1. Introduction
Metabolized drug-drug interactions (mDDIs) have been one of the main reasons for the failure of new drug research and development; a variety of drugs have been forced to withdraw from the market due to serious DDIs [
1,
2,
3,
4]. With the increasing and development of new drugs and usage, clinical combination therapy has become very common and inevitably increases the probability of occurrence of DDI. Consequently, evaluation of a potential risk of mDDIs is essential to improve safety and minimize the clinical risks associated with drug interactions [
5].
In general, metabolites are formed primarily via metabolic enzymes, which play an important role in pharmacological activity and toxicity. Compared to the parent drug, it is generally considered less likely to cause metabolized drug interactions due to more polarity. In vitro studies of parent drugs are sufficient to avoid DDI risks [
6].
However, it has recently been found that some important metabolites of inhibitors also have inhibitory effects [
6,
7,
8]. In the latest Food and Drug Administration (FDA) draft guidance [
9], it is explicitly stated that metabolites should be studied in DDI if the metabolite’s area under the plasma concentration-time curve (AUC) is greater than or equal to 25% of the parent AUC (AUCm/AUCp ≥ 0.25). The European Medicines Agency (EMA) further emphasizes that, for metabolites with AUCm/AUCp > 0.25 and represent >10% of total drug-related material [
10], it is recommended to evaluate their DDI. In addition, regulators are also strongly proposing to predict and understand potential clinical DDI from the perspective of physiologically-based pharmacokinetic (PBPK), especially those complex DDIs [
9,
10,
11]. The PBPK model provides a dynamic method for evaluation of DDI based on the physiological mechanism [
12,
13,
14,
15]. Compared with the static approach, it is reasonable to anticipate that the dynamic model is more accurate in the predication of DDIs, such as simultaneous inhibition and induction [
16,
17], the DDI of both substrate and inhibitory metabolites [
15,
18,
19] and multiple DDIs. Recently, PBPK models have been widely applied in research and development, and even some good models are accepted by regulatory agencies and can be used to exempt some clinical trials [
11,
20,
21,
22,
23].
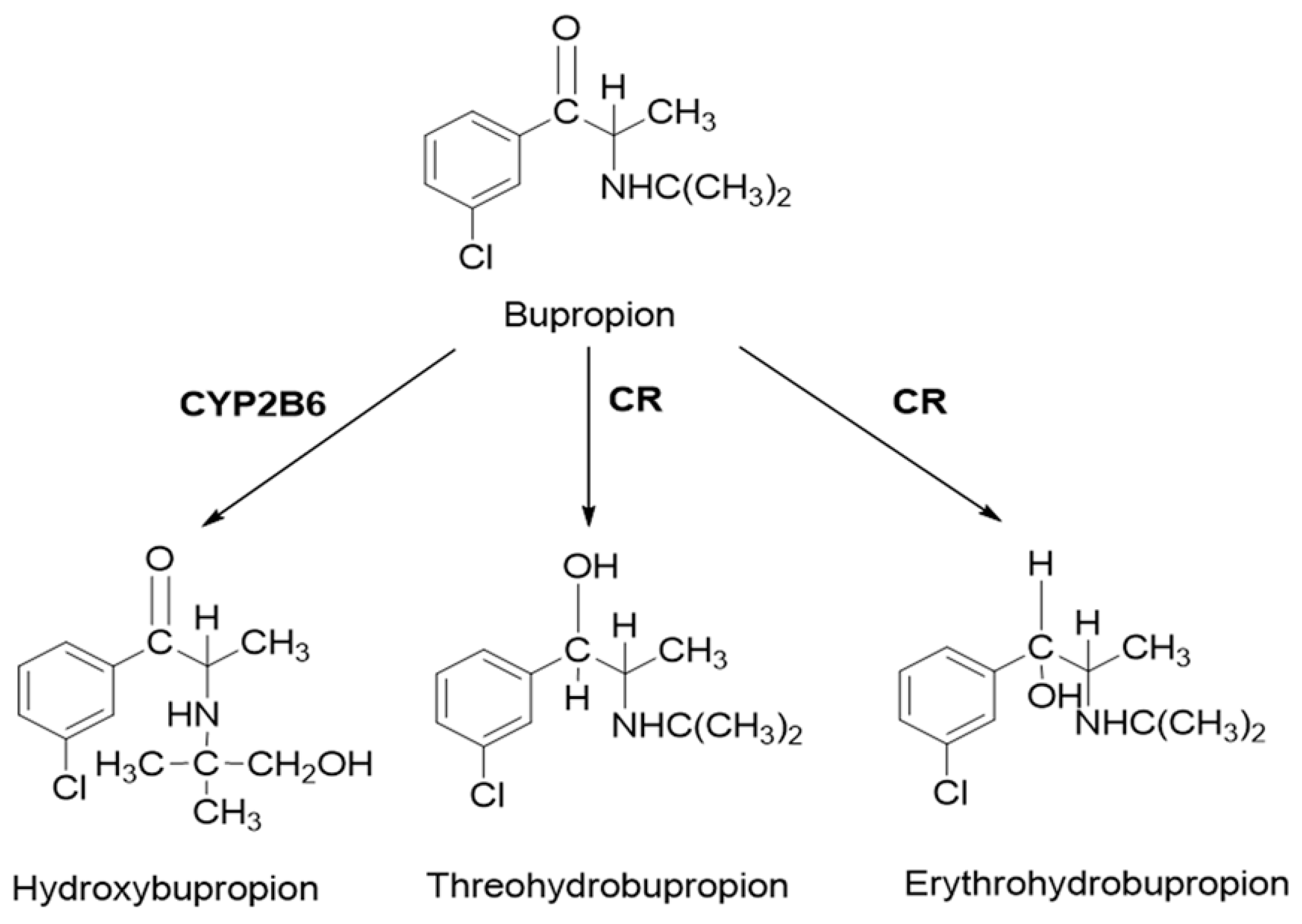
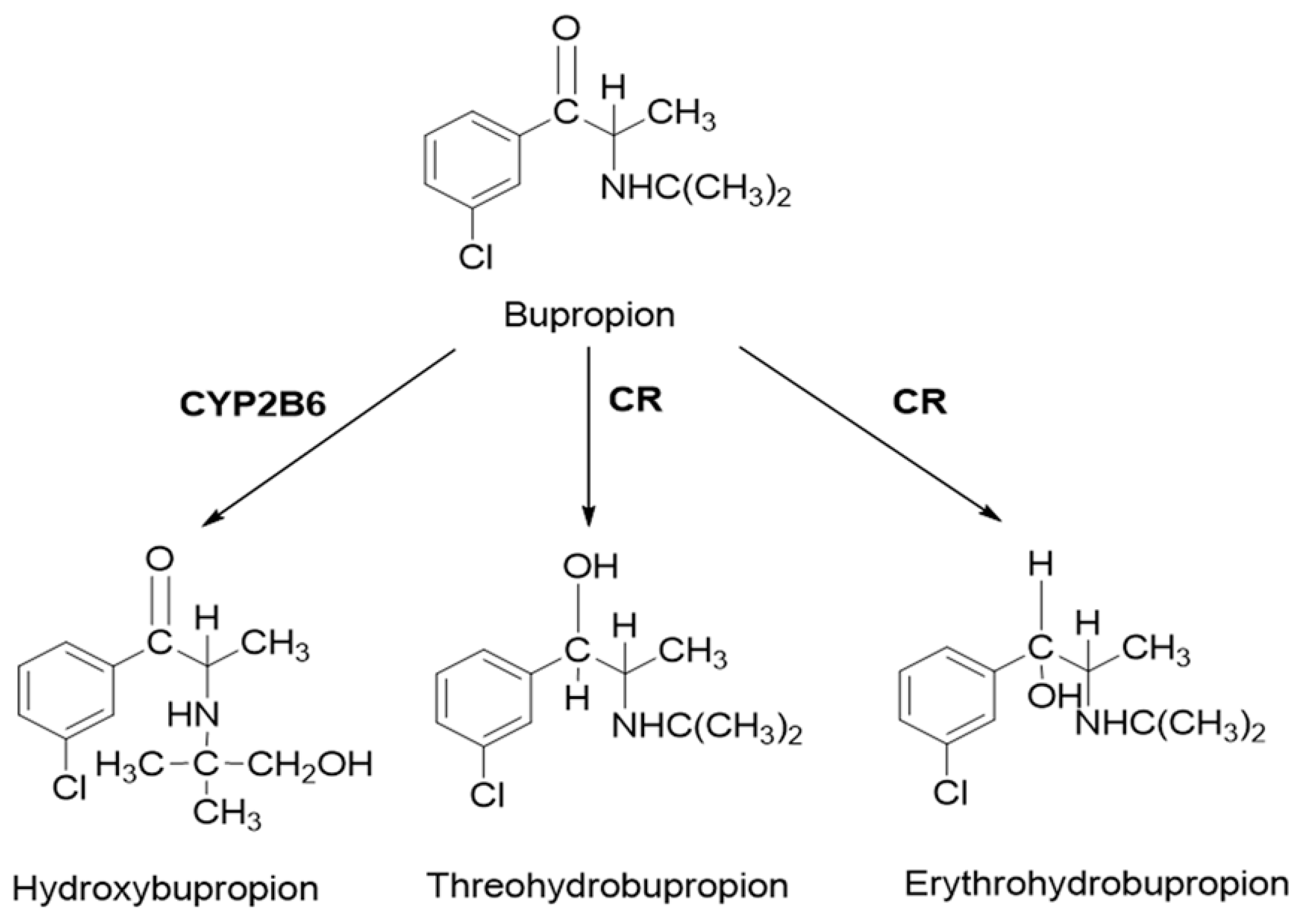
Bupropion is widely used in the treatment of major depressive disorder and smoking cessation. As a classical probe substrate for CYP2B6, it is metabolized to hydroxybupropion. In human, carbonyl reductase also plays an important role in the metabolism of bupropion. Threohydrobupropion and erythrohydrobupropion are two major metabolites produced by the reduction of the carbonyl group [
24,
25,
26,
27] (
Figure 1). Although bupropion is not a substrate for CYP2D6, it also inhibits CYP2D6 activity [
27,
28]. Clinical studies have shown that there is a significant increase in substrate exposure when bupropion was administered in combination with substrates for CYP2D6. For desipramine, a five-fold increase in exposure was caused [
29]. However, in vitro studies have shown that bupropion and hydroxybupropion are weak CYP2D6 inhibitors (IC50 = 58 and 74 μM, respectively) [
27]. Thus, bupropion was chosen as the model drug. To better understand the complex DDI, a PBPK model was taken in the present study.
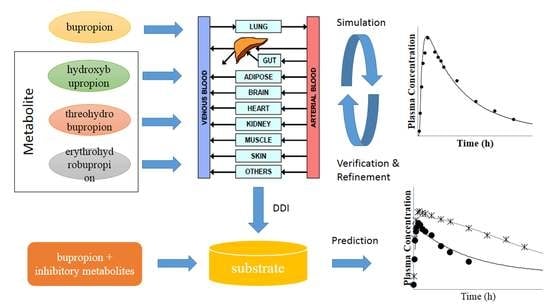
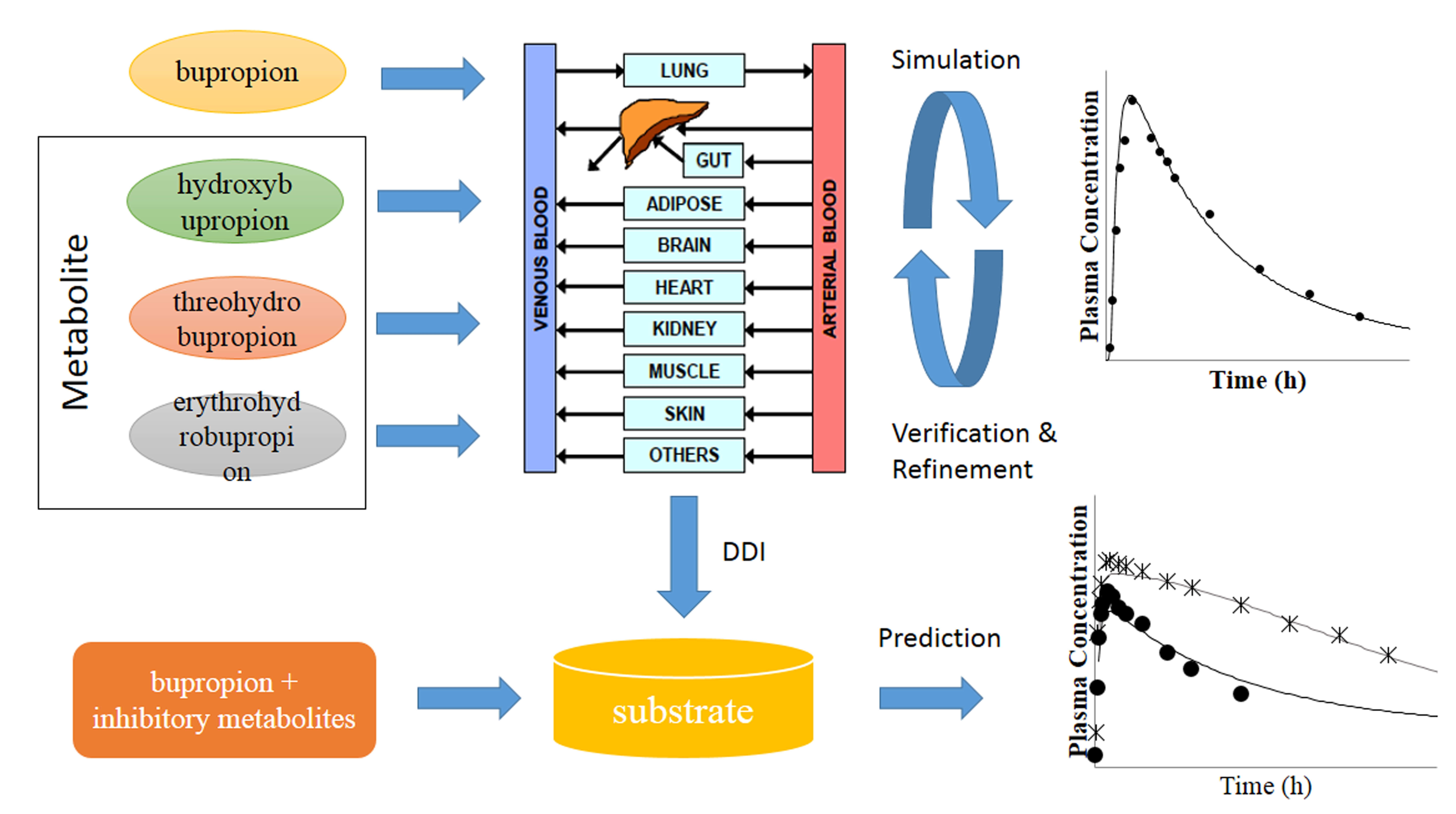
The objectives of the present work are (1) to build a PBPK model that can describe the PK profile of bupropion, hydroxybupropion, threohydrobupropion and erythrohydrobupropion; (2) to verify the bupropion PBPK model on the basis of the results of different-dose bupropion PK studies; and ultimately (3) to apply the PBPK model to predict the clinically observed DDIs with bupropion and its metabolites as the CYP2D6 inhibitors, and to better understand the involvement and impact of inhibitory metabolites for DDIs.
4. Discussion
It is common to think that the possibility of causing drug interactions for metabolites compared with the parent drug is low. However, recently, more and more studies have shown that the perpetrator drug’s metabolites may also have a significant impact on CYP-mediated DDI. With the development of the PBPK model, it has been widely used in various stages of drug development, especially in evaluation of DDIs. The PBPK model can simulate a dynamic process which is closer to the in vivo behavior based on in vitro biotransformation and physicochemical parameters. Many studies have successfully evaluated drug interactions using PBPK model [
12,
56,
57,
58]. However, only a few studies have built a PBPK model to evaluate DDI caused by inhibitory metabolite [
18,
59,
60,
61]. Many compounds, such as bupropion, have an unexpected DDI in clinic, although in vitro study showed that bupropion was a weak CYP2D6 inhibitor. It is possible that the inhibition from metabolites contributes to the observed DDI. To better address this apparent discrepancy between in vitro and in vivo studies, bupropion was chosen as an example, and the PBPK model was employed to describe the complex drug interactions involving inhibitory metabolite.
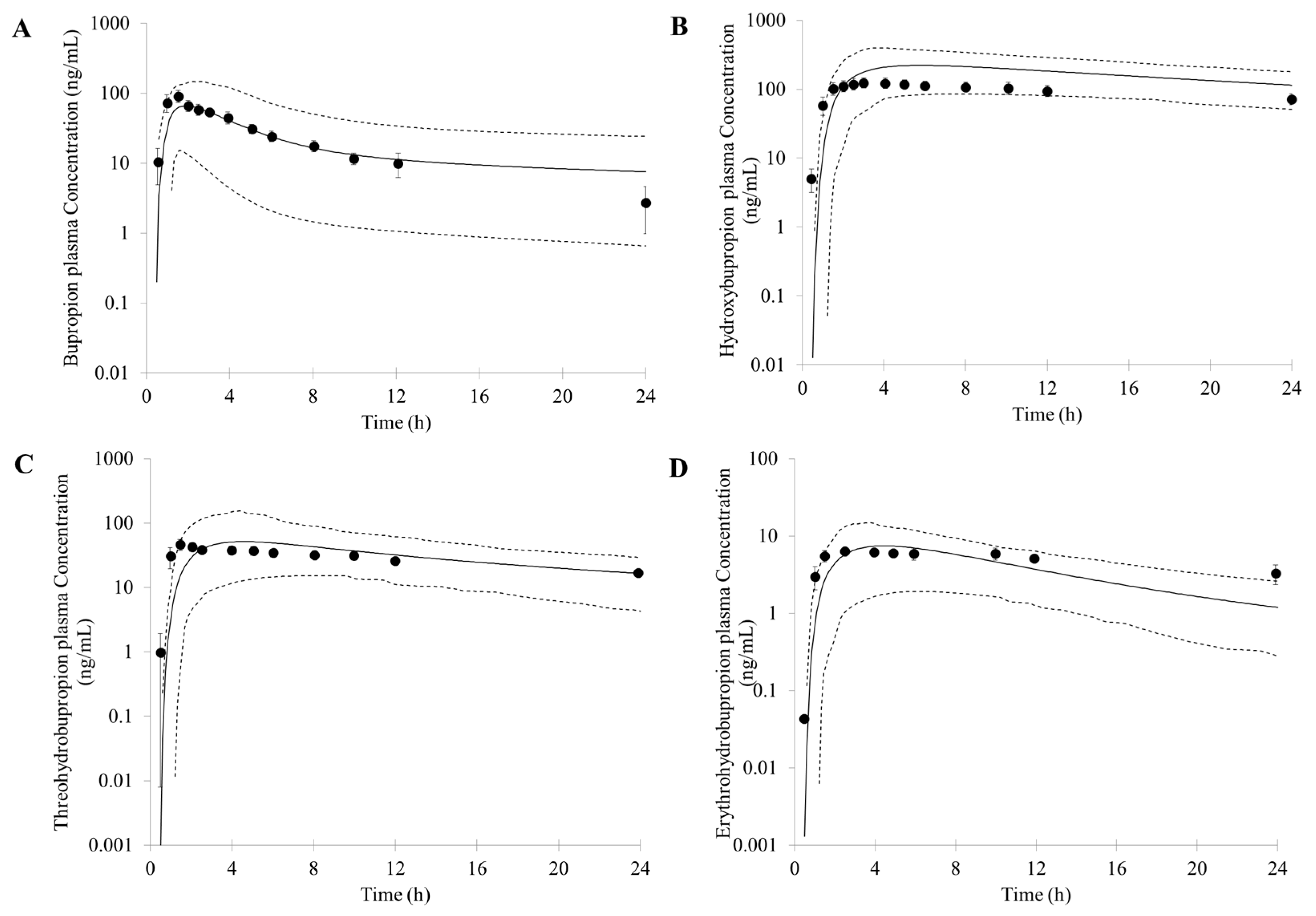
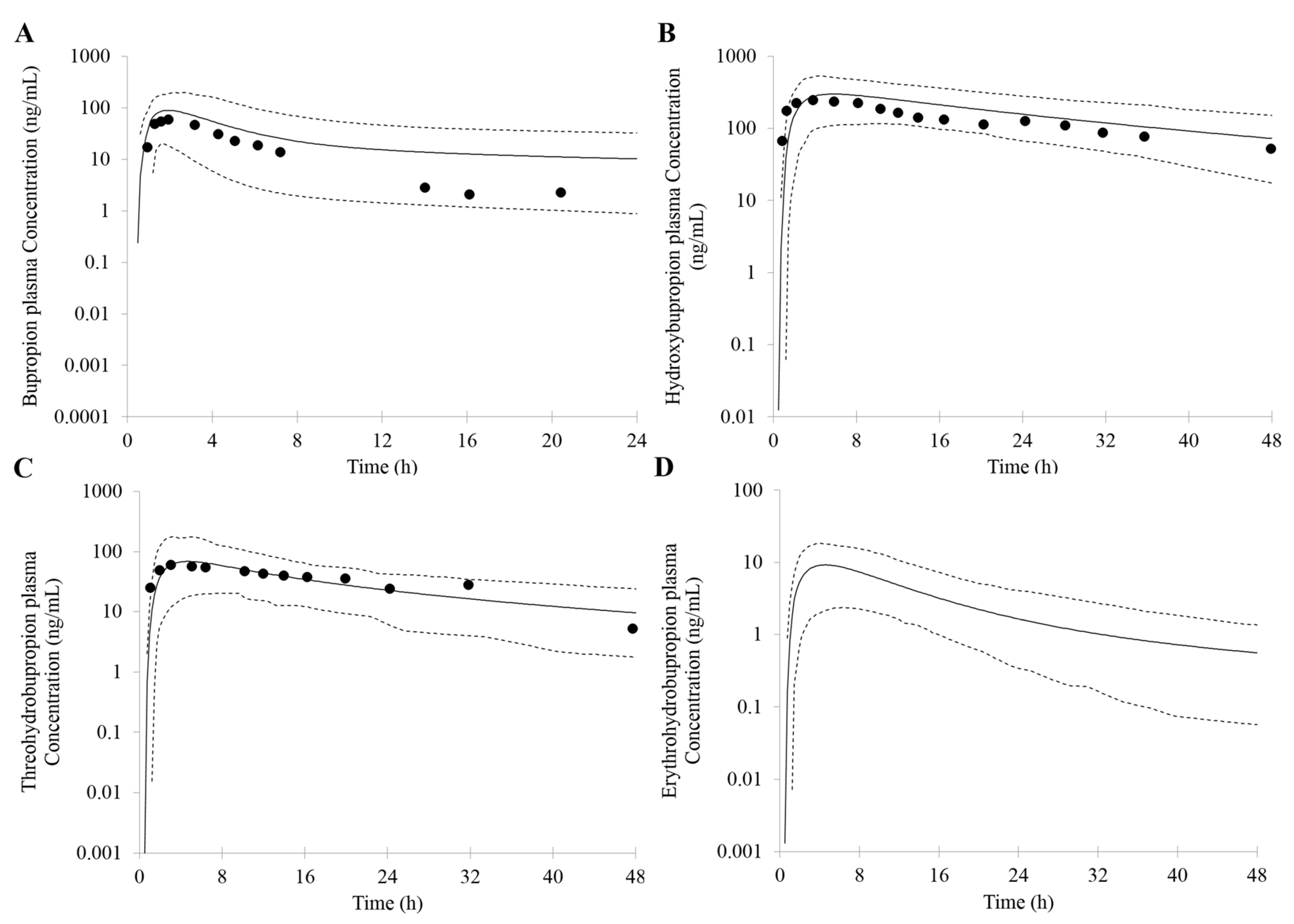
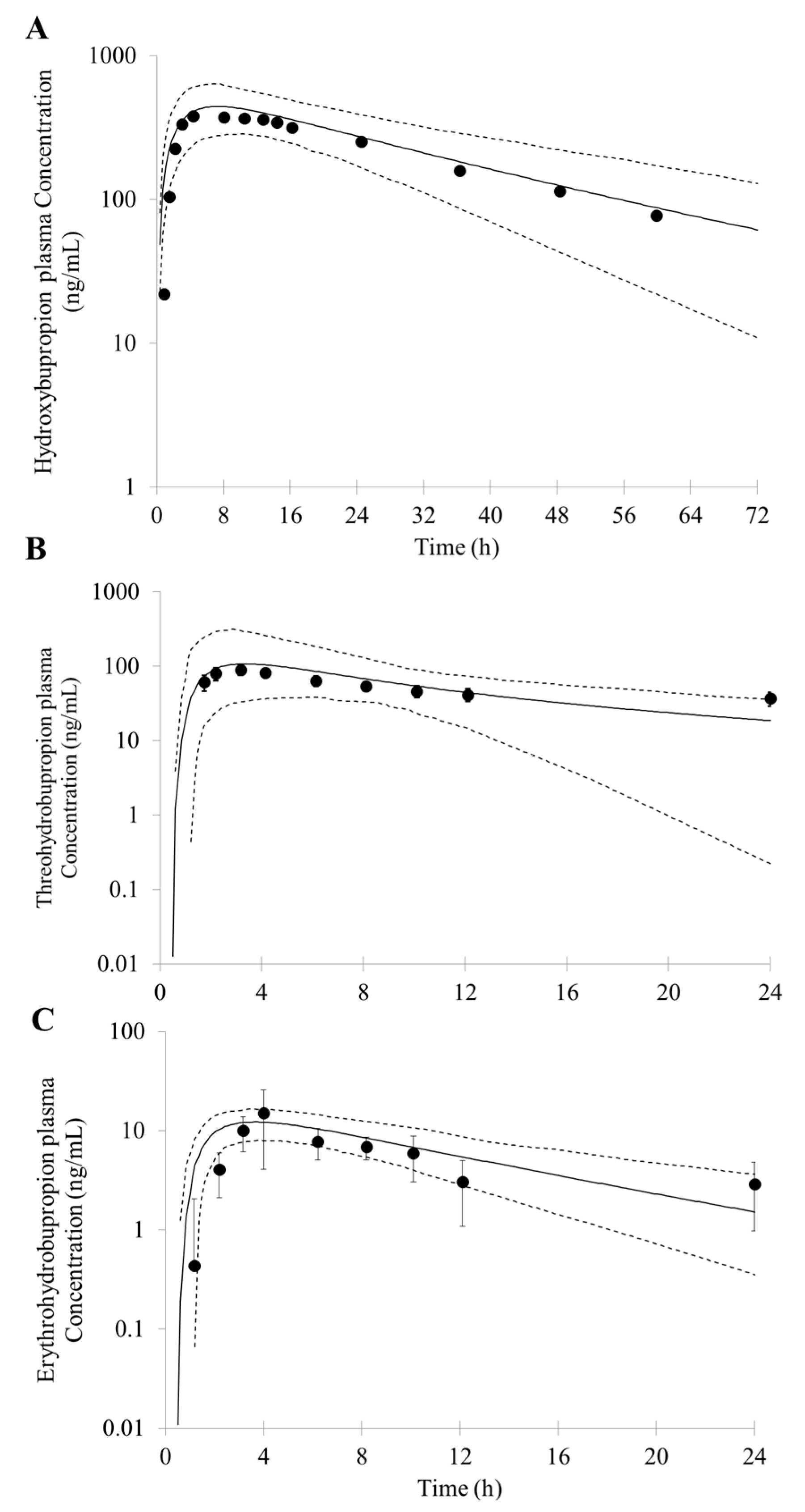
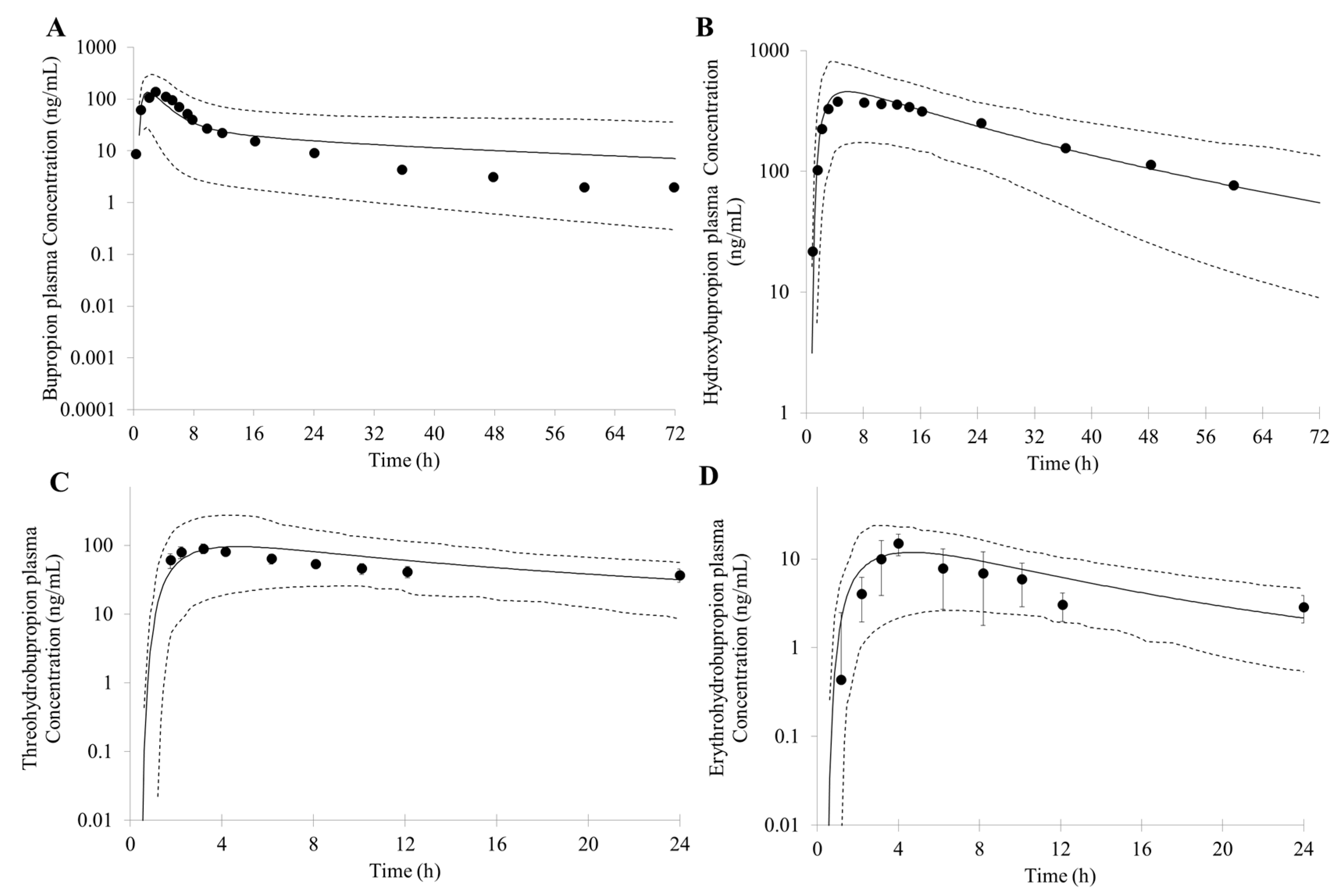
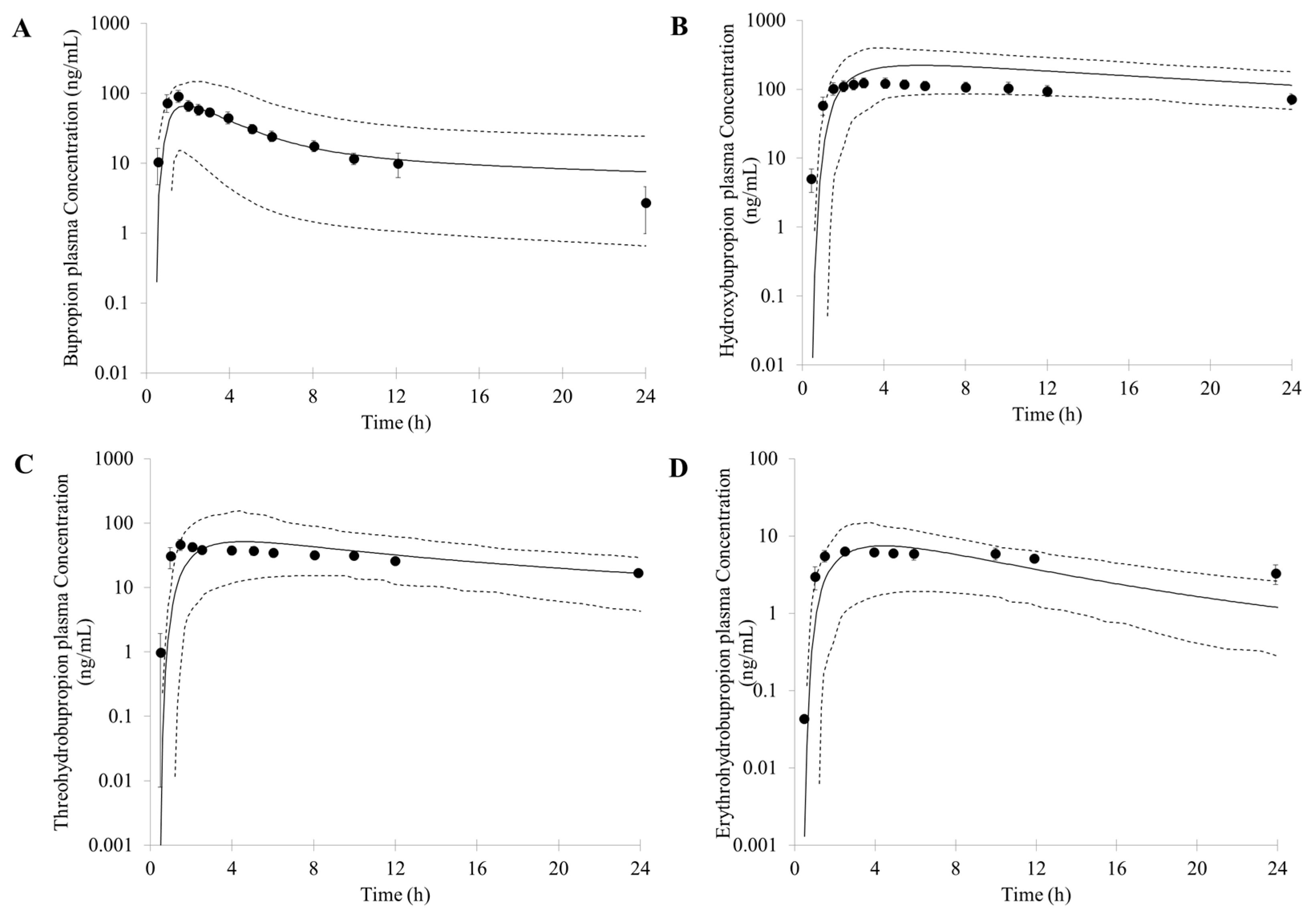
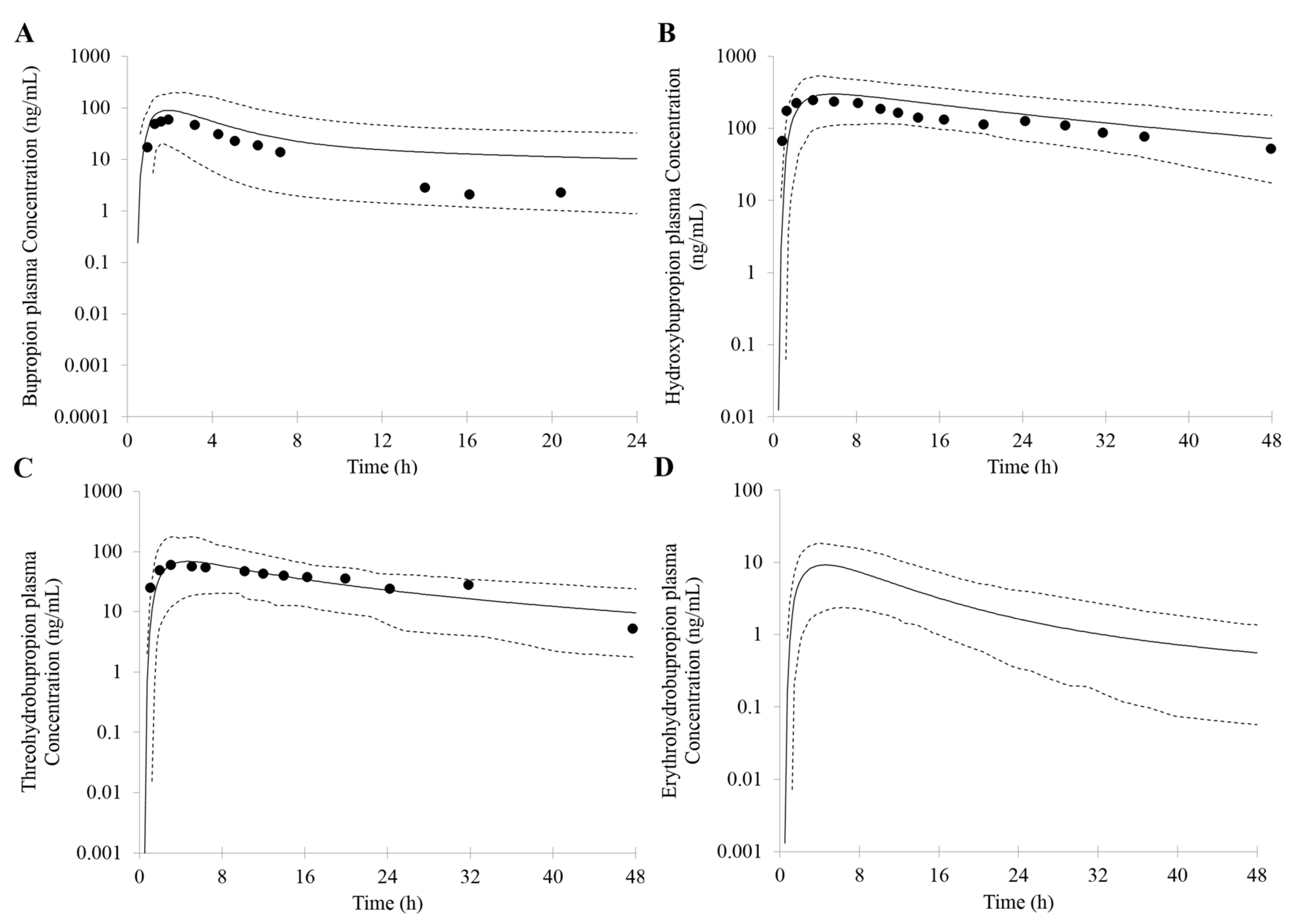
First, an accurate simulation of PK profiles of both parent and metabolite is required to maximize the confidence in the DDI prediction. Therefore, in our study, many observed PK profiles of different doses were used to verify the bupropion model. A full PBPK distribution model and first order absorption model was used for a good description of bupropion PK profile. Bupropion is mainly metabolized by the liver, and less than 1% of the parent drug is found in the urine [
26,
29]. In addition to hydroxybupropion that are mediated by CYP enzymes, bupropion is also metabolized by 11β-HSD to form threohydrobupropion and erythrohydrobupropion [
62,
63]. To better describe and build the PBPK model, CYP2B6 instead of carbonyl reductase was set in Simcyp as the metabolic enzymes of formation of threohydrobupropion and erythrohydrobupropion, and a f
u,
mic was used to correct the expression of carbonyl reductase to obtain the best simulation results compared to observed data.
For those uncertain or unknown parameters, a sensitivity analysis is performed to assess the importance and effect of these parameters in human PK and DDI prediction. In the PBPK model of bupropion, the logP, pKa, and three f
u,mic were considered for sensitivity analysis. According to the analysis, the logP, pKa and the f
u,mic for formation of erythrohydrobupropion were not sensitive to the prediction of PK. However, f
u,mic for formation of hydroxybupropion and threohydrobupropion has a certain impact on the prediction of PK and the f
u,mic for formation of erythrohydrobupropion. Thus, the logP and pKa from the drug bank were inputted into the model. The f
u,mic for formation of hydroxybupropion and threohydrobupropion were optimized at the 0.16 and 0.003, respectively. The erythrohydrobupropion and threohydrobupropion are formed via reduction of the carbonyl group. Thus, the same f
u,mic is integrated into the model. The detailed sensitivity analysis results are shown in
Supplementary Figure S1.
Based on the in vitro data and the mechanisms mentioned above, 1% of the fe (fraction of total body clearance via renal excretion) and geometric mean 174 (L/h) of CL were reasonably predicted by the PBPK model. Studies have shown that the CL for bupropion is in the range of 113 to 215 L/h [
31,
47,
49,
64,
65,
66,
67,
68]. For the PK profile of metabolites, the minimal PBPK distribution model or minimal PBPK distribution model + adjusting compartment distribution model have a good description based on in vitro data, in silico data and clinical PK data. More importantly, the developed PBPK model was well captured the PK profile after oral dose of 75 mg and 100 mg bupropion.
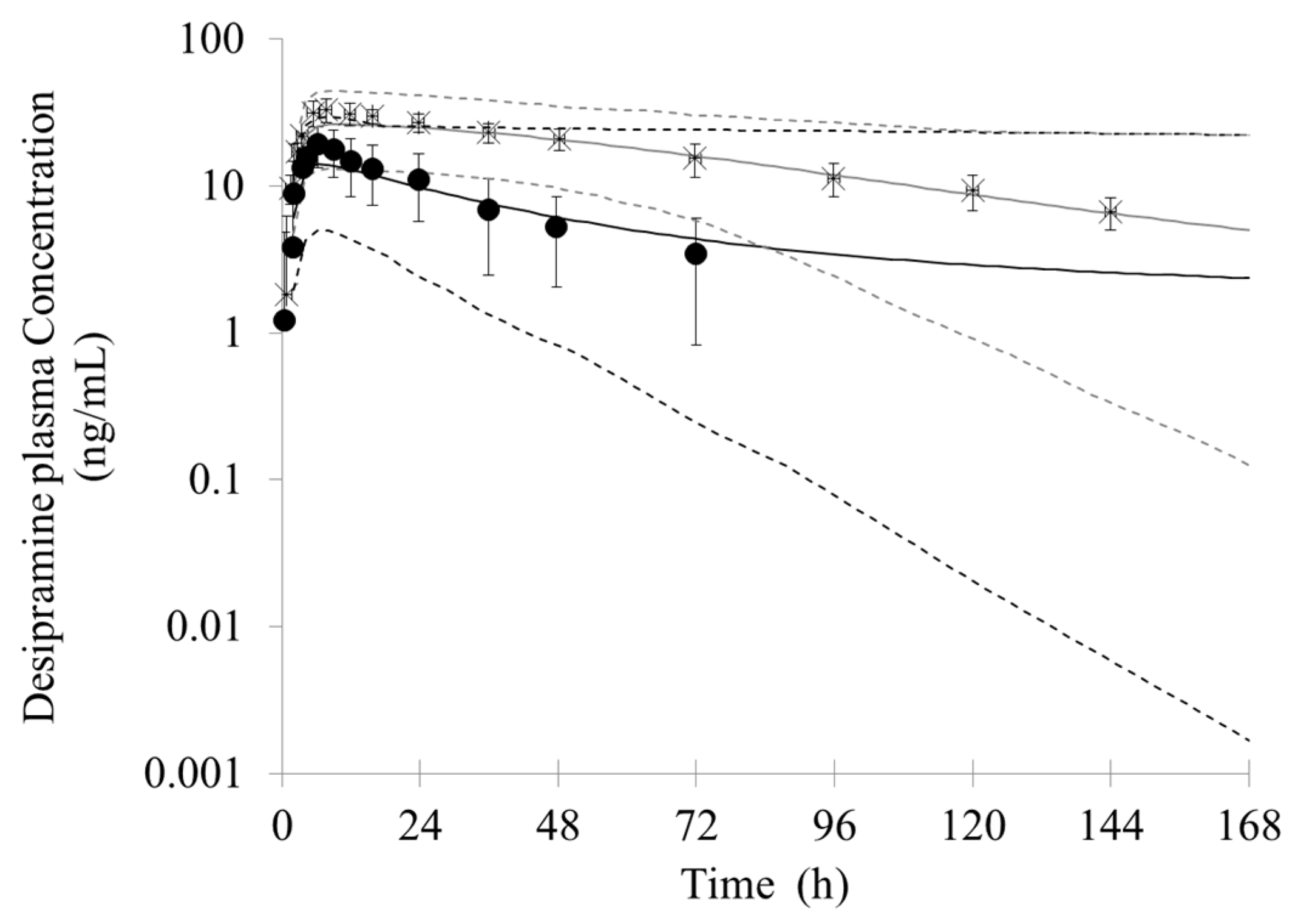
In the Simcyp, the user can only simultaneously select one specified inhibitor metabolite to simulate the interaction effects. To better describe the actual clinical DDI, metabolites were regarded as different inhibitors and combined with bupropion to simulate the complex DDI with other CYP2D6 substrates. A sensitivity analysis on the dosage of metabolites was conducted; the results predicted by the model were in good agreement with the observed PK profiles when doses of hydroxybupropion, threohydrobupropion and erythrohydrobupropion were assumed to be 90 mg, 30 mg and 4 mg, respectively. This indicates that the plasma concentration of metabolites formed by single oral 150 mg bupropion is equivalent to plasma levels in vivo after a single oral of 90 mg hydroxybupropion, 30 mg threohydrobupropion and 4 mg erythrohydrobupropion, respectively. (
Figure 6). The predicted C
max (hydroxybupropion 443 ng/mL threohydrobupropion 107 ng/mL and erythrohydrobupropion 16 ng/mL) and AUC (hydroxybupropion 15,215 ng∙h/mL, threohydrobupropion 1178 ng∙h/mL and erythrohydrobupropion 185 ng∙h/mL) were within 2-fold error of the observed values [
34,
47].
To sum up, dynamic PK process of bupropion and its metabolites were well characterized in PBPK models. The successful simulations of clinically observed PK profiles build confidence in the prediction and mechanistic understanding of the DDI caused by bupropion, and particularly the unexpected DDI potential contributed by its metabolites. On the basis of the above model, we then applied the PBPK model to predict the clinically observed DDIs involving bupropion and its metabolites as the CYP2D6 inhibitors. The result contribute to the understanding of the involvement and impact of inhibitory metabolites on DDIs observed in the clinic.
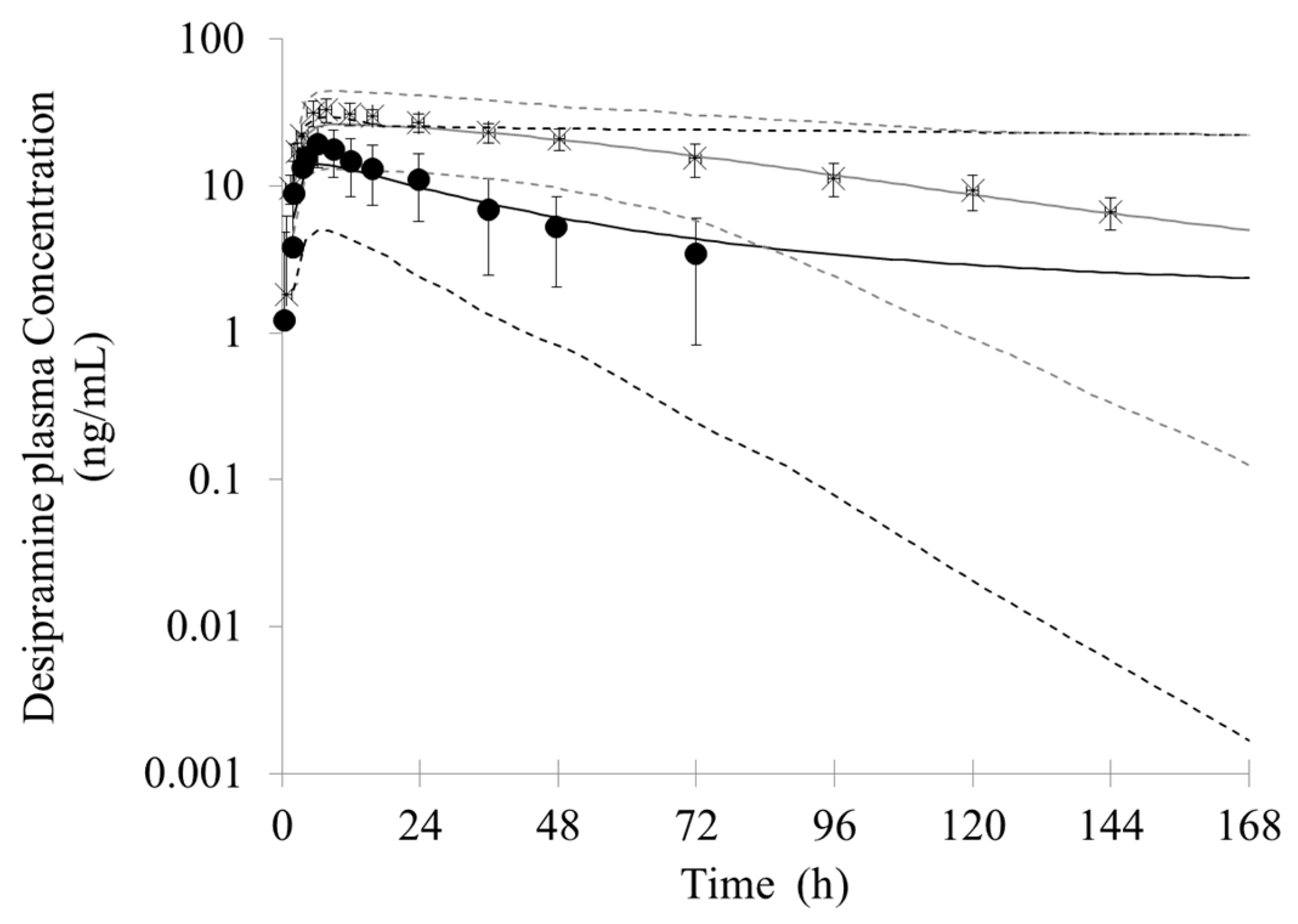
In the simulation of bupropion-desipramine interaction, the addition of the inhibitory metabolites into the PBPK model resulted in more accurate prediction of DDIs (AUC and C
max ratio) compared with that when only the inhibition of P450 from the parent drug (bupropion) was taken into account. The simulation suggests that bupropion and its metabolites contribute to the DDI between bupropion and desipramin. Although in vitro study showed that the inhibitory potency from strong to weak were erythrohydrobupropion, threohydrobupropion, hydroxybupropion and bupropion, respectively, the simulation of in vivo DDI suggests that hydroxybupropion is the most potent competitive CYP2D6 inhibitor. It can be possible due to the greater exposure of hydroxybupropion. The plasma level of hydroxybupropion is five- to ten-fold higher than the parent drug [
29,
69,
70,
71,
72]. The exposure of threohydrobupropion is similar to the parent drug; however, it has a stronger in vitro inhibition constant than parent drug and hydroxybupropion. For the erythrobupropion, it is predicted to have similar importance in in vivo DDIs as hydroxybupropion, despite the fact that its plasma concentration is much lower than hydroxybupropion. This may be related to its strongest inhibition constant. Conversely, even though the exposure of bupropion is similar to threohydrobupropion. The PBPK simulation shows bupropion is the weakest competitive CYP2D6 inhibitor. The result may attribute to the relatively weakest inhibition constant.
Consistently, a minimal effect of bupropion on venlafaxine was predicted if only the competitive inhibition from the parent drug was considered. With the addition of the inhibitory metabolites into the PBPK model, there was a more accurate prediction of DDIs. The inhibitory potency from strong to weak was hydroxybupropion, threohydrobupropion, erythrohydrobupropion, and bupropion, respectively.
In the DDI study of bupropion with other CYP2D6 substrates, the significant increase in exposure of dextromethorphan, metoprolol, bufuralol and tolterodine after coadministration of bupropion was predicted. These DDI predictions may explain the occurrence of severe sinus bradycardia after coadministration of bupropion and metoprolol and highlight the need for caution and dosage adjustment when combining bupropion with medications metabolized by CYP2D6.
To better understand the effect of stereo-selective bupropion and its metabolites on the DDI, a stereo-selective PBPK model for bupropion and its metabolites was further developed. The PBPK model considered multiple metabolic pathways including CYP2B6, 2C19, 3A4 and carbonyl reductase, and it is reasonable to describe the proportion of each metabolite in total clearance of bupropion. The simulated PK profile was a good match with the observed clinical data, although, in the simulation of DDI between bupropion with desipramine, the predicted DDI was lower than the observed. The results indicated that RR-hydroxybupropion was a major contributor to the inhibition of CYP2D6 from bupropion. The inhibitory effect of bupropion on CYP2D6 may be the result of synergistic production of all stereo-selective parent drugs and its metabolites. Currently, all inhibitors cannot be simultaneously integrated into the model for simulation. Only four inhibitors can be allowed to integrate into the model in Simcyp. In addition, the stereo-chemical threohydrobupropion and erythrohydrobupropion may have different inhibitory contributions compared to non-stereo-chemical, and the in vitro inhibition rate constants of the stereo-chemical threohydrobupropion and erythrohydrobupropion have not been reported. The PBPK model of stereo-selective bupropion and its metabolites still need to be further improved and optimized after obtaining more data in the future.
Overall, we successfully developed a PBPK model to describe the dynamic PK process of bupropion and its metabolites and understand the involvement and impact of inhibitory metabolites for DDIs observed in the clinic. The present bupropion PBPK model can be useful for predicting inhibition from bupropion in other clinical studies. However, the use of the PBPK model for a true prospective prediction of DDI caused by inhibitory metabolite is still very challenging, as the in vitro inhibition and human PK data for the metabolite are not routinely generated. To maximize confidence in the DDI prediction, more information is needed for the inhibitory potency of the metabolites towards the P450 enzymes.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}