Therapeutic Potency of Nanoformulations of siRNAs and shRNAs in Animal Models of Cancers
by
 , , and
, , and
Md. Emranul Karim
 ,
,
Kyi Kyi Tha
, 
Iekhsan Othman
,
Mohammad Borhan Uddin
and
Ezharul Hoque Chowdhury
* Jeffrey Cheah School of Medicine and Health Sciences, Monash University Malaysia, Jalan Lagoon Selatan, Bandar Sunway, 47500 Petaling Jaya, Selangor, Malaysia
*
Author to whom correspondence should be addressed.
Pharmaceutics 2018, 10(2), 65; https://doi.org/10.3390/pharmaceutics10020065
Submission received: 6 April 2018
/
Revised: 19 May 2018
/
Accepted: 22 May 2018
/
Published: 26 May 2018
Abstract
:RNA Interference (RNAi) has brought revolutionary transformations in cancer management in the past two decades. RNAi-based therapeutics including siRNA and shRNA have immense scope to silence the expression of mutant cancer genes specifically in a therapeutic context. Although tremendous progress has been made to establish catalytic RNA as a new class of biologics for cancer management, a lot of extracellular and intracellular barriers still pose a long-lasting challenge on the way to clinical approval. A series of chemically suitable, safe and effective viral and non-viral carriers have emerged to overcome physiological barriers and ensure targeted delivery of RNAi. The newly invented carriers, delivery techniques and gene editing technology made current treatment protocols stronger to fight cancer. This review has provided a platform about the chronicle of siRNA development and challenges of RNAi therapeutics for laboratory to bedside translation focusing on recent advancement in siRNA delivery vehicles with their limitations. Furthermore, an overview of several animal model studies of siRNA- or shRNA-based cancer gene therapy over the past 15 years has been presented, highlighting the roles of genes in multiple cancers, pharmacokinetic parameters and critical evaluation. The review concludes with a future direction for the development of catalytic RNA vehicles and design strategies to make RNAi-based cancer gene therapy more promising to surmount cancer gene delivery challenges.
1. Introduction
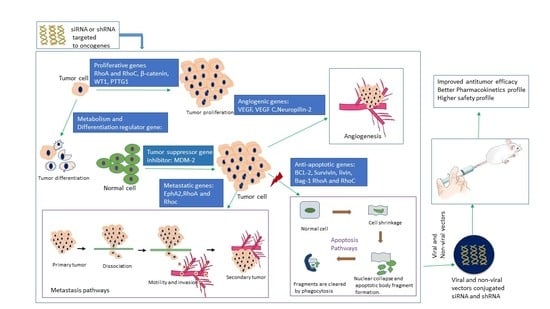
Gene therapy is the delivery of functional genes into the cell to modulate gene expression and the extent of their expression. Specificity and stability in biological media should be adequate to carry the corrected genetic information to their progeny for consistent effects. Since the discovery of gene sequencing technology, the journey from concept to clinical trial has faced a lot of hurdles to attain clinical reliability [1]. The era of catalytic RNA added a new dimension to the management of a broad range of critical human disorders like cancers and Alzheimer’s disease. The formal appearance of RNAi techniques based on a double stranded RNA (dsRNA) came into sight at the end of the nineteenth century to silence genes in nematodes [2,3]. The long dsRNA is able to silence gene expression in both nematodes and mammalian cells, but its non-specificity, sequence-independent pathways and activation of innate immune response make it incompatible in gene silencing technology [4,5]. At the beginning of the twentieth century, vigorous research on minimizing the shortcomings of dsRNA concluded that long dsRNAs can be processed into less than 30 base pair sequences, inducing the sequence-specific silencing of targeted genes in mammals [6]. This finding unfolded synthetic siRNA (small interfering RNA) as a new biological tool for silencing the genes in mammalian cells specifically. The smaller fragments of siRNA (21–23 nucleotides) are processed from long pieces of dsRNA by the enzyme dicer [7,8]. The siRNA can be produced synthetically and delivered into mammalian cells via multiple transfection methods. After reaching the cytoplasm of the target mammalian cell, siRNA is incorporated with RNA-induced silencing complex (RISC) [9]. Argonaute 2, a multi protein of Argonaute family is the catalytic core of RISC that unwinds the siRNA double strand and degrades the sense strand (passenger strand). The activated guide strand (anti-sense strand) containing RISC identifies and guides the sequence-specific cleavage at a position of nucleotides 10–11 of target mRNA by post-transcriptional gene silencing mechanisms, preventing translation and silencing specific gene expression [10,11] (Figure 1). The activated RISC then heads to other mRNA targets until their degradation. The degradation of activated RISC requires 3–7 days for fast growing cells and several weeks for slowly dividing cells [12,13]. On the other hand, short hairpin RNAs (shRNAs) are synthesized in the nucleus of a cell, transported into the cytoplasm and processed into siRNA for silencing target mRNA via binding with RISC. The shRNA expression vectors, therefore, need to be transported into the nucleus of a cell for transcription [14,15,16]. The precisely designed siRNA can specifically silence any gene in the body and, therefore, appears to be therapeutically more potent than conventional small molecular drugs.
Cancer is generally characterized by genetically abnormal signal transduction pathways that lead to heterogeneous cellular proliferation because of mutation and heritable alteration in an oncogene or tumor suppressor gene [17]. Cancer gene therapy includes gene amplification or deletion, suicide gene therapy, inactivation of oncogene by epigenetic silencing, mutation correction, chromosomal translocation, tumor suppressor activation and blocking of angiogenesis via the delivery of nucleic acids like plasmid DNA (pDNA), mRNA, short interfering or silencing RNA (siRNA), micro RNA (miRNA), short hairpin RNA (shRNA), anti-sense oligonucleotides (AONs) and DNA enzymes into the target cells [18,19,20,21]. Since the emergence of gene therapy, approximately 1300 clinical trials in different cancers have been taken place [22,23]. Cancer management still poses a major challenge with high incidence and mortality rates (Figure 2) due to the detrimental effects of conventional treatment modalities, despite the fact that the number of cancer survivors in the USA is increasing because of advancement in early cancer diagnosis and treatment [24].
Here, we review the current advancement of siRNA-based cancer therapy, emphasizing the challenges and breakthroughs of siRNA delivery especially in preclinical studies and the development of various delivery vehicles from viral vectors to inorganic nanoparticles in overcoming the biological barriers. The roles of genes in multiple cancers and the knockdown effects of those genes in tumor regression will be discussed, highlighting the pharmacokinetic parameters of the delivery vehicles in different animal models of cancers.
2. Challenges and Breakthrough of siRNA Delivery: From Concept to Clinical Trial
Gene sequencing technology has gained significant attention owing to its ability to unmask genomic data for treating disease at the genetic level. The effective and controlled intracellular delivery of siRNAs into cancer cells and cancer stem cells both in vitro and in vivo has remained a long-standing challenge, hampering siRNA-based therapeutics to move into the clinical setting. The amount of siRNA should be optimized for maximum potency. Although naked siRNA without and with chemical modification demonstrated silencing and therapeutic efficacy in the lung [26] and brain [27], respectively, following local administration, systemic administration of unmodified siRNA is subjected to nuclease degradation, phagocytic uptake, aggregation with serum protein and rapid renal clearance before reaching the cytoplasm of the target cell [28,29,30,31] (Figure 3). Once the siRNA extravasates, it faces the barriers of the dense structure of Extracellular matrix (ECM) [32], higher interstitial fluid pressure [33] and negatively charged cellular membrane. After endocytosis-facilitated entry into the cell, siRNA should escape from the endosome; otherwise, it would be subjected to lysosomal degradation [34]. Besides, naked siRNA exerts innate immune responses and off-target effects and demands repeated dosing for consistent effects [35,36,37]. To overcome these hurdles, an effective delivery vehicle is required for the delivery of siRNA to the target site by preventing the naked or modified siRNA from biological degradation and enabling it to cross the intended cellular membrane and release from the endosome [9,38,39].
Delivery vehicles are categorized into two main subclasses: viral vectors and non-viral vectors (Figure 4). The viral vectors like retroviruses, adeno viruses, and adeno associated virus (AAV) have been successfully used to carry genes in both preclinical and clinical trials (69% of total human trials are viral-assisted cancer gene therapy) [23]. The engineered viral vectors possess high specificity, unassisted entry to the cell membrane, lack of replication and pathogenicity and higher gene expression efficiency [40,41]. The activation of innate immune response, carcinogenicity and higher treatment cost of viral vectors limit their use in the clinic [20,40,42,43,44], thus motivating to develop non-viral vehicles for specific delivery of nucleic acids to the target cancer cells. The non-viral vectors are non- immunogenic, biologically compatible, higher specificity, higher packing capacity and less manufacturing cost in contrast to viral counterparts [23,45]. They are mainly subdivided into two categories depending on their composition: organic and inorganic carriers. The organic nanoparticles are widely used and are categorized into three classes based on their constituents, namely cationic lipids, cationic polymers and hybrids of both lipids and polymers. Several preclinical studies using lipid nanoparticles (LNPs), dendrimers, carbon nanotubes, graphene oxide and micelles, etc. as carriers have been accomplished for silencing mutant genes in multiple animal models of cancer. Among them, a number of organic nanocarriers are in clinical trials. The poor delivery efficiency, low rate of endosomal escape (1%) and retarded release of siRNA from the carriers and associated toxicity have slowed down the success rate of organic carrier-based RNAi therapy for cancers management [46,47,48]. On the other hand, inorganic nanoparticles, the newly introduced nanocarriers, have been widely explored to suppress the shortcomings of organic carriers in a gene silencing platform. Inorganic nanoparticles possess unique physicochemical properties, biocompatibility, flexibility to be functionalized with different ligands and surface-coating materials, such as Polyethylene glycol (PEG), controlled release pattern and compatibility with other existing therapeutic agents [49]. The inorganic nanoparticles that are frequently used in both in vitro and in vivo studies to repress multiple overexpressed cancer genes are gold NPs, magnetic NPs, up conversion NPs, mesoporous silica NPs, pH sensitive carbonate apatite NPs and other multifunctional NPs. Both organic and inorganic carriers have undergone several modifications like PEGylation for escaping RES (reticuloendothelial system) and renal clearance, attachment of targeting moiety like ligands and antibodies for specific delivery as part of active targeting, cell-penetrating peptides (CPP) for improving the cellular uptake and stimuli-responsive linker to improve the therapeutic index of antisense cancer gene therapy [50].
The emergence of multifunctional NPs and comprehensive investigation of RNAi against oncogenes led to potential NP-based cancer gene therapies and more than 40,000 (based on active targeting strategy) studies have been reported in the last decades [51]. However, the issues of biological stability and delivery of siRNA in the presence of multiple physiological barriers have yet to be fully resolved. Tumor microenvironment is another prominent barrier for successful intracellular delivery of RNAi therapeutics. Although the immature blood vessel with heterogeneous hyper permeability and the irregular lymphatic vasculature cause abnormal blood circulation and poor lymphatic drainage [52], respectively, resulting in enhanced permeability and retention of NPs in tumor tissues, higher interstitial pressure, synthesis of heterogeneous extracellular matrix molecules (e.g., glycoprotein) and excessive matrix metalloproteinase pose hindrance to insufficient delivery of the particles [53,54,55]. Poor cellular uptake by RNAi therapeutics is another hurdle arising as a result of repulsion between the negatively charged siRNA and the anionic cell membrane, and could be improved via chemical modification, e.g., by coating with cell-penetrating peptide [56] or stably complexing or conjugating with NPs. After entering a cell, the siRNA is usually transported from early endosome to late endosomes for degradation. To get better silencing outcome, siRNA could be released in the cytosol from endosomes at an early stage, for instance, by complexing with pH-sensitive nanoparticles (e.g., carbonate apatite). Exocytosis or cellular recycling of internalized siRNA via recycling routes e.g., non-secretory exosome might reduce the silencing efficiency of siRNAs, and could be prevented by using an exocytosis inhibitor [57,58]. Despite the successes in cell culture and preclinical studies, a small number of NPs-based siRNA formulations have successfully entered clinical trials (Table 1). Toxicity of vectors, immunogenicity and off-target effects of siRNA are the remaining hurdles in the clinic. Moreover, a difference between the physiology of humans and experimental animals, extracellular physiological barrier, technological barrier and inability to target metastases limit the success in clinical trials.
3. In-Vivo Delivery of siRNAs and shRNAs Directed against Different Cancer-Causing Genes in Various Cancer Models
3.1. Silencing of Bcl-2 Gene
Bcl-2 (a mitochondrial protein of 239 amino acids), is a crucial factor for promoting cell growth and survival by inhibiting apoptosis pathways of cells (Figure 5). It plays critical roles in the growth, development and metastasis of various cancers, including breast, lung, liver and gastric cancers [59,60,61,62]. Bcl-2 expression was found in 37% (10 out of 27) of biliary tract invasive cancers and the frequency of expression was observed in 90% of small cell lung cancers [63,64]. The role in cancer development and the frequency of expression make Bcl-2 as a potential target for cancer gene therapy.
The DNA vector-based Bcl-2 siRNA was synthesized under the control of H1 RNA polymerase III (pol III) promoter and mixed with liposome-protamine for evaluating anti-tumor activity of liposome-protamine-psilencer3.1 H1-Bcl-2 complexes (at a ratio of 10:3:1) in a liver cancer mouse model generated via subcutaneous injection of and H22 (liver tumor) cells into male Balb/C mice of 26 ± 2 gm. Intravenous injections of 0.4 mL of liposome-protamine-psilencer3.1 H1-Bcl-2, 0.4 mL of liposome-protamine-psilencer3.1 H1 and liposome-protamine(0.4 mL) as a control were given daily over a seven days period. There was a 66.5% decrease in tumor weight in the group of mice treated with liposome-protamine-psilencer3.1 H1-Bcl-2 in contrast to control group. Moreover, it was found that treatment with Bcl-2-silencing plasmid efficiently down regulated the expression of Bcl-2 gene in tumor cells and induced their apoptosis ratio remarkably [65]. Other researchers who used Bcl-2-targeted siRNA also observed consistent anti-cancer effects upon Bcl-2 gene silencing, indicating that the Bcl-2 antisense therapy is one of the most fruitful strategies to induce apoptosis in cancer cells [66,67].
In gallbladder carcinoma, antisense-Bcl-2-siRNA against aberrantly expressed Bcl-2 gene (positive expression rate is ~23.4–51.7%) exerted significant tumor regression effect in a mouse model study [68,69,70,71]. In an in vivo tumorigenicity study, Balb/c nude mice (4–6 weeks) of 18–22 gm were subjected to subcutaneous injection of Bcl-2-siRNA transfected human gallbladder carcinoma cells (GBC-SD) suspensions of 6 × 106 cells in 0.2 mL for experimental group and GBC-SD suspensions of 6 × 106 cells in 0.2 mL alone for control group into the left flank of the mice. After 39 days of treatment, it was found that the volumes of tumor for both Bcl-2-siRNA transfected group and the control group were 629 ± 78.9 mm3 and 1914 ± 125.0 mm3 respectively. The average weights of tumors were 0.77 ± 0.12 gm and 2.24 ± 0.33 gm for experimental and control groups, respectively, and the rate of tumor growth with Bcl-2-siRNA treatment was 14.99% compared to 45.58% in the control group. Thus, Bcl-2-siRNA treating mice group had three-fold reduction of tumor volume and weight in comparison to control group and significant lower tumor growth rate relative to control group [72]. For gene therapy evaluation studies, the antisense Bcl-2 sequences were mixed with recombinant plasmid vector and formed pSilencer™ EGFP sh515 (experimental group) and pSilencer™-EGFP shCon (negative control group) for comparative study. Subsequent to the establishment of a human gallbladder carcinoma model by implanting 6 × 106 of GBC-SD cells (human gallbladder carcinoma cells) into the Balb/C mice of 4–6 weeks, 10 µg of pSilencer™ EGFP sh515 with 30 µL of lipofectamine-2000 liposome (Invitrogen, Carlsbad, CA, USA) (experimental group) and 10 µg of pSilencer™-EGFP shCon with 30 µL of lipofectamine-2000 liposome (Invitrogen) (negative control group) were administered into multiple sites of peritumoral tissue for five times with a two-days. After 39 days of injection, the average tumor volume for pSilencer™ EGFP sh515 containing group was approximately 700 mm3 (experimental group) relative to negative control group, psilencer-EGFP shcon (1400 mm3) and control group (1400 mm3), demonstrating 50% reduction of tumor volume in recombinant DNA plasmid psilencer-EGFP sh515-treated mice [72].
In addition, cationic liposome LIC-101 and B717, a sequence-specific synthetic Bcl-2-siRNA was mixed to form B717/LIC-101 for exploring tumor inhibitory effect in a prostate cancer mouse model, developed by given 5 × 106 of PC-3 cells (prostate cancer cell) subcutaneously into 5 weeks old male Balb/C mice. Intravenous injections of 0.1 mg of B717/LIC-101 and 10% (w/v) maltose solution as a control were given for five times in a week from day 7 to day 18 after the tumor volume became palpable. The tumor volumes were measured by using Dunnett’s test at a regular time interval for 36 days of treatment. The group of mice receiving B717/LIC-101 was found to have smaller tumor volume (487 mm3) than the control group (1300 mm3), showing 63% reduction of tumor volume at the end of experiment [73].
Similarly, antitumor activity and pharmacokinetics parameter of sequence-specific Bcl-2 antisense siRNA (BO43) and liposome complex could be improved through encapsulation of PEG into the surface of liposomal–siRNA complex by enhancing blood retention time, reducing the plasma clearance by the liver and thus, increasing accumulation of siRNA in the tumor region. In a study, male Balb/c nude mice (5 weeks) were allowed to inoculate prostate tumor by injecting 3.0 × 106 of PC-3 cells (human prostate cancer cells) in 100 µL of the nutrient mixture, resulting in growth of tumor size approximately 50–80 mm3 after 10 days of subcutaneous injection. PEGylated B043/PEG-LIC (antisense Bcl-2/PEG-LIC) was given intravenously at three different doses of 1 mg/kg, 3 mg/kg, and 10 mg/kg in two five-day consecutive courses of injections and 10% maltose solution for control groups. After 24 days of post injection, the average tumor volumes for treatments with 1 mg/kg, 3 mg/kg, and 10 mg/kg of B043/PEG-LIC were 310 mm3, 220 mm3 and 160 mm3, respectively, whereas for the control group it was 450 mm3, demonstrating the tumor inhibitory effects (65% at a dose 10 mg/kg of B043/PEG-LIC) in a dose-dependent manner. Accumulation of siRNA in S.C. tumor was also improved three-fold more than non-PEGylated liposome, resulting in superior targeted antitumor action of PEGylated siRNA-LIC complex [74].
3.2. Silencing of VEGF Gene
Angiogenesis is a developmental and adult physiological process of the formation of new blood vessels from preexisting micro vessels that regulate growth and metastasis of solid tumor [75,76,77,78]. Several pathological conditions like hypoxia, ischemia and inflammation result in imbalance between proangiogenic and antiangiogenic factors, leading to interactions among endothelial cells, tumor cells and a variety of growth factors. This imbalance leads to extracellular matrix remodeling, endothelial cell migration and proliferation, and capillary differentiation of tumor cells [79]. VEGF (vascular endothelial growth factor) also known as vascular permeability factor is an endothelial cell specific mitogen that plays a vital role in stimulating angiogenesis and vascular permeability through binding with VEGF tyrosine kinase receptors VEGFR1 (Flt-1), VEGFR2 (KDR, Flk-1) and VEGFR3 [80,81,82,83]. There are six subtypes of VEGF protein including VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E and placental growth factor that regulate tumor-induced angiogenesis, vascular permeability and cell migration for the survival of cancer cells [84,85,86]. Over expression of VEGF and vascular density have been found to be higher in many human tumors including breast cancer and renal carcinoma, relative to normal cells [87,88,89,90,91]. Therefore, to stop metastasis of various cancer cells, VEGF gene silencing could be one of the most efficient therapeutic modalities [92,93,94].
A replication-deficient recombinant adenoviral vector-based antisense-VEGF-cDNA (Ad5CMV-αVEGF) was developed [95] and applied in an in vivo study for exploring the antitumor efficacy against a breast tumor induced by subcutaneous injection of 5 × 106 of human breast cancer cells (MDA251-MB) into the mammary fat pads of athymic female nude mice (4–6 weeks of age). E1-deleted adenovirus type-5 without the gene (Ad5 (d1312) [96] and Ad5CMV-αVEGF were given intratumorally with 5 × 108 plaque-forming units (PFU) into the mice bearing breast tumor after 4 days of inoculation. The mean tumor sizes for the Ad5CMV-αVEGF and Ad5 (d1312) were, respectively, 67.85 ± 34.65 mm3 and 335.23 ± 83.98 mm3, with 80% reduction in tumor size by the former (Ad5CMV-αVEGF) [97].
A non-viral polyplex-based delivery vehicle, composed of PEG-b-poly (L-lysine) di block co-polymer and PEG-grafted PEI was synthesized to deliver therapeutic genes with improved biological stability, reduced virus-associated toxicity and increased anti-tumor effects [98,99,100,101,102]. Antisense-VEGF-siRNA was encapsulated with polyplex (VEGF-siRNA-PEG) and polyethyleneimine (PEI) to form VEGF-siRNA-PEG/PEI PEC micelles to assess the tumor inhibitory effects for both intratumoral and intravenous administration in a prostate tumor model generated by subcutaneous injection of 1.5 × 106 PC-3 cells (human prostate cancer cells) into the flank of the mouse. 500 pmol of VEGF-siRNA formulation at 0, 6 and 15 days was given intratumorally after tumor size grew to 50 mm3. Tumor regression result showed 79% reduction of tumor volume in VEGF-siRNA-PEG/PEI PEC micelles-treated mice (120 mm3) compared to the untreated group (590 mm3). For systemic delivery, 1.5 nmol of VEGF-siRNA-PEG/PEI PEC was given intravenously into the tail vein of mice at 0, 4, 10, 18 and 28 days, exerting drastic tumor inhibitory effects (86%) (tumor volume, 196 mm3) relative to the control group (tumor volume, 1400 mm3) [103].
VEGF-C, a subtype of the VEGF family is an essential lymphangiogenic factor which promotes lymphogenesis, tumorigenesis and metastasis of cancer cells [104,105,106,107]. The autocrine effect of upregulated VEGF-C initiates several intracellular signaling pathways that mediate tumor progression in various cancers like ovarian cancer, breast cancer, bladder cancer and non-small cell lung cancer (NSCLC) [105,106,107,108,109,110,111,112,113,114,115,116,117,118,119]. Hifectin was employed to deliver VEGF-C-targeted siRNA (siV2) and assess potency of the delivered siRNA against the breast tumors developed by subcutaneous administration of 5 × 104 of 4T1 cells (mouse breast cancer cells) in the right-front dorsum of a Balb/C female mouse (4 weeks old) of 18–20 gm each. When the size of tumors reached at ~0.1 cm3, the mice were treated with siV2 (1 µg/gm body weight), scrambled siRNA (SCR) (1 µg/gm body weight) as a negative control and PBS (Blank control) intratumorally for every 2 days. After 3 days of post injection, the mean tumor weights for siV2, SCR and controls were 2.272 g, 3.242 g and 3.185 g, respectively, and the tumor volume for either control or SCR was approximately 2.5 cm3, whereas that for the siV2-treated group was 1.8 cm3, thus demonstrating 28% reduction of tumor volume in comparison to the control group [120].
In addition, to increase the antitumor efficacy VEGF-C-directed siRNA was mixed with lentivirus (Lv-VEGF-C-siRNA) for specific delivery to the target gene. The knockdown efficacy of virus-associated VEGF-siRNA (Lv-VEGF-C-siRNA) was assayed in a lung cancer mouse model generated via subcutaneous injection of 1 × 107 A549 cells into the left flank of a Balb/c nude mouse at 6 weeks of age. Lv-VEGF-C-siRNA, Lv-siRNA negative control and PBS at a dose of 250 µL in 24 h intervals for 3 weeks into the tail vein of the mice were given after 15 days of subcutaneous injection. The average volumes of tumors for Lv-VEGF-C-siRNA-treated group and untreated group were 87.36 ± 10.93 and 241.88 ± 34.03, respectively. The weights of tumor for untreated and Lv-VEGF-C-siRNA-treated groups were 1079 ± 168.47 mg and 565.57 ± 89.33 mg, respectively, thus showing reduced tumor weight (64%) and volume (48%) in Lv-VEGF-C-siRNA-treated group relative to the untreated group. Protein and mRNA expression analysis confirmed that the silencing of VEGF-C gene expression lowered the expression of VEGFR-2, VEGFR-3, CXCR4 and CXCR7 significantly in NSCLC and suppressed the effects of AKT, ERK and p38 signaling pathways, thereby regressing the tumor growth [121].
Neuropilin-2 (NRP-2), a non-kinase cell surface receptor binds with VEGF and regulates vascularization and lymphogenesis of various tumors. Despite its expression in normal tissues, over expression of NPR-2 is found in colorectal cancer cells and important for the development and metastasis of colorectal cancer cells [122,123,124]. Liposomal complexes of NRP-2-targeted siRNA (NPR-2-siRNA-DOPC) could be used to slow down the tumor growth. Following administration of 1×106 cells of HTC-116 cells (lenti-luc-transfected colorectal cancer cell) into the male athymic nude mice (6–8 weeks old), NPR-2-siRNA/liposomal complexes (NPR-2-siRNA-DOPC) and siRNA-DOPC (without NPR-2) as a negative control were given intraperitoneally at a dose of 5 µg of siRNA on every 5 days. The tumor volumes were measured 36 mm3 and 420 mm3 for both NPR-2-siRNA-DOPC- and control siRNA-DOPC-treated mice. Tumor growth was clearly slowed down by NPR-2-siRNA-DOPC (91.43%) compared to control siRNA-DOPC [125].
Although targeted therapeutic gene delivery has been successfully used to inhibit tumor growth by silencing specific genes, toxicity has not been diminished completely due to non-specific immune reactions and poor intracellular uptake [126,127,128,129,130]. To overcome this hurdle, self-assembled siRNA layered nanoparticles (nanoplexes) comprising PEI and PEGylated Ar3-Gly-Asp (RGD) peptide ligand were synthesized to target the tumor neovasculature expressing integrin to attenuate the VEGFR2 expression and tumor angiogenesis [131,132,133,134,135,136,137,138,139]. VEGF-R2 sequence-specific siRNA encapsulated with RGD-PEG-PEI (RPP) was subjected to an in vivo study for assessing the antitumor activity against a neuroblastoma tumor developed by administrating 1 × 106 of N2A cells (mouse neuroblastoma cells) into the flank of the female nude mice (6–8 weeks old). After 7 days of inoculation, when the mice had tumor size of 20 mm3, the mice were treated with RPP-nanoplexes-siLacZ (nonspecific siRNA) and RPP-nanoplexes-siVEGFR-2 at a dose of 40 µg into the tail vein at every 3 days. The average tumor volume measured for VEGFR-2 sequence-specific siRNA-treated group was approximately 100 mm3, whereas in siLacZ-treated and control groups, the mean volumes were 1700 mm3 and 1200 mm3 respectively, demonstrating that RPP-VEGFR-2 siRNA reduced 91.67% of tumor volume in comparison to the control groups [100].
Taken together, VEGF and its subtype VEGF-C, VEGF receptors (VEGFR) and their signaling pathways NRP-2 play vital roles in developing tumor and lymphangiogenesis. The adenoviral vector-mediated siRNA reduced 80% of tumor volume in breast cancer. However, non-viral carrier, PEC loaded siRNA was shown to lower 86% of tumor volume with a minimal toxicity in prostate cancer mouse model. In case of VEGF-C, naked anti-VEGF-C-siRNA reduced only 28% of tumor volume, but when it was mixed with lentivector the inhibition rate was increased up to 48%. NRP-2 along with liposomal complex also inhibited 91.43% of tumor volume in colorectal cancer. VEGFR 2 with nanoplexes-ligand reduced 91.67% of tumor volume without any notable toxicity in neuroblastoma. The anti-tumor effects following nanoparticle-assisted delivery of tailored siRNAs directed against VEGF, NRP-2, VEGFR 2 and EGFR genes warrant further clinical investigation like toxicity to establish VEGF targeted gene therapy either alone or in combination as successful modalities for different cancer treatments. The tissue-targeted siRNA therapy allows huge improvement in silencing technology and identification of genes as the most selective targets for cancer gene therapy.
3.3. Silencing of EGF Receptor Genes
Epidermal growth factor (EGF) receptors, EGFR-1 and EGFR-2 are cell surface proteins that initiate growth signals in a normal cell. EGF receptors stimulate tyrosine kinase activity and activate downstream signaling pathways, playing key roles in cell division and proliferation [140]. EGFR triggers the expression of VEGF gene. Overexpression of EGFR-1 and EGFR-2 has been found in many breast cancers (30%) [141]. To improve the silencing efficacy of EGFR-1 and EGFR-2, siRNAs targeting EGFR-1 and EGFR-2 were electrostatically complexed with pH-sensitive carbonate apatite (CA) nanoparticles [142] and the anti-cancer effects of the complexes were assessed in a murine syngeneic breast cancer model. Breast tumors were inoculated with subcutaneous injection of 1 × 105 4T1 cells into the mammary pads of female Balb/c mice (15–20 g) with ages of 6–8 weeks. When the tumor volume reached 13.20 ± 2.51 mm3, CA-siRNA(s) against EGFR-1(50 nM) or EGFR-2 (50 nM) were given intravenously into the tail vein of mice at 3 days with a total of two injections. CA-siRNA(s) directed EGFR-1(50 nM) or EGFR-2 (50 nM) showed no robust tumor regression after 29 days of treatment. On the other hand, concurrent delivery of EGFR-1 and EGFR-2 siRNAs with carbonate apatite exerted significant tumor volume reduction (61%) in contrast to control groups. These results suggested that combined delivery of multiple siRNAs with pH-sensitive CA is a very promising strategy in treating cancer, highlighting a huge potential of pH-responsive drug delivery to the tumor microenvironment [143].
3.4. Silencing of Survivin Gene
Survivin is a human gene that encodes a 16.5-kD wild-type protein of 142 amino acids, which is extensively expressed at G2/M phase and declines rapidly in G1 phase of the cell cycle [144,145]. Survivin is one of the subtypes of apoptosis protein inhibitor family (IAP) including X-linked inhibitor of apoptosis (XIAP), cIAP1, cIAP2, NAIP (NLR family, apoptosis inhibitory protein), Livin, ILP2 (IAP-like protein) and BRUCE [146,147,148]. It suppresses apoptosis by blocking both intrinsic and extrinsic pathways of apoptosis [149]. Survivin can be co-immune precipitated with caspases-3, caspases-7 and caspases-9 processing and blocks apoptosis [150]. It also mediates cellular division by increasing escape from G1 checkpoint arrest and facilitates to enter into S phase subsequently and influences tumor aggressiveness in cancer patients [145,151,152,153]. Survivin helps to develop chemo-resistance against various chemotherapeutics, thereby increasing tumor recurrence rate [154,155,156,157]. Survivin expression in normal tissue is low, but an aberrant expression of Survivin is found in various types of cancers, including esophageal, lung, ovarian, central nervous system, breast, colorectal, bladder, gastric, prostate, pancreatic, laryngeal, uterine, hepatocellular, renal cancers as well as sarcoma, melanoma and hematologic malignancies [158]. This abnormal expression of Survivin makes it a potential biomolecular target for different therapeutic strategies of cancer management. Several RNAi studies targeting Survivin gene in vitro and in vivo were performed by using different carriers to deliver specifically and explore the roles of Survivin in cancer development and metastasis as well as the antitumor activity of anti-Survivin-siRNA in various cancer cell lines (Table 2).
Chitosan, a non-viral vector for gene delivery, has been widely used due to their nontoxic, non-immunogenic, biodegradable and biocompatible properties [159]. The siRNA targeted to the Survivin was electrostatically binds chitosan (CS-siRNA) for improving siRNA stabilization against nuclease-mediated degradation and avoids renal clearance [160,161]. However, a poor water-soluble CS-siRNA complex is attacked by blood proteins and attributed to protonation effects in biological media. To overcome this hurdle, the CS-siRNA complex was grafted with PEG by an ionic gelation method for increasing stability and solubility in biological media [162]. The PEG provides steric stabilization effect of CS-siRNA complex against blood proteins and cells [163,164]. Mouse breast cancer cell lines (4T1) were inoculated into female Balb/C mice at the age of 4–6 weeks, and PEG-CS-siRNA (0.3 mg siRNA/Kg), free siRNA (negative control group) and saline (blank) were administered for total five times at every 2 days after the mice had a tumor volume of 100 mm3. At 28 days of post-injection, the tumor volume of PEG-CS-siRNA-treated mice was found to be approximately 500 mm3, whereas for saline and naked siRNA treatments the volumes were approximately 1500 mm3 and 1100 mm3, respectively. PEGylated-CS-siRNA demonstrated increased biological stability and selective tumor accumulation, thereby reducing tumor growth (55%) more significantly than the non-PEGylated ones [165].
Although chitosan nanoparticles (CS-NPs) are stable in biological media, the use of CS-NPs is limited due to poor cell penetrating capability and buffering capacity. Cell penetrating peptides (CPPs), 6-poly arginine and histidine (H6R6) were combined with CS-siRNA for improvising cellular internalization and mediating early endosomal escape of antisense siRNA into the cytoplasm [166,167,168]; 6-poly arginine has strong positive charge which helps to transport of nucleic acids into tumor more easily, whereas poly-histidine increases the buffering capacity of chitosan nanoparticles and thereby destabilizes endosomal membrane in the acidic environment for rapid endosomal escape [169,170,171,172,173]. The H6R6-CS and siRNA directed to Survivin (H6R6-CS-siRNA nanoparticles) was combined by complex coacervation method [174,175] before being investigated for the activity against tumor growth and metastasis. Breast tumors were induced into 4–6-week-old Balb/C female mice via subcutaneous injection of 4T1 cells prior to the evaluation of antitumor efficacy of H6R6-CS-siRNA at a concentration of 0.3 mg/kg through injections of the formulation for five times at a two days after the tumor size became 130–140 mm3. Naked siRNA, as well as saline, were also given as controls. After 28 days of treatment, the tumor volume for H6R6-CS-siRNA was approximately 600 mm3, whereas in blank and naked siRNA-treated group the volumes were, respectively, 1600 mm3 and 1200 mm3, thus demonstrating significant antitumor efficacy for H6R6-CS-siRNA complexes (63% reduction of tumor volume) compared to the control group. The improved cellular internalization and early endosomal escape capability of H6R6-CS-siRNA complexes resulted in more transfection efficiency with higher capability of silencing Survivin gene [176].
Melanoma is one of the most common malignancies characterized by strong metastasis and chemo-resistance properties, leading to higher mortality rate relative to other cancers [177,178,179]. The combined action of anti-apoptotic factors, pro-apoptotic effectors and stringent survival signal contributes to the development of chemo-resistance. The anti-apoptotic protein Survivin has a strong correlation with melanoma metastasis and is found to be expressed at a higher level in melanoma which makes Survivin a potential therapeutic target for melanoma treatment [151,152,153]. Sticky siRNA (ssiRNA) with cationic linear polyethyleneimine (PEI) was shown to promote gene silencing by improving stability and reducing conventional toxicity [180]. Murine melanoma tumors were implanted in NMRI nude female mice (5 weeks old) by subcutaneous injection of 1 × 106 B16-F10 cells into the right flank of mice. When the tumor volume grew to 50 mm3, the mice were treated with glucose (control), negative control ssiRNA and antisense Survivin-siRNA/PEI at a dose of 1 mg/kg. At 10 days of post injection, the tumor volumes for both ssiRNA-Survivin/PEI and control groups were approximately 1000 mm3 and 2000 mm3, respectively. The ssiRNA-Survivin/PEI reduced 50% of the tumor volume in comparison to the control group, via attenuating the Survivin gene expression [181].
Sialic acid (SA) is found to be overexpressed on cell surfaces of most cancer cells and aids in tumor metastasis by avoiding immune recognition of cancer cells [182,183,184]. Phenylboronic acid (PBA) grafted with polyethyleneimine (PBA-PEI) for targeting SA, and shielded with PEG (polyethylene glycol) to increase stability in systemic circulation, was shown to reduce off-target effects and carry anti-Survivin siRNA into targeted tumor cells. The PEG shell which could be detached from NP-siRNA complexes at extracellular pH of tumor enabled internalization of the NP-siRNA complexes into cancer cell. After internalization, the PBA-ribose ester bond was completely disrupted at the acidic pH of tumor microenvironment, releasing the siRNA into the cytoplasm. Tumor growth inhibition of the dual pH-responsive nanoparticle complex (PEG-CPB-PEI (PCPP)/siRNA) was studied in a breast tumor mouse model generated by subcutaneous injection of 1 × 105 of 4T1 cells in mammary fat pad of Balb/C mice. When the mice had a tumor of 80 mm3, the tumor-bearing mice were treated 6 times in every 3 days with saline, naked siSUR, PEI1.8ksiSUR, PCPPsiN.C (siRNA of nonsense sequences), and PCPPsiSUR (siRNA of anti-Survivin gene) at a dose of 1 mg/kg (siRNA). The tumor volumes of the mice treated with PCPPsiSUR and control groups were approximately 500 mm3 and 1300–1500 mm3, respectively, revealing notable tumor volume reduction (66%) in comparison to the control groups [185].
The rigorous study on Survivin demonstrated that Survivin is one of the key players to drive cancer formation by mediating apoptosis in various tumors. The issue of effective, non-toxic delivery and cellular internalization is a key challenge for successful siRNA delivery. To address this challenge, researchers are trying to establish a safe and secure carrier for the successful delivery of Survivin into target cancer cells. Sticky siRNA (ssiRNA) with Polyethyleneimine (PEI) reduced 50% of tumor volume in malignant melanoma. Additionally, when the siRNA was given with polyethylene grafted chitosan (PEG-CS), the tumor reduction rate increased to 55% along with reduction of side effects. Recently, to improve cellular internalization, cellular peptide proteins (CPPs) with chitosan were used to carry Survivin-siRNA which improved the antitumor efficacy (63%). Moreover, dual pH-responsive PCPP exerted significant tumor regression effects (66%) without hampering normal physiology of mice.
3.5. Silencing of Cyclin-B1 Gene
Cyclic-B1 is a regulatory protein, involved in mitosis. Cyclin B1, along with Ser/Thr kinase Cdc2 (cyclin-dependent kinase1, cdk1), forms the “mitosis promoting factor” and this complex triggers the cell from G2 phase to mitosis [186,187,188]. Overexpression of cyclin-B1 resulted in uncontrolled cell proliferation and hampered the stability of chromosomes [189,190,191,192,193]. Deregulated Cyclin-B1 expression has been observed in various cancers like esophageal squamous cell carcinoma, laryngeal squamous cell carcinoma, and colorectal cell carcinoma and induces resistance against radiotherapy in different tumors [194,195,196]. The silencing of the cyclin-B1 gene caused induction of growth arrest at G2 phase and stopped cancer cell division [197,198]. The inevitable role of cyclin-B1 in abnormal cellular growth makes it a striking therapeutic target for cancer management. Several studies with the siRNA targeting cyclin-B1 were accomplished in the mouse model for evaluating its role in tumorigenesis (Table 2).
Non-covalent bonding between carrier and siRNAs is a very promising technique for complexing and delivering siRNA into tumor cells as well as embryonic stem cells [199,200,201,202,203] more efficiently. Cell-penetrating peptide, MPG-8 (Primary amphipathic peptide carrier) binds with siRNA non-covalently to form stable complexes with an siRNA which can be further functionalized for improving intracellular delivery [204,205]. The MPG-siRNA complex was tailored with cholesterol for improving stability in biological fluid and overall potency. The higher stability in biological fluid and slow release pattern of MPG-siRNA to the target tumor cells make them more advantageous over the other existing strategies. A prostate cancer mouse model was developed by injecting 106 PC-3 cells into Swiss nude mice. When the mice had a tumor size of 100 mm3, they were treated with 0.1 mL of PBS (as a control), free cyclin-B1 siRNA (100 µg), control siRNA Cyc-B3(50 µg), cyclin-B1-siRNA (5 µg and 10 µg) complexes with MPG-8/chol-MPG-8 at a 1/20 molar ratio and cyclin-B1 siRNA with MPG-8(10 µg) in every 3 days. After 50 days of treatment, it was found that the Cyc-B1 siRNA-MPG-8/chol-MPG-8 (5 µg) reduced 60% of tumor size while for 10 µg of Cyc-B1 siRNA-MPG-8/chol-MPG-8 it was 92% in contrast to control groups. Cyclin-B1 protein expression was also reduced to 60% and 80% for both 5 µg and 10 µg of Cyc-B1 siRNA-chol-MPG-8 containing group at the end of 48 days. There was dose dependent tumor growth inhibition were seen for Cyclin-B1 siRNA treating group. The survival rate for 10 µg of Cyc-B1 siRNA-MPG-8/chol-MPG-8 was 70% at day 40 while in cholesterol free group it was only 20% [206]. On the other hand, intratumoral injection of cyclin-B1-siRNA (1 µg and 5 µg) complexes with MPG-8/chol-MPG-8 at a 1/20 molar ratio and cyclin-B1 siRNA with MPG (5 µg) caused 75% reduction of tumor volume for 1 µg and complete disappearance of tumor for 5 µg of Cyclin-B1-directed siRNA. The notable antitumor efficacy of Cyclin-B1-siRNA for both intravenous and Intratumoral administration and their prolonged survival rate confer new prospective for tumor management modality [206].
In addition, a sticky siRNA (ssiRNA) with cyclin-B1-antisense was encapsulated with cationic linear polyethylenimine (PEI) for improving target ability and biological stability. The murine melanoma tumor was inoculated into the NMR1 (5 weeks) nude mice via subcutaneous injection of 1 × 106 cells of B-16-F10 into the right flank of mice. When the mice had a tumor volume of 50 mm3, the mice were treated with glucose (as a control), negative control ssiRNA and cyclin-B1-ssiRNA at a dose of 1 mg/kg intravenously every alternative day. After 20 days of treatment, massive enlargement of tumor volume for both control groups (1300 mm3 and 1100 mm3) were seen in contrast to cyclin-B1-ssiRNA/PEI group (700 mm3). The ssiRNA cyclin-B1-PEI complex delivered the cyclin-B1-ssiRNA to the targeted tumor cell and down regulated the Cyclin-B1 expression which resulted in significant inhibition of tumor growth [181].
In conclusion, Cyclin-B1 has a prominent role in tumor cell proliferation and is found to be heterogeneously expressed in multiple cancers. Sticky siRNA (ssiRNA) targeting Cyclin-B1 exerted 44% reduction in tumor volume and improved biological safety profile in malignant melanoma. However, Cyclin-B1 targeted siRNA loaded with cholesterol-layered MPG-8-CPP reduced more than 90% of the tumor volume in a dose-dependent manner. The prolonged survival rate and null immune responses rendered it a promising tool for cancer treatment in a therapeutic context for the future.
3.6. Silencing of RhoA and RhoC Gene
Ras homologous A (RhoA) and Ras homologous C (RhoC) are the members of GTP/GDP-binding GTPase of the Ras superfamily [207,208]. They are low molecular weight compounds and act as molecular switches to promote cellular processes like actin and microtubule cycloskeleton organization, cell division, motility, cell adhesion, vesicular trafficking, phagocytosis, transcriptional regulation, matrix remodeling and cell mobility [209,210,211]. RhoA and RhoC are the key drivers for a series of pathologies of cancer, including cell motility, proliferation, apoptosis inhibition, cell cycle progression, invasion, metastasis and inflammation of tumor cells [212,213,214,215,216,217]. RhoA activation triggers signal transduction pathways; Rho-associated coiled coil-containing protein kinase (ROCK) activation pathway and the phosphatidylinositol-3-phospokinase/protein B (PI3-K/AKT) pathways. These activated pathways resulted cell locomotion, cell survival and expression of cell proliferative genes [218]. On the other hand, the active RhoC drives cell invasion and metastasis by increasing focal adhesion, contact information and angiogenesis [219,220]. Moreover, excessive expression of RhoA and RhoC are found to be more than 30% of all cancers which make them a vulnerable molecular target for cancer therapy.
Colorectal carcinoma is one of the fastest growing cancers all over the world [221,222,223]. There is a remarkable percentage of deaths from colorectal cancer owing to its tumor metastasis and recurrence properties [224]. Several mutant genes are responsible for proliferation, invasion and metastasis of colorectal cancer. Among them, RhoA and RhoC genes are found to be overexpressed in colorectal cancer and are considered to be potential targets for the management of colorectal cancer. Recombinant adenovirus-based shRNA-targeted RhoA and RhoC (Ad-RhoA-RhoC) were synthesized and subjected to animal studies for evaluating anti-tumor efficacy. 1.0 × 107 of HCT116 (human colon carcinoma cell line) were implanted subcutaneously into the right flank of the athymic nude male Balb/C mice (15–18 g) of 4–5 weeks old. When the tumor nodules grew to 5–7 mm, the mice were assigned to treatment. Each group of mice (n = 7) was treated with normal saline as a control group (30 µL/mouse), Ad-Hk (negative control group) at a dose of 4 × 108 pfu (30 µL/mouse) and Ad-RhoA-RhoC at a dose of 4 × 108 plaque-forming unit (pfu) (30 µL/mouse) intratumorally 4 times daily at a 1-day interval of total accumulated dose of 1.6 × 109 pfu (plaque-forming unit). After 17 days of treatment, the tumor volume for Ad-RhoA-RhoC was (444.38 ± 63.03) mm3 whereas for the control and Ad-HK groups it was (699.62 ± 190.56) mm3 and (678.81 ± 155.39) mm3, respectively. The tumor volume for control and Ad-Hk was almost five-fold higher than the starting volume. On the other hand, the Ad-RhoA-RhoC-containing group exerted a relatively slow tumor growth (2.38-fold) and reduced approximately 37% of tumor volume in comparison to the control group [225].
Aggressiveness of breast cancers is the most lethal condition that leads to death in most breast cancer patients. RhoA and RhoC are the key drivers for cancer aggressiveness e.g., increased cellular proliferation and metastasis by activating several pathways that catalyze cell survival and proliferation [207,226]. RNAi therapy targeted to RhoA and RhoC genes is supposed to be more effective to suppress tumor growth than conventional therapy. The anti-tumor effect following cytofectin-mediated delivery of anti-RhoA and anti-RhoC siRNAs was evaluated in a breast cancer mouse model, developed by employing 4 × 106 of MDA-MB-231 cells (human breast carcinoma cells) into female athymic nude mice of 6 weeks of age. After 2 weeks of implantation, when the mice had a tumor volume of 20 mm3, the mice were treated with 100 µL of anti-RhoA (85 nM), anti-RhoC (85 nM) siRNA and cytofectin containing excipient as a control intratumarally at a 3-day interval over a period of 20 days. Mice treated with anti-RhoA siRNA (tumor volume, 200 mm3) and anti-RhoC siRNA (tumor volume, 600 mm3) showed less tumor growth than in the control group (1300 mm3). The angiogenesis index values of anti-RhoA and anti-RhoC siRNA were 8.75 ± 3.30 and 22.5 ± 3.32 relative to 30.5 ± 4.12 for the control group. The above findings demonstrated that anti-RhoA and anti-RhoC siRNA reduced tumor volume by 85% and 53%, respectively, in contrast to the control by down regulating RhoA and RhoC gene expression. The remarkable gene silencing effect and antitumor effects of anti-RhoA in intratumoral delivery revealed that there is a strong correlation between RhoA, and tumorigenesis. To furnish this approach for further investigation, intravenous administration of anti-RhoA siRNA is required for assessing its efficacy and safety in the systemic circulation [227].
Chitosan, a mucopolysaccharides was combined with polyisohexylcyanoacrylate (PIHCA) to deliver siRNA to the target site as well as to protect it from enzymatic degradation. The efficacy and safety profile of chitosan-polyisohexylcyanoacrylate-antiRhoA were assayed in a mouse model study. Human breast cancer (MDA-MB-231) cells (5 × 106 cells in a volume of 250 µL) were injected subcutaneously into the right hind limb of the female athymic nude (nu/nu) mice aged 6 weeks to let the cells grow into a tumor of 20 mm3, and the mice were treated with 150 µg of siRNA/kg body weight in the low dose group and 1500 µg of siRNA/kg body weight in the high dose group, while empty chitosan nanoparticles at a 300 µg/mL were also injected as a control at every 3 days to 30 days. The anti-RhoA siRNA inhibited tumor growth in a dose-dependent manner. There was >90% decrease in mean tumor volume for 150 µg (low dose group) siRNA containing group, whereas for 1500 µg (high dose group) siRNA containing group it completely disappeared in comparison to control group [228].
Inflammatory breast cancer (IBC) is characterized by fast proliferation, metastasis, lower survival rate and local recurrences [219,229,230,231,232]. It was found that 90% of IBCs contain over-expressed RhoC GTPase gene [233] which might be involved in growth and metastasis of cancer cells. Cellular uptake and bioavailability of the anti-RhoC siRNA were increased by mixing with lipofectamine. SUM149 cells (1 × 107) were injected into each female BALB/c-nu mice (47 weeks old) subcutaneously to allow them to grow into a tumor volume of 50–70 mm3. Intratumoral injections of saline (control), control siRNA, and anti-RhoC-siRNA (0–1 mL, 80 nM) were given at 2-day-intervals for a total of 14 days. The relative tumor volume for anti-RhoC siRNA was 3.4, whereas it was approximately 5.2 for both control and untreated siRNA, demonstrating 35% reduction of tumor volume in contrast to the control group. The survival rate after 30 days of treatment was approximately 85% for anti-RhoC siRNA-treated group, indicating that lipofectamine mediated anti RhoC siRNA had a significant effect on tumor growth reduction as well as survival rate enhancement. The down regulation of RhoC gene expression results in up-regulation of metastasis suppressor gene KAll expression as well as reducing the expression of CXCR4 and MMP cardinal regulator of breast metastasis [234].
The amazing results of pre-clinical studies (Table 2) indicate that RhoA and RhoC have different clinical roles in regulating transcriptional factors, invasion and metastasis of cancer cells in multiple cancers and are overexpressed in various solid tumors. Despite the technique’s novelty, the roles of RhoA and RhoC are being searched for successful management of cancer for further advancement in the future. Adenoviral-mediated combined delivery of RhoA and RhoC in colorectal carcinoma reduced 37% of tumor volume. RhoA-directed siRNA exerted extraordinary tumor volume inhibitory effects (85% of tumor volume) in breast cancer, whereas when it was given with chitosan the tumor volume reduction was increased to 90% (to completely abolishing) for the higher dose. On the other hand, RhoC with lipofactamine in inflammatory breast cancer reduced only 35% of tumor volume and 53% in aggressive breast cancer. Although RhoC is extensively expressed in almost all cancers, its clinical role and efficient treatment modality should be investigated for using it in the clinical setting.
3.7. Silencing of β-Catenin Gene
β-Catenin is a protein that is encoded by the CTNNB1 gene in humans and a subunit of cadherin protein, which are the key drivers for the WNT pathway [235,236]. It regulates the intracellular signal of WNT pathway and exerts a dual function, like cell adhesion and gene transcription which ultimately control the cellular proliferation [237,238,239]. β-Catenin undergoes ubiquitination and proteasome degradation without WNT pathways. However, in the presence of WNT pathways, WNT ligands prevent the β-catenin degradation by knocking down the destruction complex of β-catenin [240,241]. The activated β-catenin binds to the Tcf/Let family and initiates transcription of cyclin D1, C-myc, MMP-7, Lgr5+ and CD-44 in nucleus continuously without any external stimulation [242,243]. It also acts as a proto-oncogene and accumulation of β-catenin in nucleus serves as a tumor marker for diagnosis. Mutations of β-catenin have been found in a variety of solid tumors like primary hepatocellular carcinoma, colorectal cancer, breast cancer, lung cancer, cervical cancer, skin cancer, liver cancer and glioblastoma [244]. The involvement of β-catenin in the cancer development makes β-catenin as a potential molecular target for cancer gene therapy.
Overexpression of β-catenin and APC (adenomatous polyposis coli) genes are seen in colorectal carcinoma, mediating uncontrolled cell proliferation and development of colon cancer [245,246,247,248,249,250,251]. The elevated β-catenin levels are supposed to be lowered by introducing sequence-specific post-transcriptional silencing siRNA directed against β-catenin [252]. HCT 116 cells (colon cancer adenocarcinoma cell lines) were inoculated into female nude/nu mice (4–6 weeks old) for tumor inoculation. Oligofectamine-associated siRNA (250 pmol) targeting β-catenin and tax-siRNA (control) was given, and tumor volume and survival rates were monitored. Mice treated with antisense-β-catenin siRNA showed higher tumor inhibitory effects (three-fold smaller in tumor size) than the control group. The survival rate for the β-catenin-directed group was more significant and the expression of β-catenin was also down remarkably regulated relative to the control group [253].
The stem cells of the gastric gland (Lgr5+) are responsible for the development of stomach and intestines [254,255]. Mutation and dysregulation of signaling pathways of Lgr5+ stem cells result in intestinal cancer and gastric cancer. Cell cycle and apoptosis regulators 1 (CCAR1/CARP-1) has cell growth inhibition and apoptosis promoting effects in human breast Cancer cells [256,257]. Co-delivery of CCAR1-shRNA with β-catenin-shRNA improved the transcriptional activation of β-catenin in colon cancer cells [258]. Besides CCAR1, a coactivator of β-catenin is considered to have a vital role in the tumorigenesis and metastasis of gastric cancer. Lentivirus based Sh-CCAR1 for targeting CCAR 1 and scrambled shRNA as a negative control were prepared to explore the tumor regression capability. Male athymic nude mice of 8 weeks were taken and subjected to subcutaneous injection of 5 × 106 AGS cells into the posterior leg of mice at a regular time period. After a certain time period the mice were divided into two groups (n = 3) and assigned to the treatment of shRNA-CCRA1 and scramble shRNA (shNuIIT) as a control. Tumor volume and width were measured found that, control group has a fast growing tumor size of 800 mm3 whereas CCAR1-shRNA groups has a relatively slow growing and smaller tumor size of 200 mm3, exerting 75% reduction of tumor size in CCAR 1-shRNA group in comparison control group [259].
In summary, the proto-oncogene β-catenin along with WNT pathway controls cellular proliferation by regulating transcriptional processes in various tumors. Overexpression of β-catenin is observed in a wide variety of cancers. β-catenin-targeted siRNA reduced 62% of tumor volume with a significant survival rate of colon cancer. Another most amazing finding was that the siRNA directed against β-catenin suppressed the β-catenin expression for a long period of time (6 h) even at a small dose, which is very promising for drug safety profile. On the other hand, for gastric cancer, shRNA directed against CCAR1 co-activator of β-catenin reduced 75% of tumor volume and lowered the growth rate. So β-catenin targeted siRNA and shRNA are very promising for further study to make it feasible to the clinical trial.
3.8. Silencing of EphA2 Gene
Erythropoietin-producing hepatocellular (Eph) receptors are the tyrosine kinase receptors of the Ephrin family that play a vital role in the development of malignancy [260,261]. The Ephrin family is divided into two subclasses such as Ephrin A and Ephrin B based on their attachment to the membrane [262]. The EphA2 is located on human chromosome 1p36 [263] and contribute to develop Central nervous system (CNS) in the embryonic stage. In adult tissue, EphA2 is found to be expressed smaller amount in epithelial cells, but excessive expression almost ten-fold higher is seen in various tumors, including GBM patient tumor (61%), ovarian cancer (76%), prostate adenocarcinoma (85%), gastric cancer (77%), hepatocellular carcinoma, colorectal carcinoma and endometrial cancer [264,265,266,267,268,269,270,271,272,273]. Overexpressed EphA2 elicits oncogenic effects by enhancing cell-extracellular matrix (ECM) adhesion, anchorage-dependent growth and metastasis [274]. The expression rate, localization and significant clinical role in developing tumors make it an ideal target for cancer treatment.
The pharmacokinetic profile and specific delivery of the anti-EphA2 siRNA could be improved via inclusion of neutral lipid 1,2-dioleoyl-sn-glycero-3-phosphatidyl choline (DOPC) in liposome. Ovarian cancer cell lines, HeyA8 cells (2.5 × 105) and SkOV3ip1 cells (1.0 × 106) were given through intraperitoneal injection into female athymic nude mice (Ncr-nu). Empty DOPC liposomes, control siRNA in DOPC liposomes, EphA2-targeting siRNA in DOPC liposomes, paclitaxel (100 µg) + control EphA2 siRNA in DOPC liposomes and paclitaxel+EphA2 siRNA in DOPC liposomes were administered after the mice had a tumor size of to 0.5 to 1.0 cm3. The siRNA-liposomes at a dose of 150 µg/kg siRNA were given twice in a week and paclitaxel was given once in a week. In HeyA8 cell line, the size of the tumor for EphA2 targeted siRNA group was 0.98 g relative to control siRNA 1.51 g. On the other hand, in the SkOV3ip1 cell line, the tumor volume for EphA2-targeted siRNA and control siRNA were 0.35 gm and 0.70 gm, respectively. Liposomal anti-EphA2-siRNA reduced 35–50% of tumor size, but when it was co-delivered with paclitaxel, tumor size reduction went up to 82% relative to the control group [275].
In addition, to improve the antitumor activity of EphA2-targeted siRNA in ovarian cancer, combinatorial targeting of EphA2, Src and FAK (focal adhesion kinase) genes were made to downregulate the expression of FAK and Src (non-receptor tyrosine kinase) involved in tumor growth and aggressiveness in ovarian cancer [276,277,278,279,280,281,282]. The siRNAs-loaded DOPC liposomes were employed in ovarian cancer mouse model generated by treating female athymic nude mice with 5 × 105 of HeyA8 cells and 1.0 × 106 of SKOVip1 cells. The tumor-bearing mice were treated with control siRNA-DOPC, EphA2-siRNA-DOPC, EphA2+FAK-siRNA-DOPC and EphA2+Src-siRNA-DOPC intraperitoneally twice in a week at a dose of 5 µg siRNA/200 µL suspensions. EphA2-FAK-siRNA-DOPC exerted significant tumor growth reduction (90%) in comparison to EphA2-siRNA-DOPC (67%) and FAK-siRNA-DOPC (62%) in HeyA8 model. While in SkOVi31 model, there was also notable reduction of tumor growth in EphA2-FAK-siRNA-DOPC (76%)-treated group compared to the EphA2- (50%) and FAK- (61%) treated group. Combined delivery of EphA2 and FAK siRNAs decreased 67–70% of tumor weight compared to EphA2 and FAK (single) treated group. The EphA2-Src-siRNA DOPC had a less reduction rate of tumor growth than EphA2-FAK-siRNA DOPC-treated group. Besides, EphA2-FAK-siRNA-DOPC reduced 62–82% more tumor metastasis than a single group [283].
In gastric carcinoma, erythropoietin producing hepatocellular (EphA2), a receptor tyrosine kinase (RTK) is found to be over expressed in 77.3% of gastric cancer patients [260,264,266,284] and plays a vital role in metastasis via regulation of MMP-9 gene expression. MMP-9 (gelatinase-B), member of matrix metalloproteinase (MMPs) increased cancer cell metastasis by degrading denatured collagens (gelatins) and type iv collagen which are the structural components of ECM(extra cellular matrix). The MMP-9 mediated degradation of collagen is one of the most important steps in the development of cancer cell. SGC 7901 (human gastric adenocarcinoma) cells were injected into the axillary fossa region of 3–4 weeks old Balb/C mice (18–22 g), and when the tumor size grew to 52.2 ± 6.9 mm3, intratumoral injections of 0.9% sodium chloride as a blank control group, non-silencing siRNA with liposomes and EphA2-siRNA with liposome were given twice in a week. After 21 days of treatment it was found that EphA2-targeted siRNA inhibited the tumor growth by 43.1% compared to the negative control group. Immunocytochemistry analysis of tumor xenograft demonstrated less expression of MMP-9 in the mouse treated with EphA2-siRNA, suggesting that silencing of EphA2 gene down regulated the MMP-9 gene expression and exerted tumor inhibitory effects [285].
Briefly, EphA2 is functionally vital to tumor growth and development. It is highly expressed in a variety of cancers. In ovarian cancers, anti-EphA2-siRNA with DOPC reduced the 35–50% of tumor volume and its antitumor activity was increased when it has given with anticancer drug paclitaxel (67–82%). After couple years later, dual therapy of anti-FAK-siRNA along with anti-EphA2 in DOPC exerted significant tumor reduction (90%). The tumor metastasis rate also decreased to (62–82%) which is very promising. On the other hand, intratumoral injection of liposomal anti-EphA2 reduces 43.1% of tumor volume in gastric cancer cell which is need to be further analyzed in systemic administration to make it feasible for use in human.
3.9. Silencing of MDM-2 Gene
p53, a tumor suppressor gene that is involved in cell cycle arrest, apoptosis and tumor growth inhibition. Oncoprotein mouse model minute 2 (MDM-2) inhibits the regulation of p53 tumor suppressor gene. It interacts with p53 and deactivates the p53 pathway. MDM-2 is over expressed in a various number of human tumors, including lung and prostate carcinoma [286]. Activation of p53 pathway by blocking the MDM-2 gene is considered to be an effective treatment modality of human cancers [287,288]. To attenuate the expression of MDM-2 in the tumor cell, anti-sense-MDM-2 were employed for specific tumor growth inhibition. But the delivery of siRNA in vivo is limited due to nonspecific tissue distribution, less blood circulation retention time and self-aggregation by biological salt [289]. To overcome this hurdle, pH-responsive di-block copolymer of poly (methacryloyloxy ethyl phosphorylcholine)-b-poly (diisopropanolamine ethyl methacrylate) PMPC-b-PDPA were synthesized for the delivery of siRNA for improving biological stability, tumor specific delivery and cellular uptake. The PMPC-b-PDPA/siRNA complex were prepared by precipitation method. Athymic mice (25 g) of 6–8 weeks old were taken and 5 × 106 of H2009 cells (NSCLC cells) were injected subcutaneously. After ten days of injection, when the tumor size reached at 100 mm3, the mice were treated with; siRNA-MDM-2 loaded nanoparticles (0.32 mg/kg) and siRNA-Scr with NPS as a negative control (6.4 mg/kg) into the tail veins at 2 days for total 12 days. The volume of tumor was measured for both groups, with 67% reduction of tumor growth observed for siRNA-MDM-2-loaded nanoparticles in comparison to Scr-siRNA. The down regulation of MDM-2 might induce apoptosis by activating p53 gene, exerting a remarkable tumor inhibitory effect [290].
A mixture of siRNA(s) targeting MDM-2 gene along with other oncogenes, c-myc and VEGF that initiate and flourish the metastatic behavior of a cancer, were encapsulated with nanoparticles formulated by protamine, cationic lipid and PEG. The constructed combo siRNA-NP complex showed superior anti-tumor effects over the control. In a study, female C57B216 mice of 16–18 g (6–7 weeks old) were inoculated with 2 × 105 of B16F10 cells, combo siRNA (MDM-2, c-myc and VEGF at a ratio of 1:1:1), the non-targeted NP, free siRNA and control siRNA at a dose of 0.45 mg/kg of total two consecutive doses were given. After 7 days of treatment, the mice were killed and lung of mice were collected for analysis. It was found that the control group had no notable tumor regression, whereas the combo siRNA with targeted NP exerted 20–30% reduction of tumor load. The survival rate of combo siRNA-containing group was found to be extended in comparison to control group of mice [291].
So, MDM-2 gene has a driving role in development and regulation of tumors via inhibiting expression of p53 tumor suppressor gene. MDM-2 targeted siRNA along with pH-responsive di-block copolymer of poly (methacryloyloxy ethyl phosphorylcholine)-b-poly (diisopropanolamine ethyl methacrylate) PMPC-b-PDPA reduced 67% of tumor volume in NSLSC animal model. On the other hand, combination of MDM-2, c-myc and VEGF siRNAs also reduced tumor load in lungs (20–30%) and extended survival rate were found in comparison to control group. The excellent tumor regression rate of MDM-2-siRNA and reduced lung tumor load warrant more preclinical studies (Table 2) focusing on pharmacokinetics profile and survival study to make it more reliable for clinicians.
3.10. Silencing of IGF-1R Gene
IGF-1R (Type 1 insulin-like growth factor receptor) is a tyrosine kinase receptor which is the members of the insulin receptor family including IR (a homodimer), IGF-1R (homodimer), IGF-1R/R (hybrid, heterodimeric receptors) and mannose 6-phosphate receptor (IGF-2R) [292]. The insulin receptor (IR) has two subtypes IR-α and IR-β. The ligands of IGF-1R, IGF-1 and IGF-2 interact with IR receptors, resulting in receptor oligomerization, PTK activation and other pathways. The activation of these pathways mediate gene activation, DNA synthesis and cell proliferation [293,294,295]. The IGF-1R also plays a vital role in cell metabolism, differentiation, apoptosis, chemo resistance and angiogenesis as well as protecting cells from UV irradiation, cytokine and gamma radiation-induced apoptosis [296]. It is ubiquitously expressed in normal tissues for growth and multiple physiological activities, but heterogeneous expression has been found in many solid tumors like breast cancer, prostate cancer, colorectal cancer, lung cancer and hematological malignancies [297,298,299,300]. The excessive expression in tumors and their role in tumor development makes IGF-1R is an auspicious bio molecular target for cancer treatment.
Non-small cell lung cancer (NSCLC) comprises more than 80% of the total lung cancer, which is the leading cause of death all over the world with a minimal survival time [301,302,303]. Insulin like growth factor 1 receptor (IGF-1R) is expressed abnormally in lung cancer and mediates malignant transformation of lung tumor cell. Overexpressed IGF-1R gene hampered UV irradiation-, cytokine- and gamma radiation-induced cell apoptosis and play a critical role in the development of tumors [304]. Among the other treatment options, Anti-sense-siRNA targeting IGF-1R is considered due to its high specificity. Polyethyleneimine (PEI) coated anti-sense-IGF1R-siRNA were examined for assessing antitumor efficacy in human adenocarcinoma mouse model, developed by injecting 2 × 107 of A549 into the male nude mice of 4–5 weeks of age. IGF-1R specific siRNA (0.125 µg/mm3 of tumor volume) expressing plasmids with polyethyleneimine (PEI) 3 µL/µg and negative control siRNA at a same dose were injected intratumorally 4 times every 5 days. After 40 days of treatment, the tumor volume for IGF-1R siRNA was found approximately 200 mm3, whereas for the control group it was approximately 490 mm3. The IGF-1R siRNA inhibit 60% of tumor growth in comparison to control group. The apoptotic cells were also increased remarkably in IGF-1R siRNA groups (117.6 ± 8.07) than control group (40 ± 9.11) [305].
Magnetofection is a process of delivery of nucleic acid-magnetic nanoparticles complexes to the target cells through applying magnetic field. The IGF-1R-targeted catalytic RNA was given through magnetofection for improving antitumor effects in an NSCLC animal model. Super-paramagnetic iron oxide nanoparticles (SPIONS) were complexed with cationic lipids (lipofectamine 2000) and plasmid DNA to form self-assembling magnetic lipoplexes for the targeted delivery of IGP-1R gene and evaluated the anti-cancer potential in a non-small cell lung cancer mouse model. Male Balb/CAnNcrj-nu mice of 4 weeks old were implanted with 5 × 106 of A549 cells (NSCLC cells line with overexpression of IGF-1R) in 100 µL medium and allowed to grow tumor size about 400 mm3. The group of mice was treated with; 200 µL PBS as a control, pGFPshIGF-1R (50 µg/mouse):Lp2000 (125 µL/mouse) as a lipofection group and a pGFPshIGF-IR (50 µg/mouse) combined with MAG (50 µg/mouse):Lp2000 (125 µL/mouse) under the influence of a magnetic field (400 mT), which was holding onto the subcutaneous tumor surface for 1 min to additional 14 min following the injection. The IGF-1R-shRNA of liposomal magnetofection silenced the expression of IGF-1R 85.1 ± 3% than lipofection group 56.1 ± 6%, suggesting liposomal magnetofection improve site specificity and cellular uptake which significantly down regulated the IGF-1R gene expression [306].
Taken together, IGF-1R plays a vital role in cellular proliferation and tumor growth and aberrantly expressed in multiple cancers. The anti-sense siRNA targeting IGF-1R along with PEI reduced 60% of tumor volume and increased apoptotic cell expression more than 60% in lung adenocarcinoma. After a couple of years of this experiment, IGF-1R targeted shRNA with super-paramagnetic iron-oxide nanoparticles (SPIONs) under the influence of magnetic field silenced 86% of IGF-1R gene expression in the same lung cancer model. These exciting results provide us a novel delivery method for siRNA in different cancer cell lines.
3.11. Silencing of Livin Gene
Human IAP (inhibitors of apoptosis protein) are the endogenous proteins that are thwarted both extrinsic and intrinsic apoptosis pathways by interacting with specific cysteine proteases or caspases [307,308,309,310]. There are eight members of human IAP have been reported, including NAIP, c-IAP-1 (MIHB, HIAP-2), c-IAP-2 (MIHC, HIAP-1), XIAP (hILP, MIHA, ILP-1), Survivin, Apollon (Bruce), ILP-2 and Livin (ML-IAP, KIAP) [311]. Among them Livin, a 39 kDa protein is a novel family of Human IAP and it has two analogs like Livin-α (298 amino acids) and Livin-β (280 amino acids) which protect the cells of heart, placenta, lung, spleen and ovary from tumor necrosis factor (TNF) and anti-CD95-induced apoptosis [312,313]. Livin expression is limited in most of the normal tissues, but found to be overexpressed in a variety of human malignancies such as colon cancer, gastric cancer, breast carcinomas, melanomas and lung cancer [312,314,315,316]) It plays a significant role in tumor progression, chemo resistance development and anti-apoptotic activity which makes Livin a potential therapeutic target for cancer treatment. Several siRNAs targeted to Livin gene were investigated both in vitro and in vivo (Table 2) to evaluate pharmacodynamics and pharmacokinetics properties of antisense-Livin.
Transformation of non-metastatic human melanoma to metastatic melanoma and their resistance to the current chemotherapeutics account for a huge number of deaths in both male and female in U.S.A. [317,318,319,320]. The anti-Livin-siRNA was combined with single chain antibody (anti-MM scFC-tp-siRNA) for targeted delivery in order to reduce potential side effects and enhance therapeutic efficacy in vivo [321,322,323]. LiBr, malignant melanoma (MM) cells (1 × 107) were given subcutaneously into right side of the flank region of a nude mouse to induce the tumor. PBS (200 µL) as a control, anti-MM scFv-tp-siRNA NC (Mock group) and anti-MM-ScFV-tp-siRNA (20 µg of siRNA) were given at 2 days through the tail vein. Anti-MM scFv-tp-Livin-siRNA(400 mm3) reduced 64% of the tumor volume compared to the control group (1100 mm3) [324].
Similarly, RNAi targeting Livin gene coated with lipofectamine were generated for improving apoptosis and chemo-sensitivity to chemotherapeutics and comparing the antitumor effects between treatment and control groups. In a human lung cancer mouse model generated by injecting 1 × 106 SPCA-1 (human lung cancer cell lines), plasmid vector pS-L1, PS-NS (a non-specific sequence as a control) and pS-CMV neo plasmids (250 µg each) were injected through the intratumoral route after 7 and 14 days of post tumor implantation. The mean tumor size for control groups (Mock, pS-CMV neo or pS-NS treated) was approximately 690 mm3, whereas for the group treated with siRNA targeting Livin it was approximately 190 mm3 with 73% reduction of mean tumor size. The apoptotic fraction of siRNA-Livin group (30%) was also increased significantly compared to the control groups (5%), along with extended survival rate [325].
The above data exposed that, there was a strong correlation between Livin and tumor development. Abnormal expression of Livin is found in many solid tumors. Recently intravenous administration of antibody conjugated siRNA in Livin reduces 64% of tumor volume in malignant melanoma. But in lung cancer cells, Intratumoral administration of Livin-siRNA along with lipofectamine reduces 73% of tumor volume and increased 30% of apoptotic fraction. It should be extended to systemic administration for testing pharmacokinetics parameter for further advancement.
3.12. Silencing of WT1 Gene
Wilms’ tumor gene 1 (WT1) is a 52–54 kDa protein, encodes four zinc finger transcriptional factors located at chromosome 11p13q and arises due to inactivation of the WT1 alleles gene [326,327]. It is the key drivers that control cell proliferation and apoptosis via attenuation of the expression of proliferative gene [328]. WT1 was defined as a tumor suppressor gene but recent evidence claimed that, it acts as an oncogene in leukemogenesis and tumorigenesis [329,330,331,332,333,334]. It is highly expressed in most of the acute myeloid leukemia (AML) and acute lymphoid leukemia (ALL) [335,336,337], lung, breast, thyroid and melanomas [338,339,340,341]. High rate of expression of WT1 and their significant role as a prognostic factor makes WT1 mRNA an ideal tumor marker for leukemic blast cells as well as a potential target for RNAi mediated gene therapy for various cancer treatments (Table 2).
Signal transducers and activators of transcription 3 (STAT-3) enhances the expression of anti-apoptotic gene MCI-1 and BCL-XL as well as other gene like cyclin D1/D2 and c-Myc which ultimately mediates cancer development and progression [342,343,344,345,346]. Over expression of WT1 increased transcriptional activity of phosphorylated STAT 3 (P-STAT 3) that upregulated anti-apoptotic gene and regulate the growth and differentiation of lung tumor cells [338,347]. To identify the relationship between over expressed WT1 and NSCLC (non-small cell lung cancer) development as well as to reveal the antitumor activity of WT1-targeted shRNA in vivo, NSCLC model was generated by subcutaneously injecting 5 × 106 cells into Balb/C nu/nu mice. When the tumor size reached at palpable size, the three groups of mice were subcutaneously treated with pLV-GEP-WT1, pLL3.7-WT1-shRNA (plasmid expressing WT1-shRNA) with wild type WT1 and empty plasmid (as a control). After 31 days of treatment, remarkable reduction of tumor volume was found for pLL3.7-WT1-shRNA (76%, 74% and 69%), whereas tumor growth curves for pLV-GEP-WT1 and the control group were found to be increased [348].
Transferrin (Tf), a glycoproteins over expressed on the surface of the cancer cells was used as a ligand to carry WT1-shRNA to tumor [349,350,351,352]. The WT1 shRNA-Tf was complexed with liposomes and polyethylene glycol (PEG) for improving biological stability of the shRNA in the systemic circulation. To explore the antitumor activity, B16F10 cell (5 × 105 cells) were subcutaneously injected into the female C57BL/6 mice (20–25 g) of 7–8 weeks aged, prior to intravenous administration of 50 µL of Lip-RNAi-Tf, lip-RNAi against WT1 (50 µL), 50 µL of lip-GFP-Tf (PEGFP-N3 vector, liposome and Tf complex) and 50 µL of saline solution at a 5 days until 29 days. The mean tumor weight for Lip-RNAi-Tf was 5.5 g whereas it was 8.8 g for untreated group. The mice of the other two groups were not counted because of all of the mice were dead after day 30. The liposome-WT1-shRNA-Tf reduced 34% of tumor weight in comparison to control group. Survival rate was also prolonged in liposome-WT1 shRNA-Tf containing group (62.5%) than control group (22.2%) [353].
In summary, WT1 plays key role in leukemogenesis and tumorigenesis as an oncogene but it was originally known as a tumor suppressor gene. Overexpression of WT1 has been detected in many solid tumors. In different lung cancer cell lines (A549, H1299 and H1650), anti-WT1 shRNA exerted notable tumor reduction 76%, 74% and 69% for lung cancer cell lines. On the other hand, ligand conjugated WT1-shRNA reduces 34% of tumor weight with prolonged survival rate. So this variation arises more questions about the role of WT1 and their targeting efficacy for the management of different tumors. To go for clinical setting more investigation is required to establish WT1 as a potential target for cancer management. Although clinical role of WT1 is obscured, liposome and ligand mediated delivery reduced the secondary effects and increased site specific delivery, which is very promising for the delivery of catalytic RNA.
3.13. Miscellaneous
The human Bcl-2-associated athanogene-1 (Bag-1) encodes the three major isoforms, including Bag-1S (p36), Bag-1M (p46) and Bag-1L (p50) that are located on chromosome 9 and involved in differentiation, cell cycle and apoptosis. It regulates Bcl-2 gene expression and mimics the anti-apoptotic activities via bridging between the growth factor and anti-apoptotic mechanism. The aberrant expression of Bag-1 has been found in breast, lung, cervix, esophagus and colorectal cancers [354,355,356]. Colorectal cancer has the second largest morbidity and mortality rate all over the world [357,358] and human Bcl-2 associated athanogene-1(bag-1) anti-apoptotic gene is found to be involved in the tumor genesis of colorectal cancer (CRC) by mediating progression and metastasis and acts as a positive regulator of Bcl-2 gene in CRC [359]. The anti-Bag-1-siRNA plasmid was combined with magnetic gold nanoparticles and evaluated its tumor inhibitory effects in colon cancer mouse model, developed by giving 1 × 106 of LoVo cell (human colon cancer cell lines) into the right flank of the Balb/C nude mice (14–16 g) of 4–5 weeks old. The tumor-bearing mice were divided into five groups and treated with; normal saline, nanoparticles (25 µg), plasmid (5 µg), nano-plasmid complexes (nano-plasmid-1; 5 µg plasmid/25 µg nanoparticles) and nano-plasmid complexes (nano-plasmid 2; 5 µg plasmid/25 µg nanoparticles) intratumorally. Nano-plasmid 2 was given to mice under the influence of about 5000 gauges magnetic fields. The 5 doses were given at a 3 day intervals. The tumor volume was measured after every 3 days, 28 days later of treatment, the tumor volume for nano-plasmid complex-1 was found to be approximately 200 mm3, whereas it was 700 mm3 and 650 mm3 for nanoparticle alone and control group and it was 69% reduction of tumor volume. The nano-plasmid-2 under the influence of magnetic field reduced the tumor volume at a same rate of nano-plasmid-1 without any significant reduction. Silencing of Bag-1 gene was supposed to down-regulate the expression of C-MYC protein that is clinically important for colorectal cancer. The potent anti-tumor effect makes anti-Bag-1-siRNA as a potential therapeutic for colorectal cancer management [360].
The pituitary tumor transforming gene 1 (PTTG1) is the member of PTTG family, including PTTG2 and PTTG3, that plays a vital role in several cellular processes like mitosis, DNA repair, apoptosis and gene regulation. Abnormal expression of PTTG-1 interferes in cellular processes and causes aneuploidy which is the necessary events in the tumorigenesis [361,362,363,364,365]. PTTG1 is over-expressed in various cancers and promotes tumor development and angiogenesis via triggering the expression of the fibroblast growth factor 2 and VEGF [366,367,368,369,370,371]. Tuning of PTTG1 gene expression by using anti-sense siRNA is considered to be an effective modality to reduce cancer aggressiveness. Hepatocellular carcinoma (HCC) is the most prominent tumor in humans all over the world having a higher alarming rate of incidence rate [372,373]. Over expression of PTTG-1 hampered the expression and function of intact p53, which ultimately promotes hepatocellular carcinogenesis [374].
Adenovirus vector assisted delivery of siRNA targeting PTTG-1 would be more efficient in down-regulation of PTTG-1 expression for hepatocellular carcinoma management. Five weeks old Balb/C nude mice were given with SH-J1 cells (107) in 100 µL of PBS subcutaneously into the right flank of the mice and allowed to grow to a tumor volume of 3–5 mm in diameter. Following intratumoral administration of 1 × 109 plaque-forming units (pfu) of Ad-PTTG-1-siRNA into the mice, tumor regression study demonstrated that Ad-PTTG-1-siRNA-treated groups regressed 84% of tumor volume in comparison to the control groups, suggesting that Ad-PTTG1-siRNA may serve a new paradigm for treating human cancers [375].
CD-47 is a membrane receptor of the immunoglobulin (Ig) superfamily that induces the phagocytic process of macrophages via binding with signal regulatory proteins α (SiRP α). Overexpression of CD-47 causes the tumor cells escape from immunosurveillance and results in tumor progression [376,377]. The expression level of CD-47 was reported to be higher in leukemic cell lines, bladder-tumor-initiating cells and lymphomas [378,379,380,381]. The LPH-NPs (liposome-protamine-hyaluronic acid) were constructed to carry CD-47-targeted siRNA for evaluating pharmacodynamic properties against melanoma tumors. C57B2/6 mice of 6–8 weeks were harvested with subcutaneous injection of B16F10 cells at a concentration of 2 × 105 cells/50 µL into the hind legs of mice. The mice were treated with 12 µg of CD-47 or control siRNA (0.6 mg/kg) along with LPH-NPs intravenously after the mice had a tumor volume of 50 mm3. The doses were given at a one-day interval to a total of six injections from the 8th day of tumor implantation. The tumor volume for CD-47-siRNA-LPH-NPs was found to be smaller than the control group, with approximately 93% reduction of tumor volume in comparison to the untreated group [382].
4. Future Directions
In preclinical studies, catalytic RNAs are mixed with several viral and non-viral vectors for safe, effective and targeted delivery to the cytoplasm of cancer cells. As exemplified in this review through various in vivo studies, siRNAs or shRNAs targeted to oncogenes, tumor suppressor genes and anti-angiogenic genes have been used with the help of various carriers over the past two decades for improving anticancer efficacy (Table 2). Among them, only few are translated into human trials and the others are in the pipe line. Although tremendous progress has been made, limitations still remain in the systemic applications of RNAi-based cancer nanotherapeutics. These hurdles should be addressed and resolved for the rational design of delivery vehicles for the targeted drug delivery to cancer cells in the future. Firstly, the issues of manufacturing hurdles like small size distribution, homogeneity, uniform functionalization, reproducibility at a large scale and manufacturing cost. Secondly, the poor understanding of mechanistic behavior of RNAi therapy with a view to their penetration and deposition in tumor tissue are not still clear. Thirdly, the poor cellular uptake and lower silencing effects which are the key parameters of therapeutic index along with less tumor accumulation (0.7%). These obstacles have hampered the progress of RNAi-based cancer therapy to get clinical approval.
To minimize the hurdles mentioned above, extensive research on molecular events of cancer pathogenesis and mechanisms of siRNA delivery including cellular attachment, target binding, biological interactions, intracellular trafficking and nuclease attack should be further studied to understand absolute biological phenomena. This new RNAi-based therapeutic entity should overcome the physiological barriers and differentiate between cancer and normal cells for a broader therapeutic index. From a clinical standpoint, biological readouts, bio distribution and kinetics should be analyzed to ensure safety and efficacious therapies. Future studies should, therefore, be emphasized for development of SMART nano carrier (Figure 6) and computer assisted drug delivery for evaluating pharmacokinetics and pharmacodynamics profiles before heading to clinical trials.
5. Conclusions
Although RNAi technology has advanced rapidly and become a clinical reality as this technology has the capability to reach and treat cancer at the molecular level, cancer target discovery and validation, gene editing system and cancer stem cell targeting demand further research for improved RNAi-based cancer therapy. The RNAi-based cancer therapy would, therefore, lead the next wave of cancer nanomedicine.
Author Contributions
Md. Emranul Karim prepared the initial draft of the manuscript under the supervision of Ezharul Hoque Chowdhury, while Kyi Kyi Tha, Iekhsan Othman, Mohammad Borhan Uddin and Ezharul Hoque Chowdhury were responsible for correcting the manuscript.
Funding
This work has been financially supported by FRGS/1/2016/STG05/MUSM/02/3.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Naldini, L. Gene therapy returns to centre stage. Nature 2015, 526, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Zamore, P.D. RNA interference: Big applause for silencing in Stockholm. Cell 2006, 127, 1083–1086. [Google Scholar] [CrossRef] [PubMed]
- Hannon, G.J. RNA interference. Nature 2002, 418, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Williams, B. Role of the Double-Stranded RNA-Activated Protein Kinase (PKR) in Cell Regulation; Portland Press Limited: London, UK, 1997. [Google Scholar]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Hannon, G.J.; Rossi, J.J. Unlocking the potential of the human genome with RNA interference. Nature 2004, 431, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Meister, G.; Tuschl, T. Mechanisms of gene silencing by double-stranded RNA. Nature 2004, 431, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, K.A.; Langer, R.; Anderson, D.G. Knocking down barriers: Advances in siRNA delivery. Nat. Rev. Drug Discov. 2009, 8, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Matranga, C.; Tomari, Y.; Shin, C.; Bartel, D.P.; Zamore, P.D. Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell 2005, 123, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Rand, T.A.; Petersen, S.; Du, F.; Wang, X. Argonaute2 cleaves the anti-guide strand of siRNA during RISC activation. Cell 2005, 123, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Hutvágner, G.; Zamore, P.D. A microRNA in a multiple-turnover RNAi enzyme complex. Science 2002, 297, 2056–2060. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, D.W.; Davis, M.E. Insights into the kinetics of siRNA-mediated gene silencing from live-cell and live-animal bioluminescent imaging. Nucl. Acids Res. 2006, 34, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.M.; Li, M.Z.; Chang, K.; Ge, W.; Golding, M.C.; Rickles, R.J.; Siolas, D.; Hu, G.; Paddison, P.J.; Schlabach, M.R.; et al. Second-generation shRNA libraries covering the mouse and human genomes. Nat. Genet. 2005, 37, 1281. [Google Scholar] [CrossRef] [PubMed]
- Rao, D.D.; Vorhies, J.S.; Senzer, N.; Nemunaitis, J. siRNA vs. shRNA: Similarities and differences. Adv. Drug Deliv. Rev. 2009, 61, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Cullen, B.R. RNAi the natural way. Nat. Genet. 2005, 37, 1163. [Google Scholar] [CrossRef] [PubMed]
- Karim, E.; Rosli, R.; Chowdhury, E.H. Systemic Delivery of Nanoformulations of Anti-cancer Drugs with Therapeutic Potency in Animal Models of Cancer. Curr. Cancer Ther. Rev. 2016, 12, 204–220. [Google Scholar] [CrossRef]
- Davis, M.E. Non-viral gene delivery systems. Curr. Opin. Biotechnol. 2002, 13, 128–131. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, A.; Miyata, K.; Kataoka, K. Recent progress in development of siRNA delivery vehicles for cancer therapy. Adv. Drug Deliv. Rev. 2016, 104, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Pahle, J.; Walther, W. Vectors and strategies for nonviral cancer gene therapy. Expert Opin. Biol. Ther. 2016, 16, 443–461. [Google Scholar] [CrossRef] [PubMed]
- Wirth, T.; Parker, N.; Ylä-Herttuala, S. History of gene therapy. Gene 2013, 525, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, N.; Wu, J.; Xu, X.; Kamaly, N.; Farokhzad, O.C. Cancer nanotechnology: The impact of passive and active targeting in the era of modern cancer biology. Adv. Drug Deliv. Rev. 2014, 66, 2–25. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Liu, X.; Zhu, D.; Wang, Y.; Zhang, Z.; Zhou, X.; Qiu, N.; Chen, X.; Shen, Y. Nonviral cancer gene therapy: Delivery cascade and vector nanoproperty integration. Adv. Drug Deliv. Rev. 2017, 115, 115–154. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics, 2016. CA Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Fitzmaurice, C.; Allen, C.; Barber, R.M.; Barregard, L.; Bhutta, Z.A.; Brenner, H.; Dicker, D.J.; Chimed-Orchir, O.; Dandona, R.; Dandona, L. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: A systematic analysis for the global burden of disease study. JAMA Oncol. 2017, 3, 524–548. [Google Scholar] [PubMed]
- Bitko, V.; Musiyenko, A.; Shulyayeva, O.; Barik, S. Inhibition of respiratory viruses by nasally administered siRNA. Nat. Med. 2005, 11, 50–55. [Google Scholar] [CrossRef] [PubMed]
- DiFiglia, M.; Sena-Esteves, M.; Chase, K.; Sapp, E.; Pfister, E.; Sass, M.; Yoder, J.; Reeves, P.; Pandey, R.K.; Rajeev, K.G. Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits. Proc. Natl. Acad. Sci. USA 2007, 104, 17204–17209. [Google Scholar] [CrossRef] [PubMed]
- Pecot, C.V.; Calin, G.A.; Coleman, R.L.; Lopez-Berestein, G.; Sood, A.K. RNA interference in the clinic: Challenges and future directions. Nat. Rev. Cancer 2011, 11, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Bumcrot, D.; Manoharan, M.; Koteliansky, V.; Sah, D.W. RNAi therapeutics: A potential new class of pharmaceutical drugs. Nat. Chem. Biol. 2006, 2, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Scherphof, G. In vivo behavior of liposomes: Interactions with the mononuclear phagocyte system and implications for drug targeting. In Targeted Drug Delivery; Springer: Berlin, Germany, 1991; pp. 285–327. [Google Scholar]
- Zamecnik, J.; Vargova, L.; Homola, A.; Kodet, R.; Sykova, E. Extracellular matrix glycoproteins and diffusion barriers in human astrocytic tumours. Neuropathol. Appl. Neurobiol. 2004, 30, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Decuzzi, P.; Causa, F.; Ferrari, M.; Netti, P. The effective dispersion of nanovectors within the tumor microvasculature. Ann. Biomed. Eng. 2006, 34, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Dominska, M.; Dykxhoorn, D.M. Breaking down the barriers: siRNA delivery and endosome escape. J. Cell Sci. 2010, 123, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Rossi, J.J. Strategies for silencing human disease using RNA interference. Nat. Rev. Genet. 2007, 8, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.L.; Bartz, S.R.; Schelter, J.; Kobayashi, S.V.; Burchard, J.; Mao, M.; Li, B.; Cavet, G.; Linsley, P.S. Expression profiling reveals off-target gene regulation by RNAi. Nat. Biotechnol. 2003, 21, 635–637. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.L.; Burchard, J.; Schelter, J.; Chau, B.N.; Cleary, M.; Lim, L.; Linsley, P.S. Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA 2006, 12, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Petros, R.A.; DeSimone, J.M. Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. Drug Discov. 2010, 9, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Kanasty, R.L.; Whitehead, K.A.; Vegas, A.J.; Anderson, D.G. Action and reaction: The biological response to siRNA and its delivery vehicles. Mol. Ther. 2012, 20, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003, 4, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Castanotto, D.; Rossi, J.J. The promises and pitfalls of RNA-interference-based therapeutics. Nature 2009, 457, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Walter, W.; Stein, U. Viral vectors for gene transfer a review of their use in the treatment of human disease. Drugs 2000, 60, 249–271. [Google Scholar] [CrossRef]
- Ylä-Herttuala, S. Glybera’s second act: The curtain rises on the high cost of therapy. Mol. Ther. 2015, 23, 217–218. [Google Scholar] [CrossRef] [PubMed]
- Kay, M.A. State-of-the-art gene-based therapies: The road ahead. Nat. Rev. Genet. 2011, 12, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Mintzer, M.A.; Simanek, E.E. Nonviral vectors for gene delivery. Chem. Rev. 2008, 109, 259–302. [Google Scholar] [CrossRef] [PubMed]
- Gilleron, J.; Querbes, W.; Zeigerer, A.; Borodovsky, A.; Marsico, G.; Schubert, U.; Manygoats, K.; Seifert, S.; Andree, C.; Stöter, M. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat. Biotechnol. 2013, 31, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, D.V.; Fidelman, N.A.; Dan, N.; Lauffenburger, D.A. Vector unpacking as a potential barrier for receptor-mediated polyplex gene delivery. Biotechnol. Bioeng. 2000, 67, 598–606. [Google Scholar] [CrossRef]
- Lv, H.; Zhang, S.; Wang, B.; Cui, S.; Yan, J. Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release 2006, 114, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Loh, X.J.; Lee, T.-C.; Dou, Q.; Deen, G.R. Utilising inorganic nanocarriers for gene delivery. Biomater. Sci. 2016, 4, 70–86. [Google Scholar] [CrossRef] [PubMed]
- Shim, M.S.; Kwon, Y.J. Efficient and targeted delivery of siRNA in vivo. FEBS J. 2010, 277, 4814–4827. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 2018, 9, 1410. [Google Scholar] [CrossRef] [PubMed]
- Stacker, S.A.; Williams, S.P.; Karnezis, T.; Shayan, R.; Fox, S.B.; Achen, M.G. Lymphangiogenesis and lymphatic vessel remodelling in cancer. Nat. Rev. Cancer 2014, 14, 159. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; Stylianopoulos, T. Delivering nanomedicine to solid tumors. Nat. Rev. Clin. Oncol. 2010, 7, 653. [Google Scholar] [CrossRef] [PubMed]
- Danquah, M.K.; Zhang, X.A.; Mahato, R.I. Extravasation of polymeric nanomedicines across tumor vasculature. Adv. Drug Deliv. Rev. 2011, 63, 623–639. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Mangala, L.S.; Rodriguez-Aguayo, C.; Kong, X.; Lopez-Berestein, G.; Sood, A.K. RNA interference-based therapy and its delivery systems. Cancer Metastasis Rev. 2018, 37, 107–124. [Google Scholar] [CrossRef] [PubMed]
- Layek, B.; Lipp, L.; Singh, J. Cell penetrating peptide conjugated chitosan for enhanced delivery of nucleic acid. Int. J. Mol. Sci. 2015, 16, 28912–28930. [Google Scholar] [CrossRef] [PubMed]
- Shukla, R.S.; Jain, A.; Zhao, Z.; Cheng, K. Intracellular trafficking and exocytosis of a multi-component siRNA nanocomplex. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Sahay, G.; Querbes, W.; Alabi, C.; Eltoukhy, A.; Sarkar, S.; Zurenko, C.; Karagiannis, E.; Love, K.; Chen, D.; Zoncu, R. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat. Biotechnol. 2013, 31, 653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pezzella, F.; Turley, H.; Kuzu, I.; Tungekar, M.F.; Dunnill, M.S.; Pierce, C.B.; Harris, A.; Gatter, K.C.; Mason, D.Y. bcl-2 protein in non-small-cell lung carcinoma. N. Engl. J. Med. 1993, 329, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, Y. Antisense bcl-2 oligodeoxynucleotide enhancing 5-fluorouracil induced apoptosis in human gastric cancer cell line SGC 7901. Tumor 2002, 5, 15. [Google Scholar]
- Joensuu, H.; Pylkkänen, L.; Toikkanen, S. Bcl-2 protein expression and long-term survival in breast cancer. Am. J. Pathol. 1994, 145, 1191. [Google Scholar] [PubMed]
- Sinicrope, F.A.; Hart, J.; Michelassi, F.; Lee, J.J. Prognostic value of bcl-2 oncoprotein expression in stage II colon carcinoma. Clin. Cancer Res. 1995, 1, 1103–1110. [Google Scholar] [PubMed]
- Li, S.-M.; Yao, S.-K.; Yamamura, N.; Nakamura, T. Expression of Bcl-2 and Bax in extrahepatic biliary tract carcinoma and dysplasia. World J. Gastroenterol. 2003, 9, 2579. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.X.; Sato, Y.; Kuwao, S.; Kameya, T. Expression of bcl-2 oncogene protein is prevalent in small cell lung carcinomas. J. Pathol. 1995, 177, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Fu, G.-F.; Lin, X.-H.; Han, Q.-W.; Xu, Y.-F.; Guo, D.; Xu, G.-X.; Hou, Y.-Y. RNA interference remarkably suppresses bcl-2 gene expression in cancer cells in vitro and in vivo. Cancer Biol. Ther. 2005, 4, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Koty, P.P.; Zhang, H.; Levitt, M.L. Antisense bcl-2 treatment increases programmed cell death in non-small cell lung cancer cell lines. Lung Cancer 1999, 23, 115–127. [Google Scholar] [CrossRef]
- Lima, R.T.; Martins, L.M.; Guimaraes, J.E.; Sambade, C.; Vasconcelos, M.H. Specific downregulation of bcl-2 and xIAP by RNAi enhances the effects of chemotherapeutic agents in MCF-7 human breast cancer cells. Cancer Gene ther. 2004, 11, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Weyhenmeyer, B.; Murphy, A.; Prehn, J.; Murphy, B. Targeting the anti-apoptotic Bcl-2 family members for the treatment of cancer. Exp. Oncol. 2012, 34, 192–199. [Google Scholar] [PubMed]
- Garcia-Saez, A. The secrets of the Bcl-2 family. Cell Death Differ. 2012, 19, 1733–1740. [Google Scholar] [CrossRef] [PubMed]
- Sasatomi, E.; Tokunaga, O.; Miyazaki, K. Spontaneous apoptosis in gallbladder carcinoma: Relationships with clinicopathologic factors, expression of E-cadherin, bcl-2 protooncogene, and p53 oncosuppressor gene. Cancer 1996, 78, 2101–2110. [Google Scholar] [CrossRef]
- Mikami, T.; Yanagisawa, N.; Baba, H.; Koike, M.; Okayasu, I. Association of Bcl-2 protein expression with gallbladder carcinoma differentiation and progression and its relation to apoptosis. Cancer 1999, 85, 318–325. [Google Scholar] [CrossRef]
- Geng, Z.-M.; Zhang, M.; Pan, X.-T.; Wang, L. Bcl-2 gene silencing by RNA interference inhibits the growth of the human gallbladder carcinoma cell line GBC-SD in vitro and in vivo. Oncol. Rep. 2013, 30, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.; Hirabayashi, K.; Nakagawa, S.-I.; Yamaguchi, T.; Nogawa, M.; Kashimori, I.; Naito, H.; Kitagawa, H.; Ishiyama, K.; Ohgi, T. Antitumor activity of small interfering RNA/cationic liposome complex in mouse models of cancer. Clin. Cancer Res. 2004, 10, 7721–7726. [Google Scholar] [CrossRef] [PubMed]
- Sonoke, S.; Ueda, T.; Fujiwara, K.; Sato, Y.; Takagaki, K.; Hirabayashi, K.; Ohgi, T.; Yano, J. Tumor regression in mice by delivery of Bcl-2 small interfering RNA with pegylated cationic liposomes. Cancer Res. 2008, 68, 8843–8851. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, M.; Avraham, I.; Dor, Y.; Bachar-Lustig, E.; Itin, A.; Yung, S.; Chimenti, S.; Landsman, L.; Abramovitch, R.; Keshet, E. VEGF-induced adult neovascularization: Recruitment, retention, and role of accessory cells. Cell 2006, 124, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Saint-Geniez, M.; D’amore, P.A. Development and pathology of the hyaloid, choroidal and retinal vasculature. Int. J. Dev. Biol. 2004, 48, 1045–1058. [Google Scholar] [CrossRef] [PubMed]
- Stefater, J.A., III; Lewkowich, I.; Rao, S.; Mariggi, G.; Carpenter, A.C.; Burr, A.R.; Fan, J.; Ajima, R.; Molkentin, J.D.; Williams, B.O. Regulation of angiogenesis by a non-canonical Wnt-Flt1 pathway in myeloid cells. Nature 2011, 474, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folkman, J. Angiogenesis and breast cancer. J. Clin. Oncol. 1994, 12, 441–443. [Google Scholar] [CrossRef] [PubMed]
- Blood, C.H.; Zetter, B.R. Tumor interactions with the vasculature: Angiogenesis and tumor metastasis. Biochim. Biophys. Acta Rev. Cancer 1990, 1032, 89–118. [Google Scholar] [CrossRef]
- Ferrara, N.; Gerber, H.-P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F.; Detmar, M.; Claffey, K.P.; Nagy, J.A.; van de Water, L.; Senger, D.R. Vascular permeability factor/vascular endothelial growth factor: An important mediator of angiogenesis in malignancy and inflammation. Int. Arch. Allergy Immunol. 1995, 107, 233–235. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Kerbel, R.S. Angiogenesis as a therapeutic target. Nature 2005, 438, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. VEGF and the quest for tumour angiogenesis factors. Nat. Rev. Cancer 2002, 2, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.H.; Lee, W.M. Modeling antiangiogenesis gene therapy. Cancer Biol. Ther. 2002, 1, 554–555. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-P.; Feng, G.-S.; Liang, H.-M.; Zheng, C.-S.; Li, X. Vascular endothelial growth factor antisense oligodeoxynucleotides with lipiodol in arterial embolization of liver cancer in rats. World J. Gastroenterol. 2004, 10, 813. [Google Scholar] [CrossRef] [PubMed]
- Namiecińska, M.; Marciniak, K.; Nowak, J.Z. VEGF jako czynnik angiogenny, neurotroficzny i neuroprotekcyjny* VEGF as an angiogenic, neurotrophic, and neuroprotective factor. Postep. Hig. Med. Dosw. 2005, 59, 573–583. [Google Scholar]
- Takahashi, Y.; Kitadai, Y.; Bucana, C.D.; Cleary, K.R.; Ellis, L.M. Expression of vascular endothelial growth factor and its receptor, KDR, correlates with vascularity, metastasis, and proliferation of human colon cancer. Cancer Res. 1995, 55, 3964–3968. [Google Scholar] [PubMed]
- Toi, M.; Inada, K.; Suzuki, H.; Tominaga, T. Tumor angiogenesis in breast cancer: Its importance as a prognostic indicator and the association with vascular endothelial growth factor expression. Breast Cancer Res. Treat. 1995, 36, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Weidner, N.; Folkman, J.; Pozza, F.; Bevilacqua, P.; Allred, E.N.; Moore, D.H.; Meli, S.; Gasparini, G. Tumor angiogenesis: A new significant and independent prognostic indicator in early-stage breast carcinoma. JNCI J. Natl. Cancer Inst. 1992, 84, 1875–1887. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Chung, Y.; Takatsuka, S.; Ogawa, Y.; Sawada, T.; Yamashita, Y.; Onoda, N.; Kato, Y.; Nitta, A.; Arimoto, Y. Tumor angiogenesis as a predictor of recurrence in gastric carcinoma. J. Clin. Oncol. 1995, 13, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Algire, G.H.; Chalkley, H.W.; Legallais, F.Y.; Park, H.D. Vasculae reactions of normal and malignant tissues in vivo. I. vascular reactions of mice to wounds and to normal and neoplastic transplants. JNCI J. Natl. Cancer Inst. 1945, 6, 73–85. [Google Scholar] [CrossRef]
- Rhee, J.; Hoff, P.M. Angiogenesis inhibitors in the treatment of cancer. Expert Opin. Pharmacother. 2005, 6, 1701–1711. [Google Scholar] [CrossRef] [PubMed]
- Takei, Y.; Kadomatsu, K.; Matsuo, S.; Itoh, H.; Nakazawa, K.; Kubota, S.; Muramatsu, T. Antisense oligodeoxynucleotide targeted to Midkine, a heparin-binding growth factor, suppresses tumorigenicity of mouse rectal carcinoma cells. Cancer Res. 2001, 61, 8486–8491. [Google Scholar] [PubMed]
- Lu, P.Y.; Xie, F.Y.; Woodle, M.C. Modulation of angiogenesis with siRNA inhibitors for novel therapeutics. Trends Mol. Med. 2005, 11, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Fang, X.; Branch, C.; Mazur, W.; French, B.; Roth, J. Generation and identification of recombinant adenovirus by liposome-mediated transfection and PCR analysis. Biotechniques 1993, 15, 868–872. [Google Scholar] [PubMed]
- Jones, N.; Shenk, T. An adenovirus type 5 early gene function regulates expression of other early viral genes. Proc. Natl. Acad. Sci. USA 1979, 76, 3665–3669. [Google Scholar] [CrossRef] [PubMed]
- Im, S.; Kim, J.; Gomez-Manzano, C.; Fueyo, J.; Liu, T.; Cho, M.; Seong, C.; Lee, S.; Hong, Y.; Yung, W. Inhibition of breast cancer growth in vivo by antiangiogenesis gene therapy with adenovirus-mediated antisense-VEGF. Br. J. Cancer 2001, 84, 1252. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Togawa, H.; Harada, A.; Yasugi, K.; Matsumoto, T.; Katayose, S. Spontaneous formation of polyion complex micelles with narrow distribution from antisense oligonucleotide and cationic block copolymer in physiological saline. Macromolecules 1996, 29, 8556–8557. [Google Scholar] [CrossRef]
- Vinogradov, S.V.; Bronich, T.K.; Kabanov, A.V. Self-Assembly of Polyamine—Poly (ethylene glycol) Copolymers with Phosphorothioate Oligonucleotides. Bioconjugate Chem. 1998, 9, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Schiffelers, R.M.; Ansari, A.; Xu, J.; Zhou, Q.; Tang, Q.; Storm, G.; Molema, G.; Lu, P.Y.; Scaria, P.V.; Woodle, M.C. Cancer siRNA therapy by tumor selective delivery with ligand-targeted sterically stabilized nanoparticle. Nucl. Acids Res. 2004, 32, e149. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Jeong, J.H.; Cho, K.C.; Kim, S.W.; Park, T.G. Target-specific gene silencing by siRNA plasmid DNA complexed with folate-modified poly (ethylenimine). J. Control. Release 2005, 104, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Kataoka, K. Formation of polyion complex micelles in an aqueous milieu from a pair of oppositely-charged block copolymers with poly (ethylene glycol) segments. Macromolecules 1995, 28, 5294–5299. [Google Scholar] [CrossRef]
- Kim, S.H.; Jeong, J.H.; Lee, S.H.; Kim, S.W.; Park, T.G. Local and systemic delivery of VEGF siRNA using polyelectrolyte complex micelles for effective treatment of cancer. J. Control. Release 2008, 129, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Joukov, V.; Kaipainen, A.; Jeltsch, M.; Pajusola, K.; Olofsson, B.; Kumar, V.; Eriksson, U.; Alitalo, K. Vascular endothelial growth factors VEGF-B and VEGF-C. J. Cell. Physiol. 1997, 173, 211–215. [Google Scholar] [CrossRef]
- Cohen, B.; Addadi, Y.; Sapoznik, S.; Meir, G.; Kalchenko, V.; Harmelin, A.; Ben-Dor, S.; Neeman, M. Transcriptional regulation of vascular endothelial growth factor C by oxidative and thermal stress is mediated by lens epithelium-derived growth factor/p75. Neoplasia 2009, 11, 921. [Google Scholar] [CrossRef] [PubMed]
- Su, J.-L.; Yen, C.; Chen, P.; Chuang, S.; Hong, C.; Kuo, I.; Chen, H.; Hung, M.-C.; Kuo, M. The role of the VEGF-C/VEGFR-3 axis in cancer progression. Br. J. Cancer 2007, 96, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, S.; Brown, L.F.; Kodama, S.; Paavonen, K.; Alitalo, K.; Detmar, M. VEGF-C–induced lymphangiogenesis in sentinel lymph nodes promotes tumor metastasis to distant sites. Blood 2007, 109, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Jennbacken, K.; Vallbo, C.; Wang, W.; Damber, J.E. Expression of vascular endothelial growth factor C (VEGF-C) and VEGF receptor-3 in human prostate cancer is associated with regional lymph node metastasis. Prostate 2005, 65, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Jenny, B.; Harrison, J.; Baetens, D.; Tille, J.C.; Burkhardt, K.; Mottaz, H.; Kiss, J.Z.; Dietrich, P.Y.; De Tribolet, N.; Pizzolato, G. Expression and localization of VEGF-C and VEGFR-3 in glioblastomas and haemangioblastomas. J. Pathol. 2006, 209, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H. AZD6244 (ARRY-142886) enhances the antitumor activity of rapamycin in mouse models of human hepatocellular carcinoma. Cancer 2010, 116, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Morelli, M.P.; Brown, A.M.; Pitts, T.M.; Tentler, J.J.; Ciardiello, F.; Ryan, A.; Jürgensmeier, J.M.; Eckhardt, S.G. Targeting vascular endothelial growth factor receptor-1 and-3 with cediranib (AZD2171): Effects on migration and invasion of gastrointestinal cancer cell lines. Mol. Cancer Ther. 2009, 8, 2546–2558. [Google Scholar] [CrossRef] [PubMed]
- Svensson, S.; Jirström, K.; Rydén, L.; Roos, G.; Emdin, S.; Ostrowski, M.C.; Landberg, G. ERK phosphorylation is linked to VEGFR2 expression and Ets-2 phosphorylation in breast cancer and is associated with tamoxifen treatment resistance and small tumours with good prognosis. Oncogene 2005, 24, 4370–4379. [Google Scholar] [CrossRef] [PubMed]
- Brand, S.; Dambacher, J.; Beigel, F.; Olszak, T.; Diebold, J.; Otte, J.-M.; Göke, B.; Eichhorst, S.T. CXCR4 and CXCL12 are inversely expressed in colorectal cancer cells and modulate cancer cell migration, invasion and MMP-9 activation. Exp. Cell Res. 2005, 310, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.-B.; Peek, V.; Zhai, Y.; Paul, D.C.; Lou, Q.; Xia, X.; Eessalu, T.; Kohn, W.; Tang, S. Akt Activation, but not Extracellular Signal–Regulated Kinase Activation, Is Required for SDF-1α/CXCR4–Mediated Migration of Epitheloid Carcinoma Cells. Mol. Cancer Res. 2005, 3, 227–236. [Google Scholar] [PubMed]
- Fournier, E.; Birnbaum, D.; Borg, J.-P. Receptors for factors of the VEGF (vascular endothelial growth family). Bull. Cancer 1997, 84, 397–405. [Google Scholar] [PubMed]
- Deckers, M.M.; Karperien, M.; van der Bent, C.; Yamashita, T.; Papapoulos, S.E.; Löwik, C.W. Expression of vascular endothelial growth factors and their receptors during osteoblast differentiation. Endocrinology 2000, 141, 1667–1674. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.-L.; Zhang, X.; Zhang, J.-Y.; Hou, L.; Tian, R.-H. The mechanisms on apoptosis by inhibiting VEGF expression in human breast cancer cells. Int. Immunopharmacol. 2009, 9, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Kodama, M.; Kitadai, Y.; Tanaka, M.; Kuwai, T.; Tanaka, S.; Oue, N.; Yasui, W.; Chayama, K. Vascular endothelial growth factor C stimulates progression of human gastric cancer via both autocrine and paracrine mechanisms. Clin. Cancer Res. 2008, 14, 7205–7214. [Google Scholar] [CrossRef] [PubMed]
- Jüttner, S.; Wiβmann, C.; Jöns, T.; Vieth, M.; Hertel, J.; Gretschel, S.; Schlag, P.M.; Kemmner, W.; Höcker, M. Vascular endothelial growth factor-D and its receptor VEGFR-3: Two novel independent prognostic markers in gastric adenocarcinoma. J. Clin. Oncol. 2006, 24, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Ma, W.H.; Ge, Y.L.; Xue, M.L.; Zhang, Z.; Zhang, J.Y.; Hou, L.; Mu, R.H. RNAi-mediated gene silencing of vascular endothelial growth factor C suppresses growth and induces apoptosis in mouse breast cancer in vitro and in vivo. Oncol. Lett. 2016, 12, 3896–3904. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Hu, J.; Ma, J.; Feng, K.; Zhang, X.; Yang, S.; Wang, W.; Zhang, J.; Zhang, Y. RNAi-mediated silencing of VEGF-C inhibits non-small cell lung cancer progression by simultaneously down-regulating the CXCR4, CCR7, VEGFR-2 and VEGFR-3-dependent axes-induced ERK, p38 and AKT signalling pathways. Eur. J. Cancer 2011, 47, 2353–2363. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.M. The role of neuropilins in cancer. Mol. Cancer Ther. 2006, 5, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
- Bielenberg, D.R.; Pettaway, C.A.; Takashima, S.; Klagsbrun, M. Neuropilins in neoplasms: Expression, regulation, and function. Exp. Cell Res. 2006, 312, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Kärpänen, T.; Heckman, C.A.; Keskitalo, S.; Jeltsch, M.; Ollila, H.; Neufeld, G.; Tamagnone, L.; Alitalo, K. Functional interaction of VEGF-C and VEGF-D with neuropilin receptors. FASEB J. 2006, 20, 1462–1472. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.J.; Van Buren, G.; Dallas, N.A.; Xia, L.; Wang, X.; Yang, A.D.; Somcio, R.J.; Lin, Y.G.; Lim, S.; Fan, F. Therapeutic targeting of neuropilin-2 on colorectal carcinoma cells implanted in the murine liver. J. Natl. Cancer Inst. 2008, 100, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Hillan, K.J.; Gerber, H.-P.; Novotny, W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat. Rev. Drug Discov. 2004, 3, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Elbashir, S.M.; Harborth, J.; Weber, K.; Tuschl, T. Analysis of gene function in somatic mammalian cells using small interfering RNAs. Methods 2002, 26, 199–213. [Google Scholar] [CrossRef]
- Paddison, P.; Hannon, G. siRNAs and shRNAs: Skeleton keys to the human genome. Curr. Opin. Mol. Ther. 2003, 5, 217–224. [Google Scholar] [PubMed]
- Carpenter, A.E.; Sabatini, D.M. Systematic genome-wide screens of gene function. Nat. Rev. Genet. 2004, 5, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Ganju, P.; Hall, J. Potential applications of siRNA for pain therapy. Expert Opin. Biol. Ther. 2004, 4, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Woodle, M.; Scaria, P.; Ganesh, S.; Subramanian, K.; Titmas, R.; Cheng, C.; Yang, J.; Pan, Y.; Weng, K.; Gu, C. Sterically stabilized polyplex: Ligand-mediated activity. J. Control. Release 2001, 74, 309–311. [Google Scholar] [CrossRef]
- Langer, R. Drugs on target. Science 2001, 293, 58–59. [Google Scholar] [CrossRef] [PubMed]
- Suh, W.; Han, S.-O.; Yu, L.; Kim, S.W. An angiogenic, endothelial-cell-targeted polymeric gene carrier. Mol. Ther. 2002, 6, 664–672. [Google Scholar] [CrossRef]
- Verbaan, F.; Oussoren, C.; Snel, C.; Crommelin, D.; Hennink, W.; Storm, G. Steric stabilization of poly (2-(dimethylamino) ethyl methacrylate)-based polyplexes mediates prolonged circulation and tumor targeting in mice. J. Gene Med. 2004, 6, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Ogris, M.; Walker, G.; Blessing, T.; Kircheis, R.; Wolschek, M.; Wagner, E. Tumor-targeted gene therapy: Strategies for the preparation of ligand–polyethylene glycol–polyethylenimine/DNA complexes. J. Control. Release 2003, 91, 173–181. [Google Scholar] [CrossRef]
- Wagner, E.; Kircheis, R.; Walker, G.F. Targeted nucleic acid delivery into tumors: New avenues for cancer therapy. Biomed. Pharmacother. 2004, 58, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Hart, S.L.; Knight, A.M.; Harbottle, R.P.; Mistry, A.; Hunger, H.; Cutler, D.F.; Williamson, R.; Coutelle, C. Cell binding and internalization by filamentous phage displaying a cyclic Arg-Gly-Asp-containing peptide. J. Biol. Chem. 1994, 269, 12468–12474. [Google Scholar] [PubMed]
- Janssen, M.L.; Oyen, W.J.; Dijkgraaf, I.; Massuger, L.F.; Frielink, C.; Edwards, D.S.; Rajopadhye, M.; Boonstra, H.; Corstens, F.H.; Boerman, O.C. Tumor targeting with radiolabeled αvβ3 integrin binding peptides in a nude mouse model. Cancer Res. 2002, 62, 6146–6151. [Google Scholar] [PubMed]
- Zitzmann, S.; Ehemann, V.; Schwab, M. Arginine-glycine-aspartic acid (RGD)-peptide binds to both tumor and tumor-endothelial cells in vivo. Cancer Res. 2002, 62, 5139–5143. [Google Scholar] [PubMed]
- Voldborg, B.R.; Damstrup, L.; Spang-Thomsen, M.; Poulsen, H.S. Epidermal growth factor receptor (EGFR) and EGFR mutations, function and possible role in clinical trials. Ann. Oncol. 1997, 8, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, E. pH-sensitive nano-crystals of carbonate apatite for smart and cell-specific transgene delivery. Expert Opin. Drug Deliv. 2007, 4, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Tiash, S.; Kamaruzman, N.I.B.; Chowdhury, E.H. Carbonate apatite nanoparticles carry siRNA (s) targeting growth factor receptor genes EGFR1 and ErbB2 to regress mouse breast tumor. Drug Deliv. 2017, 24, 1721–1730. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Fang, F.; Ludewig, G.; Iones, G.; Jones, D. A mutation found in the promoter region of the human survivin gene is correlated to overexpression of survivin in cancer cells. DNA Cell Biol. 2004, 23, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Wheatley, S.P.; McNeish, I.A. Survivin: A protein with dual roles in mitosis and apoptosis. Int. Rev. Cytol. 2005, 247, 35–88. [Google Scholar] [CrossRef]
- Altieri, D.C. Survivin, cancer networks and pathway-directed drug discovery. Nat. Rev. Cancer 2008, 8, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Hunter, A.M.; LaCasse, E.C.; Korneluk, R.G. The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis 2007, 12, 1543–1568. [Google Scholar] [CrossRef] [PubMed]
- Schimmer, A.D. Inhibitor of apoptosis proteins: Translating basic knowledge into clinical practice. Cancer Res. 2004, 64, 7183–7190. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C. Targeted therapy by disabling crossroad signaling networks: The survivin paradigm. Mol. Cancer Ther. 2006, 5, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, S.; Pelus, L.M. Survivin, a cancer target with an emerging role in normal adult tissues. Mol. Cancer Ther. 2006, 5, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Liu, T.; Cotter, M.A.; Florell, S.R.; Robinette, K.; Hanks, A.N.; Grossman, D. Melanocyte expression of survivin promotes development and metastasis of UV-induced melanoma in HGF-transgenic mice. Cancer Res. 2007, 67, 5172–5178. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, S.; Languino, L.R.; Raskett, C.M.; Mercurio, A.M.; Dohi, T.; Altieri, D.C. IAP regulation of metastasis. Cancer Cell 2010, 17, 53–64. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, J.A.; Liu, T.; Goodson, A.; Grossman, D. Survivin enhances motility of melanoma cells by supporting Akt activation and α5 integrin upregulation. Cancer Res. 2010, 194. [Google Scholar] [CrossRef]
- Kawasaki, H.; Altieri, D.C.; Lu, C.-D.; Toyoda, M.; Tenjo, T.; Tanigawa, N. Inhibition of apoptosis by survivin predicts shorter survival rates in colorectal cancer. Cancer Res. 1998, 58, 5071–5074. [Google Scholar] [PubMed]
- Tanaka, K.; Iwamoto, S.; Gon, G.; Nohara, T.; Iwamoto, M.; Tanigawa, N. Expression of survivin and its relationship to loss of apoptosis in breast carcinomas. Clin. Cancer Res. 2000, 6, 127–134. [Google Scholar] [PubMed]
- Zaffaroni, N.; Pennati, M.; Colella, G.; Perego, P.; Supino, R.; Gatti, L.; Pilotti, S.; Zunino, F.; Daidone, M. Expression of the anti-apoptotic gene survivin correlates with taxol resistance in human ovarian cancer. Cell. Mol. Life Sci. 2002, 59, 1406–1412. [Google Scholar] [CrossRef] [PubMed]
- Tran, J.; Master, Z.; Joanne, L.Y.; Rak, J.; Dumont, D.J.; Kerbel, R.S. A role for survivin in chemoresistance of endothelial cells mediated by VEGF. Proc. Natl. Acad. Sci. USA 2002, 99, 4349–4354. [Google Scholar] [CrossRef] [PubMed]
- Cong, X.L.; Han, Z.C. Survivin and leukemia. Int. J. Hematol. 2004, 80, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.; Sun, W.; Kissel, T. Chitosan-based formulations for delivery of DNA and siRNA. Adv. Drug Deliv. Rev. 2010, 62, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Jeong, E.J.; Lee, J.; Rhim, T.; Lee, S.K.; Lee, K.Y. Preparation and characterization of nonaarginine-modified chitosan nanoparticles for siRNA delivery. Carbohydr. Polym. 2013, 92, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Somavarapu, S.; Colombani, A.; Govind, N.; Taylor, K.M. Nebulised siRNA encapsulated crosslinked chitosan nanoparticles for pulmonary delivery. Int. J. Pharm. 2013, 455, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Yun, K.-S.; Ban, H.-S.; Choe, W.; Lee, S.K.; Lee, K.Y. Preparation and characterization of chitosan/polyguluronate nanoparticles for siRNA delivery. J. Control. Release 2009, 139, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Guţoaia, A.; Schuster, L.; Margutti, S.; Laufer, S.; Schlosshauer, B.; Krastev, R.; Stoll, D.; Hartmann, H. Fine-tuned PEGylation of chitosan to maintain optimal siRNA-nanoplex bioactivity. Carbohydr. Polym. 2016, 143, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.N.; Xie, H.G.; Yu, W.T.; Liu, X.D.; Xie, W.Y.; Zhu, J.; Ma, X.J. Chitosan-g-MPEG-modified alginate/chitosan hydrogel microcapsules: A quantitative study of the effect of polymer architecture on the resistance to protein adsorption. Langmuir 2010, 26, 17156–17164. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Huang, W.; Jin, M.; Wang, Q.; Fan, B.; Kang, L.; Gao, Z. Chitosan-based nanoparticles for survivin targeted siRNA delivery in breast tumor therapy and preventing its metastasis. Int. J. Nanomed. 2016, 11, 4931. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Lins, L.; Divita, G.; Brasseur, R. Realistic modeling approaches of structure–function properties of CPPs in non-covalent complexes. Biochim. Biophys. Acta Biomembr. 2010, 1798, 2217–2222. [Google Scholar] [CrossRef] [PubMed]
- Veiman, K.-L.; Künnapuu, K.; Lehto, T.; Kiisholts, K.; Pärn, K.; Langel, Ü.; Kurrikoff, K. PEG shielded MMP sensitive CPPs for efficient and tumor specific gene delivery in vivo. J. Control. Release 2015, 209, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Bass, J. CPP magnetoresistance of magnetic multilayers: A critical review. J. Magn. Magn. Mater. 2016, 408, 244–320. [Google Scholar] [CrossRef]
- Ronca, F.; Raggi, A. Structure-function relationships in mammalian histidine-proline-rich glycoprotein. Biochimie 2015, 118, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.-T.; Hom, K.; Zhang, D.; Leng, Q.; Tricoli, L.J.; Hustedt, J.M.; Lee, A.; Shapiro, M.J.; Seog, J.; Kahn, J.D. Enhanced silencing and stabilization of siRNA polyplexes by histidine-mediated hydrogen bonds. Biomaterials 2014, 35, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Guo, Z.; Du, Z.; Fang, R.; Wu, H.; Zeng, X.; Wang, C.; Feng, M.; Pan, S. Serum tolerance and endosomal escape capacity of histidine-modified pDNA-loaded complexes based on polyamidoamine dendrimer derivatives. Biomaterials 2012, 33, 8111–8121. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.R.; Huang, Y.-W.; Winiarz, J.G.; Chiang, H.-J.; Lee, H.-J. Intracellular delivery of quantum dots mediated by a histidine-and arginine-rich HR9 cell-penetrating peptide through the direct membrane translocation mechanism. Biomaterials 2011, 32, 3520–3537. [Google Scholar] [CrossRef] [PubMed]
- Moreira, C.; Oliveira, H.; Pires, L.R.; Simões, S.; Barbosa, M.A.; Pêgo, A.P. Improving chitosan-mediated gene transfer by the introduction of intracellular buffering moieties into the chitosan backbone. Acta Biomater. 2009, 5, 2995–3006. [Google Scholar] [CrossRef] [PubMed]
- Corbet, C.; Ragelle, H.; Pourcelle, V.; Vanvarenberg, K.; Marchand-Brynaert, J.; Préat, V.; Feron, O. Delivery of siRNA targeting tumor metabolism using non-covalent PEGylated chitosan nanoparticles: Identification of an optimal combination of ligand structure, linker and grafting method. J. Control. Release 2016, 223, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Huang, W.; Li, Y.; Liu, S.; Jin, M.; Wang, Y.; Jia, L.; Gao, Z. Anti-tumor effects in mice induced by survivin-targeted siRNA delivered through polysaccharide nanoparticles. Biomaterials 2013, 34, 5689–5699. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Huang, W.; Kang, L.; Jin, M.; Fan, B.; Jin, H.; Wang, Q.-M.; Gao, Z. siRNA-loaded poly (histidine-arginine) 6-modified chitosan nanoparticle with enhanced cell-penetrating and endosomal escape capacities for suppressing breast tumor metastasis. Int. J. Nanomed. 2017, 12, 3221. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.X.; Bos, P.D.; Massagué, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Fernando, H.C. Surgical and nonresectional therapies for pulmonary metastasis. Surg. Clin. N. Am. 2010, 90, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Soengas, M.S.; Lowe, S.W. Apoptosis and melanoma chemoresistance. Oncogene 2003, 22, 3138–3151. [Google Scholar] [CrossRef] [PubMed]
- Bolcato-Bellemin, A.-L.; Bonnet, M.-E.; Creusat, G.; Erbacher, P.; Behr, J.-P. Sticky overhangs enhance siRNA-mediated gene silencing. Proc. Natl. Acad. Sci. USA 2007, 104, 16050–16055. [Google Scholar] [CrossRef] [PubMed]
- Kedinger, V.; Meulle, A.; Zounib, O.; Bonnet, M.-E.; Gossart, J.-B.; Benoit, E.; Messmer, M.; Shankaranarayanan, P.; Behr, J.-P.; Erbacher, P. Sticky siRNAs targeting survivin and cyclin B1 exert an antitumoral effect on melanoma subcutaneous xenografts and lung metastases. BMC Cancer 2013, 13, 338. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.; Sukumar, S. Distant metastasis in breast cancer: Molecular mechanisms and therapeutic targets. Cancer Biol. Ther. 2003, 2, 13–22. [Google Scholar] [CrossRef]
- Matsumoto, A.; Cabral, H.; Sato, N.; Kataoka, K.; Miyahara, Y. Assessment of Tumor Metastasis by the Direct Determination of Cell-Membrane Sialic Acid Expression. Angew. Chem. Int. Ed. 2010, 49, 5494–5497. [Google Scholar] [CrossRef] [PubMed]
- Büll, C.; Boltje, T.J.; van Dinther, E.A.; Peters, T.; de Graaf, A.M.; Leusen, J.H.; Kreutz, M.; Figdor, C.G.; den Brok, M.H.; Adema, G.J. Targeted delivery of a sialic acid-blocking glycomimetic to cancer cells inhibits metastatic spread. ACS Nano 2015, 9, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Fan, B.; Kang, L.; Chen, L.; Sun, P.; Jin, M.; Wang, Q.; Bae, Y.H.; Huang, W.; Gao, Z. Systemic siRNA delivery with a dual pH-responsive and tumor-targeted nanovector for inhibiting tumor growth and spontaneous metastasis in orthotopic murine model of breast carcinoma. Theranostics 2017, 7, 357. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.O. Cyclin-dependent kinases: Engines, clocks, and microprocessors. Ann. Rev. Cell Dev. Biol. 1997, 13, 261–291. [Google Scholar] [CrossRef] [PubMed]
- Krek, W.; Nigg, E. Differential phosphorylation of vertebrate p34cdc2 kinase at the G1/S and G2/M transitions of the cell cycle: Identification of major phosphorylation sites. EMBO J. 1991, 10, 305. [Google Scholar] [PubMed]
- Castedo, M.; Perfettini, J.; Roumier, T.; Kroemer, G. Cyclin-dependent kinase-1: Linking apoptosis to cell cycle and mitotic catastrophe. Cell Death Differ. 2002, 9, 1287. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Hardy, S.; Morgan, D.O. Nuclear localization of cyclin B1 controls mitotic entry after DNA damage. J. Cell Biol. 1998, 141, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Chae, H.-D.; Yun, J.; Jung, M.; Kim, Y.-S.; Kim, S.-H.; Han, M.H.; Shin, D.Y. Constitutive activation of cyclin B1-associated cdc2 kinase overrides p53-mediated G2-M arrest. Cancer Res. 2000, 60, 542–545. [Google Scholar] [PubMed]
- Yin, X.-Y.; Grove, L.; Datta, N.S.; Katula, K.; Long, M.W.; Prochownik, E.V. Inverse regulation of cyclin B1 by c-Myc and p53 and induction of tetraploidy by cyclin B1 overexpression. Cancer Res. 2001, 61, 6487–6493. [Google Scholar] [PubMed]
- Santana, C.; Ortega, E.; García-Carrancá, A. Oncogenic H-ras induces cyclin B1 expression in a p53-independent manner. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2002, 508, 49–58. [Google Scholar] [CrossRef]
- Sarafan-Vasseur, N.; Lamy, A.; Bourguignon, J.; Le Pessot, F.; Hieter, P.; Sesboue, R.; Bastard, C.; Frebourg, T.; Flaman, J.-M. Overexpression of B-type cyclins alters chromosomal segregation. Oncogene 2002, 21, 2051–2057. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Sui, L.; Watanabe, Y.; Sugimoto, K.; Tokuda, M. Clinical relevance of cyclin B1 overexpression in laryngeal squamous cell carcinoma. Cancer Lett. 2002, 177, 13–19. [Google Scholar] [CrossRef]
- Hassan, K.A.; Ang, K.K.; El-Naggar, A.K.; Story, M.D.; Lee, J.I.; Liu, D.; Hong, W.K.; Mao, L. Cyclin B1 overexpression and resistance to radiotherapy in head and neck squamous cell carcinoma. Cancer Res. 2002, 62, 6414–6417. [Google Scholar] [PubMed]
- Takeno, S.; Noguchi, T.; Kikuchi, R.; Uchida, Y.; Yokoyama, S.; Müller, W. Prognostic value of cyclin B1 in patients with esophageal squamous cell carcinoma. Cancer 2002, 94, 2874–2881. [Google Scholar] [CrossRef] [PubMed]
- Goga, A.; Yang, D.; Tward, A.D.; Morgan, D.O.; Bishop, J.M. Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nat. Med. 2007, 13, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Gros, E.; Aldrian-Herrada, G.; Choob, M.; Archdeacon, J.; Heitz, F.; Divita, G. A non-covalent peptide-based carrier for in vivo delivery of DNA mimics. Nucl. Acids Res. 2007, 35, e49. [Google Scholar] [CrossRef] [PubMed]
- Simeoni, F.; Morris, M.C.; Heitz, F.; Divita, G. Insight into the mechanism of the peptide-based gene delivery system MPG: Implications for delivery of siRNA into mammalian cells. Nucl. Acids Res. 2003, 31, 2717–2724. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.V.; Chan, S.W.-L.; Jacobsen, S.E.; Looney, D.J. Small interfering RNA-induced transcriptional gene silencing in human cells. Science 2004, 305, 1289–1292. [Google Scholar] [CrossRef] [PubMed]
- Zeineddine, D.; Papadimou, E.; Chebli, K.; Gineste, M.; Liu, J.; Grey, C.; Thurig, S.; Behfar, A.; Wallace, V.A.; Skerjanc, I.S. Oct-3/4 dose dependently regulates specification of embryonic stem cells toward a cardiac lineage and early heart development. Dev. Cell 2006, 11, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Depollier, J.; Mery, J.; Heitz, F.; Divita, G. A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat. Biotechnol. 2001, 19, 1173–1176. [Google Scholar] [CrossRef] [PubMed]
- Crombez, L.; Morris, M.C.; Deshayes, S.; Heitz, F.; Divita, G. Peptide-based nanoparticle for ex vivo and in vivo dug delivery. Curr. Pharm. Des. 2008, 14, 3656–3665. [Google Scholar] [CrossRef] [PubMed]
- Zorko, M.; Langel, Ü. Cell-penetrating peptides: Mechanism and kinetics of cargo delivery. Adv. Drug Deliv. Rev. 2005, 57, 529–545. [Google Scholar] [CrossRef] [PubMed]
- Deshayes, S.; Morris, M.; Divita, G.; Heitz, F. Cell-penetrating peptides: Tools for intracellular delivery of therapeutics. Cell. Mol. Life Sci. 2005, 62, 1839–1849. [Google Scholar] [CrossRef] [PubMed]
- Crombez, L.; Morris, M.C.; Dufort, S.; Aldrian-Herrada, G.; Nguyen, Q.; Mc Master, G.; Coll, J.-L.; Heitz, F.; Divita, G. Targeting cyclin B1 through peptide-based delivery of siRNA prevents tumour growth. Nucl. Acids Res. 2009, 37, 4559–4569. [Google Scholar] [CrossRef] [PubMed]
- Aznar, S.; Lacal, J.C. Rho signals to cell growth and apoptosis. Cancer Lett. 2001, 165, 1–10. [Google Scholar] [CrossRef]
- Schmitz, A.A.; Govek, E.-E.; Böttner, B.; Van Aelst, L. Rho GTPases: Signaling, migration, and invasion. Exp. Cell Res. 2000, 261, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Fritz, G.; Just, I.; Kaina, B. Rho GTPases are over-expressed in human tumors. Int. J. Cancer 1999, 81, 682–687. [Google Scholar] [CrossRef]
- Ridley, A.J. Rho GTPases and cell migration. J. Cell Sci. 2001, 114, 2713–2722. [Google Scholar] [PubMed]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; DiVito, M.M.; Merajver, S.D.; Boyanapalli, M.; Van Golen, K.L. Regulation of pancreatic cancer cell migration and invasion by RhoC GTPase and caveolin-1. Mol. Cancer 2005, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, I.P.; Zohn, I.E.; Der, C.J. Rho GTPase-dependent transformation by G protein-coupled receptors. Oncogene 2001, 20, 1547. [Google Scholar] [CrossRef] [PubMed]
- Gur, S.; Kadowitz, P.J.; Hellstrom, W.J. RhoA/Rho-kinase as a therapeutic target for the male urogenital tract. J. Sex. Med. 2011, 8, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, R.; Berrier, A.; Alahari, S.K. Role of Rho GTPases and their regulators in cancer progression. Front. Biosci. 2011, 16, 2561–2571. [Google Scholar] [CrossRef]
- Kwiatkowska, A.; Symons, M. Signaling determinants of glioma cell invasion. In Glioma Signaling; Springer: Berlin, Germany, 2013; pp. 121–141. [Google Scholar]
- Oh, H.K.; Sin, J.-I.; Choi, J.; Park, S.H.; Lee, T.S.; Choi, Y.S. Overexpression of CD73 in epithelial ovarian carcinoma is associated with better prognosis, lower stage, better differentiation and lower regulatory T cell infiltration. J. Gynecol. Oncol. 2012, 23, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Denoyelle, C.; Albanese, P.; Uzan, G.; Hong, L.; Vannier, J.-P.; Soria, J.; Soria, C. Molecular mechanism of the anti-cancer activity of cerivastatin, an inhibitor of HMG-CoA reductase, on aggressive human breast cancer cells. Cell. Signal. 2003, 15, 327–338. [Google Scholar] [CrossRef]
- Van Golen, K.L.; Wu, Z.-F.; Qiao, X.T.; Bao, L.W.; Merajver, S.D. RhoC GTPase, a novel transforming oncogene for human mammary epithelial cells that partially recapitulates the inflammatory breast cancer phenotype. Cancer Res. 2000, 60, 5832–5838. [Google Scholar] [PubMed]
- Ma, L.; Teruya-Feldstein, J.; Weinberg, R.A. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature 2007, 449, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Siegel, R.; Ward, E.; Murray, T.; Xu, J.; Thun, M.J. Cancer statistics, 2007. CA Cancer J. Clin. 2007, 57, 43–66. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Murray, T.; Thun, M.J. Cancer statistics, 2008. CA Cancer J. Clin. 2008, 58, 71–96. [Google Scholar] [CrossRef] [PubMed]
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global cancer statistics, 2002. CA Cancer J. Clin. 2005, 55, 74–108. [Google Scholar] [CrossRef] [PubMed]
- Wilke, H.-J.; Van Cutsem, E. Current treatments and future perspectives in colorectal and gastric cancer. Ann. Oncol. 2003, 14, ii49–ii55. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, G.; Liu, X.; Sui, A.; Yang, K.; Yao, R.; Wang, Z.; Shi, Q. Silencing of RhoA and RhoC expression by RNA interference suppresses human colorectal carcinoma growth in vivo. J. Exp. Clin. Cancer Res. 2010, 29, 123. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, J.-R.; Pottier, M.; Vekris, A.; Opolon, P.; Maksimenko, A.; Malvy, C. Comparison of antisense oligonucleotides and siRNAs in cell culture and in vivo. Biochem. Biophys. Res. Commun. 2002, 296, 1000–1004. [Google Scholar] [CrossRef]
- Pillé, J.-Y.; Denoyelle, C.; Varet, J.; Bertrand, J.-R.; Soria, J.; Opolon, P.; Lu, H.; Pritchard, L.-L.; Vannier, J.-P.; Malvy, C. Anti-RhoA and anti-RhoC siRNAs inhibit the proliferation and invasiveness of MDA-MB-231 breast cancer cells in vitro and in vivo. Mol. Ther. 2005, 11, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Pillé, J.-Y.; Li, H.; Blot, E.; Bertrand, J.-R.; Pritchard, L.-L.; Opolon, P.; Maksimenko, A.; Lu, H.; Vannier, J.-P.; Soria, J. Intravenous delivery of anti-RhoA small interfering RNA loaded in nanoparticles of chitosan in mice: Safety and efficacy in xenografted aggressive breast cancer. Hum. Gene Ther. 2006, 17, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Suwa, H.; Ohshio, G.; Imamura, T.; Watanabe, G.; Arii, S.; Imamura, M.; Narumiya, S.; Hiai, H.; Fukumoto, M. Overexpression of the RhoC gene correlates with progression of ductal adenocarcinoma of the pancreas. Br. J. Cancer 1998, 77, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Zhu, M.; Lv, G.; Zhang, Q.; Wang, G. The role of RhoC in the proliferation and apoptosis of hepatocellular carcinoma cells. Med. Oncol. 2012, 29, 1802–1809. [Google Scholar] [CrossRef] [PubMed]
- Robertson, F.M.; Bondy, M.; Yang, W.; Yamauchi, H.; Wiggins, S.; Kamrudin, S.; Krishnamurthy, S.; Le-Petross, H.; Bidaut, L.; Player, A.N. Inflammatory breast cancer: The disease, the biology, the treatment. CA Cancer J. Clin. 2010, 60, 351–375. [Google Scholar] [CrossRef] [PubMed]
- Anderson, W.F.; Schairer, C.; Chen, B.E.; Hance, K.W.; Levine, P.H. Epidemiology of inflammatory breast cancer (IBC) 1. Breast Dis. 2006, 22, 9–23. [Google Scholar] [CrossRef]
- Van Golen, K.L.; Bao, L.W.; Pan, Q.; Miller, F.R.; Wu, Z.F.; Merajver, S.D. Mitogen activated protein kinase pathway is involved in RhoC GTPase induced motility, invasion and angiogenesis in inflammatory breast cancer. Clin. Exp. Metastasis 2002, 19, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.-D.; Shen, H.-B.; Zhu, L.; Lu, J.-Q.; Zhang, L.; Luo, Z.-Y.; Wu, Y.-Q. Anti-rhoc sirnas inhibit the proliferation and invasiveness of breast cancer cells via modulating the Kai1, MMP9, and CXCR4 expression. OncoTargets Ther. 2017, 10, 1827. [Google Scholar] [CrossRef] [PubMed]
- Kraus, C.; Liehr, T.; Hülsken, J.; Behrens, J.; Birchmeier, W.; Grzeschik, K.-H.; Ballhausen, W.G. Localization of the human β-catenin gene (CTNNB1) to 3p21: A region implicated in tumor development. Genomics 1994, 23, 272–274. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Peifer, M.; Rauskolb, C.; Williams, M.; Riggleman, B.; Wieschaus, E. The segment polarity gene armadillo interacts with the wingless signaling pathway in both embryonic and adult pattern formation. Development 1991, 111, 1029–1043. [Google Scholar] [PubMed]
- Noordermeer, J.; Klingensmith, J.; Perrimonl, N. Dishevelled and armadillo act in the Wingless signalling pathway in Drosophila. Nature 1994, 367, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Peifer, M.; Berg, S.; Reynolds, A.B. A repeating amino acid motif shared by proteins with diverse cellular roles. Cell 1994, 76, 789–791. [Google Scholar] [CrossRef]
- Bilić, J.; Huang, Y.-L.; Davidson, G.; Zimmermann, T.; Cruciat, C.-M.; Bienz, M.; Niehrs, C. Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science 2007, 316, 1619–1622. [Google Scholar] [CrossRef] [PubMed]
- Schwarz-Romond, T.; Fiedler, M.; Shibata, N.; Butler, P.J.G.; Kikuchi, A.; Higuchi, Y.; Bienz, M. The DIX domain of Dishevelled confers Wnt signaling by dynamic polymerization. Nat. Struct. Mol. Biol. 2007, 14, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Behrens, J.; von Kries, J.P.; Kühl, M.; Bruhn, L.; Wedlich, D.; Grosschedl, R.; Birchmeier, W. Functional interaction of β-catenin with the transcription factor LEF-1. Nature 1996, 382, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, M.; van de Wetering, M.; Oosterwegel, M.; Peterson-Maduro, J.; Godsave, S.; Korinek, V.; Roose, J.; Destrée, O.; Clevers, H. XTcf-3 transcription factor mediates β-catenin-induced axis formation in Xenopus embryos. Cell 1996, 86, 391–399. [Google Scholar] [CrossRef]
- Grigoryan, T.; Wend, P.; Klaus, A.; Birchmeier, W. Deciphering the function of canonical Wnt signals in development and disease: Conditional loss-and gain-of-function mutations of β-catenin in mice. Genes Dev. 2008, 22, 2308–2341. [Google Scholar] [CrossRef] [PubMed]
- Hajra, K.M.; Fearon, E.R. Cadherin and catenin alterations in human cancer. Genes Chromosom. Cancer 2002, 34, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Ilyas, M.; Tomlinson, I.; Rowan, A.; Pignatelli, M.; Bodmer, W. β-Catenin mutations in cell lines established from human colorectal cancers. Proc. Natl. Acad. Sci. USA 1997, 94, 10330–10334. [Google Scholar] [CrossRef] [PubMed]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef] [PubMed]
- Polakis, P. Wnt signaling and cancer. Genes Dev. 2000, 14, 1837–1851. [Google Scholar] [CrossRef] [PubMed]
- Sparks, A.B.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Mutational analysis of the APC/β-catenin/Tcf pathway in colorectal cancer. Cancer Res. 1998, 58, 1130–1134. [Google Scholar] [PubMed]
- Su, L.-K.; Vogelstein, B.; Kinzler, K.W. Association of the APC tumor suppressor protein with catenins. Science 1993, 262, 1734–1738. [Google Scholar] [CrossRef] [PubMed]
- Korinek, V.; Barker, N.; Morin, P.J.; Van Wichen, D.; De Weger, R.; Kinzler, K.W.; Vogelstein, B.; Clevers, H. Constitutive transcriptional activation by a β-catenin-Tcf complex in APC−/− colon carcinoma. Science 1997, 275, 1784–1787. [Google Scholar] [CrossRef] [PubMed]
- Bass, B.L. Double-stranded RNA as a template for gene silencing. Cell 2000, 101, 235–238. [Google Scholar] [CrossRef]
- Verma, U.N.; Surabhi, R.M.; Schmaltieg, A.; Becerra, C.; Gaynor, R.B. Small interfering RNAs directed against β-catenin inhibit the in vitro and in vivo growth of colon cancer cells. Clin. Cancer Res. 2003, 9, 1291–1300. [Google Scholar] [PubMed]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Van Es, J.H.; Kuipers, J.; Kujala, P.; Van Den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Rishi, A.K.; Zhang, L.; Yu, Y.; Jiang, Y.; Nautiyal, J.; Wali, A.; Fontana, J.A.; Levi, E.; Majumdar, A.P. Cell cycle-and apoptosis-regulatory protein-1 is involved in apoptosis signaling by epidermal growth factor receptor. J. Biol. Chem. 2006, 281, 13188–13198. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Yang, C.K.; Heo, K.; Roeder, R.G.; An, W.; Stallcup, M.R. CCAR1, a key regulator of mediator complex recruitment to nuclear receptor transcription complexes. Mol. Cell 2008, 31, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Ou, C.-Y.; Kim, J.H.; Yang, C.K.; Stallcup, M.R. Requirement of cell cycle and apoptosis regulator 1 for target gene activation by Wnt and β-catenin and for anchorage-independent growth of human colon carcinoma cells. J. Biol. Chem. 2009, 284, 20629–20637. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.-S.; Wei, K.-L.; Lu, C.-K.; Chen, Y.-H.; Cheng, Y.-T.; Tung, S.-Y.; Wu, C.-S.; Chiang, M.-K. Inhibition of CCAR1, a Coactivator of β-Catenin, Suppresses the Proliferation and Migration of Gastric Cancer Cells. Int. J. Mol. Sci. 2017, 18, 460. [Google Scholar] [CrossRef] [PubMed]
- Amit, I.; Wides, R.; Yarden, Y. Evolvable signaling networks of receptor tyrosine kinases: Relevance of robustness to malignancy and to cancer therapy. Mol. Syst. Biol. 2007, 3, 151. [Google Scholar] [CrossRef] [PubMed]
- Zelinski, D.P.; Zantek, N.D.; Stewart, J.C.; Irizarry, A.R.; Kinch, M.S. EphA2 overexpression causes tumorigenesis of mammary epithelial cells. Cancer Res. 2001, 61, 2301–2306. [Google Scholar] [PubMed]
- Kullander, K.; Klein, R. Mechanisms and functions of Eph and ephrin signalling. Nat. Rev. Mol. Cell Biol. 2002, 3, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Hafner, C.; Schmitz, G.; Meyer, S.; Bataille, F.; Hau, P.; Langmann, T.; Dietmaier, W.; Landthaler, M.; Vogt, T. Differential gene expression of Eph receptors and ephrins in benign human tissues and cancers. Clin. Chem. 2004, 50, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-F.; Fokas, E.; Bieker, M.; Rose, F.; Rexin, P.; Zhu, Y.; Pagenstecher, A.; Engenhart-Cabillic, R.; An, H.-X. Increased expression of EphA2 correlates with adverse outcome in primary and recurrent glioblastoma multiforme patients. Oncol. Rep. 2008, 19, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Brannan, J.M.; Dong, W.; Prudkin, L.; Behrens, C.; Lotan, R.; Bekele, B.N.; Wistuba, I.; Johnson, F.M. Expression of the receptor tyrosine kinase EphA2 is increased in smokers and predicts poor survival in non–small cell lung cancer. Clin. Cancer Res. 2009, 15, 4423–4430. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.D.; Lee, M.J.; Yu, G.R.; Kim, I.H.; Yu, H.C.; Song, E.Y.; Kim, D.G. EFNA1 ligand and its receptor EphA2: Potential biomarkers for hepatocellular carcinoma. Int. J. Cancer 2010, 126, 940–949. [Google Scholar] [CrossRef] [PubMed]
- Baeten, C.I.; Hillen, F.; Pauwels, P.; de Bruine, A.P.; Baeten, C.G. Prognostic role of vasculogenic mimicry in colorectal cancer. Dis. Colon Rectum 2009, 52, 2028–2035. [Google Scholar] [CrossRef] [PubMed]
- Merritt, W.M.; Kamat, A.A.; Hwang, J.-Y.; Bottsford-Miller, J.; Lu, C.; Lin, Y.G.; Coffey, D.; Spannuth, W.A.; Nugent, E.; Han, L.Y. Clinical and biological impact of EphA2 overexpression and angiogenesis in endometrial cancer. Cancer Biol. Ther. 2010, 10, 1306–1314. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.-J.; Ge, J.; Chen, Z.-K.; Wu, S.-B.; Shen, H.; Yang, P.; Hu, B.; Zhang, G.-W.; Chen, Z.-H. Over-expression of EphA2 and EphrinA-1 in human gastric adenocarcinoma and its prognostic value for postoperative patients. Dig. Dis. Sci. 2009, 54, 2410. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Maru, Y.; Hagiwara, K.; Nishida, J.; Takaku, F. A novel putative tyrosine kinase receptor encoded by the eph gene. Science 1987, 238, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Thaker, P.H.; Deavers, M.; Celestino, J.; Thornton, A.; Fletcher, M.S.; Landen, C.N.; Kinch, M.S.; Kiener, P.A.; Sood, A.K. EphA2 expression is associated with aggressive features in ovarian carcinoma. Clin. Cancer Res. 2004, 10, 5145–5150. [Google Scholar] [CrossRef] [PubMed]
- Zeng, G.; Hu, Z.; Kinch, M.S.; Pan, C.-X.; Flockhart, D.A.; Kao, C.; Gardner, T.A.; Zhang, S.; Li, L.; Baldridge, L.A. High-level expression of EphA2 receptor tyrosine kinase in prostatic intraepithelial neoplasia. Am. J. Pathol. 2003, 163, 2271–2276. [Google Scholar] [CrossRef]
- Wykosky, J.; Gibo, D.M.; Stanton, C.; Debinski, W. Interleukin-13 receptor α2, EphA2, and Fos-related antigen 1 as molecular denominators of high-grade astrocytomas and specific targets for combinatorial therapy. Clin. Cancer Res. 2008, 14, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Lazar, D.F.; Saltiel, A.R.; Dixit, V.M. Activation of the Eck receptor protein tyrosine kinase stimulates phosphatidylinositol 3-kinase activity. J. Biol. Chem. 1994, 269, 30154–30157. [Google Scholar] [PubMed]
- Landen, C.N.; Chavez-Reyes, A.; Bucana, C.; Schmandt, R.; Deavers, M.T.; Lopez-Berestein, G.; Sood, A.K. Therapeutic EphA2 gene targeting in vivo using neutral liposomal small interfering RNA delivery. Cancer Res. 2005, 65, 6910–6918. [Google Scholar] [CrossRef] [PubMed]
- Ayaki, M.; Komatsu, K.; Mukai, M.; Murata, K.; Kameyama, M.; Ishiguro, S.; Miyoshi, J.; Tatsuta, M.; Nakamura, H. Reduced expression of focal adhesion kinase in liver metastases compared with matched primary human colorectal adenocarcinomas. Clin. Cancer Res. 2001, 70, 3106–3112. [Google Scholar]
- Cance, W.G.; Harris, J.E.; Iacocca, M.V.; Roche, E.; Yang, X.; Chang, J.; Simkins, S.; Xu, L. Immunohistochemical analyses of focal adhesion kinase expression in benign and malignant human breast and colon tissues: correlation with preinvasive and invasive phenotypes. Clin. Cancer Res. 2000, 6, 2417–2423. [Google Scholar] [PubMed]
- Owens, L.V.; Xu, L.; Craven, R.J.; Dent, G.A.; Weiner, T.M.; Kornberg, L.; Liu, E.T.; Cance, W.G. Overexpression of the focal adhesion kinase (p125FAK) in invasive human tumors. Cancer Res. 1995, 55, 2752–2755. [Google Scholar] [PubMed]
- Owens, L.V.; Xu, L.; Dent, G.A.; Yang, X.; Sturge, G.C.; Craven, R.J.; Cance, W.G. Focal adhesion kinase as a marker of invasive potential in differentiated human thyroid cancer. Ann. Surg. Oncol. 1996, 3, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, L.; Hauck, W.; Aprikian, A.G.; Begin, L.R.; Chapdelaine, A.; Chevalier, S. Focal adhesion kinase (pp125FAK) expression, activation and association with paxillin and p50CSK in human metastatic prostate carcinoma. Int. J. Cancer 1996, 68, 164–171. [Google Scholar] [CrossRef]
- Sood, A.K.; Coffin, J.E.; Schneider, G.B.; Fletcher, M.S.; DeYoung, B.R.; Gruman, L.M.; Gershenson, D.M.; Schaller, M.D.; Hendrix, M.J. Biological significance of focal adhesion kinase in ovarian cancer: role in migration and invasion. Am. J. Pathol. 2004, 165, 1087–1095. [Google Scholar] [CrossRef]
- Ishizawar, R.; Parsons, S.J. c-Src and cooperating partners in human cancer. Cancer Cell 2004, 6, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Shahzad, M.M.; Lu, C.; Lee, J.-W.; Stone, R.L.; Mitra, R.; Mangala, L.S.; Lu, Y.; Baggerly, K.A.; Danes, C.G.; Nick, A.M. Dual targeting of EphA2 and FAK in ovarian carcinoma. Cancer Biol. Ther. 2009, 8, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Sulman, E.P.; Tang, X.X.; Allen, C.; Biegel, J.A.; Pleasure, D.E.; Brodeur, G.M.; Ikegaki, N. ECK, a HumanEPH-Related Gene, Maps to 1p36. 1, a Common Region of Alteration in Human Cancers. Genomics 1997, 40, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Chen, Z.; Wu, S.; Guo, J.; Ge, J.; Yang, P.; Huang, J. Silencing of EphA2 inhibits invasion of human gastric cancer SGC-7901 cells in vitro and in vivo. Neoplasma 2012, 59, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Higashiyama, M.; Doi, O.; Kodama, K.; Yokouchi, H.; Kasugai, T.; Ishiguro, S.; Takami, K.; Nakayama, T.; Nishisho, I. MDM2 gene amplification and expression in non-small-cell lung cancer: Immunohistochemical expression of its protein is a favourable prognostic marker in patients without p53 protein accumulation. Br. J. Cancer 1997, 75, 1302–1308. [Google Scholar] [CrossRef] [PubMed]
- Graves, B.; Thompson, T.; Xia, M.; Janson, C.; Lukacs, C.; Deo, D.; Di Lello, P.; Fry, D.; Garvie, C.; Huang, K.-S. Activation of the p53 pathway by small-molecule-induced MDM2 and MDMX dimerization. Proc. Natl. Acad. Sci. USA 2012, 109, 11788–11793. [Google Scholar] [CrossRef] [PubMed]
- Koster, R.; Timmer-Bosscha, H.; Bischoff, R.; Gietema, J.A.; de Jong, S. Disruption of the MDM2–p53 interaction strongly potentiates p53-dependent apoptosis in cisplatin-resistant human testicular carcinoma cells via the Fas/FasL pathway. Cell Death Dis. 2011, 2, e148. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Wagner, E. Bioresponsive polymers for nonviral gene delivery. Curr. Opin. Mol. Ther. 2009, 11, 165–178. [Google Scholar] [PubMed]
- Yu, H.; Zou, Y.; Jiang, L.; Yin, Q.; He, X.; Chen, L.; Zhang, Z.; Gu, W.; Li, Y. Induction of apoptosis in non-small cell lung cancer by downregulation of MDM2 using pH-responsive PMPC-b-PDPA/siRNA complex nanoparticles. Biomaterials 2013, 34, 2738–2747. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-D.; Chono, S.; Huang, L. Efficient oncogene silencing and metastasis inhibition via systemic delivery of siRNA. Mol. Ther. 2008, 16, 942–946. [Google Scholar] [PubMed]
- Pollak, M. Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 2008, 8, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Beech, D.J.; Parekh, N.; Pang, Y. Insulin-like growth factor-I receptor antagonism results in increased cytotoxicity of breast cancer cells to doxorubicin and taxol. Oncol. Rep. 2001, 8, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Cantley, L.C.; Auger, K.R.; Carpenter, C.; Duckworth, B.; Graziani, A.; Kapeller, R.; Soltoff, S. Oncogenes and signal transduction. Cell 1991, 64, 281–302. [Google Scholar] [CrossRef]
- Chernicky, C.L.; Yi, L.; Tan, H.; Gan, S.U.; Ilan, J. Treatment of human breast cancer cells with antisense RNA to the type I insulin-like growth factor receptor inhibits cell growth, suppresses tumorigenesis, alters the metastatic potential, and prolongs survival in vivo. Cancer Gene Ther. 2000, 7, 384. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Tewari, M.; Cui, S.; Rubin, R. Activation of the insulin-like growth factor-I receptor inhibits tumor necrosis factor-induced cell death. J. Cell. Physiol. 1996, 168, 499–509. [Google Scholar] [CrossRef]
- Baserga, R. The IGF-I receptor in cancer research. Exp. Cell Res. 1999, 253, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Yee, D. Insulin-like growth factors and breast cancer. Biomed. Pharmacother. 1995, 49, 415–421. [Google Scholar] [CrossRef]
- Iravani, S.; Zhang, H.Q.; Yuan, Z.Q.; Cheng, J.Q.; Karl, R.C.; Jove, R.; Coppola, D. Modification of insulin-like growth factor 1 receptor, c-Src, and Bcl-X L protein expression during the progression of barrett’s neoplasia. Hum. Pathol. 2003, 34, 975–982. [Google Scholar] [CrossRef]
- LeRoith, D.; Baserga, R.; Helman, L.; Roberts, C.T., Jr. Insulin-like growth factors and cancer. Ann. Intern. Med. 1995, 122, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Siegel, R.; Xu, J.; Ward, E. Cancer statistics, 2010. CA Cancer J. Clin. 2010, 60, 277–300. [Google Scholar] [CrossRef] [PubMed]
- Gridelli, C. Targeted therapy developments in the treatment of non-small cell lung cancer: A promising but long and winding road. Curr. Opin. Oncol. 2008, 20, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.H. Treatment of advanced non-small cell lung cancer: Should include short courses of radiation, with palliation as the aim. BMJ Br. Med. J. 2002, 325, 452. [Google Scholar] [CrossRef]
- Singh, P. Insulin-like growth factor system in growth, development and carcinogenesis. J. Clin. Ligand Assay 2000, 23, 214–232. [Google Scholar]
- Dong, A.-Q.; Kong, M.-J.; Ma, Z.-Y.; Qian, J.-F.; Xu, X.-H. Down-regulation of IGF-IR using small, interfering, hairpin RNA (siRNA) inhibits growth of human lung cancer cell line A549 in vitro and in nude mice. Cell Biol. Int. 2007, 31, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ding, C.; Kong, M.; Dong, A.; Qian, J.; Jiang, D.; Shen, Z. Tumor-targeting magnetic lipoplex delivery of short hairpin RNA suppresses IGF-1R overexpression of lung adenocarcinoma A549 cells in vitro and in vivo. Biochem. Biophys. Res. Commun. 2011, 410, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Deveraux, Q.L.; Stennicke, H.R.; Salvesen, G.S.; Reed, J.C. Endogenous inhibitors of caspases. J. Clin. Immunol. 1999, 19, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, A.; Yang, A.Y.-P.; Srivastava, M. Regulators of IAP function: Coming to grips with the grim reaper. Curr. Opin. Cell Biol. 2003, 15, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Yagihashi, A.; Asanuma, K.; Tsuji, N.; Torigoe, T.; Sato, N.; Hirata, K.; Watanabe, N. Detection of anti-livin antibody in gastrointestinal cancer patients. Clin. Chem. 2003, 49, 1206–1208. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, G.; Adida, C.; Altieri, D.C. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat. Med. 1997, 3, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Richter, B.W.; Duckett, C.S. The IAP proteins: Caspase inhibitors and beyond. Sci. Signal. 2000, 2000, pe1. [Google Scholar] [CrossRef] [PubMed]
- Ashhab, Y.; Alian, A.; Polliack, A.; Panet, A.; Yehuda, D.B. Two splicing variants of a new inhibitor of apoptosis gene with different biological properties and tissue distribution pattern. FEBS Lett. 2001, 495, 56–60. [Google Scholar] [CrossRef]
- Ka, H.; Hunt, J.S. Temporal and spatial patterns of expression of inhibitors of apoptosis in human placentas. Am. J. Pathol. 2003, 163, 413–422. [Google Scholar] [CrossRef]
- Lv, J.; Chen, Z. Resent research about Livin in cancer. Chin. J. Cancer Prev. Treat. 2006, 13, 1347–1350. [Google Scholar]
- Yagihashi, A.; Ohmura, T.; Asanuma, K.; Kobayashi, D.; Tsuji, N.; Torigoe, T.; Sato, N.; Hirata, K.; Watanabe, N. Detection of autoantibodies to survivin and livin in sera from patients with breast cancer. Clin. Chim. Acta 2005, 362, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Hariu, H.; Hirohashi, Y.; Torigoe, T.; Asanuma, H.; Hariu, M.; Tamura, Y.; Aketa, K.; Nabeta, C.; Nakanishi, K.; Kamiguchi, K. Aberrant expression and potency as a cancer immunotherapy target of inhibitor of apoptosis protein family, Livin/ML-IAP in lung cancer. Clin. Cancer Res. 2005, 11, 1000–1009. [Google Scholar] [PubMed]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2015. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef] [PubMed]
- Bar-Eli, M. Role of AP-2 in tumor growth and metastasis of human melanoma. Cancer Metastasis Rev. 1999, 18, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Bar-Eli, M. Gene regulation in melanoma progression by the AP-2 transcription factor. Pigment. Cell Melanoma Res. 2001, 14, 78–85. [Google Scholar] [CrossRef]
- Soutschek, J.; Akinc, A.; Bramlage, B.; Charisse, K.; Constien, R.; Donoghue, M.; Elbashir, S.; Geick, A.; Hadwiger, P.; Harborth, J. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 2004, 432, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Dykxhoorn, D.M.; Lieberman, J. The silent revolution: RNA interference as basic biology, research tool, and therapeutic. Annu. Rev. Med. 2005, 56, 401–423. [Google Scholar] [CrossRef] [PubMed]
- Ryther, R.; Flynt, A.; Phillips, J.; Patton, J. siRNA therapeutics: Big potential from small RNAs. Gene Ther. 2005, 12, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, Y.; Wang, W.; Guan, B.; Xun, M.; Zhang, H.; Wang, Z.; Zhao, Y. Single-chain antibody–delivered Livin siRNA inhibits human malignant melanoma growth in vitro and in vivo. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Lin, F.; Wang, X.; Gao, P.; Dong, K.; Zou, A.; Cheng, S.; Wei, S.; Zhang, H. Silencing Livin gene expression to inhibit proliferation and enhance chemosensitivity in tumor cells. Cancer Gene Ther. 2008, 15, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Call, K.M.; Glaser, T.; Ito, C.Y.; Buckler, A.J.; Pelletier, J.; Haber, D.A.; Rose, E.A.; Kral, A.; Yeger, H.; Lewis, W.H. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell 1990, 60, 509–520. [Google Scholar] [CrossRef]
- Gessler, M.; Poustka, A.; Cavenee, W.; Neve, R.L.; Orkin, S.H.; Bruns, G.A. Homozygous deletion in Wilms tumours of a zinc-finger gene identified by chromosome jumping. Nature 1990, 343, 774. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, H. Wilms’ tumor gene WT1: Its oncogenic function and clinical application. Int. J. Hematol. 2001, 73, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Coppes, M.J.; Campbell, C.E.; Williams, B. The role of WT1 in Wilms tumorigenesis. FASEB J. 1993, 7, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Rauscher, F. The WT1 Wilms tumor gene product: A developmentally regulated transcription factor in the kidney that functions as a tumor suppressor. FASEB J. 1993, 7, 896–903. [Google Scholar] [CrossRef] [PubMed]
- Haber, D.; Park, S.; Maheswaran, S.; Englert, C.; Re, G.; Hazen-Martin, D.; Sens, D.; Garvin, A. WT1-mediated growth suppression of Wilms’ tumor cells expressing a WT1 splicing variant. Science 1993, 262, 2057–2059. [Google Scholar] [CrossRef] [PubMed]
- Algar, E.M.; Kenney, M.T.; Simms, L.A.; Smith, S.I.; Kida, Y.; Smith, P.J. Homozygous intragenic deletion in the WT1 gene in a sporadic Wilms’ tumour associated with high levels of expression of a truncated transcript. Hum. Mutat. 1995, 5, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Little, M.; Wells, C. A clinical overview of WT1 gene mutations. Hum. Mutat. 1997, 9, 209–225. [Google Scholar] [CrossRef]
- Sugiyama, H. Cancer immunotherapy targeting Wilms’ tumor gene WT1 product. Expert Rev. Vaccines 2005, 4, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Miwa, H.; Beran, M.; Saunders, G. Expression of the Wilms’ tumor gene (WT1) in human leukemias. Leukemia 1992, 6, 405–409. [Google Scholar] [PubMed]
- Moore, A.W.; McInnes, L.; Kreidberg, J.; Hastie, N.D.; Schedl, A. YAC complementation shows a requirement for Wt1 in the development of epicardium, adrenal gland and throughout nephrogenesis. Development 1999, 126, 1845–1857. [Google Scholar] [PubMed]
- Inoue, K.; Sugiyama, H.; Ogawa, H.; Nakagawa, M.; Yamagami, T.; Miwa, H.; Kita, K.; Hiraoka, A.; Masaoka, T.; Nasu, K. WT1 as a new prognostic factor and a new marker for the detection of minimal residual disease in acute leukemia. Blood 1994, 84, 3071–3079. [Google Scholar] [PubMed]
- Oji, Y.; Miyoshi, S.; Maeda, H.; Hayashi, S.; Tamaki, H.; Nakatsuka, S.I.; Yao, M.; Takahashi, E.; Nakano, Y.; Hirabayashi, H. Overexpression of the Wilms’ tumor gene WT1 in de novo lung cancers. Int. J. Cancer 2002, 100, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Loeb, D.M.; Evron, E.; Patel, C.B.; Sharma, P.M.; Niranjan, B.; Buluwela, L.; Weitzman, S.A.; Korz, D.; Sukumar, S. Wilms’ tumor suppressor gene (WT1) is expressed in primary breast tumors despite tumor-specific promoter methylation. Cancer Res. 2001, 61, 921–925. [Google Scholar] [PubMed]
- Miyoshi, Y.; Ando, A.; Egawa, C.; Taguchi, T.; Tamaki, Y.; Tamaki, H.; Sugiyama, H.; Noguchi, S. High expression of Wilms’ tumor suppressor gene predicts poor prognosis in breast cancer patients. Clin. Cancer Res. 2002, 8, 1167–1171. [Google Scholar] [PubMed]
- Oji, Y.; Suzuki, T.; Nakano, Y.; Maruno, M.; Nakatsuka, S.i.; Jomgeow, T.; Abeno, S.; Tatsumi, N.; Yokota, A.; Aoyagi, S. Overexpression of the Wilms’ tumor gene WT1 in primary astrocytic tumors. Cancer Sci. 2004, 95, 822–827. [Google Scholar] [CrossRef] [PubMed]
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. STATs in oncogenesis. Oncogene 2000, 19, 2474. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J. Stat proteins and oncogenesis. J. Clin. Investig. 2002, 109, 1139. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E. Validating Stat3 in cancer therapy. Nat. Med. 2005, 11, 595–596. [Google Scholar] [CrossRef] [PubMed]
- Sinibaldi, D.; Wharton, W.; Turkson, J.; Bowman, T.; Pledger, W.J.; Jove, R. Induction of p21 WAF1/CIP1 and cyclin D1 expression by the Src oncoprotein in mouse fibroblasts: Role of activated STAT3 signaling. Oncogene 2000, 19, 5419–5427. [Google Scholar] [CrossRef] [PubMed]
- Catlett-Falcone, R.; Dalton, W.S.; Jove, R. STAT proteins as novel targets for cancer therapy. Curr. Opin. Oncol. 1999, 11, 490. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Hassan, R. Immunotherapies for non-small-cell lung cancer and mesothelioma. Lancet Oncol. 2012, 13, e301–e310. [Google Scholar] [CrossRef]
- Xu, C.; Wu, C.; Xia, Y.; Zhong, Z.; Liu, X.; Xu, J.; Cui, F.; Chen, B.; Røe, O.D.; Li, A. WT1 promotes cell proliferation in non-small cell lung cancer cell lines through up-regulating cyclin D1 and p-pRb in vitro and in vivo. PLoS ONE 2013, 8, e68837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derycke, A.S.; De Witte, P.A. Transferrin-mediated targeting of hypericin embedded in sterically stabilized PEG-liposomes. Int. J. Oncol. 2002, 20, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Stavridis, J.; Deliconstantinos, G.; Psallidopoulos, M.; Armenakas, N.; Hadjiminas, D.; Hadjiminas, J. Construction of transferrin-coated liposomes for in vivo transport of exogenous DNA to bone marrow erythroblasts in rabbits. Exp. Cell Res. 1986, 164, 568–572. [Google Scholar] [CrossRef]
- Voinea, M.; Dragomir, E.; Manduteanu, I.; Simionescu, M. Binding and uptake of transferrin-bound liposomes targeted to transferrin receptors of endothelial cells. Vasc. Pharmacol. 2002, 39, 13–20. [Google Scholar] [CrossRef]
- Kobayashi, T.; Ishida, T.; Okada, Y.; Ise, S.; Harashima, H.; Kiwada, H. Effect of transferrin receptor-targeted liposomal doxorubicin in P-glycoprotein-mediated drug resistant tumor cells. Int. J. Pharm. 2007, 329, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Saavedra-Alonso, S.; Zapata-Benavides, P.; Chavez-Escamilla, A.K.; Manilla-Muñoz, E.; Zamora-Avila, D.E.; Franco-Molina, M.A.; Rodriguez-Padilla, C. WT1 shRNA delivery using transferrin-conjugated PEG liposomes in an in vivo model of melanoma. Exp. Ther. Med. 2016, 12, 3778–3784. [Google Scholar] [CrossRef] [PubMed]
- Hinitt, C.; Wood, J.; Lee, S.; Williams, A.; Howarth, J.; Glover, C.; Uney, J.; Hague, A. BAG-1 enhances cell–cell adhesion, reduces proliferation and induces chaperone-independent suppression of hepatocyte growth factor-induced epidermal keratinocyte migration. Exp. Cell Res. 2010, 316, 2042–2060. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Lu, S.; Gu, L.; Gao, Y.; Wang, T.; Zhao, J.; Rao, J.; Chen, J.; Hao, X.; Tang, S.-C. Modulation of BAG-1 expression alters the sensitivity of breast cancer cells to tamoxifen. Cell. Physiol. Biochem. 2014, 33, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Ozfiliz, P.; Arisan, E.D.; Coker-Gurkan, A.; Obakan, P.; Eralp, T.N.; Dinler-Doganay, G.; Palavan-Unsal, N. Bag-1L is a stress-withstand molecule prevents the downregulation of Mcl-1 and c-Raf under control of heat shock proteins in cisplatin treated HeLa cervix cancer cells. Asian Pac. J. Cancer Prev. 2014, 15, 4475–4482. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- El-Shami, K.; Oeffinger, K.C.; Erb, N.L.; Willis, A.; Bretsch, J.K.; Pratt-Chapman, M.L.; Cannady, R.S.; Wong, S.L.; Rose, J.; Barbour, A.L. American Cancer Society colorectal cancer survivorship care guidelines. CA Cancer J. Clin. 2015, 65, 427–455. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Meng, Q.; Tian, A. Expressions of the anti-apoptotic genes Bag-1 and Bcl-2 in colon cancer and their relationship. Am. J. Surg. 2010, 200, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zhou, G.; Ling, J.; Tian, A.; Sun, N. Silencing Bag-1 gene via magnetic gold nanoparticle-delivered siRNA plasmid for colorectal cancer therapy in vivo and in vitro. Tumor Biol. 2016, 37, 10365–10374. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; McGarry, T.J.; Bernal, T.; Kirschner, M.W. Identification of a vertebrate sister-chromatid separation inhibitor involved in transformation and tumorigenesis. Science 1999, 285, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Jallepalli, P.V.; Waizenegger, I.C.; Bunz, F.; Langer, S.; Speicher, M.R.; Peters, J.-M.; Kinzler, K.W.; Vogelstein, B.; Lengauer, C. Securin is required for chromosomal stability in human cells. Cell 2001, 105, 445–457. [Google Scholar] [CrossRef]
- Yu, R.; Lu, W.; Chen, J.; McCabe, C.J.; Melmed, S. Overexpressed pituitary tumor-transforming gene causes aneuploidy in live human cells. Endocrinology 2003, 144, 4991–4998. [Google Scholar] [CrossRef] [PubMed]
- Christopoulou, L.; Moore, J.D.; Tyler-Smith, C. Over-expression of wild-type Securin leads to aneuploidy in human cells. Cancer Lett. 2003, 202, 213–218. [Google Scholar] [CrossRef]
- Weaver, B.A.; Cleveland, D.W. Decoding the links between mitosis, cancer, and chemotherapy: The mitotic checkpoint, adaptation, and cell death. Cancer Cell 2005, 8, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Horwitz, G.A.; Heaney, A.P.; Nakashima, M.; Prezant, T.R.; Bronstein, M.D.; Melmed, S. Pituitary tumor transforming gene (PTTG) expression in pituitary adenomas. J. Clin. Endocrinol. Metab. 1999, 84, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Heaney, A.P.; Singson, R.; McCabe, C.J.; Nelson, V.; Nakashima, M.; Melmed, S. Expression of pituitary-tumour transforming gene in colorectal tumours. Lancet 2000, 355, 716–719. [Google Scholar] [CrossRef]
- Puri, R.; Tousson, A.; Chen, L.; Kakar, S.S. Molecular cloning of pituitary tumor transforming gene 1 from ovarian tumors and its expression in tumors. Cancer Lett. 2001, 163, 131–139. [Google Scholar] [CrossRef]
- Solbach, C.; Roller, M.; Fellbaum, C.; Nicoletti, M.; Kaufmann, M. PTTG mRNA expression in primary breast cancer: A prognostic marker for lymph node invasion and tumor recurrence. Breast 2004, 13, 80–81. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, A.; Ramos-Morales, F.; Romero, F.; Rios, R.M.; Dreyfus, F.; Tortolero, M.; Pintor-Toro, J.A. hpttg, a human homologue of rat pttg, is overexpressed in hematopoietic neoplasms. Evidence for a transcriptional activation function of hPTTG. Oncogene 1998, 17, 2187. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Melmed, S. Pituitary tumor transforming gene: An update. In Molecular Pathology of the Pituitary; Karger Publishers: Basel, Switzerland, 2004; Volume 32, pp. 175–185. [Google Scholar]
- Thorgeirsson, S.S.; Grisham, J.W. Molecular pathogenesis of human hepatocellular carcinoma. Nat. Genet. 2002, 31, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Llovet, J.M. Prognostic prediction and treatment strategy in hepatocellular carcinoma. Hepatology 2002, 35, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Bemal, J.; Luna, R.; Espina, A. Human securin interacts with p53 and modulates p53–mediated transcriptional activity and apoptosis. Nat. Genet. 2002, 32, 306–311. [Google Scholar]
- Jung, C.R.; Yoo, J.; Jang, Y.J.; Kim, S.; Chu, I.S.; Yeom, Y.I.; Choi, J.Y.; Im, D.S. Adenovirus-mediated transfer of siRNA against PTTG1 inhibits liver cancer cell growth in vitro and in vivo. Hepatology 2006, 43, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- Frazier, W.A.; Gao, A.-G.; Dimitry, J.; Chung, J.; Brown, E.J.; Lindberg, F.P.; Linder, M.E. The thrombospondin receptor integrin-associated protein (CD47) functionally couples to heterotrimeric Gi. J. Biol. Chem. 1999, 274, 8554–8560. [Google Scholar] [CrossRef] [PubMed]
- Oldenborg, P.-A.; Zheleznyak, A.; Fang, Y.-F.; Lagenaur, C.F.; Gresham, H.D.; Lindberg, F.P. Role of CD47 as a marker of self on red blood cells. Science 2000, 288, 2051–2054. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D.; van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.S.; Espinosa, I.; Chao, M.; Wong, D.; Ailles, L.; Diehn, M.; Gill, H.; Presti, J.; Chang, H.Y.; van de Rijn, M. Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2009, 106, 14016–14021. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Myklebust, J.H.; Varghese, B.; Gill, S.; Jan, M.; Cha, A.C.; Chan, C.K.; Tan, B.T. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell 2010, 142, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, Z.; Guo, S.; Zhang, L.; Sharma, A.; Robertson, G.P.; Huang, L. Intravenous delivery of siRNA targeting CD47 effectively inhibits melanoma tumor growth and lung metastasis. Mol. Ther. 2013, 21, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
RNAi-mediated gene silencing mechanism. Cellular internalization of siRNA-nanoparticle complex via endocytosis is followed by siRNA release from both the particle and the endosome. In cytoplasm, the released siRNA is loaded into RNA-induced silencing complex (RISC), degrades the passenger strand and activates anti-sense strand. The activated anti-sense strand subsequently cleaves target mRNA. The catalytic RNAs that are unable to escape endosome are subjected to lysosomal degradation.
Figure 1.
RNAi-mediated gene silencing mechanism. Cellular internalization of siRNA-nanoparticle complex via endocytosis is followed by siRNA release from both the particle and the endosome. In cytoplasm, the released siRNA is loaded into RNA-induced silencing complex (RISC), degrades the passenger strand and activates anti-sense strand. The activated anti-sense strand subsequently cleaves target mRNA. The catalytic RNAs that are unable to escape endosome are subjected to lysosomal degradation.

Figure 2.
The 2015 global incidence and deaths for different type of cancers, (A) represents the incident rate (thousand) per 100,000 person-years; (B) represents the death rate (thousand) per 100,000 person-years [25].
Figure 2.
The 2015 global incidence and deaths for different type of cancers, (A) represents the incident rate (thousand) per 100,000 person-years; (B) represents the death rate (thousand) per 100,000 person-years [25].

Figure 3.
Extracellular and cellular barriers faced by unmodified siRNA to reach target site after intravenous (IV) administration. A. After IV administration, naked siRNA is subjected to extracellular barriers like nuclease degradation, macrophage-facilitated hepato-biliary excretion and rapid renal clearance. B. To enter into cancer cell, naked siRNA is to fight with cellular barriers like abnormal tumor vasculature, dense and irregular structure of extracellular matrix (ECM), high interstitial fluid pressure (IFP), hypoxia and poor lymphatic drainage, and intracellular barriers like negatively charged cell membrane and lysosomal lysis.
Figure 3.
Extracellular and cellular barriers faced by unmodified siRNA to reach target site after intravenous (IV) administration. A. After IV administration, naked siRNA is subjected to extracellular barriers like nuclease degradation, macrophage-facilitated hepato-biliary excretion and rapid renal clearance. B. To enter into cancer cell, naked siRNA is to fight with cellular barriers like abnormal tumor vasculature, dense and irregular structure of extracellular matrix (ECM), high interstitial fluid pressure (IFP), hypoxia and poor lymphatic drainage, and intracellular barriers like negatively charged cell membrane and lysosomal lysis.

Figure 4.
Various viral and non-viral vectors used for the delivery of genetic materials such as DNA, RNA, siRNA and shRNA.
Figure 4.
Various viral and non-viral vectors used for the delivery of genetic materials such as DNA, RNA, siRNA and shRNA.

Figure 5.
Roles of various genes in cancer development; A. Genes that regulate differentiation and metabolism of a cancer cell e.g., IGF-1R, B. Proliferative genes like RhoA and RhoC, β-catenin, WT1, PTTG1 via activating cellular process like mitosis, C. Angiogenic genes like VEGF, VEGF-C, EGFR, Neuropilin-2 that regulate tumor-induced angiogenesis, D. Anti-apoptotic genes including BCL-2, Survivin, livin, Bag-1, Cyclin D1, RhoA and RhoC which block the normal apoptosis pathways of a cell, E. Metastasis regulating genes like EphA2, RhoA and RhoC which trigger cellular motility and invasion for metastasis of tumor cell, F. Tumor suppressor gene (p53) inhibitor e.g., MDM-2, deactivates the p53 pathways.
Figure 5.
Roles of various genes in cancer development; A. Genes that regulate differentiation and metabolism of a cancer cell e.g., IGF-1R, B. Proliferative genes like RhoA and RhoC, β-catenin, WT1, PTTG1 via activating cellular process like mitosis, C. Angiogenic genes like VEGF, VEGF-C, EGFR, Neuropilin-2 that regulate tumor-induced angiogenesis, D. Anti-apoptotic genes including BCL-2, Survivin, livin, Bag-1, Cyclin D1, RhoA and RhoC which block the normal apoptosis pathways of a cell, E. Metastasis regulating genes like EphA2, RhoA and RhoC which trigger cellular motility and invasion for metastasis of tumor cell, F. Tumor suppressor gene (p53) inhibitor e.g., MDM-2, deactivates the p53 pathways.

Figure 6.
A proposal of a tailored made SMART nanocarrier (S = specific size; M = modified surface chemistry; A = accelerated cellular internalization; R = rapid endosomal escape T = targeted tumor cells).
Figure 6.
A proposal of a tailored made SMART nanocarrier (S = specific size; M = modified surface chemistry; A = accelerated cellular internalization; R = rapid endosomal escape T = targeted tumor cells).


{kind=link}
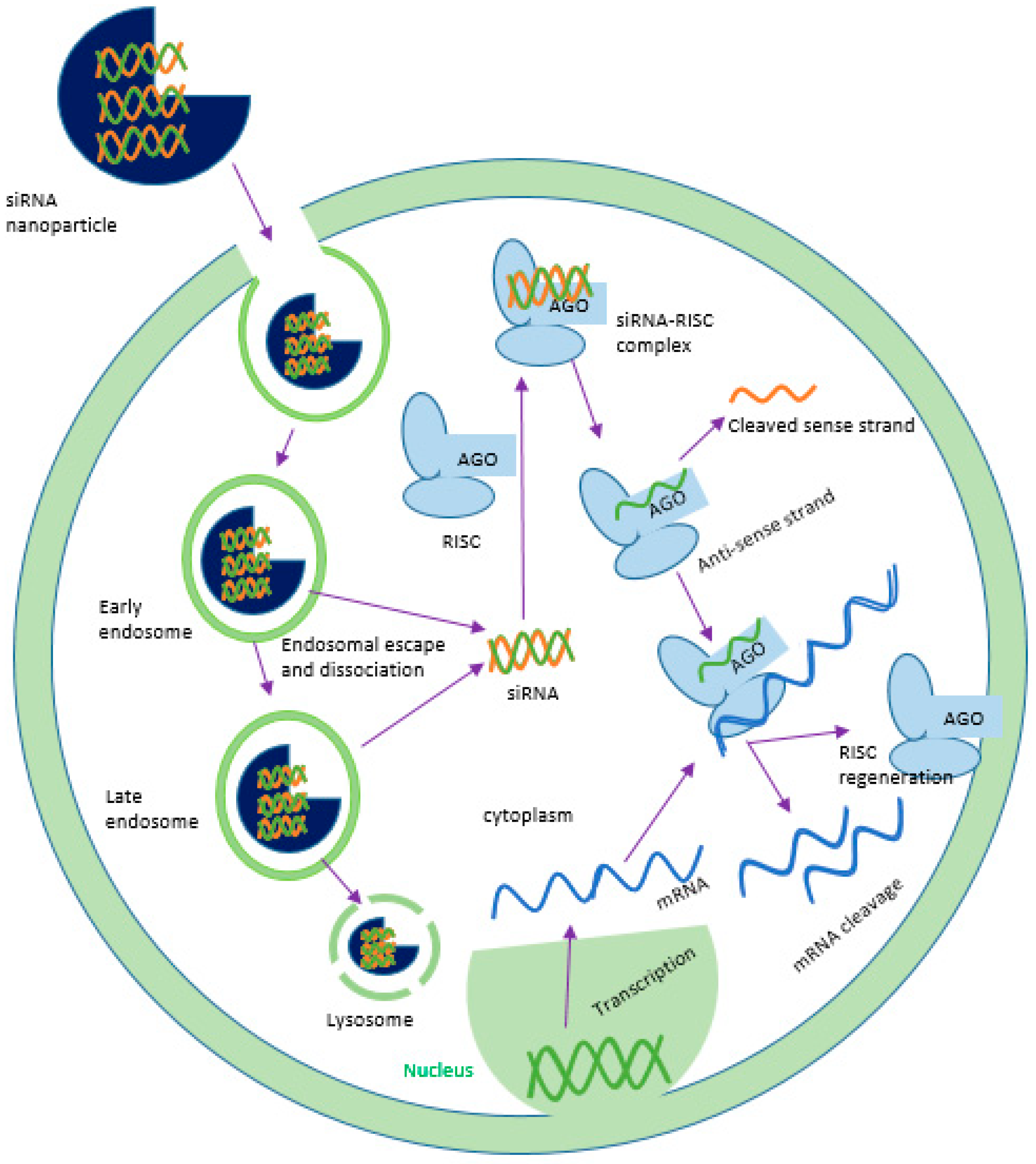{kind=link}
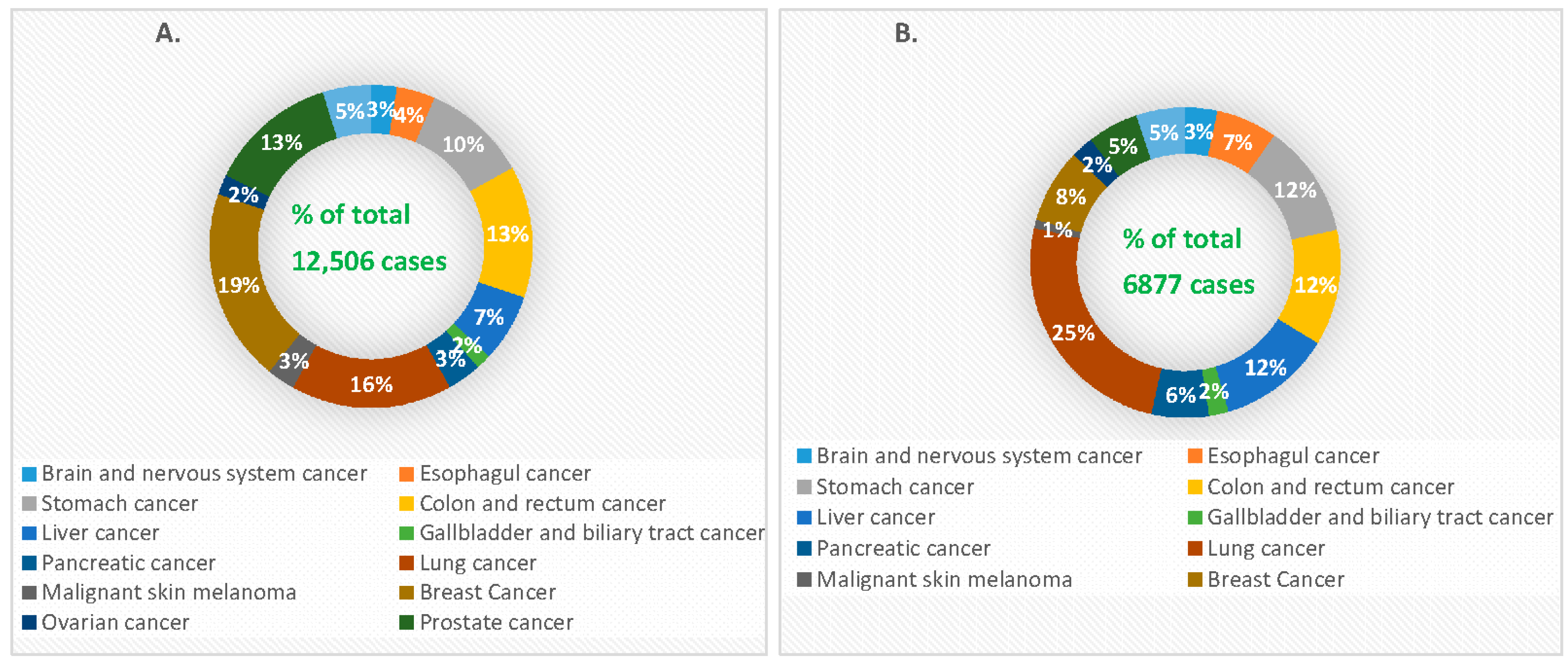{kind=link}
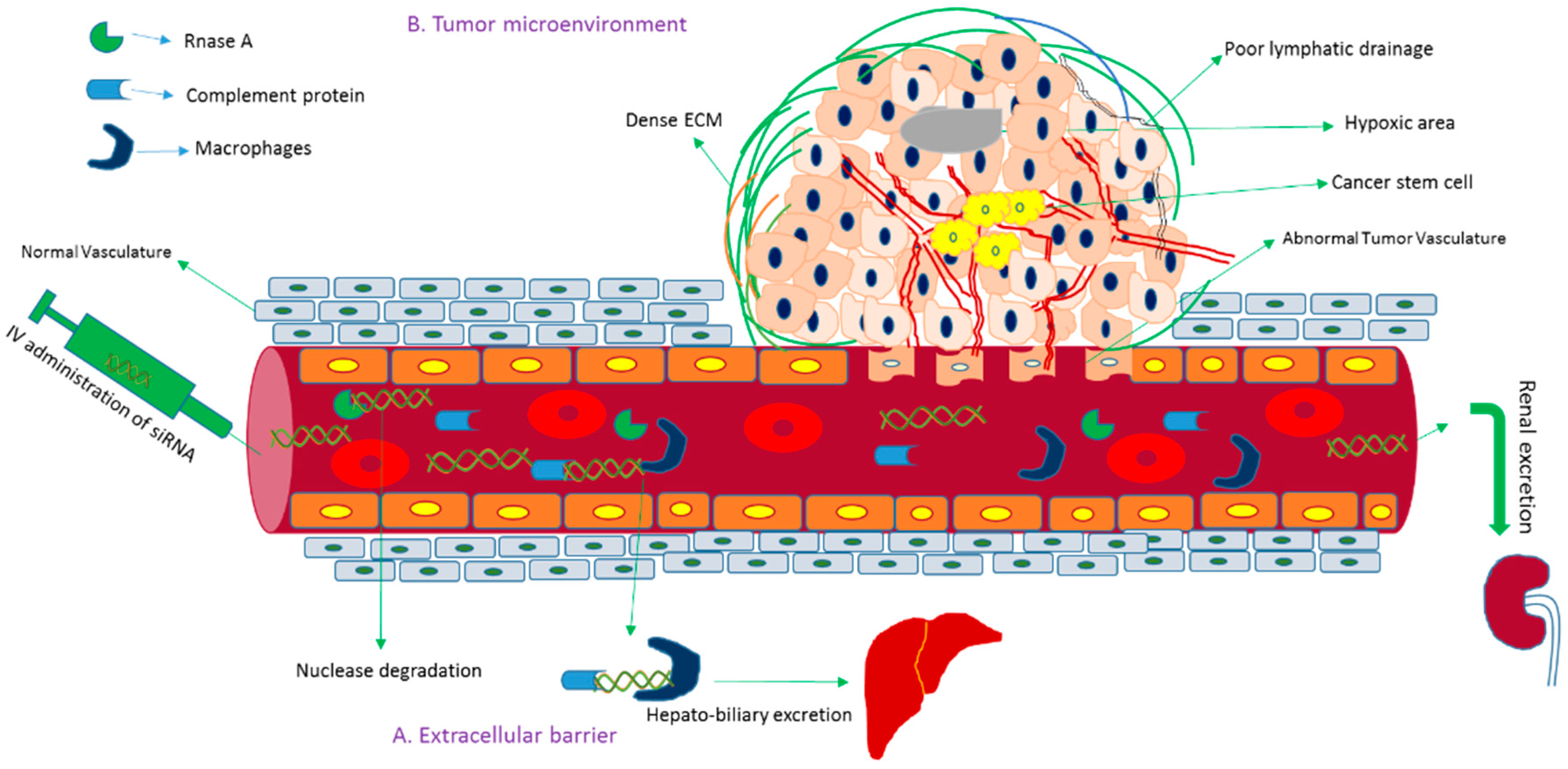{kind=link}
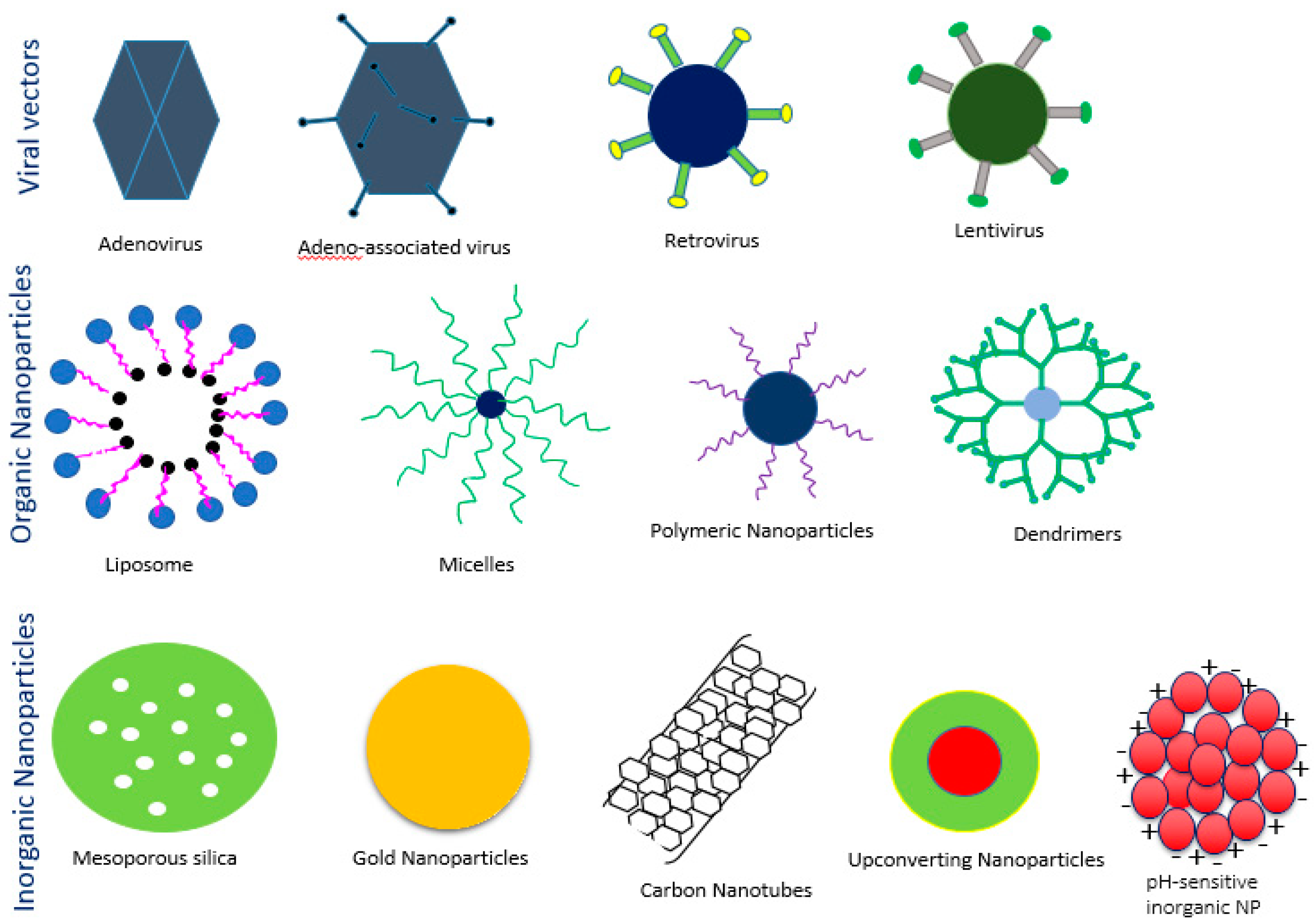{kind=link}
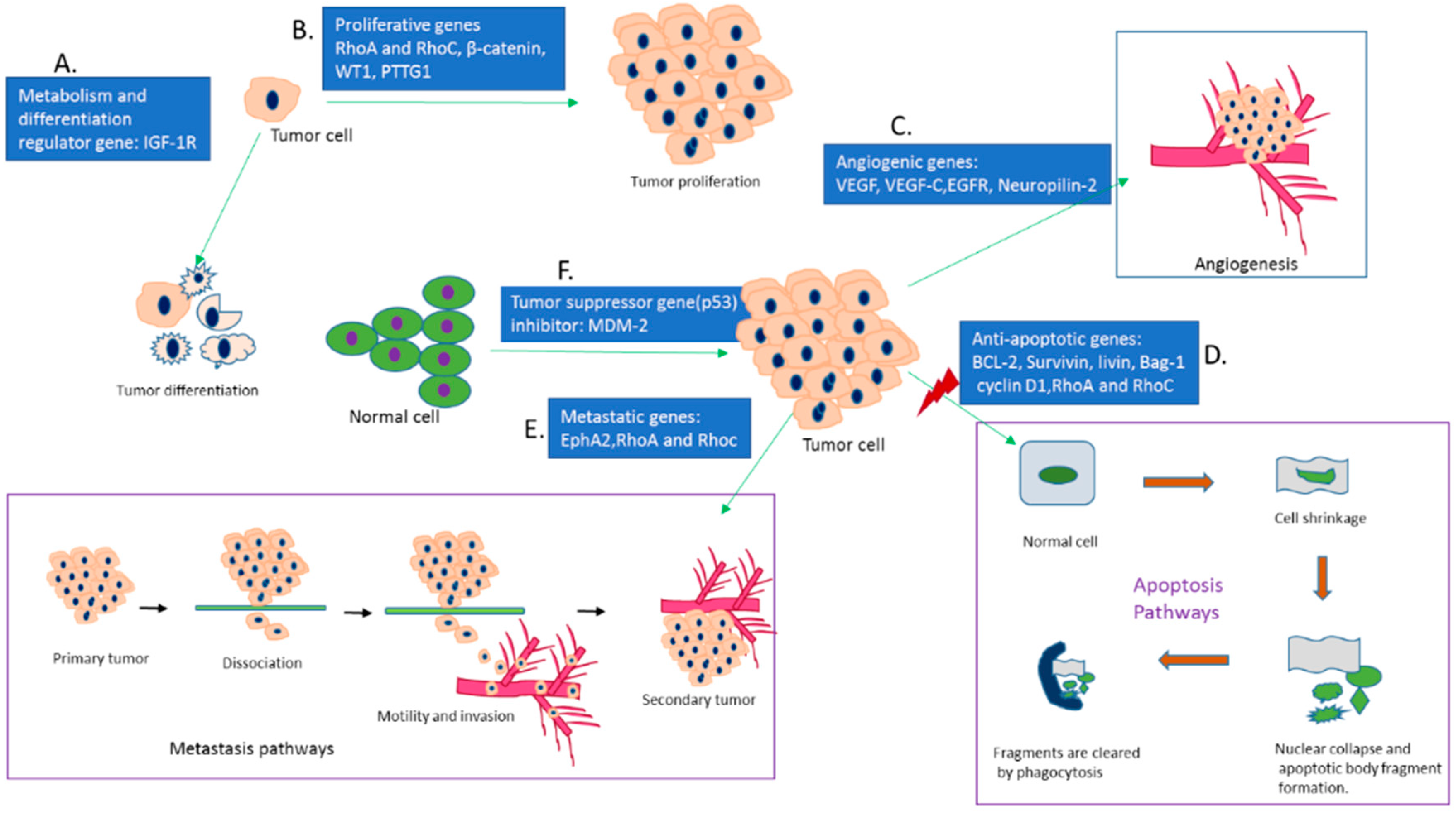{kind=link}
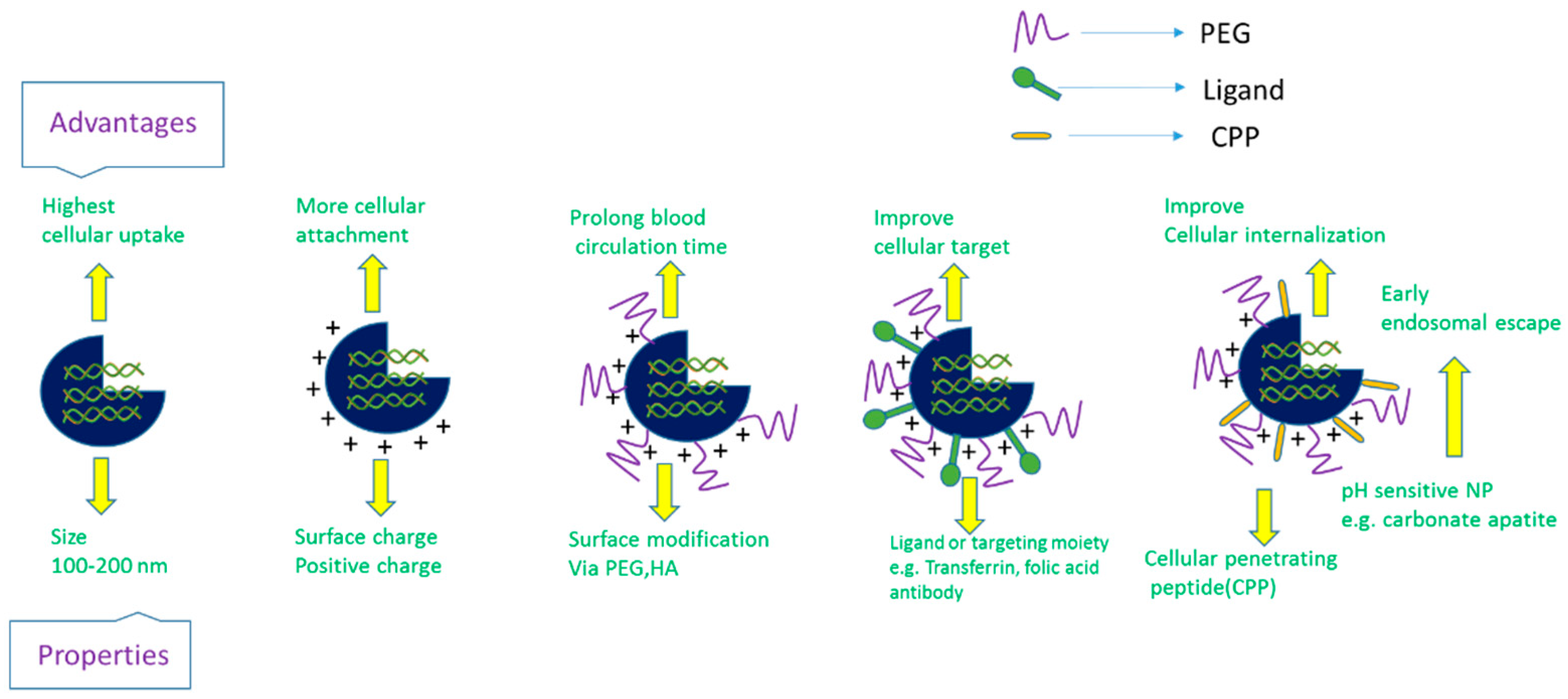{kind=link}
Table 1.
siRNAs-based clinical trials for cancer therapy.
| Drug Formulation | Target Gene | NPs | Treatment | Diseases | Phase | Status | Identifier Trial Number (https://clinicaltrails.gov) |
|---|---|---|---|---|---|---|---|
| DCR-MYC | MYC | Lipid | siRNAs | Hepatocellular carcinoma | 1/2 | Ongoing, not recruiting 2014-present | NCT02314052 |
| DCR-MYC | MYC | Lipid | siRNAs | Solid tumors, multiple myeloma, non-Hodgkin lymphoma, or pancreatic neuroendocrine tumors | 1 | Ongoing, not recruiting 2014-present | NCT02110563 |
| ALN-VSP02 | KSP and VEGF | Lipid | siRNAs | Solid tumors | 1 | Completed | NCT00882180 |
| Atu 027 | PKN3 | Lipid Nanoparticles | siRNAs | Advanced cancers | 1 | Completed | NCT00938574 |
| TKM-080301 | PLK1 | Lipid | siRNAs | Primary and secondary liver cancer | 1 | completed | NCT01437007 |
| PLK1 | Lipid | siRNAs | Neuroendocrine tumors | 1/2 | completed | NCT01262235 | |
| PLK1 | Lipid | siRNAs | Advanced hepatocellular carcinoma | 1/2 | completed | NCT02191878 | |
| siRNA-EphA2-DOPC | EphA2 | Lipid | siRNAs | Advanced solid tumors | 1 | Recruiting | NCT01591356 |
| siG12D-LODER | KRAS | LODER polymer | siRNAs | Ductal adenocarcinoma or pancreatic cancer | 1 | completed | NCT01188785 |
| siG12D-LODER | KRAS | LODER polymer | siRNAs | pancreatic cancer | 2 | Not yet recruiting | NCT01676259 |
| SNS01-T | eIF5A | polyethyleneimine | siRNAs plasmids | Multiple myeloma | 1/2 | unknown | NCT01435720 |
Table 2.
Summary of animal studies of siRNA- and shRNA-based cancer gene therapy.
| Target Genes | Role of Genes | Delivery Vehicle | Treatment | Preclinical Studies Application | Preclinical Studies Outcome | Refs. |
|---|---|---|---|---|---|---|
| Bcl-2 | Inhibits apoptosis pathways and promote cellular growth and survival in breast, lung, liver and gastric cancer | Liposome-protamine | siRNA | Balb/c mouse model inoculated with H22 liver tumor cells | 66.5% reduction of tumor growth by suppressing Bcl-2 gene expression | 65 |
| Bcl-2 | pSilencerTM-EGFP sh515 | shRNA | Balb/C mouse model inoculated with GBC-SD, gallbladder carcinoma cells | 50% reduction of tumor volume and decreased tumor growth rate | 72 | |
| Bcl-2 | Cationic liposome, LIC-101 | siRNA | Balb/C nu++ mouse model inoculated with PC-3 prostate cancer cells | 63% reduction of tumor volume | 73 | |
| Bcl-2 | PEG-LIC complex | siRNA | Balb/C mouse model inoculated with PC-3 human prostate cancer cells | Increased siRNA uptake, 65% tumor reduction without any systemic toxicity | 74 | |
| VEGF | Stimulates angiogenesis and vascular permeability | Adenoviral vector (Ad5CMV) | siRNA | Athymic female mouse model inoculated with MDA251-MB, human breast cancer cells | Reduced 80% of tumor through anti-angiogenesis mechanism | 97 |
| VEGF | Polyelectrolyte complex (PEG/PEI PEC micelles) | siRNA | Female nude mice (nu/nu) model inoculated with PC-3 human prostate cancer cells | Intratumoral injection caused 79% tumor inhibition; Intravenous administration reduced 86% of tumor volume | 103 | |
| VEGF-C | Promotes lymphogenesis, tumorigenesis and initiates metastasis | Hifectin-mediated transfection | siRNA | Balb/C mouse model inoculated with 4T1 cells, mouse breast cancer cells | Reduced 28% of tumor volume. | 120 |
| VEGF-C | Lentivirus vector (Lv) | siRNA | Balb/C mouse model inoculated with A549, human NSCLC cells | 64% tumor inhibition and 48% reduction of tumor volume by decreasing VEGF-C expression | 121 | |
| NRP-2 | Binds with VEGF and regulates vascularization and lymphogenesis of various tumors | DOPC (neutral lipid 1,2-dioleoyl-sn-glycero-3-phosphatidyl choline) | siRNA | Male athymic nude mouse model inoculated with HTC-116, human colorectal carcinoma cell lines | Reduced 91.3% of tumor volume via increasing anti-angiogenic mechanism | 125 |
| VEGF R2 | Regulates angiogenesis and tumor growth | RGD (Ar3-Gly-Asp peptides)-PEG-PEI nanoplexes | siRNA | Female nude mouse model inoculated with N2A, mouse neuroblastoma cells | Enabled tissue-specific delivery and inhibited more than 90% of tumor volume | 100 |
| EGFR 1 & ERBB2 | Activate downstream signaling pathways and play key role in cell division and proliferation | Carbonate apatite Nano-particle | siRNA | Female Balb/C mouse model inoculated with 4T1 cells, mouse breast cancer cells | 61% reduction of tumor volume without any toxicity | 143 |
| Survivin | Suppresses apoptosis by inhibiting both intrinsic and extrinsic pathways of apoptosis, as well as improves chemo-resistance to various chemotherapeutics and increases tumor recurrence rate | PEGylated chitosan (PEG-CS) | siRNA | Female Balb/C mouse model inoculated with 4T1 cells, mouse breast cancer cells | Increased biological stability and targeted gene delivery, reduced 55% of tumor volume | 165 |
| Survivin | Chiosan-6-poly arginine and histidine (H6R6-CS) | siRNA | Female Balb/C mouse model inoculated with 4T1 cells, mouse breast cancer cells | Improved cellular uptake and endosomal escape with 63% tumor inhibition | 176 | |
| Survivin | Cationic linear polyethyleneimine (PEI) | Sticky siRNA (ssiRNA) | NMRI nude female mouse model inoculated with B16-F10 cells, murine melanoma cell lines | Reduced 50% of tumor volume through silencing of Survivin gene | 181 | |
| Survivin | PCPP (PEG-CPB-PEI) nano-particle | siRNA | Balb/C mouse model inoculated with 4T1 cells, mouse breast cancer cells | Increased tumor accumulation and improved cellular uptake with 66% reduction of tumor volume | 185 | |
| Cyclin-B1 | As a mitosis promoting factor triggers uncontrolled cell proliferation and hampers the stability of chromosomes | MPG-8 (Primary amphipathic peptide carrier)-cholesterol (MPG-8/chol) | siRNA | Swiss nude mouse model inoculated with PC-3 cells, human prostate cancer cells | 90% tumor size inhibition for maximum dose, 60–80% reduction of Cyclin B1 expression and extended survival rate | 206 |
| Cyclin-B1 | Cationic linear polyethyleneimine (PEI) | Sticky siRNA (ssiRNA) | NMRI nude female mouse model inoculated with B16-F10 cells, Murine melanoma cell lines | Reduced 44% of tumor volume via down regulating Cyclin B1 expression | 181 | |
| RhoA & RhoC | Triggers signal transduction and drives a series of pathologies of cancer including cell motility, proliferation, apoptosis inhibition, cell cycle progression, invasion, metastasis and inflammation | Adenoviral vector | shRNA | Male Balb/C mouse model inoculated with HTC-116, human colorectal carcinoma cell lines | Slowed tumor growth (2.38 fold) and reduced 37% of tumor volume | 225 |
| RhoA & RhoC | Cytofectin-mediated transfection | siRNA | Athymic female mouse model inoculated with MDA251-MB, human breast cancer cells | Reduced tumor volume 85% (anti-RhoA) and 53% (anti-RhoC), lowered angiogenesis index | 227 | |
| RhoA | Chitosan-PIHCA (polyisohexylcyanoacrylate) | siRNA | Athymic female mouse model inoculated with MDA251-MB, human breast cancer cells | At higher dose the tumor were completely removed | 228 | |
| RhoC | Lipofectamine-mediated transfection | siRNA | Balb/C-nu mouse model inoculated with SUM149, human IBC cells | Reduced tumor volume by 35%, increased survival rate to 85%, up-regulated metastasis suppressor gene KAll | 234 | |
| β-Catenin | Regulates cell-cell adhesion and gene transcription, ultimately controlling cellular proliferation | Oligofectamine-mediated transfection | siRNA | Female nude/nu mouse model inoculated with HTC-116, human colorectal carcinoma cell lines | Three-fold smaller in size of tumor in comparison to control with extended survival rate | 253 |
| β-Catenin | Lentivirus vector | shRNA | Male athymic nude mouse model inoculated with AGS cells, human gastric cancer cells | 75% reduction of tumor volume by inhibiting CCAR1 gene expression | 259 | |
| EphA2 | Enhances cell-extracellular matrix (ECM) adhesion, anchorage-dependent growth and metastasis | DOPC (neutral lipid 1,2-dioleoyl-sn-glycero-3-phosphatidyl choline) | siRNA | Female athymic nude (Ncr-nu) mouse model inoculated with SkOV3ip1 cells, ovarian cancer cell lines | Reduced 35–50% of tumor size | 275 |
| EphA2 and FAK | DOPC (neutral lipid 1,2-dioleoyl-sn-glycero-3-phosphatidyl choline) | siRNA | Female athymic nude (Ncr-nu) mouse model inoculated with SkOV3ip1 cells, ovarian cancer cell lines | Reduced 62–82% of tumor metastasis and slowed down tumor growth rate | 283 | |
| EphA2 | Liposome | siRNA | Balb/C mouse model inoculated with SGC 7901, human gastric adenocarcinoma cells | 43.1% inhibition of tumor growth, with reduction in expression of metastatic gene MMP-9 | 285 | |
| MDM-2 | Inhibits the regulation of p53 tumor suppressor gene | PMPC-b-PDPA (di-block copolymer of poly (methacryloyloxy ethyl phosphorylcholine)-b-poly (diisopropanolamine ethyl methacrylate) | siRNA | Athymic mouse model inoculated with H2009 cells, NSCLC cells | 67% reduction of tumor growth via down regulation of MDM-2 gene expression without any systemic toxicity | 290 |
| MDM-2, c-myc and VEGF | cationic lipid-PEG | siRNA | Female C57B216 mouse model inoculated with B16-F10 cells, murine melanoma cell lines | 20–30% reduction of tumor load with extended survival rate | 291 | |
| IGF-1R | Promotes cellular metabolism, differentiation, apoptosis, chemo resistance and angiogenesis as well as protecting cells from UV irradiation, cytokine and gamma radiation-induced apoptosis | Plasmids-PEI | siRNA | Male nude mouse model inoculated with A549 cells, human lung adenocarcinoma cell lines | 60% of reduction of tumor volume, Increasing apoptotic cells | 305 |
| IGF-1R | Magnetic lipoplexes | shRNA | Male Balb/C AnNcrj-nu mouse model inoculated with A549 cells (NSCLC cells line with overexpression of IGF-1R) | Improved site specificity and cellular uptake, and reduced 85.1 ± 3% of IGF-1R gene expression | 306 | |
| Livin | Thwarts both extrinsic and intrinsic apoptosis pathways by interacting with specific cysteine proteases or caspases, while playing a significant role in tumor progression and chemo resistance development | Single chain antibody | siRNA | Nude mouse model inoculated with LiBr cells, malignant melanoma cell lines. | Reduces approximately 64% of tumor size. | 324 |
| Livin | Plasmid vector | siRNA | Balb/C nu/nu mouse model inoculated with SPCA-1 cells, human lung cancer cell lines. | 73% reduction of mean tumor size, with increased apoptotic fraction and improved survival rate | 325 | |
| WT1 | The key drivers that control cell proliferation and apoptosis via regulation of the expression of proliferative genes | Plasmid vector | shRNA | Balb/C nu/nu mouse model inoculated with A549, H1299 and H1650 cells | 69–76% reduction of tumor volume without any systemic toxicity | 348 |
| WT1 | Liposome-PEG | shRNA | Female C57BL/6 mouse model inoculated with B16F10 cell, murine melanoma cell lines | Reduced 34% of tumor weight and extended survival rate (62.5%) | 353 | |
| Bag-1 | Regulates Bcl-2 gene expression and mimics the anti-apoptotic activities via bridging between the growth factor and anti-apoptotic mechanisms | Magnetic gold nanoparticles | siRNA | Balb/C nude mouse model inoculated with LoVo cell, human colon cancer cell lines | 69% of tumor inhibition without toxicity | 360 |
| PTTG1 | Plays a vital role in several cellular processes like mitosis, DNA repair, apoptosis and gene regulation and causes aneuploidy. | Adenoviral vector | siRNA | Balb/C nude mouse model inoculated with SH-J1 cells, hepatoma cell lines | Significant tumor inhibition efficacy (84%) | 375 |
| CD-47 | Causes the tumor cells escape from immunosurveillance with the result of tumor progression | Liposome-protamine-hyaluronic acid | siRNA | C57B2/6 mouse model inoculated with B16F10 cell, murine melanoma cell lines | Above 90% tumor inhibition without toxicity | 382 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Karim, M.E.; Tha, K.K.; Othman, I.; Borhan Uddin, M.; Chowdhury, E.H. Therapeutic Potency of Nanoformulations of siRNAs and shRNAs in Animal Models of Cancers. Pharmaceutics 2018, 10, 65. https://doi.org/10.3390/pharmaceutics10020065
AMA Style
Karim ME, Tha KK, Othman I, Borhan Uddin M, Chowdhury EH. Therapeutic Potency of Nanoformulations of siRNAs and shRNAs in Animal Models of Cancers. Pharmaceutics. 2018; 10(2):65. https://doi.org/10.3390/pharmaceutics10020065
Chicago/Turabian StyleKarim, Md. Emranul, Kyi Kyi Tha, Iekhsan Othman, Mohammad Borhan Uddin, and Ezharul Hoque Chowdhury. 2018. "Therapeutic Potency of Nanoformulations of siRNAs and shRNAs in Animal Models of Cancers" Pharmaceutics 10, no. 2: 65. https://doi.org/10.3390/pharmaceutics10020065
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.