1. Introduction
In recent years the amorphous state has gained increasing interest in pharmaceutics due to its favourable properties compared to its crystalline counterpart [
1,
2]. These advantages include increased solubility and potential higher bioavailability, making the amorphous state a promising approach for delivering poorly soluble drugs.
However, major issues when dealing with the amorphous state are physical and chemical instabilities, and to date the prediction of physical and chemical stability of drugs in the amorphous state still proves challenging. Often it is observed that the amorphous state of a compound that had been prepared by different methods, shows differing physico-chemical properties and stability [
3]. In order to increase the stability of the amorphous state of a drug, these are often formulated with a hydrophilic polymer using a variety of preparation techniques, creating a solid dispersion or glass solution [
4]. In a glass solution the drug is molecularly dispersed in a polymer matrix.
Compared to the crystalline form of a drug, the amorphous form is in a state of higher energy. This is due to the fact that the amorphous state possesses excess thermodynamic properties such as enthalpy, entropy and Gibbs free energy.
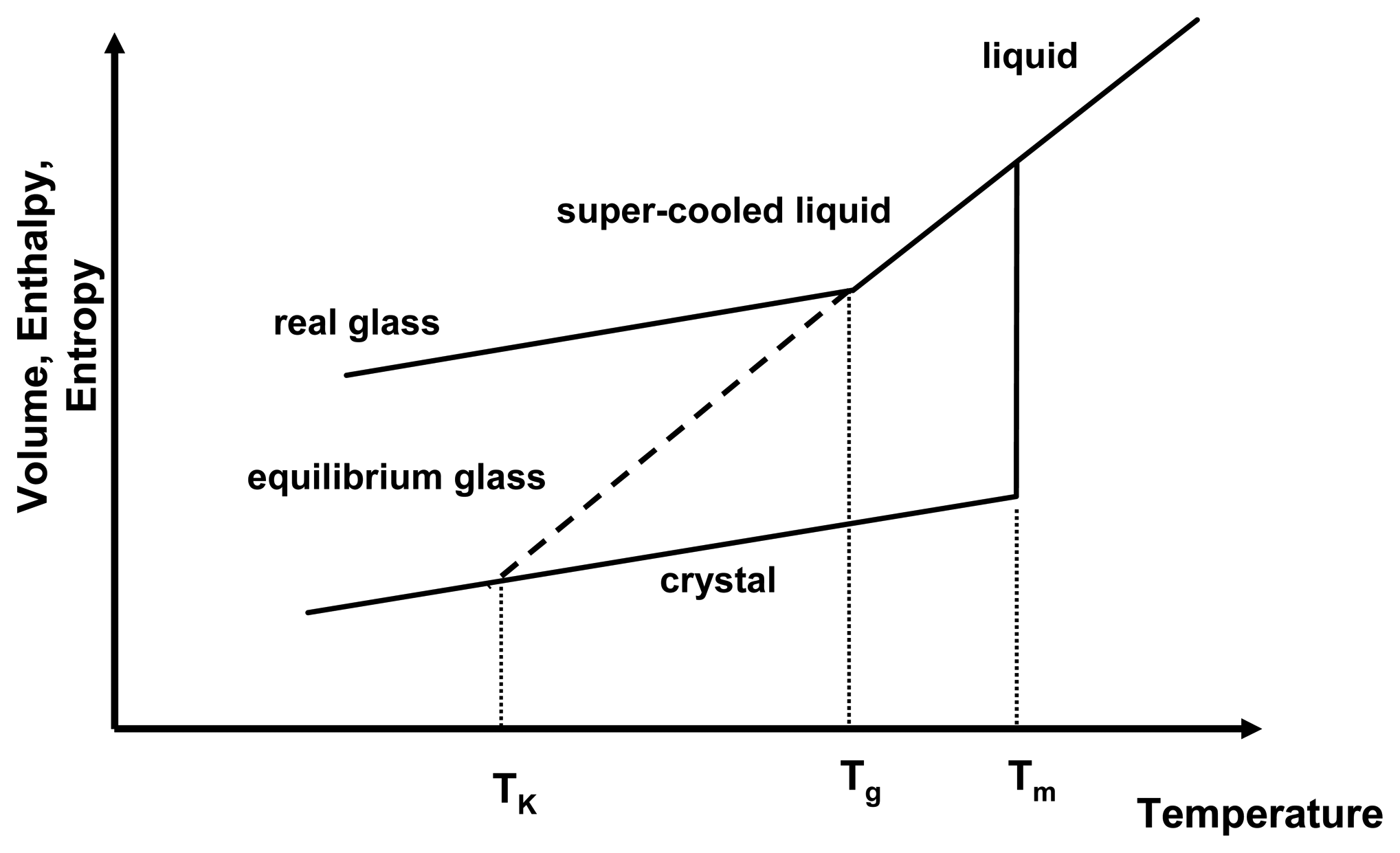
Figure 1 depicts the relationship of the thermodynamic properties and temperature for the amorphous and crystalline state. As a liquid melt of a crystal is cooled rapidly, recrystallisation may be prevented and the slope of the equilibrium liquid line may be followed below the melting temperature,
Tm, resulting in a gradual decrease of thermodynamic properties below
Tm. This is the super-cooled liquid state, in which viscosity is low (typically around 10
−3 − 10
12 Pa s) and mobility of the molecules is high and they are able to follow any further decrease of temperature to attain equilibrium conditions. However, upon further cooling, at the glass transition temperature,
Tg, the molecules cannot follow the decrease in temperature any longer and the systems solidifies, falling out of equilibrium. This is represented by the change of the slope in
Figure 1. Below the
Tg, the system is in the glassy state, exhibiting high viscosities of > 10
12 Pa s.
Figure 1.
Thermodynamic relationship of crystalline and amorphous state as a function of temperature. Shown are the melting temperature (
Tm), the glass transition temperature (
Tg) and the Kauzmann temperature (
TK). Adapted from [
1].
Figure 1.
Thermodynamic relationship of crystalline and amorphous state as a function of temperature. Shown are the melting temperature (
Tm), the glass transition temperature (
Tg) and the Kauzmann temperature (
TK). Adapted from [
1].
If the Tg did not occur and the system remained in equilibrium throughout the cooling process, the super-cooled liquid line would be followed (now as an equilibrium glass) and it would intersect the crystal line at the Kauzmann temperature, TK. Below the TK, the amorphous system would have lower entropy and enthalpy than the crystalline state, which presents a violation of the laws of thermodynamics. Therefore, the occurrence of the Tg has two implications for amorphous systems: firstly glasses are in a non-equilibrium state and equilibrium thermodynamics cannot be applied below that temperature and secondly the physico-chemical properties of amorphous systems above and below the Tg are different.
During storage, it is often observed that the amorphous state reduces its excess enthalpy and entropy without recrystallising. This is called “relaxation” as the amorphous state relaxes towards a lower energetic state, still remaining in the amorphous form. The real glass relaxes asymptotically towards the equilibrium glassy state. This relaxation behaviour can be seen as an indication that below the Tg mobility is low, but still existent.
The higher energetic state is beneficial in terms of improving the solubility of a compound, however, it is detrimental to the physical stability [
5,
6]. Due to the enhanced thermodynamic properties of the amorphous state compared to the crystalline state, recrystallisation provides a means of reducing this excess free energy but in doing so, the solubility advantages are negated.
Crystallisation is a result of a nucleation process, where stable nuclei are created, followed by crystal growth. The recrystallisation of an amorphous form is governed by the same factors as the crystallisation from a melt [
1], and therefore the crystallisation processes have been described on the basis of the classical nucleation theory (CNT) for homogenous nucleation [
7].
Nucleation is a process which involves the overcoming of a potential barrier and was first described by Gibbs [
8]. The change of Gibbs free energy (Δ
G), due to the formation of a cluster of the new phase (crystal) is given by
Equation 1:
where Δ
GS is the change in surface free energy (in J mol
−1) and Δ
GV is the volume free energy change (in J mol
−1).
The value of Δ
GS is associated with the formation of the cluster and represents a positive quantity. The value of Δ
GV is associated with the phase transition of liquid to solid and represents a negative quantity [
9]. According to the CNT, when nucleation occurs, clusters grow and decay until a stable nucleus is formed. After a cluster of a critical size has been formed, it will grow in size and hence recrystallisation of the amorphous form takes place.
Nucleation and crystal growth are not solely governed by thermodynamics but also require the individual molecules to move via diffusion. This kinetic proportion of the recrystallisation effect is usually considered as the molecular mobility or its reciprocal, the relaxation time
τ. It has long been suggested that molecules exhibit sufficient mobility only at temperatures close to or above the
Tg, and storage at temperatures below
Tg would ensure physical stability. It has however been found that nucleation and crystal growth also occur at temperatures well below the
Tg, however, the relative rate may be much slower than in the temperature region above the
Tg [
10,
11].
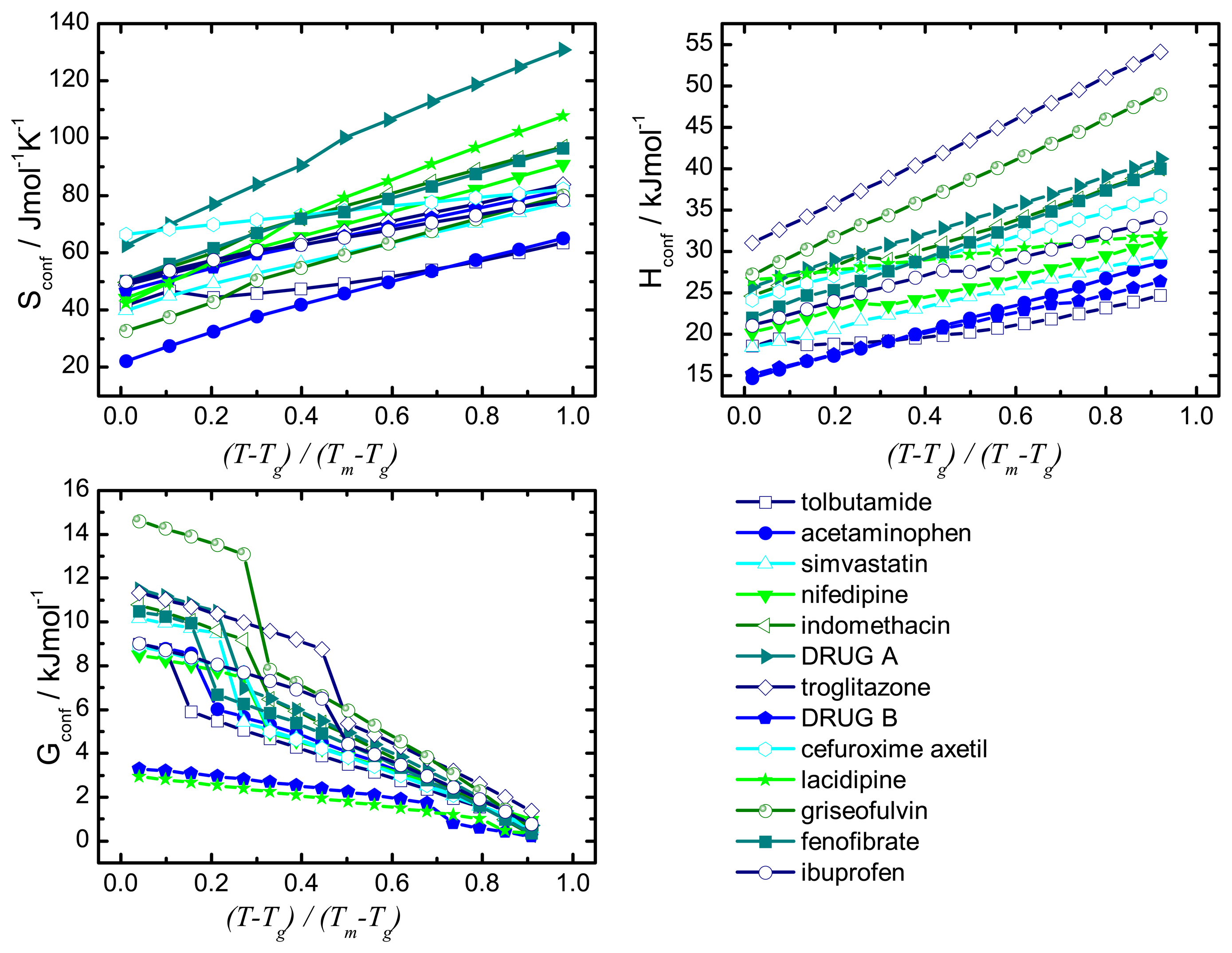
Recrystallisation should therefore be influenced by thermodynamic (such as enthalpy, entropy and Gibbs free energy) and kinetic parameters (such as mobility). A commonality of the thermodynamic and kinetic parameters is the involvement of the configurational entropy, Sconf, which is the difference in entropy between the amorphous and the crystalline state.
1.1. Thermodynamic involvement of Sconf
The desired properties of the amorphous state (higher solubility and dissolution rate compared to the crystalline form) have been attributed, at least in part, to an increase in thermodynamic properties, e.g., free energy, entropy and enthalpy. This change in free energy is regarded as a driving factor for recrystallisation: the larger the difference in free energy between the amorphous and crystalline state the more thermodynamically favorable the situation will be upon recrystallisation.
The difference in Gibbs free energy between the amorphous and the crystalline states can be calculated using enthalpic and entropic values for the amorphous and crystalline state as shown in
Equation 2:
The term “configurational” denotes the difference between the amorphous and the crystalline state and the parameters Hconf and Sconf may be calculated from their relationship with the heat capacity.
where
Hconf is the configurational enthalpy (in J mol
−1),
Sconf the configurational entropy (in J mol
−1 K
−1), Δ
Hm the melting enthalpy of the crystal (in J mol
−1) and Δ
Sm the melting entropy of the crystal (in J mol
−1 K
−1).
The melting entropy can be obtained from the following relationship:
The configurational thermodynamic values give an indication of the relationship between the amorphous state and the crystalline state of a compound. The larger the configurational values are the greater are the differences between the crystalline and the amorphous states.
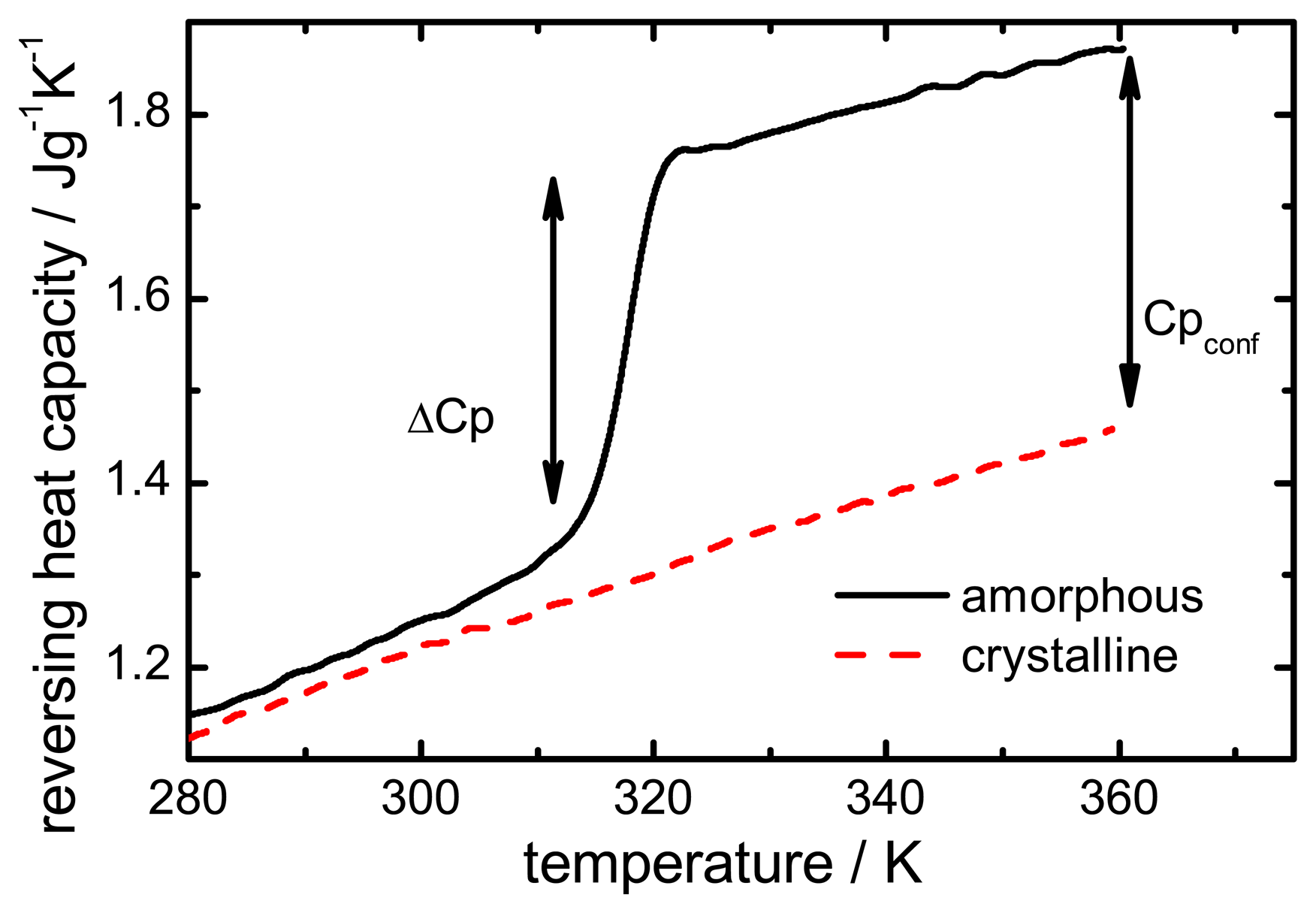
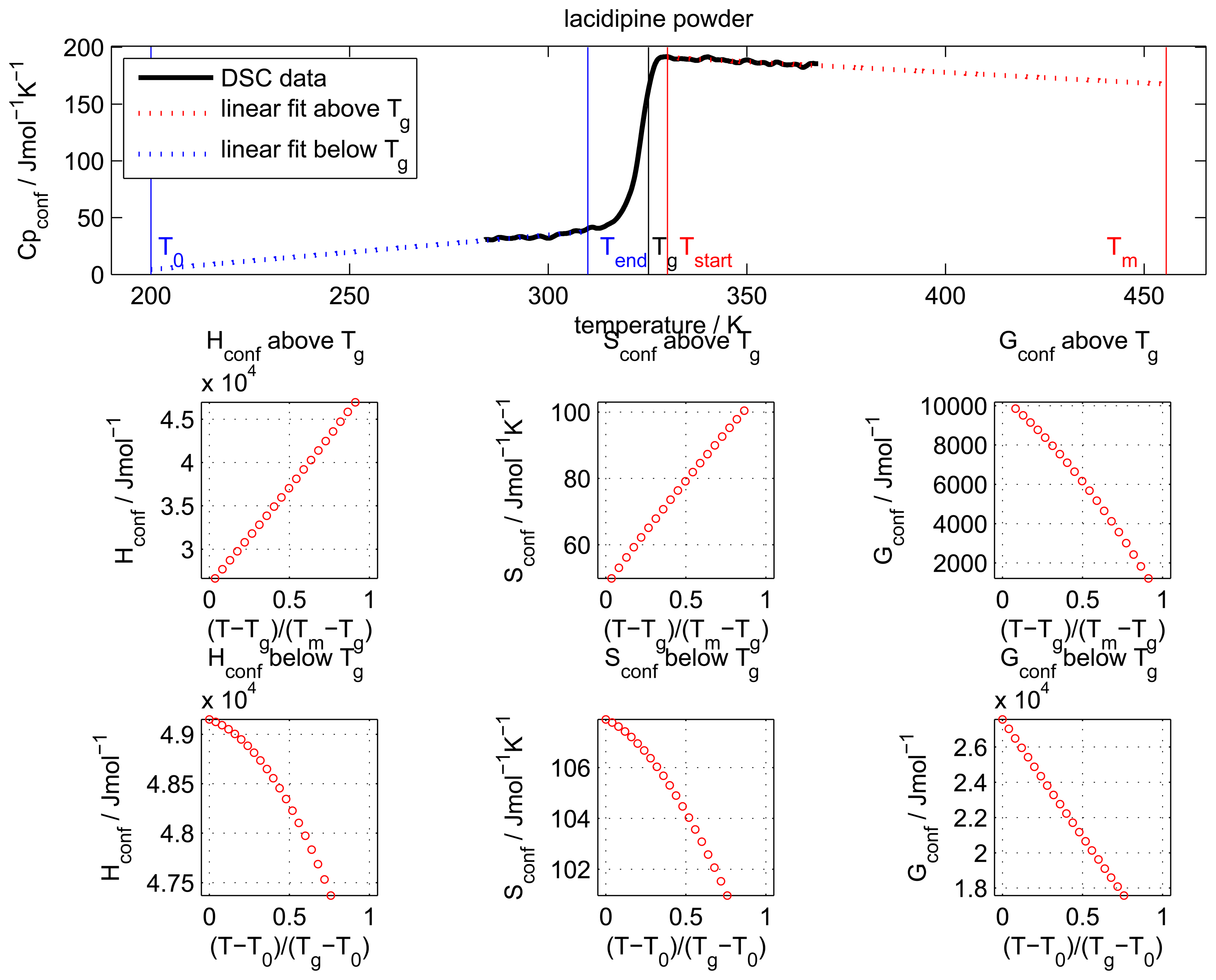
The configurational heat capacity
Cpconf, required to calculate the configurational properties, is the difference between the amorphous and the crystalline heat capacities.
Cpconf is not identical to Δ
Cp, which denotes the heat capacity change of the amorphous state at
Tg as shown in
Figure 2.
Below the Tg the heat capacity values of the glass and the crystal may be similar, however, the heat capacities are not identical, therefore, Cpconf is never zero.
After passing through the
Tg the
Cpconf of an amorphous compound may increase or decrease with temperature or follow a specific temperature dependence, depending on the properties of the material [
12,
13]. The temperature dependence of the configurational heat capacity above
Tg has been described by the hyperbolic relation presented in the following equation:
Figure 2.
Heat capacity difference (ΔCp) and configurational heat capacity (Cpconf) of indomethacin.
Figure 2.
Heat capacity difference (ΔCp) and configurational heat capacity (Cpconf) of indomethacin.
1.2. Kinetic involvement of Sconf
The amorphous state not only possesses higher thermodynamic properties compared to the crystalline state, it also shows enhanced molecular mobility [
5,
14] and this is considered an important factor for the subsequent physical and chemical instabilities [
1,
15]. Crystal nuclei formation, the first step of the recrystallisation process, is a result of the localized faster mobility of molecules [
16] and degradation reactions such as hydrolysis or protein degradation have been attributed to increased molecular mobility within the amorphous state [
17].
Molecular mobility has therefore been the topic of numerous investigations, however, due to the complex nature of the amorphous state and the still poorly understood relaxation properties, to date no single equation can be used to estimate the relaxation time of the amorphous state. Among those generally used are the Kohlrausch-Williams-Watts equation (KWW) [
18], the Adam-Gibbs equation (AG) [
19] and the Vogel–Tamman–Fulcher equation (VTF) [
20,
21,
22].
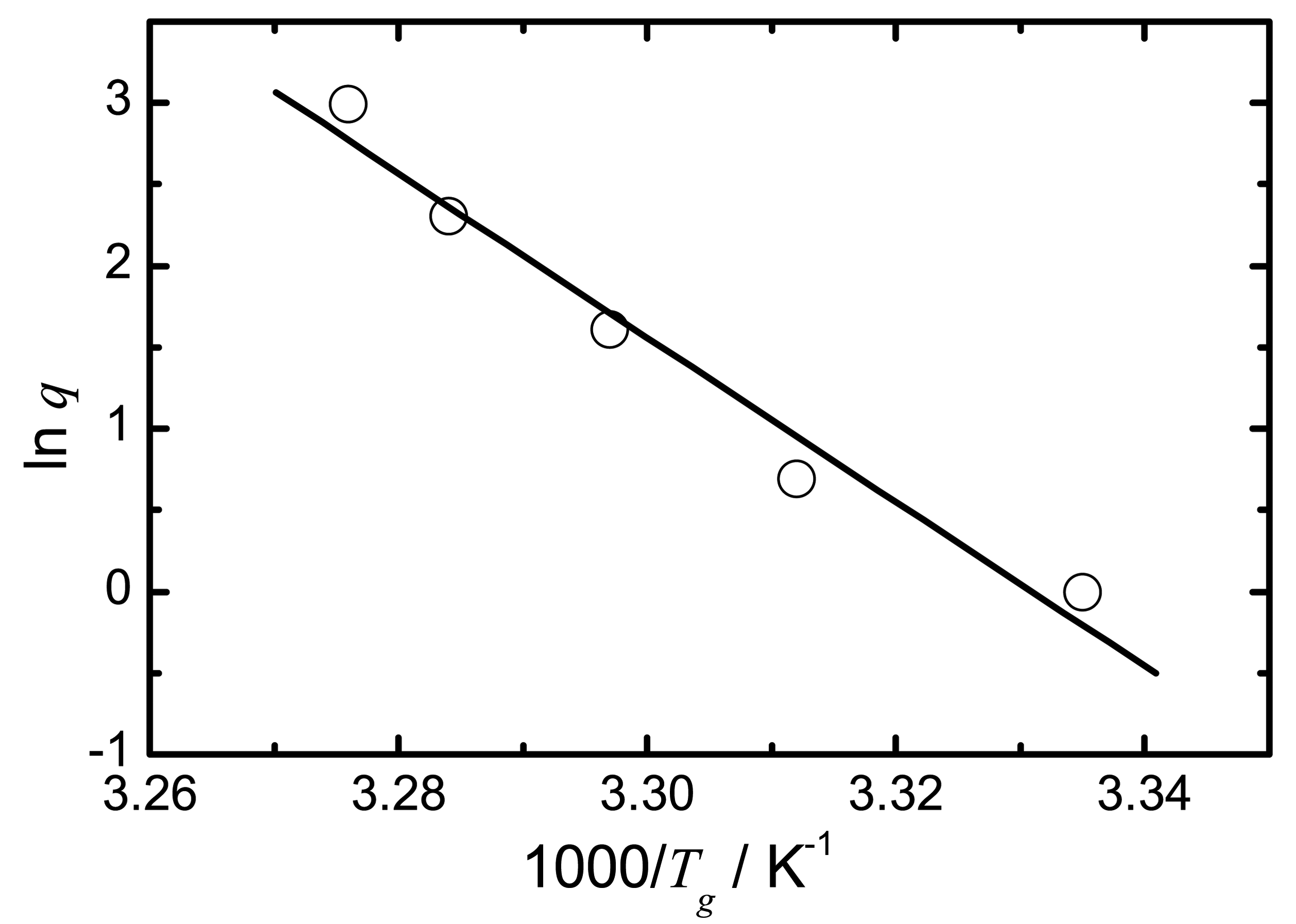
These equations all show advantages and disadvantages as they address different issues, but the most commonly used equation to estimate the molecular mobility for the temperature range below the Tg is the AG.
The governing thought of the AG equation is that a liquid consists of regions that rearrange themselves in units, the so-called cooperatively rearranging regions (CRR). Upon cooling from the super-cooled liquid state, these CRR become progressively larger. The size of the CRR is determined by the difference in the configurational entropy,
Sconf, of the liquid which varies with temperature. When the temperature is high, the Sconf is large and the size of the CRR is small. Upon cooling, the
Sconf decreases and in return the size of the CRR increases. In the AG theory, this increasing cooperativity is believed to be due to a loss of configurational entropy, which enables the calculation of molecular mobility [
5].
with
τ relaxation time below
Tg (in s),
τ0 pre-exponential parameter (lifetime of the atomic vibrations, 10-14 s), Δ
μ activation energy of cooperative rearrangement (J mol
−1),
entropy of smallest cooperative molecular region (J mol
−1 K
−1),
kB Boltzmann constant (1.38 J K
−1) and
Sconf(
T) configurational entropy at temperature T (J mol
−1 K
−1).
Equation 9 simplifies to the following expression if the properties of the glass forming liquid (Δ
μ and
) are considered constant:
where
C is a constant.
By applying
Equation 10 it has to be considered that the entropic contributions are entirely due to
Sconf and any other influence (e.g., vibrational entropy) is neglected [
23]. As a result,
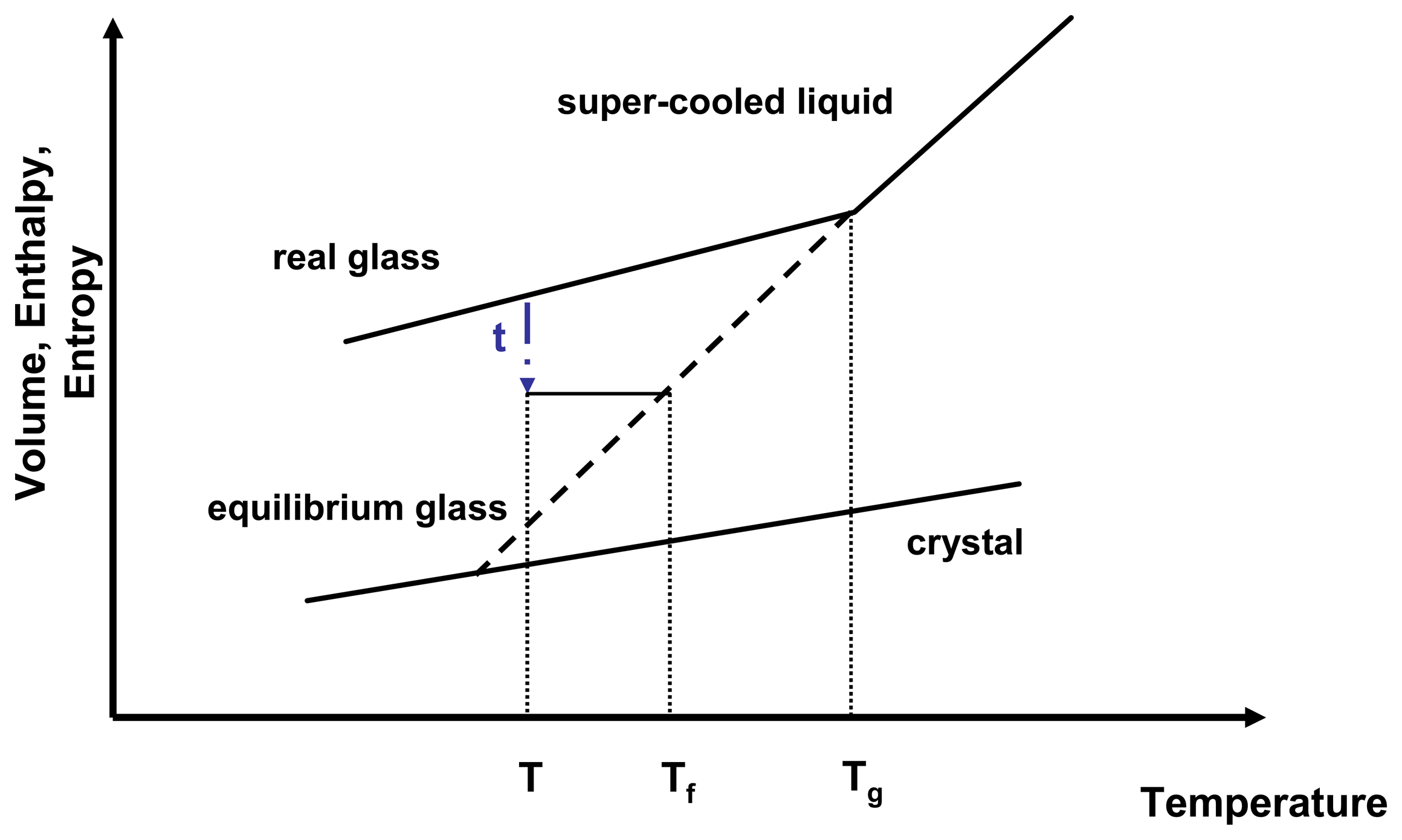
Sconf has a direct influence on the relaxation time and therefore on molecular mobility. As a glass relaxes isothermally, it reduces a portion of its excess entropy which in return leads to an increase of relaxation time. The molecular mobility of an amorphous state therefore is not only temperature dependent but also time dependent. A convenient way to express the temperature and time dependence of the molecular mobility is to introduce the fictive temperature,
Tf (
Figure 3).
Figure 3.
Relationship of enthalpy and temperature for an amorphous system.
τ represents the relaxation time,
T is the isothermal relaxation temperature,
Tf is the fictive temperature and
Tg is the glass transition temperature. Adapted from [
24].
Figure 3.
Relationship of enthalpy and temperature for an amorphous system.
τ represents the relaxation time,
T is the isothermal relaxation temperature,
Tf is the fictive temperature and
Tg is the glass transition temperature. Adapted from [
24].
A real glass is not in equilibrium, therefore applying thermodynamic principles proves challenging. However, thermodynamics can be estimated by relating the real glass to the theoretical equilibrium glass at the same temperature.
The fictive temperature is defined as the temperature at which the system under investigation has the same thermodynamic properties as its equilibrium state at that temperature and that time. The configurational entropy can therefore be described by
Equation 12.
with
configurational entropy of the real glass (in J mol
−1 K
−1),
configurational entropy of the equilibrium super-cooled liquid (in J mol
−1 K
−1) and
Tf fictive temperature (in K).
The fictive temperature is a convenient way of describing the temperature and time dependence of Sconf for real glasses and enables calculation of molecular mobility in these systems.
Through a number of rearrangements and substitutions of
Sconf in
Equation 10 [
15] the AG equation can be written as:
where
D is the dimensionless Angell's strength parameter and
T0 is the temperature of zero
Sconf (in K).
The relaxation time τ may therefore be expressed by the fictive temperature which contains the contributions from the configurational entropy.
1.3. Involvement of Sconf in solubility prediction
Amorphous compounds show a higher apparent solubility than their crystalline counterparts [
25] due to their higher energetic state and the disordered structure that does not require the crystal lattice to be broken upon dissolution. According to studies by Parks et al. the theoretical solubility ratio (
σamorph/σcrystal) of the amorphous and crystalline form at a given temperature can be described through the free energy difference between the two forms [
26,
27]:
with the gas constant
R = 8.314 J mol
−1 K
−1.
The value of
Gconf can be calculated as presented in
Equation 2, via
Cpconf and hence
Sconf. This approach has already been successfully applied for estimating the solubility differences between different crystalline polymorphs [
28,
29].
In the literature experimental solubility increases of up to 10 fold have been reported [
30] for amorphous systems, which in return can increase the bioavailability of a poorly soluble compound considerably. However, for amorphous compounds the calculated solubility may increase by up to 1,600 fold [
30,
31]. However, this potential increase usually is not observed in vitro and the estimated values are significantly smaller. This is partly due to the fact that the amorphous state is far from equilibrium which poses difficulties in determining their equilibrium thermodynamic properties and partly due to the tendency of the amorphous state to revert back to the crystalline state upon exposure to solvents such as water or biorelevant media [
30]. It is also observed that the amorphous state does not recrystallise to its original crystalline state but may crystallise to a different, potentially less soluble, polymorph [
32]. This is not trivial and can pose challenges during development of drug formulations.
Despite their inaccurate values for absolute solubility, the estimated solubility ratios are a useful tool for estimating the theoretical maximal solubility and serve as an indication of the theoretical driving force for dissolution.
These considerations regarding the Sconf highlight the importance of this parameter in terms of amorphous stability and solubility, the major factors of interest in pharmaceutics.
This paper will focus on the calculation of Sconf from simple calorimetric measurements and will give details of the methods used. It goes on to calculate the important thermodynamic, kinetic and solubility parameters. It is not in the scope of this article to compare calculated values to experimentally determined values or provide in depth interpretation of results.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
