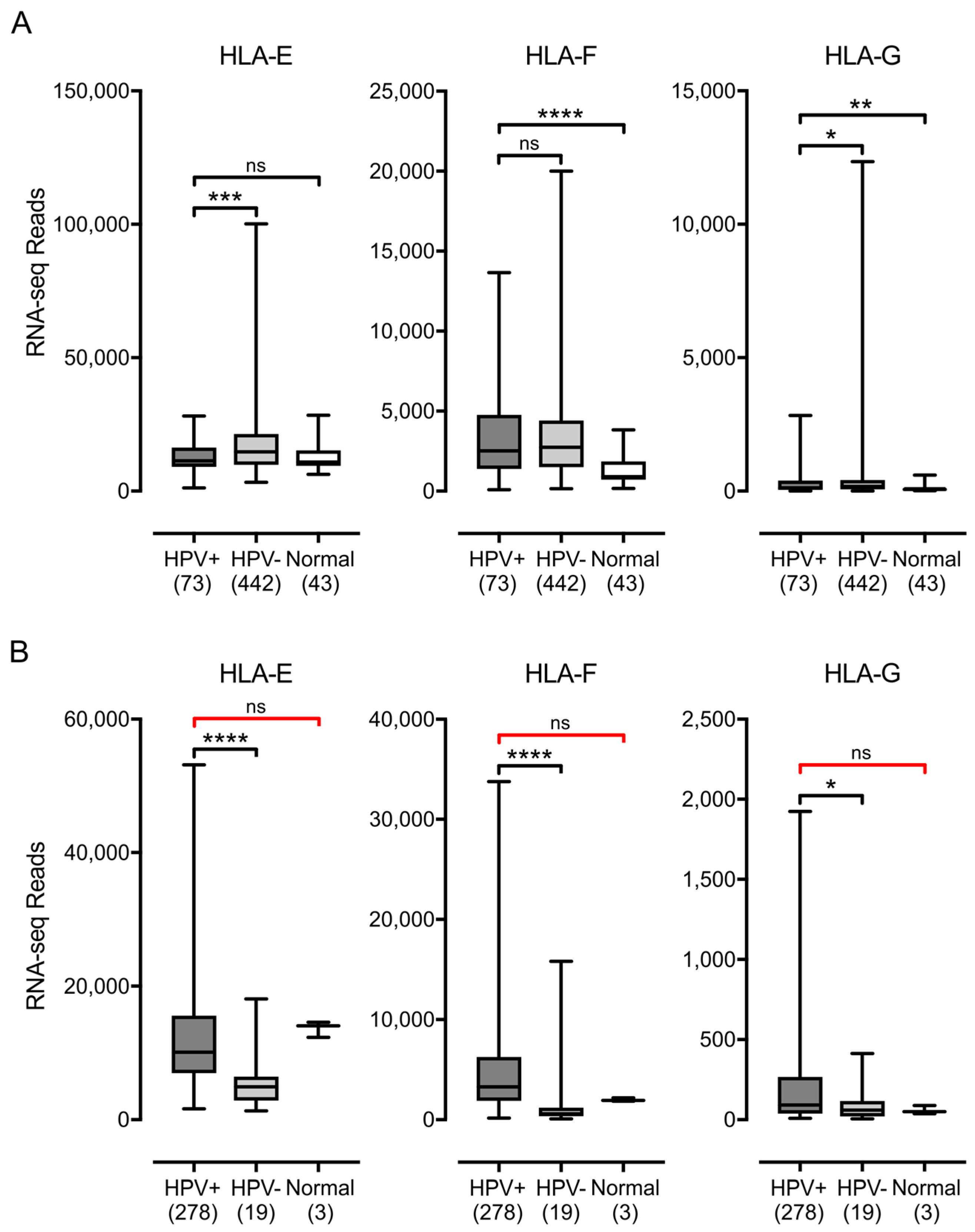
3.1. Impact of HPV Status on MHC-I Heavy Chain Expression in Human Tumors
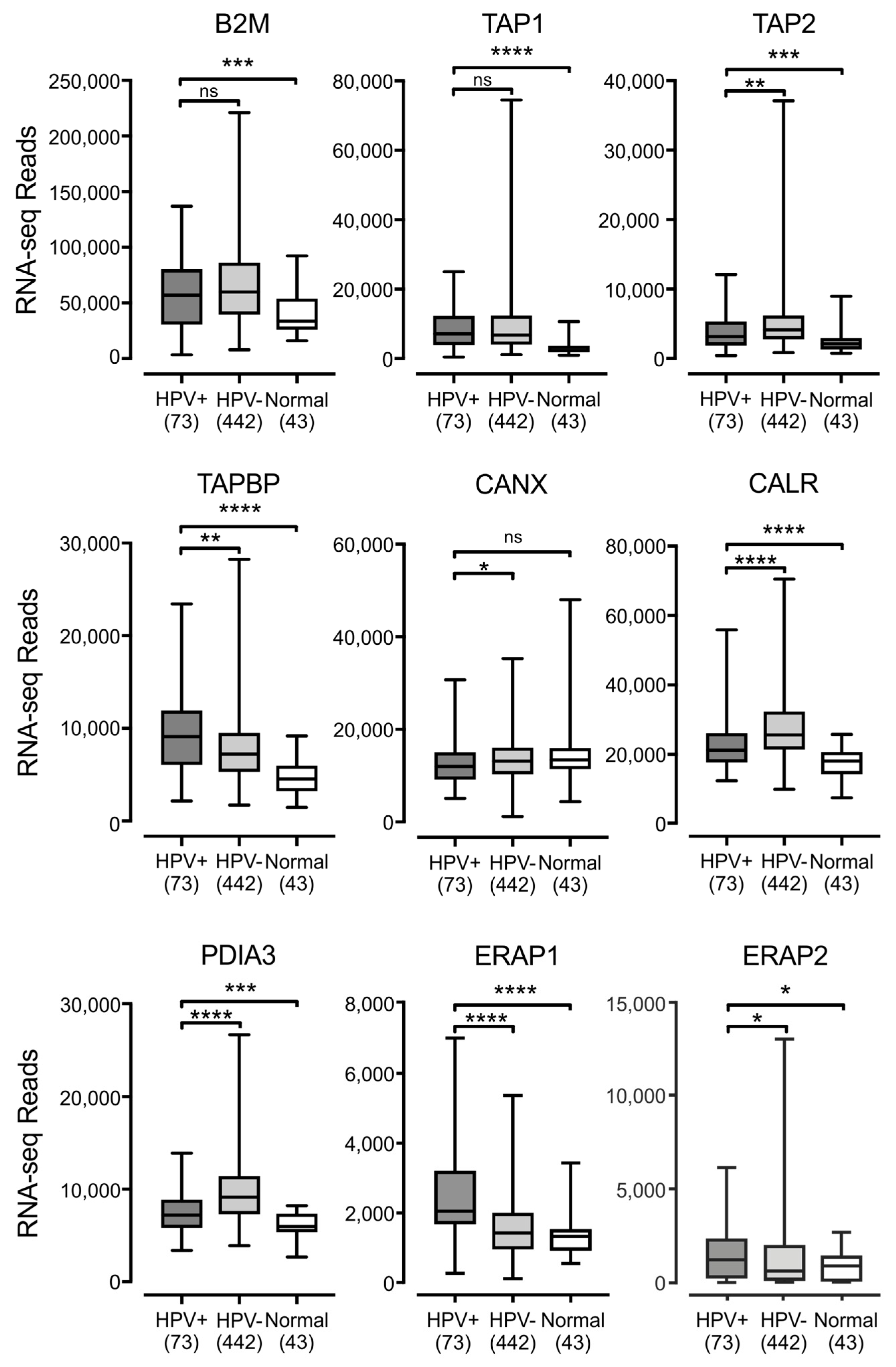
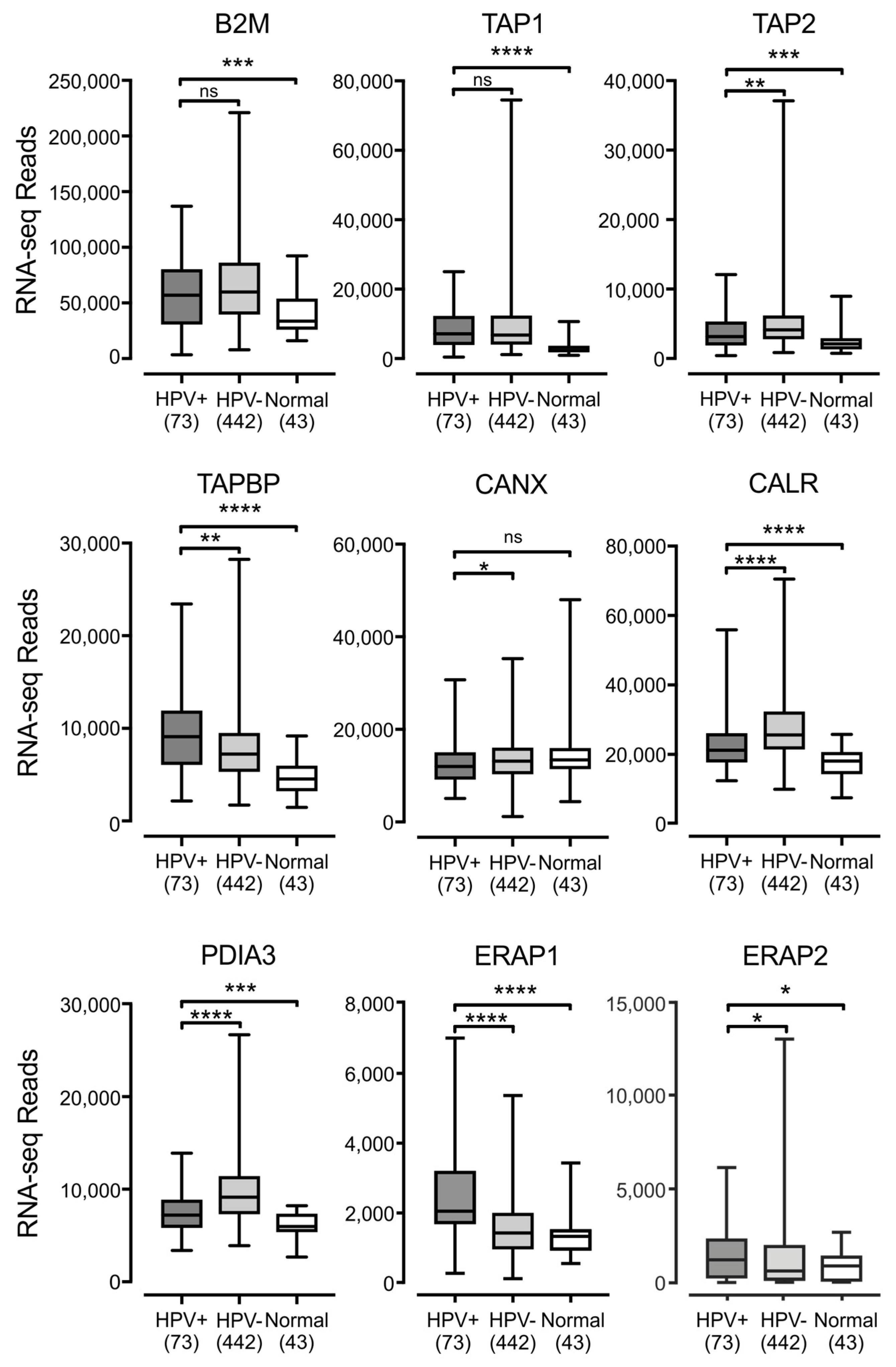
3.2. Impact of HPV Status on the Expression of other Components of the Antigen Presentation Apparatus in Human Tumors
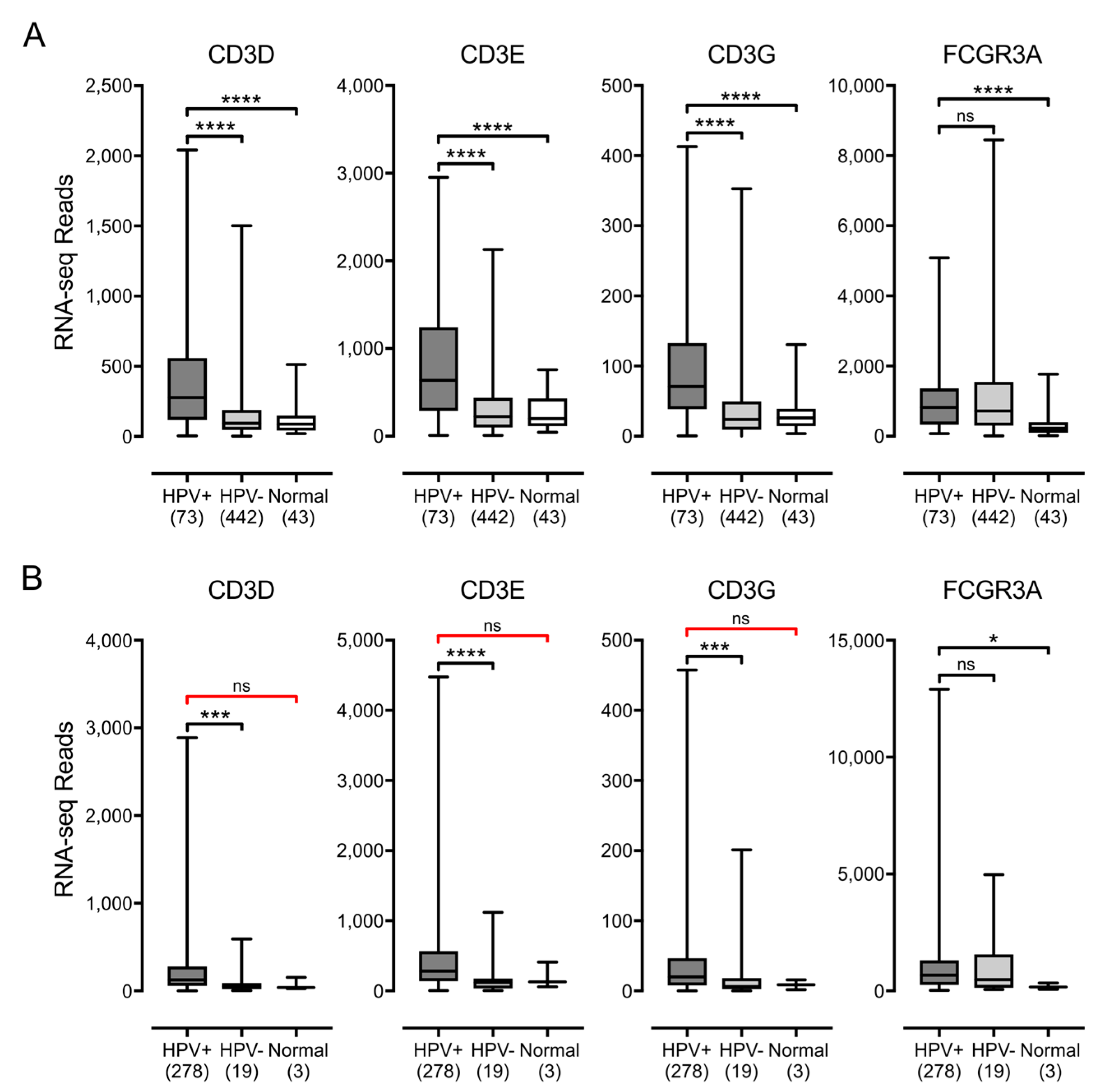
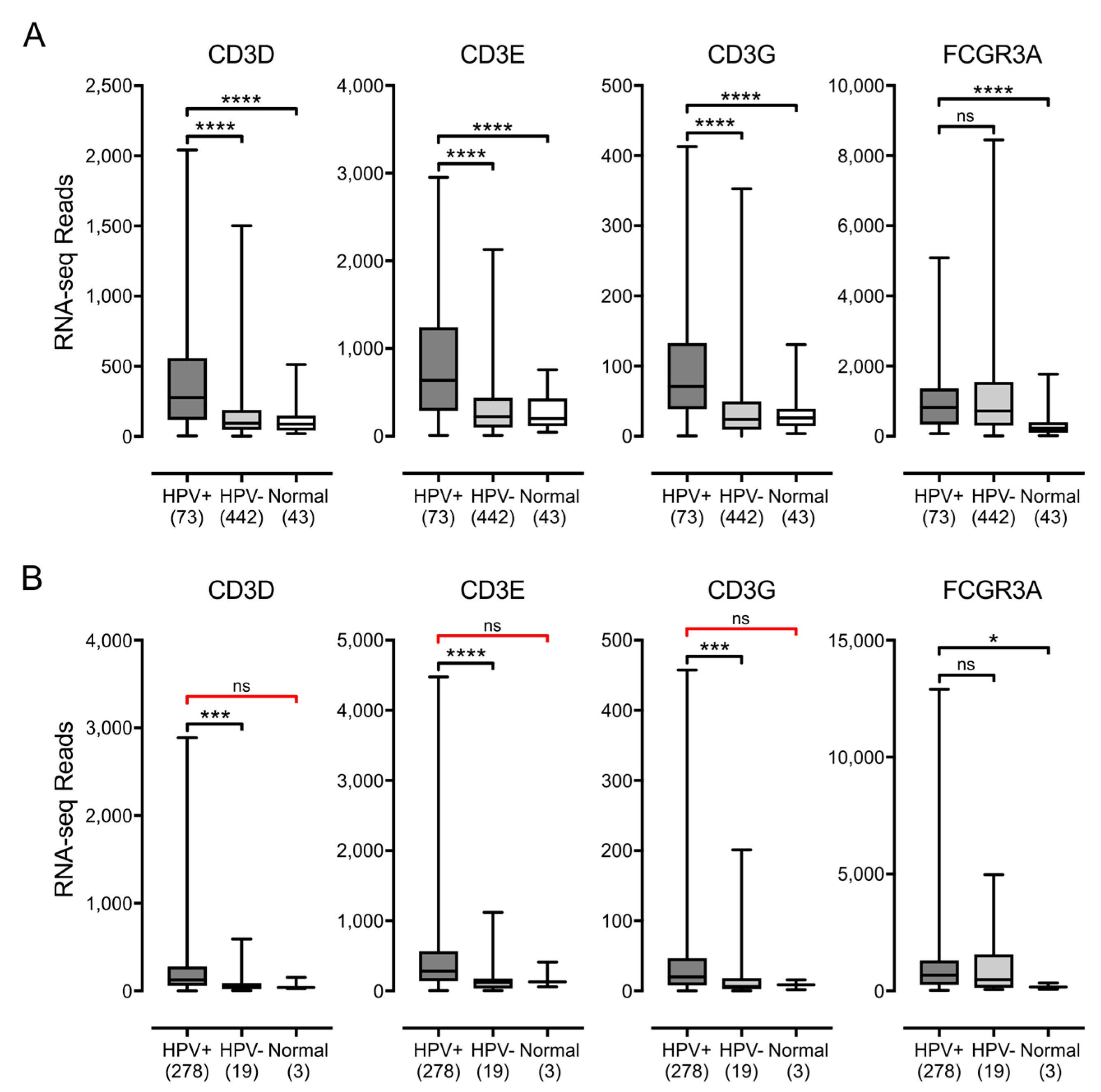
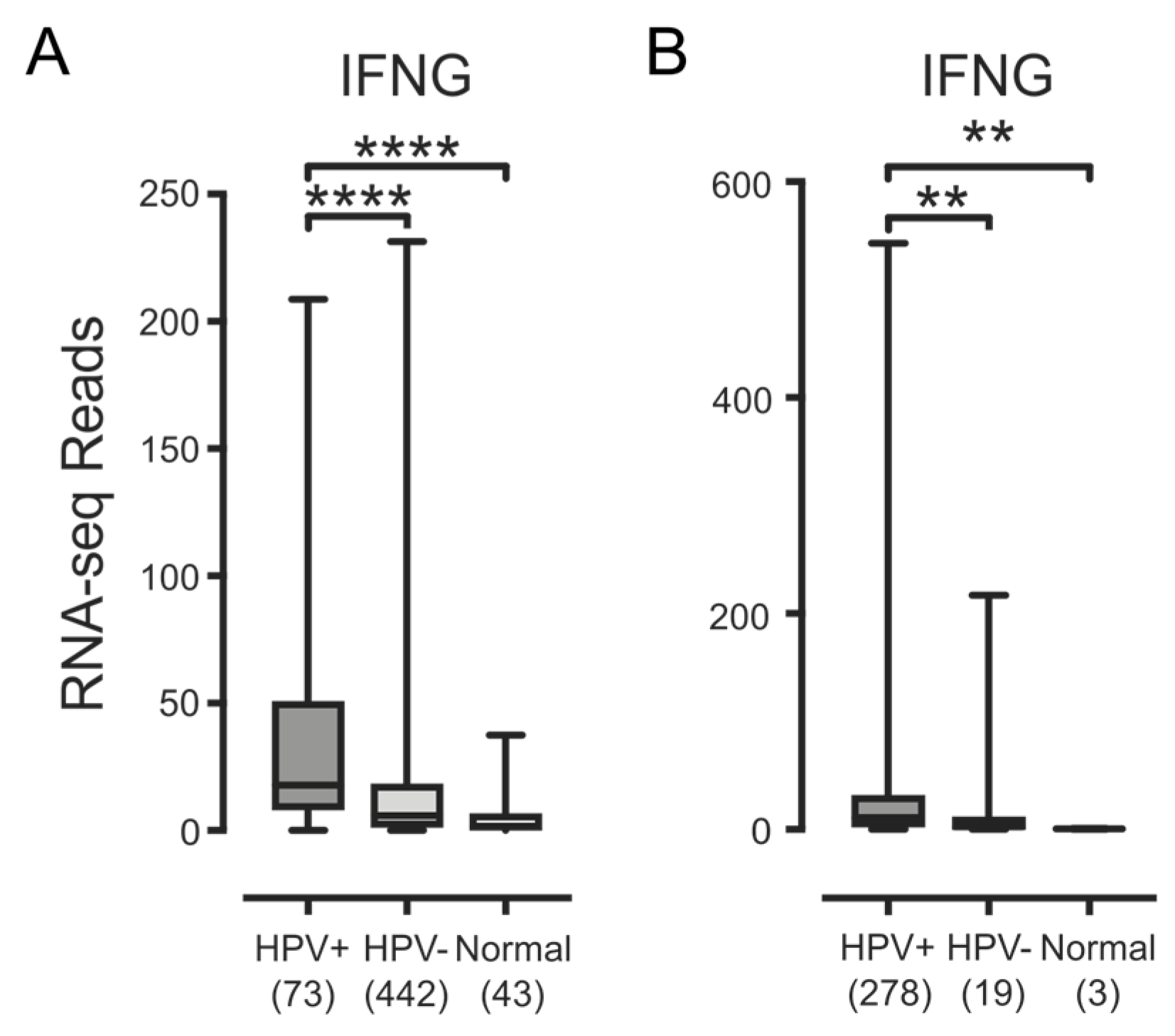
3.3. Higher Levels of Lymphocytes and Interferon γ Are Present in HPV+ Human Tumors
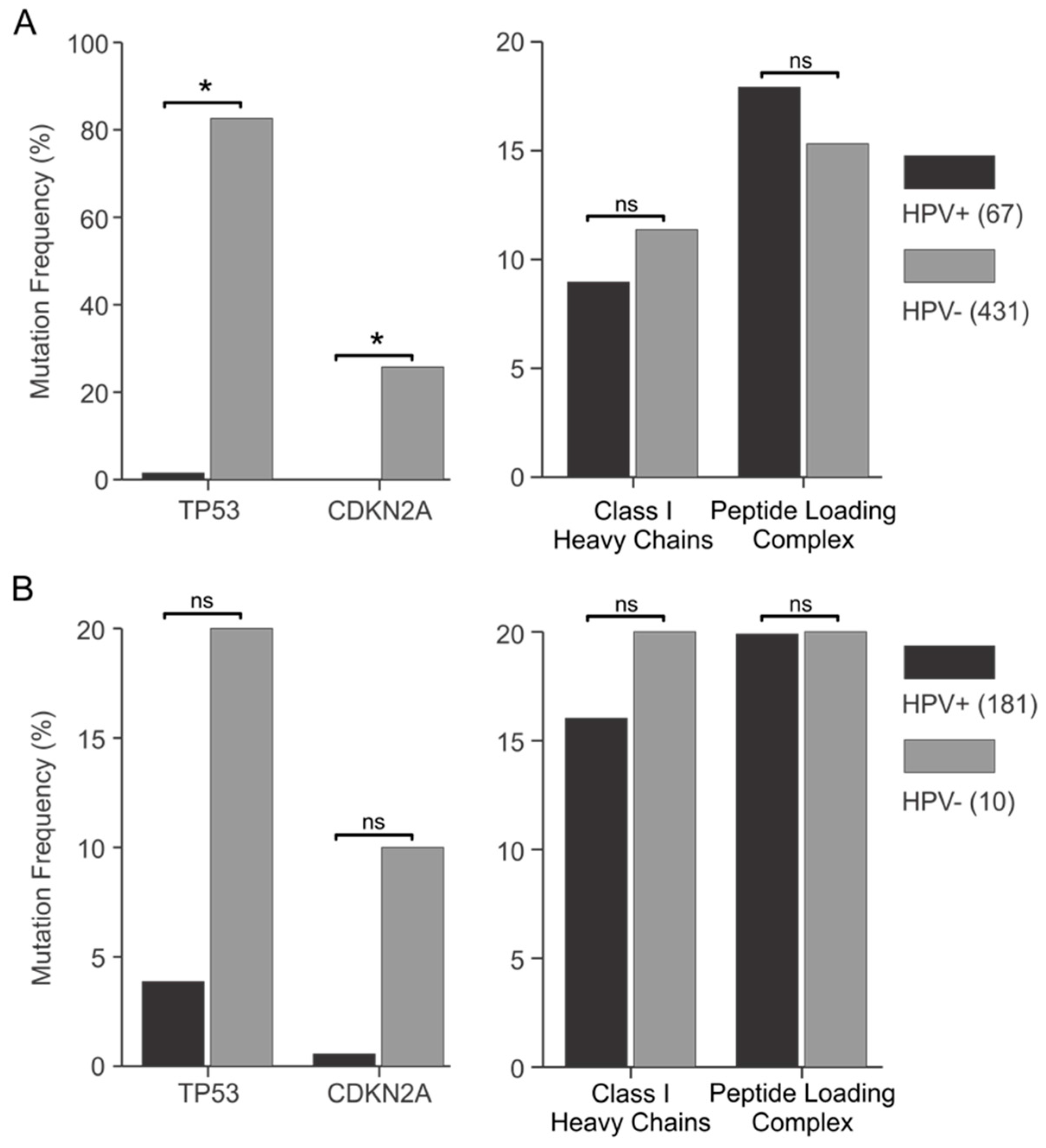
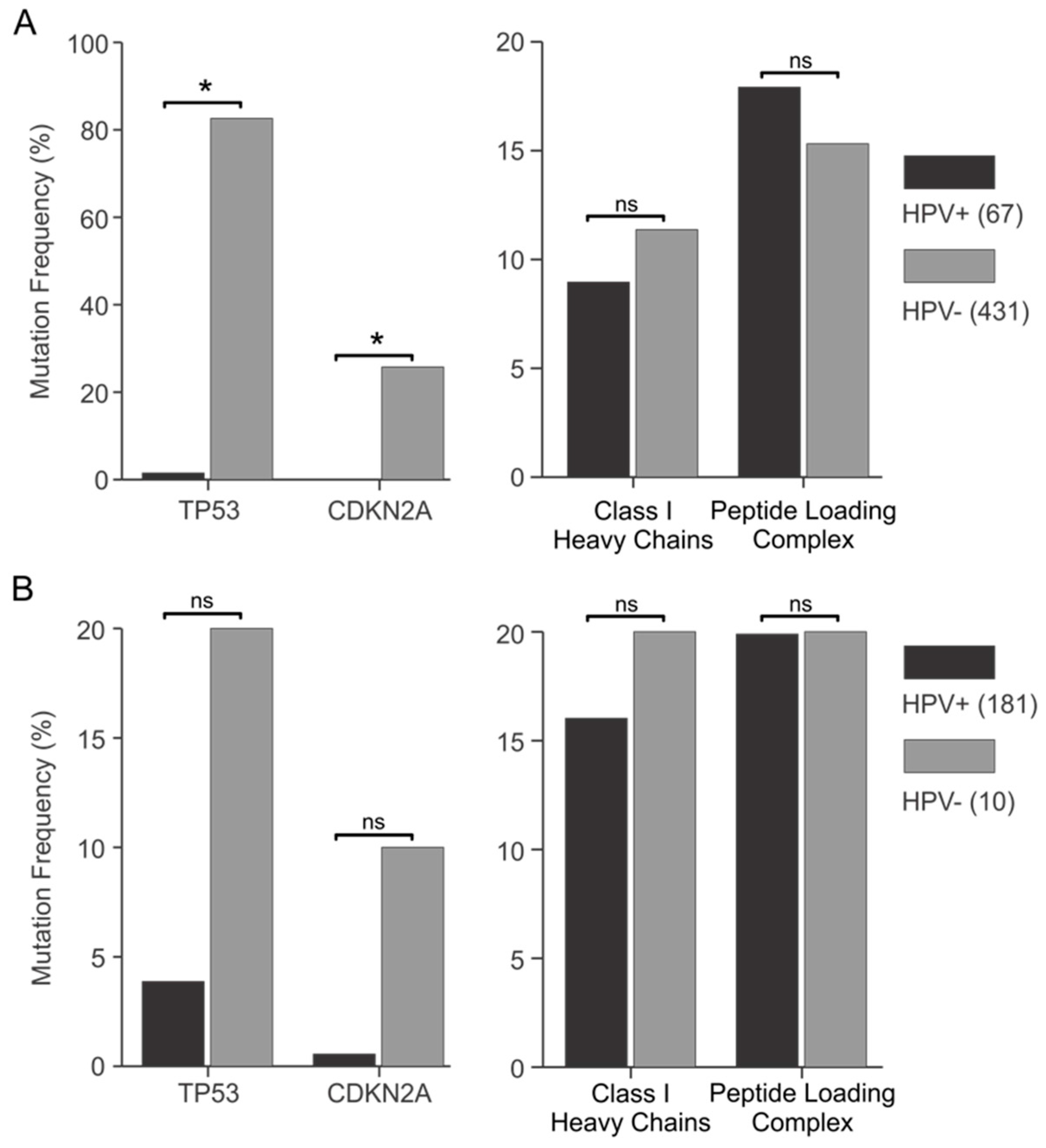
3.4. Genes Encoding Components of the Antigen Presentation Apparatus Are frequently Mutated in HPV+ Human Tumors
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yan, N.; Chen, Z.J. Intrinsic antiviral immunity. Nat. Immunol. 2012, 13, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Innate immunity to virus infection. Immunol. Rev. 2009, 227, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Tscharke, D.C.; Croft, N.P.; Doherty, P.C.; la Gruta, N.L. Sizing up the key determinants of the CD8(+) T cell response. Nat. Rev. Immunol. 2015, 15, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Dörner, T.; Radbruch, A. Antibodies and B cell memory in viral immunity. Immunity 2007, 27, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.H.; Bouvier, M. MHC class I antigen presentation: Learning from viral evasion strategies. Nat. Rev. Immunol. 2009, 9, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2015, 25, 2–23. [Google Scholar] [CrossRef] [PubMed]
- Munoz, N.; Hernandez-Suarez, G.; Méndez, F.; Molano, M.; Posso, H.; Moreno, V.; Murillo, R.; Ronderos, M.; Meijer, C.; Muñoz, A.; et al. Persistence of HPV infection and risk of high-grade cervical intraepithelial neoplasia in a cohort of Colombian women. Br. J. Cancer 2009, 100, 1184–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crosbie, E.J.; Einstein, M.H.; Franceschi, S.; Kitchener, H.C. Human papillomavirus and cervical cancer. Lancet 2013, 382, 889–899. [Google Scholar] [CrossRef]
- Alemany, L.; Cubilla, A.; Halec, G.; Kasamatsu, E.; Quirós, B.; Masferrer, E.; Tous, S.; Lloveras, B.; Hernández-Suarez, G.; Lonsdale, R.; et al. Role of Human Papillomavirus in Penile Carcinomas Worldwide. Eur. Urol. 2016, 69, 953–961. [Google Scholar] [CrossRef] [PubMed]
- De Vuyst, H.; Clifford, G.M.; Nascimento, M.C.; Madeleine, M.M.; Franceschi, S. Prevalence and type distribution of human papillomavirus in carcinoma and intraepithelial neoplasia of the vulva, vagina and anus: A meta-analysis. Int. J. Cancer 2009, 124, 1626–1636. [Google Scholar] [CrossRef] [PubMed]
- Guimerà, N.; Alemany, L.; Halec, G.; Pawlita, M.; Wain, G.V.; Vailén, J.S.S.; Azike, J.E.; Jenkins, D.; de Sanjosé, S.; Quint, W.; et al. Human papillomavirus 16 is an aetiological factor of scrotal cancer. Br. J. Cancer 2017, 146, 1299. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Koch, W.M.; Capone, R.B.; Spafford, M.; Westra, W.H.; Wu, L.; Zahurak, M.L.; Daniel, R.W.; Viglione, M.; Symer, D.E.; et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J. Natl. Cancer Inst. 2000, 92, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Syrjänen, S. Human papillomavirus (HPV) in head and neck cancer. J. Clin. Virol. 2005, 32, S59–66. [Google Scholar] [CrossRef] [PubMed]
- Mehanna, H.; Beech, T.; Nicholson, T.; El-Hariry, I.; McConkey, C.; Paleri, V.; Roberts, S. Prevalence of human papillomavirus in oropharyngeal and nonoropharyngeal head and neck cancer—systematic review and meta-analysis of trends by time and region. Head Neck 2013, 35, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Forman, D.; de Martel, C.; Lacey, C.J.; Soerjomataram, I.; Lortet-Tieulent, J.; Bruni, L.; Vignat, J.; Ferlay, J.; Bray, F.; Plummer, M.; et al. Global burden of human papillomavirus and related diseases. Vaccine 2012, 30, F12–23. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30, F55–F70. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Dyson, N.; Guida, P.; Munger, K.; Harlow, E. Homologous sequences in adenovirus E1A and human papillomavirus E7 proteins mediate interaction with the same set of cellular proteins. J. Virol. 1992, 66, 6893–6902. [Google Scholar] [PubMed]
- Brehm, A.; Miska, E.A.; McCance, D.J.; Reid, J.L.; Bannister, A.J.; Kouzarides, T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature 1998, 391, 597–601. [Google Scholar] [PubMed]
- Boyer, S.N.; Wazer, D.E.; Band, V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996, 56, 4620–4624. [Google Scholar] [PubMed]
- Westrich, J.A.; Warren, C.J.; Pyeon, D. Evasion of host immune defenses by human papillomavirus. Virus Res. 2017, 231, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.H.; Haghshenas, M.; Marchetti, B.; Campo, M.S. E5 protein of human papillomavirus 16 downregulates HLA class I and interacts with the heavy chain via its first hydrophobic domain. Int. J. Cancer 2006, 119, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Kim, E.-M.; Lee, E.-H.; Ji, K.-Y.; Yi, J.; Park, M.; Kim, K.D.; Cho, Y.-Y.; Kang, H.-S. Human papillomavirus 16E6 suppresses major histocompatibility complex class I by upregulating lymphotoxin expression in human cervical cancer cells. Biochem. Biophys. Res. Commun. 2011, 409, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Georgopoulos, N.T.; Proffitt, J.L.; Blair, G.E. Transcriptional regulation of the major histocompatibility complex (MHC) class I heavy chain, TAP1 and LMP2 genes by the human papillomavirus (HPV) type 6b, 16 and 18 E7 oncoproteins. Oncogene 2000, 19, 4930–4935. [Google Scholar] [CrossRef] [PubMed]
- Heller, C.; Weisser, T.; Mueller-Schickert, A.; Rufer, E.; Hoh, A.; Leonhardt, R.M.; Knittler, M.R. Identification of key amino acid residues that determine the ability of high risk HPV16-E7 to dysregulate major histocompatibility complex class I expression. J. Biol. Chem. 2011, 286, 10983–10997. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Deng, X.-M.; Wang, C.-X.; Zhang, X.; Zheng, G.-X.; Zhang, J.; Feng, J.-B. Down-regulation of HLA class I antigen in human papillomavirus type 16 E7 expressing HaCaT cells: Correlate with TAP-1 expression. Int. J. Gynecol. Cancer 2010, 20, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Bottley, G.; Watherston, O.G.; Hiew, Y.L.; Norrild, B.; Cook, G.P.; Blair, G.E. High-risk human papillomavirus E7 expression reduces cell-surface MHC class I molecules and increases susceptibility to natural killer cells. Oncogene 2008, 27, 1794–1799. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ou, X.; Xiong, J.; Wang, T. HPV16E7 mediates HADC chromatin repression and downregulation of MHC class I genes in HPV16 tumorigenic cells through interaction with an MHC class I promoter. Biochem. Biophys. Res. Commun. 2006, 349, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhan, T.; Li, C.; Liu, M.; Wang, Q.K. Repression of MHC class I transcription by HPV16E7 through interaction with a putative RXRβ motif and NF-κB cytoplasmic sequestration. Biochem. Biophys. Res. Commun. 2009, 388, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Kim, E.J.; Kwon, H.J.; Hwang, E.S.; Namkoong, S.E.; Um, S.J. Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV E7 oncoprotein. Implication for the E7-mediated immune evasion mechanism in cervical carcinogenesis. J. Biol. Chem. 2000, 275, 6764–6769. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Leggatt, G.R.; Frazer, I.H. Human papillomavirus 16 E7 protein inhibits interferon-γ-mediated enhancement of keratinocyte antigen processing and T-cell lysis. FEBS J. 2011, 278, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Um, S.-J.; Rhyu, J.-W.; Kim, E.-J.; Jeon, K.-C.; Hwang, E.-S.; Park, J.-S. Abrogation of IRF-1 response by high-risk HPV E7 protein in vivo. Cancer Lett. 2002, 179, 205–212. [Google Scholar] [CrossRef]
- Zhou, F.; Chen, J.; Zhao, K.-N. Human papillomavirus 16-encoded E7 protein inhibits IFN-γ-mediated MHC class I antigen presentation and CTL-induced lysis by blocking IRF-1 expression in mouse keratinocytes. J. Gen. Virol. 2013, 94, 2504–2514. [Google Scholar] [CrossRef] [PubMed]
- Perea, S.E.; Massimi, P.; Banks, L. Human papillomavirus type 16 E7 impairs the activation of the interferon regulatory factor. Int. J. Mol. Med. 2000, 5, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Jochmus, I.; Durst, M.; Reid, R.; Altmann, A.; Bijward, K.E.; Gissmann, L.; Jenson, A.B. Major histocompatibility complex and human papillomavirus type 16 E7 expression in high-grade vulvar lesions. Hum. Pathol. 1993, 24, 519–524. [Google Scholar] [CrossRef]
- Torres, L.M.; Cabrera, T.; Concha, A.; Oliva, M.R.; Ruiz-Cabello, F.; Garrido, F. HLA class I expression and HPV-16 sequences in premalignant and malignant lesions of the cervix. Tissue Antigens 1993, 41, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Keating, P.J.; Cromme, F.V.; Duggan-Keen, M.; Snijders, P.J.; Walboomers, J.M.; Hunter, R.D.; Dyer, P.A.; Stern, P.L. Frequency of down-regulation of individual HLA-A and -B alleles in cervical carcinomas in relation to TAP-1 expression. Br. J. Cancer 1995, 72, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Cromme, F.V.; Meijer, C.J.; Snijders, P.J.; Uyterlinde, A.; Kenemans, P.; Helmerhorst, T.; Stern, P.L.; van den Brule, A.J.; Walboomers, J.M. Analysis of MHC class I and II expression in relation to presence of HPV genotypes in premalignant and malignant cervical lesions. Br. J. Cancer 1993, 67, 1372–1380. [Google Scholar] [CrossRef] [PubMed]
- Ritz, U.; Momburg, F.; Pilch, H.; Huber, C.; Maeurer, M.J.; Seliger, B. Deficient expression of components of the MHC class I antigen processing machinery in human cervical carcinoma. Int. J. Oncol. 2001, 19, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Van Esch, E.M.G.; Tummers, B.; Baartmans, V.; Osse, E.M.; Ter Haar, N.; Trietsch, M.D.; Hellebrekers, B.W.J.; Holleboom, C.A.G.; Nagel, H.T.C.; Tan, L.T.; et al. Alterations in classical and nonclassical HLA expression in recurrent and progressive HPV-induced usual vulvar intraepithelial neoplasia and implications for immunotherapy. Int. J. Cancer 2014, 135, 830–842. [Google Scholar] [CrossRef] [PubMed]
- Ramqvist, T.; Mints, M.; Tertipis, N.; Näsman, A.; Romanitan, M.; Dalianis, T. Studies on human papillomavirus (HPV) 16 E2, E5 and E7 mRNA in HPV-positive tonsillar and base of tongue cancer in relation to clinical outcome and immunological parameters. Oral Oncol. 2015, 51, 1126–1131. [Google Scholar] [CrossRef] [PubMed]
- Djajadiningrat, R.S.; Horenblas, S.; Heideman, D.A.M.; Sanders, J.; de Jong, J.; Jordanova, E.S. Classic and nonclassic HLA class I expression in penile cancer and relation to HPV status and clinical outcome. J. Urol. 2015, 193, 1245–1251. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.M.; Jordanova, E.S.; Kenter, G.G.; Ferrone, S.; Fleuren, G.-J. Association of antigen processing machinery and HLA class I defects with clinicopathological outcome in cervical carcinoma. Cancer Immunol. Immunother. 2008, 57, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Lora, A.; Algarra, I.; Garrido, F. MHC class I antigens, immune surveillance, and tumor immune escape. J. Cell. Physiol. 2003, 195, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Gotwals, P.; Cameron, S.; Cipolletta, D.; Cremasco, V.; Crystal, A.; Hewes, B.; Mueller, B.; Quaratino, S.; Sabatos-Peyton, C.; Petruzzelli, L.; et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat. Rev. Cancer 2017, 17, 286–301. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. [Google Scholar]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar]
- Bratman, S.V.; Bruce, J.P.; O’Sullivan, B.; Pugh, T.J.; Xu, W.; Yip, K.W.; Liu, F.-F. Human Papillomavirus Genotype Association With Survival in Head and Neck Squamous Cell Carcinoma. JAMA Oncol. 2016, 2, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Banister, C.E.; Liu, C.; Pirisi, L.; Creek, K.E.; Buckhaults, P.J. Identification and characterization of HPV-independent cervical cancers. Oncotarget 2017, 8, 13375–13386. [Google Scholar] [CrossRef] [PubMed]
- Faul, F.; Erdfelder, E.; Lang, A.-G.; Buchner, A. G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods 2007, 39, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Mayakonda, A.; Koeffler, H.P. Maftools: Efficient analysis, visualization and summarization of MAF files from large-scale cohort based cancer studies. BioRxiv 2016, 052662. [Google Scholar] [CrossRef]
- Cicchini, L.; Blumhagen, R.Z.; Westrich, J.A.; Myers, M.E.; Warren, C.J.; Siska, C.; Raben, D.; Kechris, K.J.; Pyeon, D. High-Risk Human Papillomavirus E7 Alters Host DNA Methylome and Represses HLA-E Expression in Human Keratinocytes. Sci. Rep. 2017, 7, 3633. [Google Scholar] [CrossRef] [PubMed]
- Boehm, U.; Klamp, T.; Groot, M.; Howard, J.C. Cellular responses to interferon-γ. Annu. Rev. Immunol. 1997, 15, 749–795. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautès-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Sirota, M.; Butte, A.J. Systematic pan-cancer analysis of tumour purity. Nat. Commun. 2015, 6, 8971. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; McCormick, F. The RB and p53 pathways in cancer. Cancer Cell 2002, 2, 103–112. [Google Scholar] [CrossRef]
- Zhao, K.-N.; Chen, J. Codon usage roles in human papillomavirus. Rev. Med. Virol. 2011, 21, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, A.; Winter, J.; Reuschenbach, M.; Blatnik, R.; Klevenz, A.; Bertrand, M.; Hoppe, S.; von Knebel Doeberitz, M.; Grabowska, A.K.; Riemer, A.B. ERAP1 overexpression in HPV-induced malignancies: A possible novel immune evasion mechanism. Oncoimmunology 2017, 6, e1336594. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.; Borysiewicz, L.K.; Evans, A.S.; Rowe, M.; Jones, M.; Gileadi, U.; Cerundolo, V.; Man, S. Antigen processing defects in cervical carcinomas limit the presentation of a CTL epitope from human papillomavirus 16 E6. J. Immunol. 2001, 167, 5420–5428. [Google Scholar] [CrossRef] [PubMed]
- De Lourdes Mora-García, M.; Duenas-González, A.; Hernández-Montes, J.; De la Cruz-Hernández, E.; Pérez-Cárdenas, E.; Weiss-Steider, B.; Santiago-Osorio, E.; Ortíz-Navarrete, V.F.; Rosales, V.H.; Cantú, D.; et al. Up-regulation of HLA class-I antigen expression and antigen-specific CTL response in cervical cancer cells by the demethylating agent hydralazine and the histone deacetylase inhibitor valproic acid. J. Transl. Med. 2006, 4, 55. [Google Scholar]
- Bornstein, J.; Lahat, N.; Kinarty, A.; Revel, M.; Abramovici, H.; Shapiro, S. Interferon-β and -γ, but not tumor necrosis factor-α, demonstrate immunoregulatory effects on carcinoma cell lines infected with human papillomavirus. Cancer 1997, 79, 924–934. [Google Scholar] [CrossRef]
- Sikorski, M.; Bobek, M.; Zrubek, H.; Marcinkiewicz, J. Dynamics of selected MHC class I and II molecule expression in the course of HPV positive CIN treatment with the use of human recombinant IFN-γ. Acta Obstet. Gynecol. Scand. 2004, 83, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Gameiro, S.F.; Kolendowski, B.; Zhang, A.; Barrett, J.W.; Nichols, A.C.; Torchia, J.; Mymryk, J.S. Human papillomavirus dysregulates the cellular apparatus controlling the methylation status of H3K27 in different human cancers to consistently alter gene expression regardless of tissue of origin. Oncotarget 2017, in press. [Google Scholar] [CrossRef]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Ang, K.K.; Harris, J.; Wheeler, R.; Weber, R.; Rosenthal, D.I.; Nguyen-Tân, P.F.; Westra, W.H.; Chung, C.H.; Jordan, R.C.; Lu, C.; et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N. Engl. J. Med. 2010, 363, 24–35. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gameiro, S.F.; Zhang, A.; Ghasemi, F.; Barrett, J.W.; Nichols, A.C.; Mymryk, J.S. Analysis of Class I Major Histocompatibility Complex Gene Transcription in Human Tumors Caused by Human Papillomavirus Infection. Viruses 2017, 9, 252. https://doi.org/10.3390/v9090252
Gameiro SF, Zhang A, Ghasemi F, Barrett JW, Nichols AC, Mymryk JS. Analysis of Class I Major Histocompatibility Complex Gene Transcription in Human Tumors Caused by Human Papillomavirus Infection. Viruses. 2017; 9(9):252. https://doi.org/10.3390/v9090252
Chicago/Turabian StyleGameiro, Steven F., Ali Zhang, Farhad Ghasemi, John W. Barrett, Anthony C. Nichols, and Joe S. Mymryk. 2017. "Analysis of Class I Major Histocompatibility Complex Gene Transcription in Human Tumors Caused by Human Papillomavirus Infection" Viruses 9, no. 9: 252. https://doi.org/10.3390/v9090252