Induction of Drug Transporters Alters Disposition of Risperidone - A Study in Mice

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Materials and Methods
2.1. Drugs
2.2. Animals
2.3. Study design and drug administration
2.4. Sample analysis
2.5. Statistical analysis
3. Results and Discussion
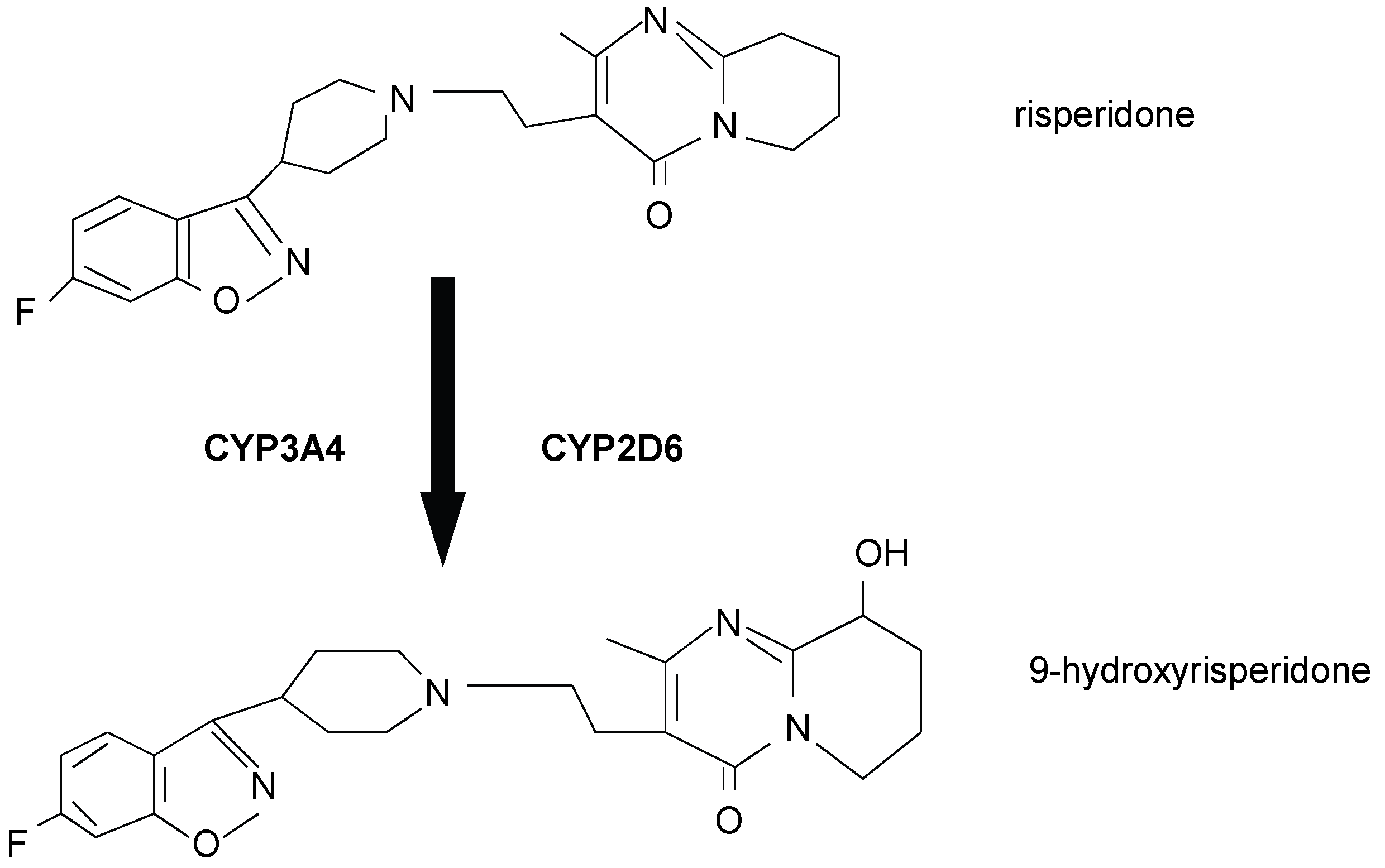
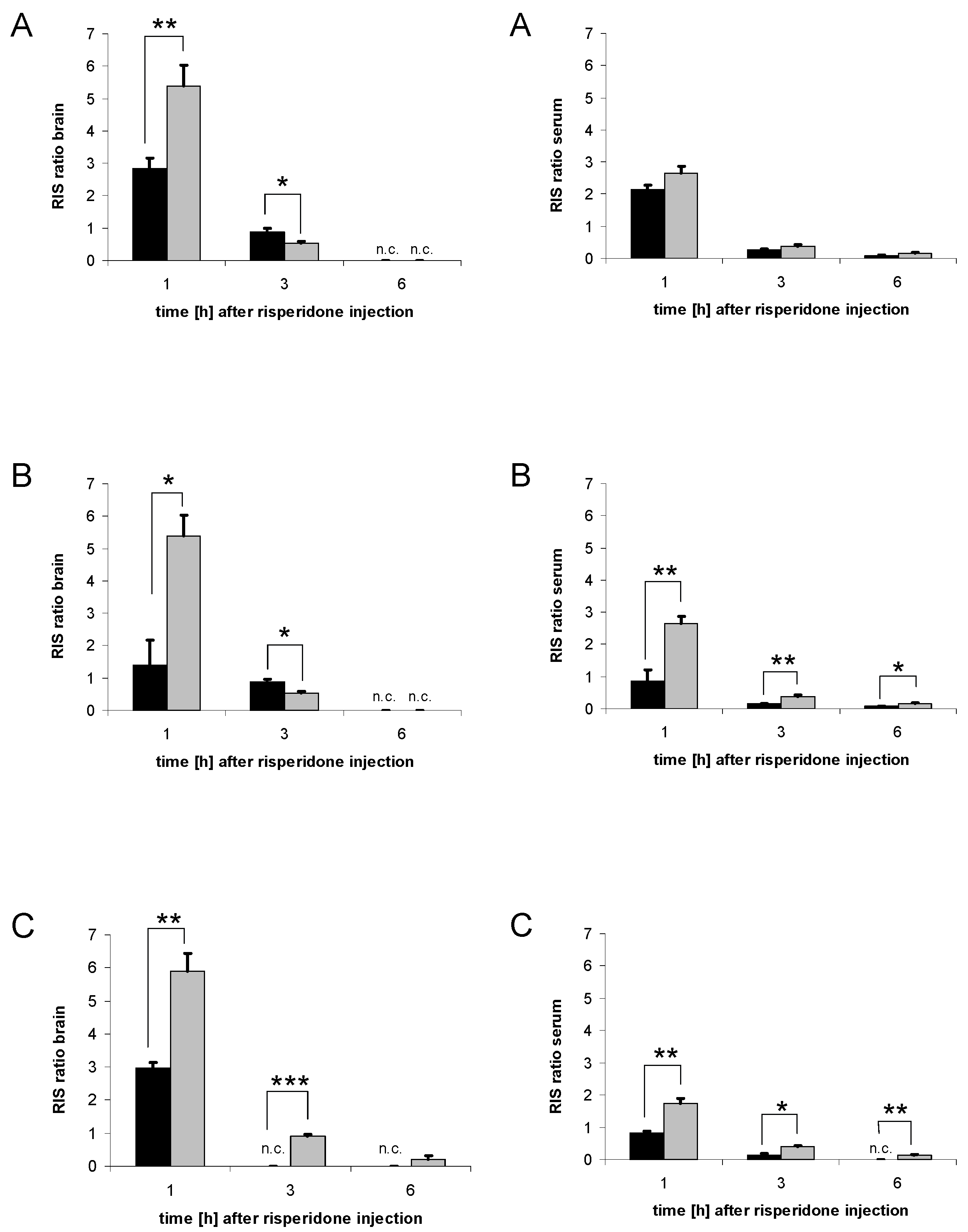
3.1. Brain concentrations of risperidone and 9-hydroxyrisperidone

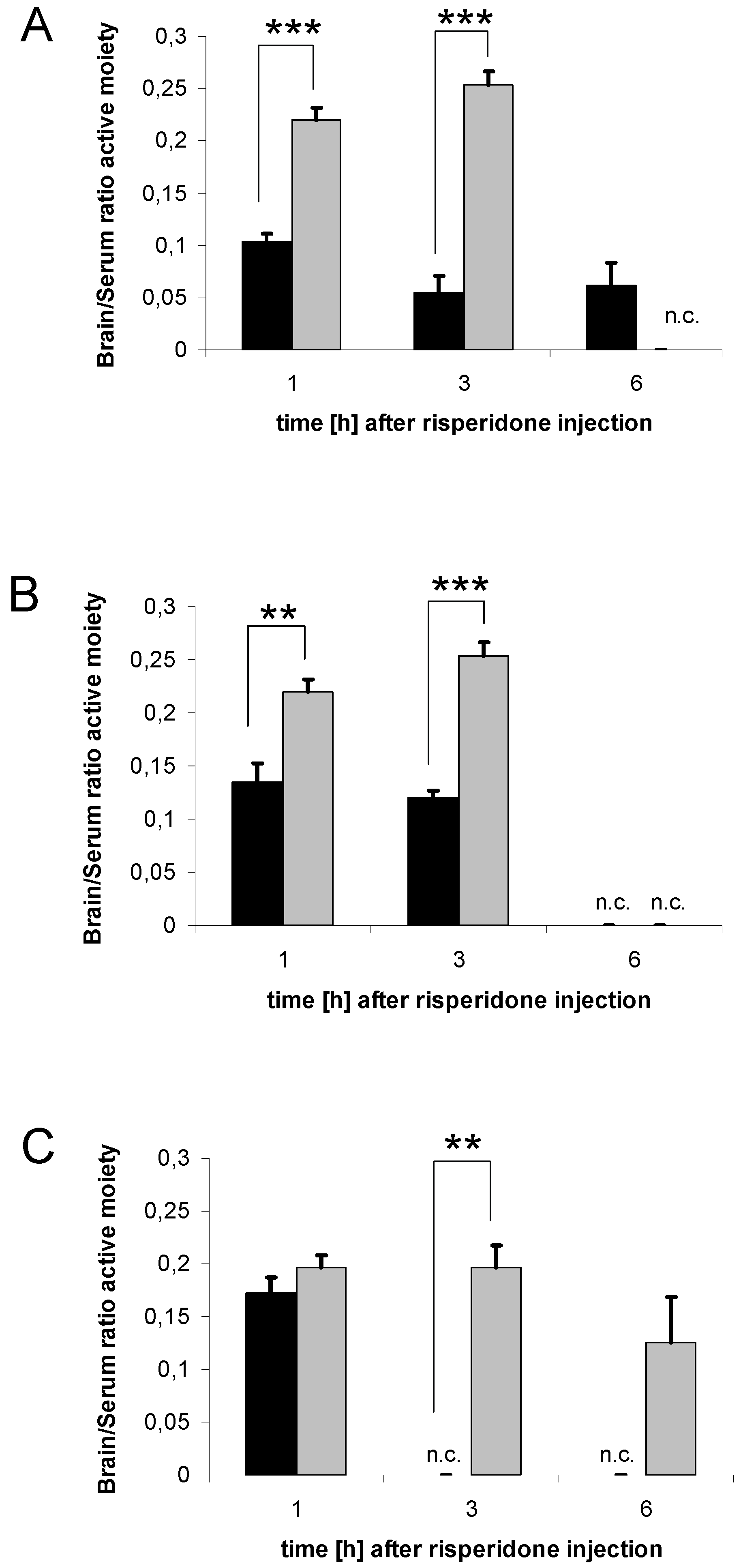
3.2. Brain concentrations of risperidone active moiety

3.3. Serum concentrations of risperidone and 9-hydroxyrisperidone

3.4. Serum concentrations of risperidone active moiety

3.5. Effects of Drug Transporter Inducers on metabolism

3.6. Distribution of risperidone and 9-hydroxyrisperidone

3.7. Distribution of risperidone active moiety

3.8. Discussion
4. Conclusions
Acknowledgements
References and Notes
- Dean, M.; Hamon, Y.; Chimini, G. The human ATP-binding cassette (ABC) transporter superfamily. J. Lipid Res. 2001, 42, 1007–1017. [Google Scholar]
- Schinkel, A.H.; Mayer, U.; Wagenaar, E.; Mol, C.A.; van Deemter, L.; Smit, J.J.; van der Valk, M.A.; Voordouw, A.C.; Spits, H.; van Tellingen, O.; Zijlmans, J.M.; Fibbe, W.E.; Borst, P. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoprotein. Proc. Natl. Acad. Sci. USA 1997, 94, 4028–4033. [Google Scholar] [CrossRef]
- Bauer, B.; Hartz, A.; Fricker, G.; Miller, D. Modulation of P-glycoprotein transport function at the blood-brain barrier. Exp. Biol. Med. 2005, 230, 118–127. [Google Scholar]
- Abou El-Ela, A.; Härtter, S.; Schmitt, U.; Hiemke, C.; Spahn-Langguth, H.; Langguth, P. Identification of P-glycoprotein substrates and inhibitors among psychoactive compounds-implications for pharmacokinetics of selected substrates. J. Pharm. Pharmacol. 2004, 56, 1–9. [Google Scholar]
- Boulton, D.W.; DeVane, C.L.; Liston, H.L.; Markowitz, J.S. In vitro P-glycoprotein affinity for atypical and conventional antipsychotics. Life Sci. 2002, 71, 163–169. [Google Scholar] [CrossRef]
- Doan, K.M.M.; Humphreys, J.E.; Webster, L.O.; Wring, S.A.; Shampine, L.J.; Serabjit-Singh, C.J.; Adkinson, K.K.; Polli, J.W. Passive permeability and P-glycoprotein-mediated efflux differentiate central nervous system (CNS) and non-CNS marketed drugs. J. Pharmacol. Exp. Ther. 2002, 303, 1029–1037. [Google Scholar] [CrossRef]
- Doran, A.; Obach, S.; Smith, B.J.; Hosea, N.A.; Becker, S.; Callegari, E.; Chen, C.; Chen, X.; Choo, E.; Cianfrogna, J.; Cox, L.M.; Gibbs, J.P.; Gibbs, M.A.; Hatch, H.; Hop, C.; Kasman, I.N.; LaPerle, J.; Liu, J.H.; Liu, X.; Logman, M.; Maclin, D.; Nedza, F.M.; Nelson, F.; Olson, E.; Rahematpura, S.; Raunig, D.; Rogers, S.; Schmidt, K.; Spracklin, D.K.; Szewc, M.; Troutman, M.; Tseng, E.; Tu, M.; van Deusen, J.W.; Venkatakrishnan, K.; Walens, G.; Wang, E.Q.; Wong, D.; Yasgar, A.S.; Zhang, C. The impact of P-glycoprotein on the disposition of drugs targeted for indications of the central nervous system: Evaluation using the MDR1a/1b knockout mouse model. Drug Metab. Dispos. 2005, 33, 165–174. [Google Scholar]
- Uhr, M.; Grauer, M.T.; Holsboer, F. Differential enhancement of antidepressant penetration into the brain in mice with abcb1 ab (mdr1ab) P-glycoprotein gene disruption. Biol. Psychiatry 2003, 54, 840–846. [Google Scholar] [CrossRef]
- Kirschbaum, K.M.; Henken, S.; Hiemke, C.; Schmitt, U. Pharmacodynamic consequences of P-glycoprotein-dependent pharmacokinetics of risperidone and haloperidol in mice. Behav. Brain Res. 2008, 188, 298–303. [Google Scholar] [CrossRef]
- Fang, J.; Bourin, M.; Baker, G.B. Metabolism of risperidone to 9-hydroxyrisperidone by human cytochromes P450 2D6 and 3A4. Naunyn Schmiedebergs Arch. Pharmacol. 1999, 359, 147–151. [Google Scholar] [CrossRef]
- Martignoni, M.; Groothuis, G.M.M.; de Kanter, R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin. Drug Metab. Toxicol. 2006, 2, 875–894. [Google Scholar] [CrossRef]
- Bauer, B.; Hartz, A.M.S.; Fricker, G.; Miller, D.S. Pregnane X receptor up-regulation of P-glycoprotein expression and transport function at the blood-brain-barrier. Mol. Pharmacol. 2004, 66, 413–419. [Google Scholar]
- Kageyama, M.; Fukushima, K.; Togawa, T.; Fujimoto, K.; Taki, M.; Nishimura, A.; Ito, Y.; Sugioka, N.; Shibata, N.; Takada, K. Relationship between excretion clearance of rhodamine 123 and p-glycoprotein (P-gp) expression induced by representative P-gp inducers. Biol. Pharm. Bull. 2006, 29, 779–784. [Google Scholar] [CrossRef]
- Medori, R.; Mannaert, E.; Gruender, G. Plasma antipsychotic concentration and receptor occupancy, with special focus on risperidone long-acting injectable. Eur. Neuropsychopharmacol. 2005, 16, 233–240. [Google Scholar] [CrossRef]
- Nyberg, S.; Dahl, M.J.; Halldin, C. A PET study of D2 and 5-HT2 receptor occupancy induced by risperidone in poor metabolizers of debrisoquin and risperidone. Psychopharmacology 1995, 119, 345–348. [Google Scholar] [CrossRef]
- Gunes, A.; Spina, E.; Dahl, M.L.; Scordo, M.G. ABCB1 polymorphisms influence steady-state plasma levels of 9-hydroxyrisperidone and risperidone active moiety. Ther. Drug Monit. 2008, 30, 628–633. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Goodwin, B.; Willson, T.M. The nuclear pregnane X receptor: A key role of xenobiotic metabolism. Endocr. Rev. 2002, 23, 687–702. [Google Scholar] [CrossRef]
- Kim, K.A.; Park, P.W.; Liu, K.H.; Kim, K.B.; Lee, H.J.; Shin, J.K.; Park, J.Y. Effect of rifampicin, an inducer of CYP3A and P-glycoprotein, on the pharmacokinetics of risperidone. J. Clin. Pharmacol. 2008, 48, 66–72. [Google Scholar] [CrossRef]
- Sonoda, J.; Rosenfeld, J.M.; Xu, L.; Evans, R.M.; Xie, W. A nuclear receptor-mediated xenobiotic response and its implication in drug metabolism and host protection. Curr. Drug Metab. 2003, 4, 59–72. [Google Scholar] [CrossRef]
- Teng, S.; Jekerle, V.; Piquette-Miller, M. Induction of ABCC3 (MRP3) by pregnane X receptor activators. Drug Metab. Dispos. 2003, 31, 1296–1299. [Google Scholar] [CrossRef]
- Breedveld, P.; Beijnen, J.S.; Schellens, J.H.M. Use of P-glycoprotein and BCRP inhibitors to improve oral bioavailability and CNS penetration of anticancer drugs. Trends Pharmacol. Sci. 2006, 27, 17–24. [Google Scholar] [CrossRef]
- Hoffmann, K.; Löscher, W. Upregulation of brain expression of P-glycoprotein in MRP2-deficient TR-rats resembles seizure-induced up-regulation of this drug efflux transporter in normal rats. Epilepsia. 2007, 48, 631–645. [Google Scholar] [CrossRef]
- Kast, H.R.; Goodwin, B.; Tarr, P.T.; Jones, S.A.; Anisfeld, A.M.; Stoltz, C.M.; Tontonoz, P.; Kliewer, S.; Willson, T.M.; Edwards, P.A. Regulation of multidrug resistance-associated protein 2 (ABCC2) by the nuclear receptors pregnane X receptor, farnesoid X-activated receptor, and constitutive androstane receptor. J. Biol. Chem. 2002, 277, 2908–2915. [Google Scholar] [CrossRef]
- Köhle, C.; Bock, K.W. Coordinate regulation of human drug-metabolizing enzymes, and conjugate transporters by the Ah receptor, pregnane X receptor and constitutive androstane receptor. Biochem. Pharmacol. 2009, 77, 689–699. [Google Scholar] [CrossRef]
- Narang, V.S.; Fraga, C.; Kumar, N.; Shen, J.; Throm, S.; Stewart, C.F.; Maters, C.M. Dexamethasone increases expression and activity of multidrug resistance transporters at the rat blood-brain barrier. Am. J. Physiol. Cell Physiol. 2008, 295, 440–450. [Google Scholar] [CrossRef]
- Matheny, C.J.; Ali, R.Y.; Yang, X.; Pollack, G.M. Effect of prototypical inducing agents on P-glycoprotein and CYP3A4 expression in mouse tissues. Drug Metab. Dispos. 2004, 32, 1008–1014. [Google Scholar]
- Cheng, X.; Klaassen, C.D. Regulation of mRNA expression of xenobiotic transporters by the pregnane X receptor in mouse liver, kidney, and intestine. Drug Metab. Dispos. 2006, 34, 1863–1867. [Google Scholar] [CrossRef]
- Guzelian, J.; Barwick, J.L.; Hunter, L.; Phang, T.L.; Quattrochi, L.C.; Guzelian, P.S. Identification of genes controlled by the pregnane X receptor by microarray analysis of mRNAs from pregnenolone16α-carbonitrile-treated rats. Toxicol Sci. 2006, 94, 379–387. [Google Scholar] [CrossRef]
- Martin, P.; Riley, R.; Thompson, P.; Williams, D.; Back, D.; Owen, A. Effect of prototypical inducers on ligand activated nuclear receptor regulated drug disposition genes in rodent hepatic and intestinal cells. Acta Pharmacol. Sin. 2010, 31, 51–65. [Google Scholar] [CrossRef]
- Yadav, S.; Dhawan, A.; Seth, P.K.; Singh, R.L.; Parmar, D. Cytochrome P4503A: Evidence for mRNA expression and catalytic activity in rat brain. Mol. Cell. Biochem. 2006, 287, 91–99. [Google Scholar] [CrossRef]
- Kirn, R.B.; Wandel, C.; Leake, B.; Cvetkovic, M.; Fromm, M.F.; Dempsey, P.J.; Roden, M.M.; Belas, F.; Chaudhary, A.K.; Roden, D.M.; Wood, A.J.J.; Wilkinson, G.R. Interrelationship between substrates and inhibitors of human CYP3A and P-glycoprotein. Pharm. Res. 1999, 16, 408–414. [Google Scholar] [CrossRef]
- Xu, C.; Li, Y.; Kong, A. Induction of phase I, II and III drug metabolism/ transport by xenobiotics. Arch. Pharm. Res. 2005, 28, 249–268. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Holthoewer, D.; Hiemke, C.; Schmitt, U. Induction of Drug Transporters Alters Disposition of Risperidone - A Study in Mice. Pharmaceutics 2010, 2, 258-274. https://doi.org/10.3390/pharmaceutics2020258
Holthoewer D, Hiemke C, Schmitt U. Induction of Drug Transporters Alters Disposition of Risperidone - A Study in Mice. Pharmaceutics. 2010; 2(2):258-274. https://doi.org/10.3390/pharmaceutics2020258
Chicago/Turabian StyleHolthoewer, David, Christoph Hiemke, and Ulrich Schmitt. 2010. "Induction of Drug Transporters Alters Disposition of Risperidone - A Study in Mice" Pharmaceutics 2, no. 2: 258-274. https://doi.org/10.3390/pharmaceutics2020258