Optimization of the Factors Affecting the Absorption of Vardenafil from Oral Disintegrating Tablets: A Clinical Pharmacokinetic Investigation

 ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Pre-Formulation Studies
2.2.1. Preparation of VRD-β-CDs Inclusion Complexes
2.2.2. Solubility Study
2.3. Formulation of ODTs
2.3.1. Application of Box Behnken Experimental Design
2.3.2. Preparation of ODTs
2.4. Evaluation of the Prepared VRD-ODTs
2.5. In Vitro Disintegration of VRD-ODTs
2.6. In Vitro Dissolution of VRD-ODTs
2.7. VRD-ODTs Formulation Data Analysis by BBD
2.8. In Vivo Evaluation of the Optimized VRD-ODTs on Human Volunteers
2.8.1. In Vivo Taste Masking and Disintegration Time Evaluation
2.8.2. Pharmacokinetic Parameters Evaluation
Population and Sampling
Chromatographic Conditions
Pharmacokinetic Analysis
3. Results and Discussion
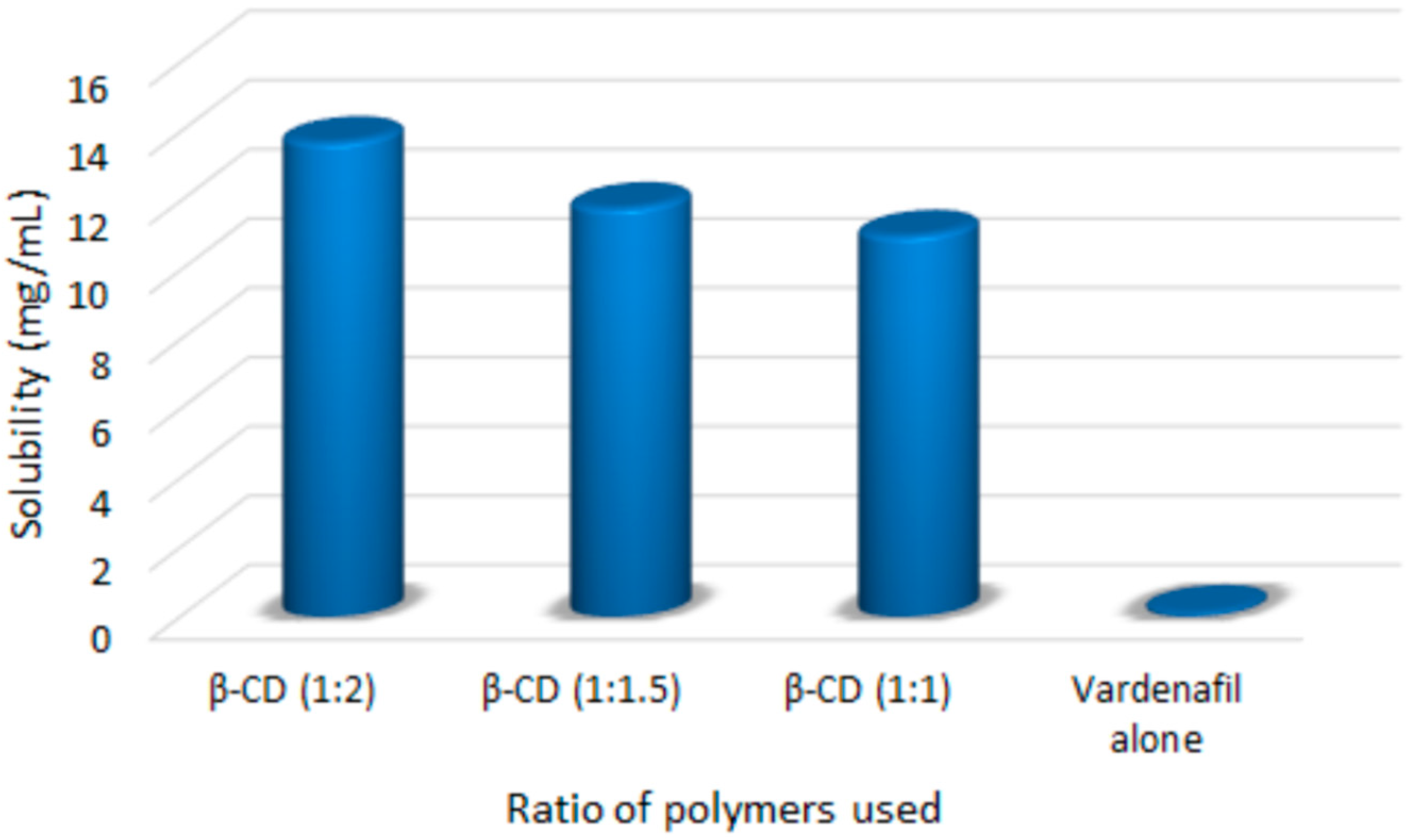
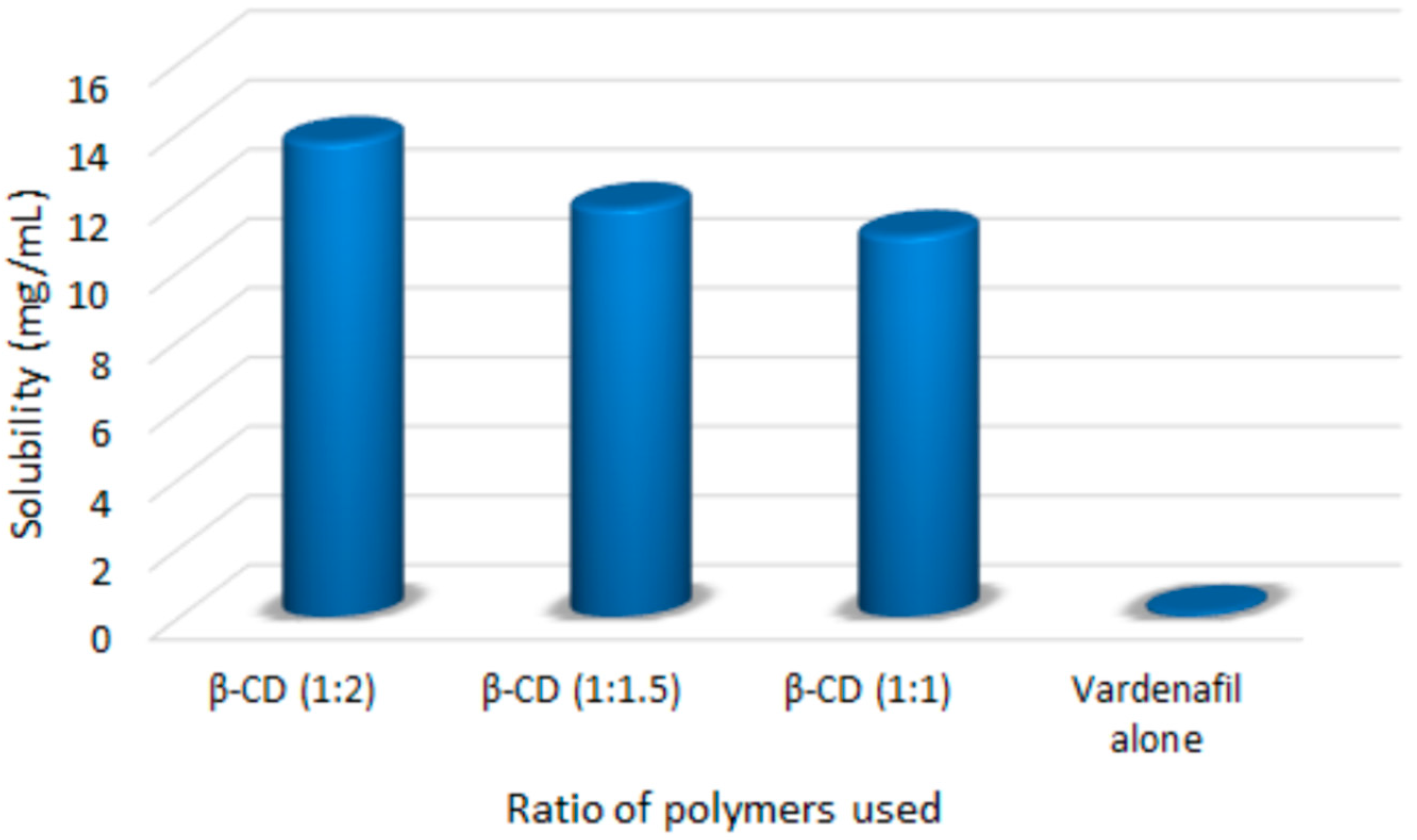
3.1. Saturation Solubility Studies of the Prepared Complexes
3.2. Development of VRD–ODTs
3.3. Evaluation of VRD-ODTs
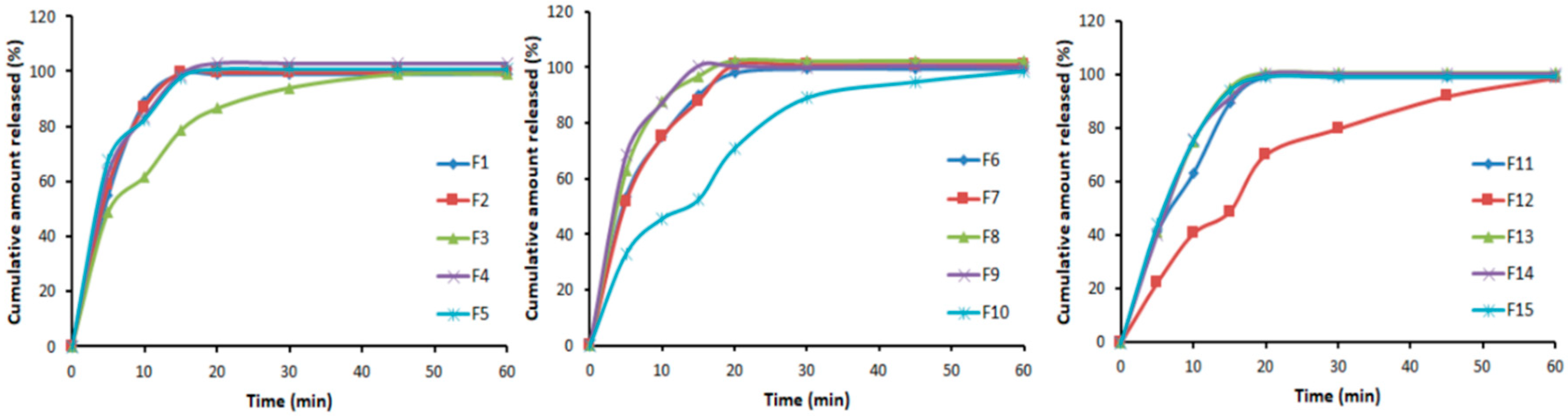
3.4. In Vitro Dissolution Studies
3.5. Quantitative Estimation of the Factors Affecting VRD-ODTs
3.5.1. Mathematical Modeling of the Data
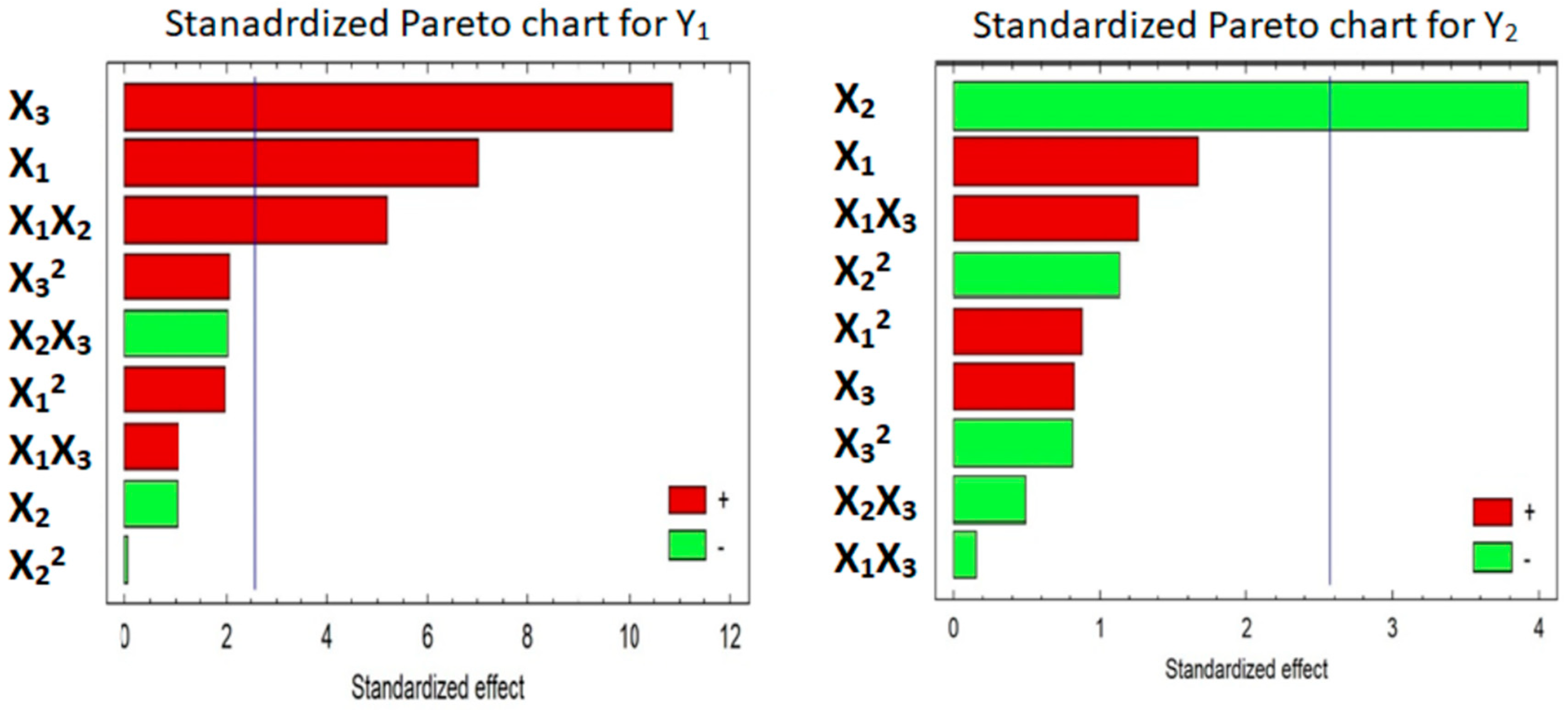
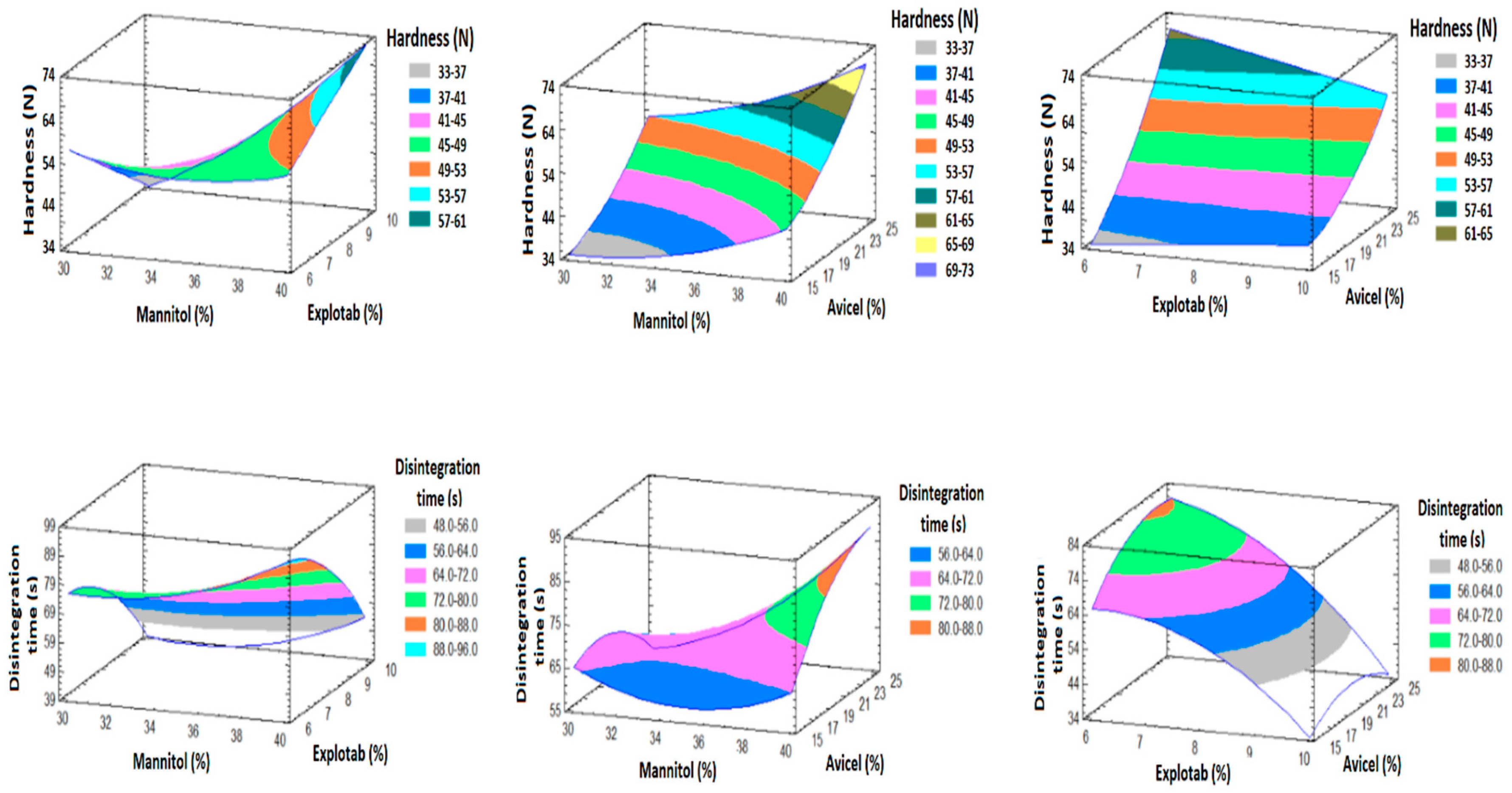
3.5.2. Effect of the Factors on Y1 and Y2
3.6. Prediction of the Optimized Formulation
3.7. In Vivo Evaluation of the Optimized VRD-ODTs on Human Volunteers
3.7.1. In Vivo Taste Masking and Disintegration Time
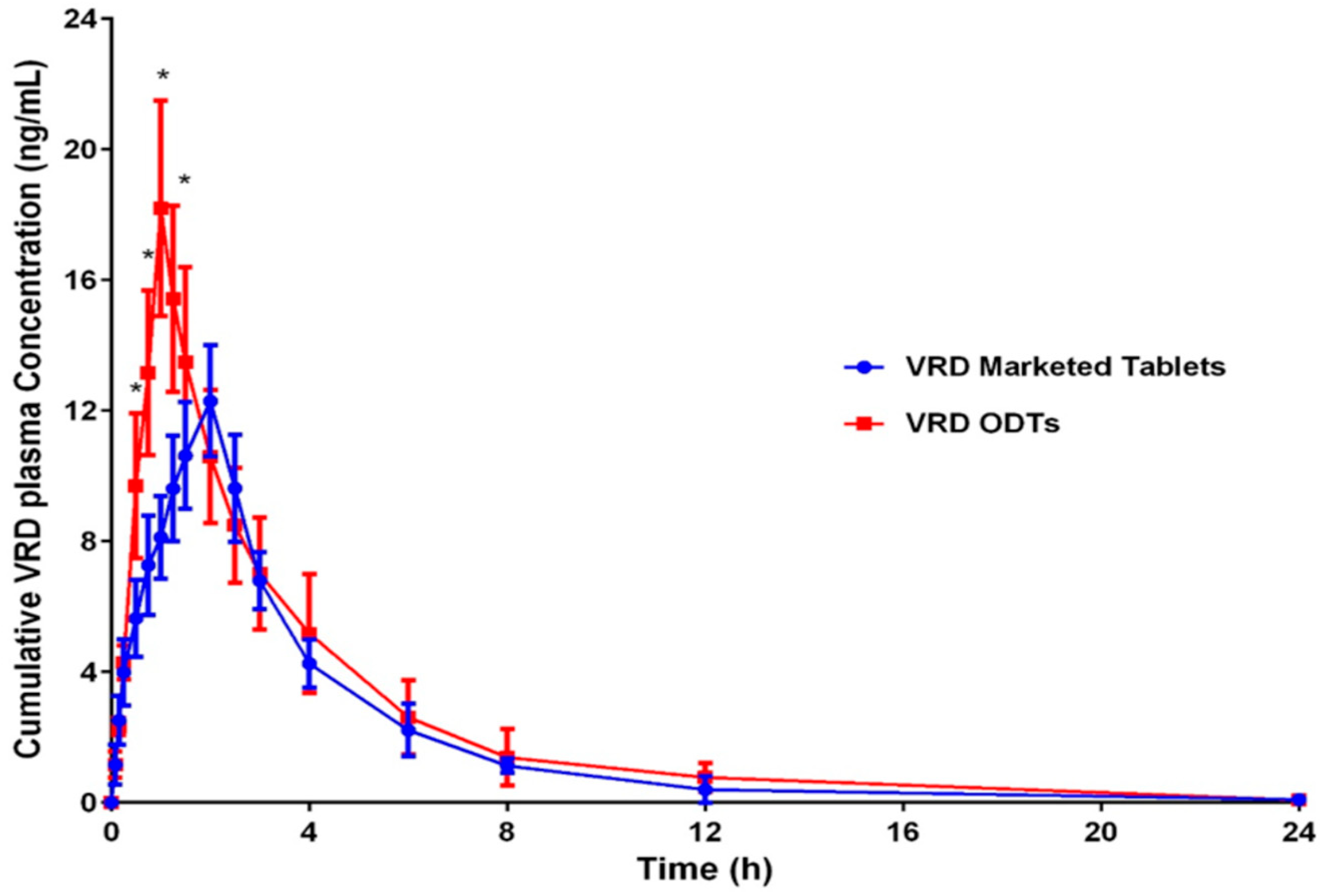
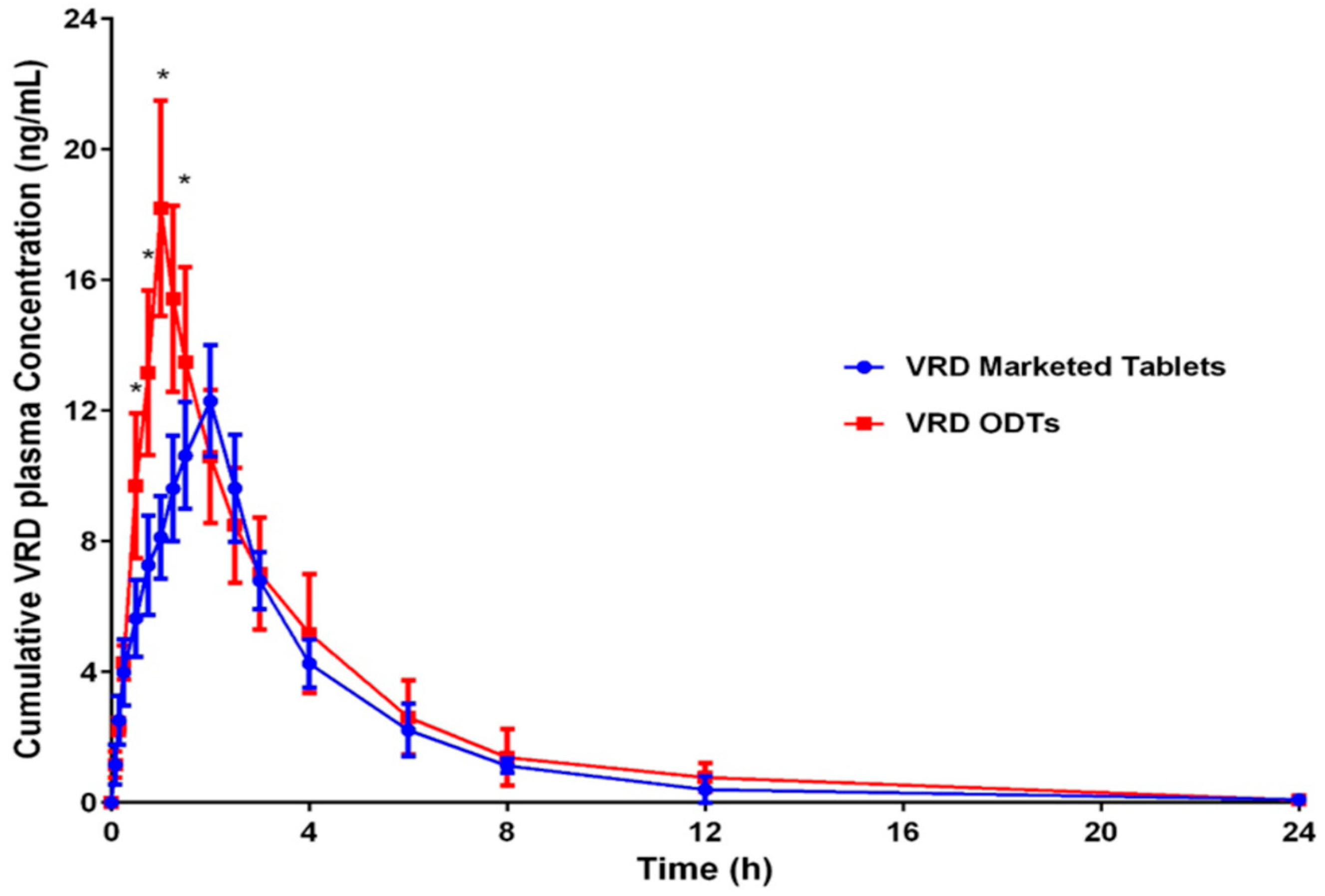
3.7.2. Pharmacokinetic Parameters Evaluation
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gittelman, M.; McMahon, C.G.; Rodríguez-Rivera, J.A.; Beneke, M.; Ulbrich, E.; Ewald, S. The POTENT II randomised trial: Efficacy and safety of an orodispersible vardenafil formulation for the treatment of erectile dysfunction. Int. J. Clin. Pract. 2010, 64, 594–603. [Google Scholar] [CrossRef]
- Wright, P.J. Comparison of phosphodiesterase type 5 (PDE5) inhibitors. Int. J. Clin. Pract. 2006, 60, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Mishra, A.; Gupta, V.; Bansal, P.; Singh, R.; Singh, A. Recent trends of fast dissolving tablet—An overview of formulation technology. Int. J. Pharm. Biol. Arch. 2010, 1, 1–10. [Google Scholar]
- Bischoff, E. Vardenafil preclinical trial data: Potency, pharmacodynamics, pharmacokinetics, and adverse events. Int. J. Impot. Res. 2004, 16, S34–S37. [Google Scholar] [CrossRef]
- Valleri, M.; Mura, P.; Maestrelli, F.; Cirri, M.; Ballerini, R. Development and Evaluation of Glyburide Fast Dissolving Tablets Using Solid Dispersion Technique. Drug Dev. Ind. Pharm. 2004, 30, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Dave, V.; Yadav, R.B.; Ahuja, R.; Yadav, S. Formulation design and optimization of novel fast dissolving tablet of chlorpheniramine maleate by using lyophilization techniques. Bull. Fac. Pharm. Cairo Univ. 2017, 55, 31–39. [Google Scholar] [CrossRef]
- Parkash, V.; Maan, S.; Deepika; Yadav, S.; Hemlata; Jogpal, V. Fast disintegrating tablets: Opportunity in drug delivery system. J. Adv. Pharm. Technol. Res. 2011, 2, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Cirri, M.; Righi, M.F.; Maestrelli, F.; Mura, P.; Valleri, M. Development of glyburide fast-dissolving tablets based on the combined use of cyclodextrins and polymers. Drug Dev. Ind. Pharm. 2009, 35, 73–82. [Google Scholar] [CrossRef]
- Bandari, S.; Mittapalli, R.K.; Gannu, R.; Rao, Y.M. Orodispersible tablets: An overview. Asian J. Pharm. 2008, 2, 2–11. [Google Scholar]
- Jeong, S.H.; Fu, Y.; Park, K. Frosta: A new technology for making fast-melting tablets. Expert Opin. Drug Deliv. 2005, 2, 1107–1116. [Google Scholar] [CrossRef]
- Kawano, Y.; Ito, A.; Sasatsu, M.; Machida, Y.; Onishi, H. Preparation and Evaluation of Taste Masked Orally Disintegrating Tablets with Granules Made by the Wet Granulation Method. Yakugaku Zasshi 2010, 130, 1737–1742. [Google Scholar] [CrossRef] [Green Version]
- Bhise, K.; Shaikh, S.; Bora, D. Taste Mask, Design and Evaluation of an Oral Formulation Using Ion Exchange Resin as Drug Carrier. AAPS PharmSciTech 2008, 9, 557–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douroumis, D. Orally disintegrating dosage forms and taste-masking technologies; 2010. Expert Opin. Drug Deliv. 2011, 8, 665–675. [Google Scholar] [CrossRef]
- Khan, S.; Kataria, P.; Nakhat, P.; Yeole, P. Taste masking of ondansetron hydrochloride by polymer carrier system and formulation of rapid-disintegrating tablets. AAPS PharmSciTech 2007, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Szejtli, J.; Szente, L. Elimination of bitter, disgusting tastes of drugs and foods by cyclodextrins. Eur. J. Pharm. Biopharm. 2005, 61, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.Y.; Borodkin, S.; Woodward, L.; Li, P.; Diesner, C.; Hernandez, L.; Vadnere, M. A Polymer Carrier System for Taste Masking of Macrolide Antibiotics. Pharm. Res. 1991, 8, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Cravotto, G.; Binello, A.; Baranelli, E.; Carraro, P.; Trotta, F. Cyclodextrins as food additives and in food processing. Curr. Nutr. Food Sci. 2006, 2, 343–350. [Google Scholar] [CrossRef]
- Sohi, H.; Sultana, Y.; Khar, R.K. Taste masking technologies in oral pharmaceuticals: Recent developments and approaches. Drug Dev. Ind. Pharm. 2004, 30, 429–448. [Google Scholar] [CrossRef] [PubMed]
- Szejtli, J. Past, present, and future of cyclodextrin research. Pure Appl. Chem. 2004, 76, 1825–1845. [Google Scholar] [CrossRef]
- Carrier, R.L.; Miller, L.A.; Ahmed, I. The utility of cyclodextrins for enhancing oral bioavailability. J. Control. Release 2007, 123, 78–99. [Google Scholar] [CrossRef]
- Mady, F.M.; Abou-Taleb, A.E.; Khaled, K.A.; Yamasaki, K.; Iohara, D.; Taguchi, K.; Anraku, M.; Hirayama, F.; Uekama, K.; Otagiri, M. Evaluation of carboxymethyl-??-cyclodextrin with acid function: Improvement of chemical stability, oral bioavailability and bitter taste of famotidine. Int. J. Pharm. 2010, 397, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Hreinsdóttir, D.; Másson, M. Evaluation of cyclodextrin solubilization of drugs. Int. J. Pharm. 2005, 302, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Szejtli, J. Medicinal Applications of Cyclodextrins. Med. Res. Rev. 1994, 14, 353–386. [Google Scholar] [CrossRef] [PubMed]
- Dungarwal, U.N.; Patil, S.B. Development of orodispersible tablets of taste masked rizatriptan benzoate using hydroxypropyl β cyclodextrin. J. Pharm. Investig. 2016, 46, 537–545. [Google Scholar] [CrossRef]
- Lenik, J.; Wesoły, M.; Ciosek, P.; Wróblewski, W. Evaluation of taste masking effect of diclofenac using sweeteners and cyclodextrin by a potentiometric electronic tongue. J. Electroanal. Chem. 2016, 780, 153–159. [Google Scholar] [CrossRef]
- Madhavi, B.R.; Murthy, V.S.N.; Rani, A.P.; Kumar, Y.M. Formulation and evaluation of taste masked oral disintegrating tablet of cefixime based on cyclodextrin binary systems. J. Glob. Trends Pharm. Sci. 2014, 5, 1738–1746. [Google Scholar]
- Cirri, M.; Rangoni, C.; Maestrelli, F.; Corti, G.; Mura, P. Development of Fast-Dissolving Tablets of Flurbiprofen-Cyclodextrin Complexes. Drug Dev. Ind. Pharm. 2005, 31, 697–707. [Google Scholar] [CrossRef]
- Fernandes, C.M.; Veiga, F.J.B. Effect of the hydrophobic nature of triacetyl-beta-cyclodextrin on the complexation with nicardipine hydrochloride: Physicochemical and dissolution properties of the kneaded and spray-dried complexes. Chem. Pharm. Bull. 2002, 50, 1597–1602. [Google Scholar] [CrossRef]
- Modi, A.; Tayade, P. Enhancement of dissolution profile by solid dispersion (kneading) technique. AAPS PharmSciTech 2006, 7, 68. [Google Scholar] [CrossRef]
- Tenjarla, S.; Puranajoti, P.; Kasina, R.; Mandal, T. Preparation, characterization, and evaluation of miconazole-cyclodextrin complexes for improved oral and topical delivery. J. Pharm. Sci. 1998, 87, 425–429. [Google Scholar] [CrossRef]
- Higuchi, T.; Connors, A. Phase-Solubility Techniques. In Advances in Analytical Chemistry Instrumentation; Wiley Interscience: New York, NY, USA, 1965; pp. 117–211. [Google Scholar]
- Lake, S.T.; Altman, P.M.; Vaisman, J.; Addison, S.R. Validated LC-MS/MS assay for the quantitative determination of vardenafi l in human plasma and its application to a pharmacokinetic study. Biomed. Chromatogr. 2010, 24, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Tan, F.; Jing, Z.; Liu, Z. Preparation and study the 1:2 inclusion complex of carvedilol with β-cyclodextrin. J. Pharm. Biomed. Anal. 2004, 34, 517–523. [Google Scholar] [CrossRef]
- Dalmora, M.E.A.; Oliveira, A.G. Inclusion complex of piroxicam with b-cyclodextrin and incorporation in hexadecyltrimethylammonium bromide based microemulsion. Int. J. Pharm. 1999, 184, 157–164. [Google Scholar] [CrossRef]
- Esclusa-Diaz, M.T.; Gayo-Otero, M.; Perez-Marcos, M.B.; Vila-Jato, J.L.; Torres-Labandeira, J.J. Preparation and evaluation of ketoconazole-β-cyclodextrin multicomponent complexes. Int J. Pharm. 1996, 142, 183–187. [Google Scholar] [CrossRef]
- Hiremath, S.; Raghavendra, R.; Sunil, F.; Danki, L.; Rampure, M.; Swamy, P.; Bhosale, U. Dissolution enhancement of gliclazide by preparation of inclusion complexes with Î2-cyclodextrin. Asian J. Pharm. 2008, 2, 73–76. [Google Scholar] [CrossRef]
- Madhavi, B.; Devi, N.; Rani, A. Preparation and characterization of Zafirlukast-Î2-cyclodextrin complexes using Solid dispersion techniques. Int. J. Pharm. Sci. Rev. Res. 2010, 4, 88–93. [Google Scholar]
- Badr-eldin, S.M.; Ahmed, T.A.; Ismail, H.R. Aripiprazole-Cyclodextrin Binary Systems for Dissolution Enhancement: Effect of Preparation Technique, Cyclodextrin Type and Molar Ratio. Iran. J. Basic Med. Sci. 2013, 16, 1223–1231. [Google Scholar]
- Puttewar, T.Y.; Kshirsagar, M.D.; Chandewar, A.V.; Chikhale, R.V. Formulation and evaluation of orodispersible tablet of taste masked doxylamine succinate using ion exchange resin. J. King Saud Univ. Sci. 2010, 22, 229–240. [Google Scholar] [CrossRef]
- Badawy, S.I.F.; Shah, K.R.; Surapaneni, M.S.; Szemraj, M.M.; Hussain, M. Effect of spray-dried mannitol on the performance of microcrystalline cellulose-based wet granulated tablet formulation. Pharm. Dev. Technol. 2010, 15, 339–345. [Google Scholar] [CrossRef]
- De Bastos, M.O.; Friedrich, R.B.; Beck, R.C. Effects of Filler-Binders and Lubricants on Physicochemical Properties of Tablets Obtained by Direct Compression: A 22 Factorial Design. Lat. Am. J. Pharm. 2008, 27, 578–583. [Google Scholar]
- Fatohy, H.A.; Abdul-rasool, A.A. Effect of Different Diluent and Binder Types on the Preparation of Bisoprolol Fumarate as Tablet Dosage Form. Iraqi J. Pharm. Sci. 2013, 22, 32–39. [Google Scholar]
- Mehta, S.; De Beer, T.; Remon, J.P.; Vervaet, C. Effect of disintegrants on the properties of multiparticulate tablets comprising starch pellets and excipient granules. Int. J. Pharm. 2012, 422, 310–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazzal, S.; Zaghloul, A.-A.; Khan, M.A. Effect of extragranular microcrystalline cellulose on compaction, surface roughness, and in vitro dissolution of a self-nanoemulsified solid dosage form of ubiquinone. Pharm. Technol. 2002, 26, 86–98. [Google Scholar]
- Shu, T.; Suzuki, H.; Hironaka, K.; Ito, K. Studies of Rapidly Disintegrating Tablets in the Oral Cavity Using Co-Ground Mixtures of Mannitol with Crospovidone. Chem. Pharm. Bull. 2002, 2, 193–198. [Google Scholar] [CrossRef]
- Zulfiker, A.H.M.; Islam, K.M.; Islam, R.; Azam, K.R.; Reza, M.S. Formulation Development Using Maize Starch & Avicel Ph101 As Disintegrating Agents and Their Effect on Physical Characteristics & in Vitro Release Profile. Int. J. Pharm. Sci. Res. 2011, 2, 2136–2141. [Google Scholar]
- Muñoz, N.; Ferrero, C.; Muñoz-Ruiz, A.; Velasco, M.V.; Jiménez-Castellanos, M.R. Effect of explotab on the tabletability of a poorly soluble drug. Drug Dev. Ind. Pharm. 1998, 24, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Vraníková, B.; Gajdziok, J.; Doležel, P. The effect of superdisintegrants on the properties and dissolution profiles of liquisolid tablets containing rosuvastatin. Pharm. Dev. Technol. 2017, 22, 138–147. [Google Scholar] [CrossRef]
- Kawabata, Y.; Wada, K.; Nakatani, M.; Yamada, S.; Onoue, S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: Basic approaches and practical applications. Int. J. Pharm. 2011, 420, 1–10. [Google Scholar] [CrossRef]




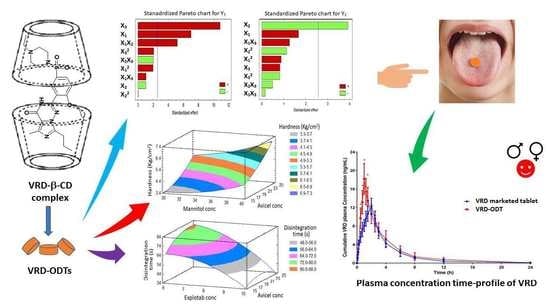
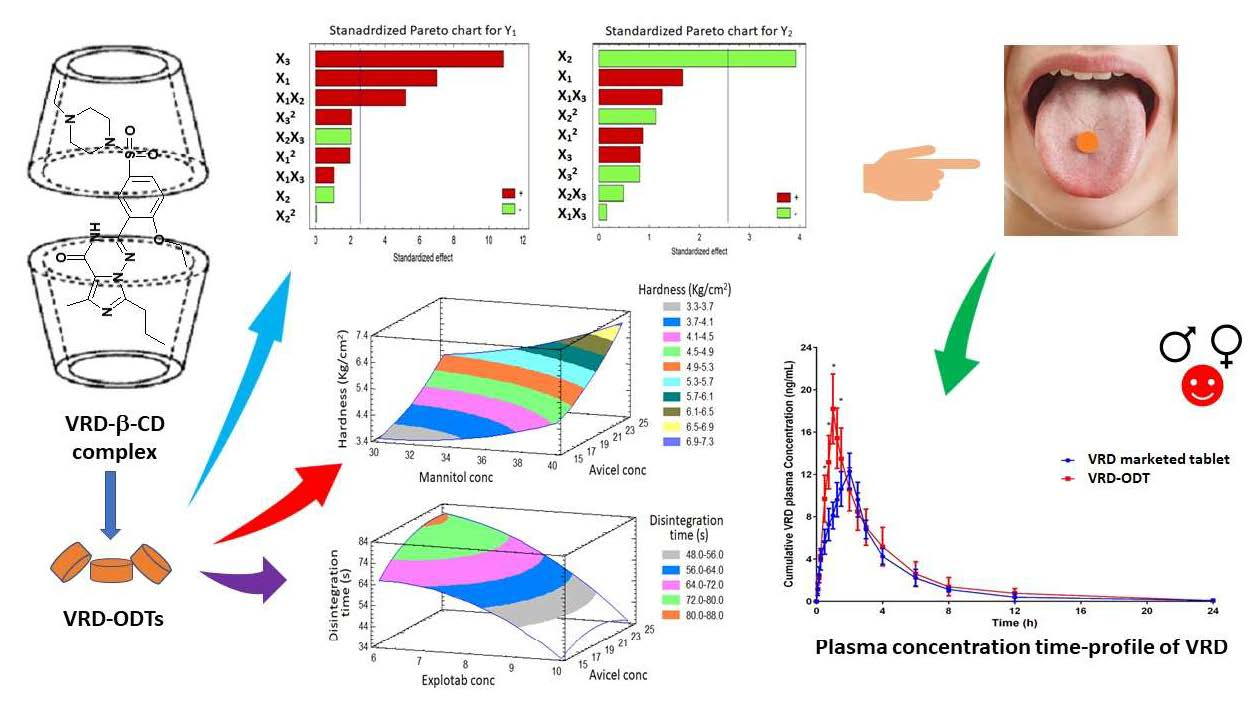{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factors | −1 | 0 | +1 | Response | Goal | Desirability |
|---|---|---|---|---|---|---|
| Mannitol (%) | 30 | 35 | 40 | Hardness (N) | Maximize | 70 N |
| Explotab (%) | 6 | 8 | 10 | Disintegration time (s) | Minimize | 30 s |
| Avicel (%) | 15 | 20 | 25 |
| Formula # | Drug Complex * | Mannitol | Explotab | Avicel | Crospovidone | Lactose | Magnesium Stearate | Talc |
|---|---|---|---|---|---|---|---|---|
| (mg) | ||||||||
| F1 | 30 | 70 | 20 | 30 | 10 | 36 | 2 | 2 |
| F2 | 30 | 70 | 20 | 50 | 10 | 16 | 2 | 2 |
| F3 | 30 | 60 | 12 | 40 | 10 | 44 | 2 | 2 |
| F4 | 30 | 70 | 12 | 30 | 10 | 44 | 2 | 2 |
| F5 | 30 | 60 | 16 | 50 | 10 | 30 | 2 | 2 |
| F6 | 30 | 80 | 16 | 30 | 10 | 30 | 2 | 2 |
| F7 | 30 | 70 | 12 | 50 | 10 | 24 | 2 | 2 |
| F8 | 30 | 60 | 20 | 40 | 10 | 36 | 2 | 2 |
| F9 | 30 | 80 | 20 | 40 | 10 | 16 | 2 | 2 |
| F10 | 30 | 80 | 16 | 50 | 10 | 10 | 2 | 2 |
| F11 | 30 | 60 | 16 | 30 | 10 | 50 | 2 | 2 |
| F12 | 30 | 80 | 12 | 40 | 10 | 24 | 2 | 2 |
| F13 | 30 | 70 | 16 | 40 | 10 | 30 | 2 | 2 |
| F14 | 30 | 70 | 16 | 40 | 10 | 30 | 2 | 2 |
| F15 | 30 | 70 | 16 | 40 | 10 | 30 | 2 | 2 |
| Formula # | Weight (mg) | Thickness (mm) | Hardness (N) | Friability (%) | Drug Content (%) | Disintegration Time (s) |
|---|---|---|---|---|---|---|
| F1 | 203.0 ± 3.06 | 2.64 ± 0.015 | 36.482 ± 0.26 | 0.635 | 97.77 ± 0.04 | 38.32 ± 5.77 |
| F2 | 201.6 ± 0.57 | 2.62 ± 0.035 | 54.723 ± 0.21 | 0.496 | 101.07 ± 0.08 | 48.67 ± 7.64 |
| F3 | 203.3 ± 1.15 | 2.60 ± 0.003 | 46.485 ± 0.15 | 0.147 | 100.98± 0.01 | 88.33 ± 10.4 |
| F4 | 204.3 ± 0.57 | 2.60 ± 0.001 | 33.736 ± 0.10 | 0.645 | 99.37 ± 0.04 | 52.67 ± 2.51 |
| F5 | 202.3 ± 1.53 | 2.69 ± 0.006 | 52.075 ± 0.21 | 0.494 | 101.7 ± 0.04 | 46.67 ± 5.77 |
| F6 | 202.6 ± 0.57 | 2.69 ± 0.006 | 44.817 ± 0.31 | 0.493 | 98.7 ± 0.08 | 72.67 ± 2.51 |
| F7 | 200.0 ± 1.53 | 2.60 ± 0.000 | 63.059 ± 0.12 | 0.301 | 96.84 ± 0.05 | 76.33 ± 3.21 |
| F8 | 201.6 ± 1.15 | 2.60 ± 0.001 | 31.088 ± 0.06 | 0.297 | 99.86 ± 0.05 | 36.33 ± 3.21 |
| F9 | 203.0 ± 1.0 | 2.57 ± 0.025 | 61.489 ± 0.50 | 0.197 | 99.73 ± 0.01 | 41.43 ± 2.88 |
| F10 | 203.0 ± 1.0 | 2.59 ± 0.010 | 65.903 ± 0.06 | 0.492 | 100.09 ± 0.04 | 88.49 ± 7.63 |
| F11 | 199.3 ± 0.57 | 2.60 ± 0.001 | 36.776 ± 0.21 | 0.051 | 100.45 ± 0.04 | 65.0 ± 10.0 |
| F12 | 203.0 ± 2.08 | 2.60 ± 0.001 | 48.447 ± 0.38 | 0.977 | 97.59 ± 0.01 | 97.67 ± 8.73 |
| F13 | 201.0 ± 1.53 | 2.60 ± 0.341 | 42.170 ± 0.16 | 0.299 | 95.43 ± 0.04 | 66.43 ± 5.13 |
| F14 | 202.0 ± 1.53 | 2.60 ± 0.001 | 44.524 ± 0.11 | 0.098 | 98.84 ± 0.04 | 73.33 ± 5.77 |
| F15 | 202.0 ± 1.0 | 2.61 ± 0.001 | 45.799 ± 0.10 | 0.198 | 100.36 ± 0.08 | 63.45 ± 2.88 |
| Factor | Hardness (Y1) | Disintegration Time (Y2) | ||||
|---|---|---|---|---|---|---|
| Estimate | F-Ratio | p-Value | Estimate | F-Ratio | p-Value | |
| A: Mannitol | 1.3825 | 49.05 | 0.0009 * | 15.9825 | 2.78 | 0.1565 |
| B: Explotab | −0.2025 | 1.05 | 0.3520 | −37.5625 | 15.34 | 0.0112 * |
| C: Avicel | 2.14 | 117.53 | 0.0001 * | 7.875 | 0.67 | 0.4490 |
| AA | 0.5717 | 3.87 | 0.1063 | 12.4133 | 0.77 | 0.4195 |
| AB | 1.45 | 26.98 | 0.0035 * | −2.12 | 0.02 | 0.8819 |
| AC | 0.295 | 1.12 | 0.3390 | 17.075 | 1.58 | 0.2637 |
| BB | −0.0183 | 0.00 | 0.9521 | −16.0067 | 1.29 | 0.3083 |
| BC | −0.565 | 4.10 | 0.0989 | −6.655 | 0.24 | 0.6445 |
| CC | 0.5967 | 4.22 | 0.0952 | −11.4717 | 0.66 | 0.4534 |
| R2 (%) | 97.647 | 82.457 | ||||
| Adjusted R2 (%) | 93.411 | 50.879 | ||||
| Analysis of Variance | ||||||
| F-ratio | 23.2920 | 2.6039 | ||||
| p-value | 0.0015 | 0.1522 | ||||
| Lack of Fit | ||||||
| F ratio | 2.8493 | 11.2529 | ||||
| p value | 0.2705 | 0.0827 | ||||
| R2 | 0.9956 | 0.9902 | ||||
| Volunteer No. | Disintegration Time (s) | Taste Masking (0–3) |
|---|---|---|
| V1 | 65 | 0 |
| V2 | 60 | 1 |
| V3 | 62 | 1 |
| V4 | 63 | 0 |
| V5 | 59 | 0 |
| V6 | 65 | 0 |
| Mean | 62.33 ± 2.503 |
| Pharmacokinetic Parameter | VRD Marketed Tablet | Optimized VRD-ODTs |
|---|---|---|
| Cmax (ng/mL) | 12.29383 ± 2.55 | 18.191 ± 1.95 * |
| tmax (h) | 2.0 ± 0.13 | 1.0 ± 0.21 * |
| AUC(0–24) (ng·h/mL) | 46.68801 ± 4.63 | 58.81263 ± 5.15 |
| AUC(24–∞) (ng·h/mL) | 0.460347 ± 0.34 | 0.332505 ± 0.17 |
| AUC(0–∞) (ng·h/mL) | 47.14836 ± 3.61 | 59.14514 ± 17.35 |
| AUMC(0–24) ng·hr2/mL | 184.5833 | 239.0714 |
| AUMC(24–end) ng·hr2/mL | 11.04833 | 7.980117 |
| AUMC(0–end) ng·hr2/mL | 195.6317 | 247.0515 |
| Kel (h−1) | 0.209 ± 0.01 | 0.230573 |
| t1/2 (h) | 3.317 ± 0.63 | 3.005555 ± 0.53 |
| MRT (h) | 4.149 ± 0.93 | 4.174615 ± 1.33 |
| CL (mL/h) | 3.824974 | 2.929219 |
| Relative bioavailability (%) | 100 | 125.445 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Gethmy, H.A.; Fahmy, U.A.; Alhakamy, N.A.; Ahmed, O.A.A.; El-Say, K.M. Optimization of the Factors Affecting the Absorption of Vardenafil from Oral Disintegrating Tablets: A Clinical Pharmacokinetic Investigation. Pharmaceutics 2019, 11, 11. https://doi.org/10.3390/pharmaceutics11010011
Al-Gethmy HA, Fahmy UA, Alhakamy NA, Ahmed OAA, El-Say KM. Optimization of the Factors Affecting the Absorption of Vardenafil from Oral Disintegrating Tablets: A Clinical Pharmacokinetic Investigation. Pharmaceutics. 2019; 11(1):11. https://doi.org/10.3390/pharmaceutics11010011
Chicago/Turabian StyleAl-Gethmy, Hytham A., Usama A. Fahmy, Nabil A. Alhakamy, Osama A. A. Ahmed, and Khalid M. El-Say. 2019. "Optimization of the Factors Affecting the Absorption of Vardenafil from Oral Disintegrating Tablets: A Clinical Pharmacokinetic Investigation" Pharmaceutics 11, no. 1: 11. https://doi.org/10.3390/pharmaceutics11010011