Dietary Iron Supplementation Alters Hepatic Inflammation in a Rat Model of Nonalcoholic Steatohepatitis

Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
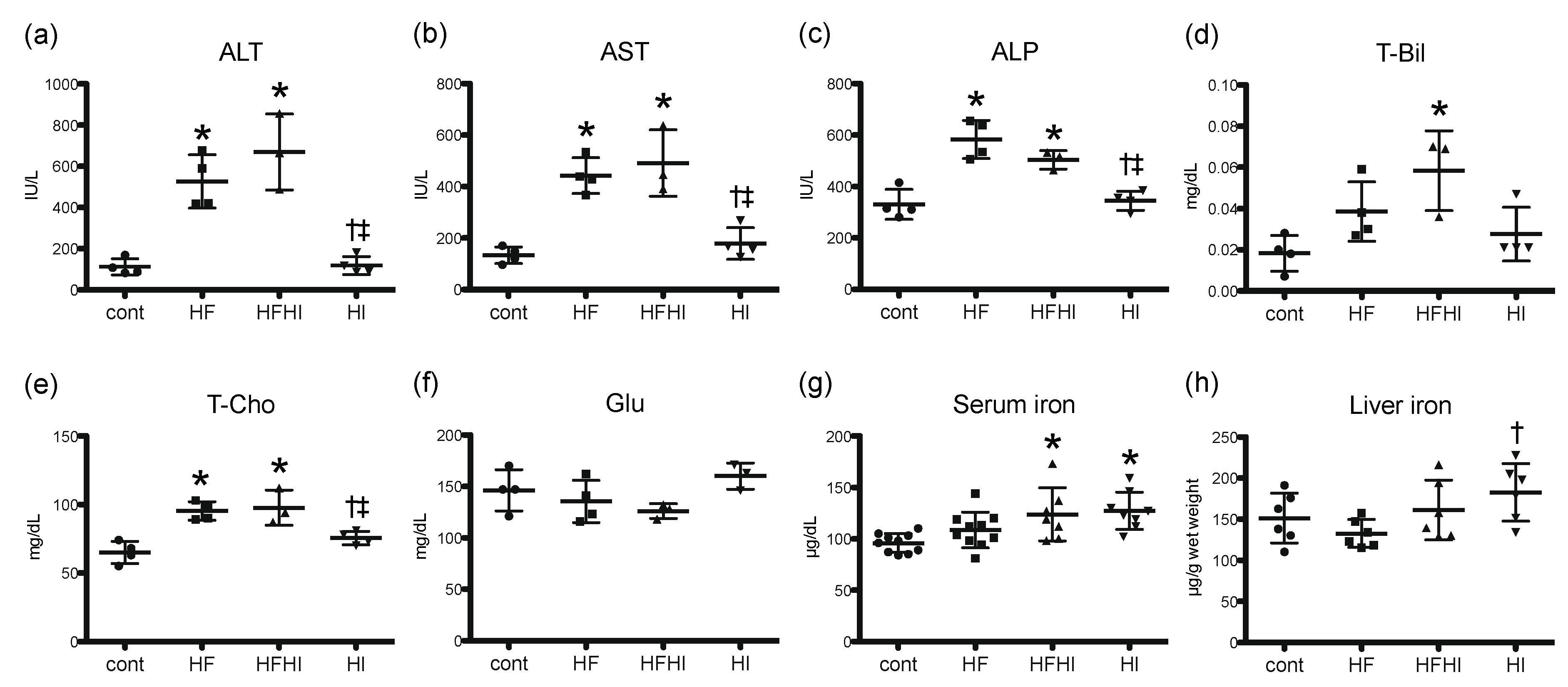
2.2. Biochemical Analyses
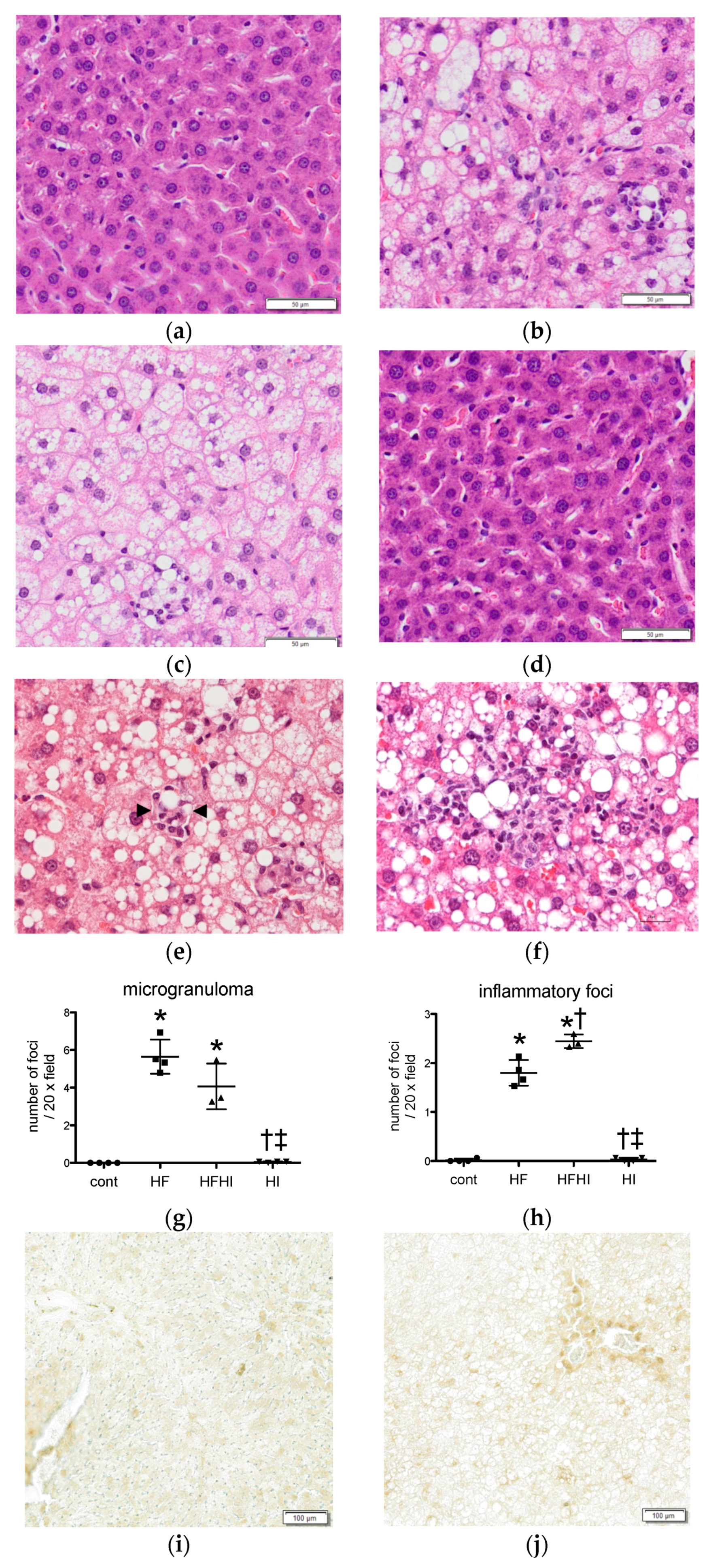
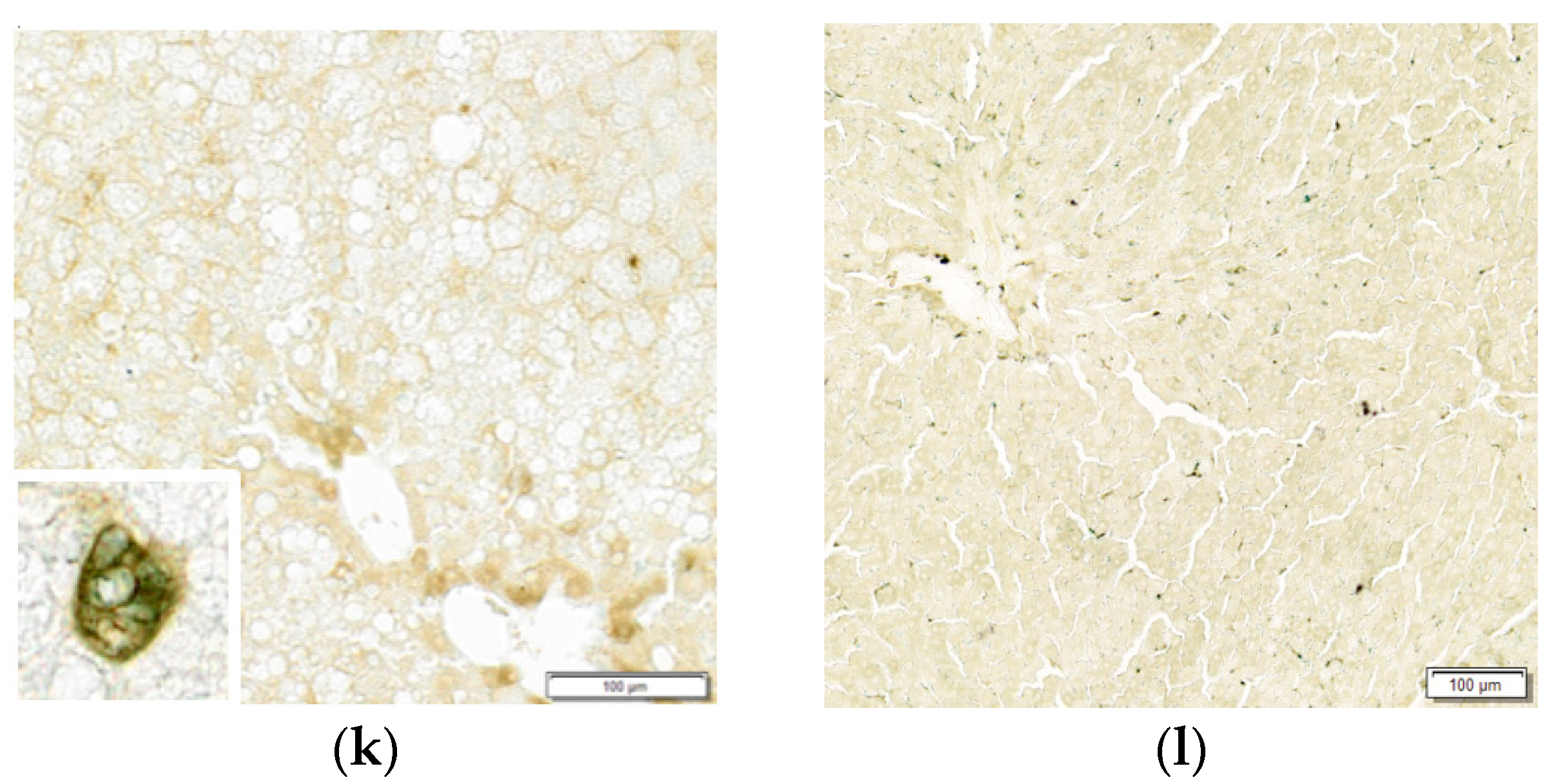
2.3. Histopathology
2.4. Cell Counts
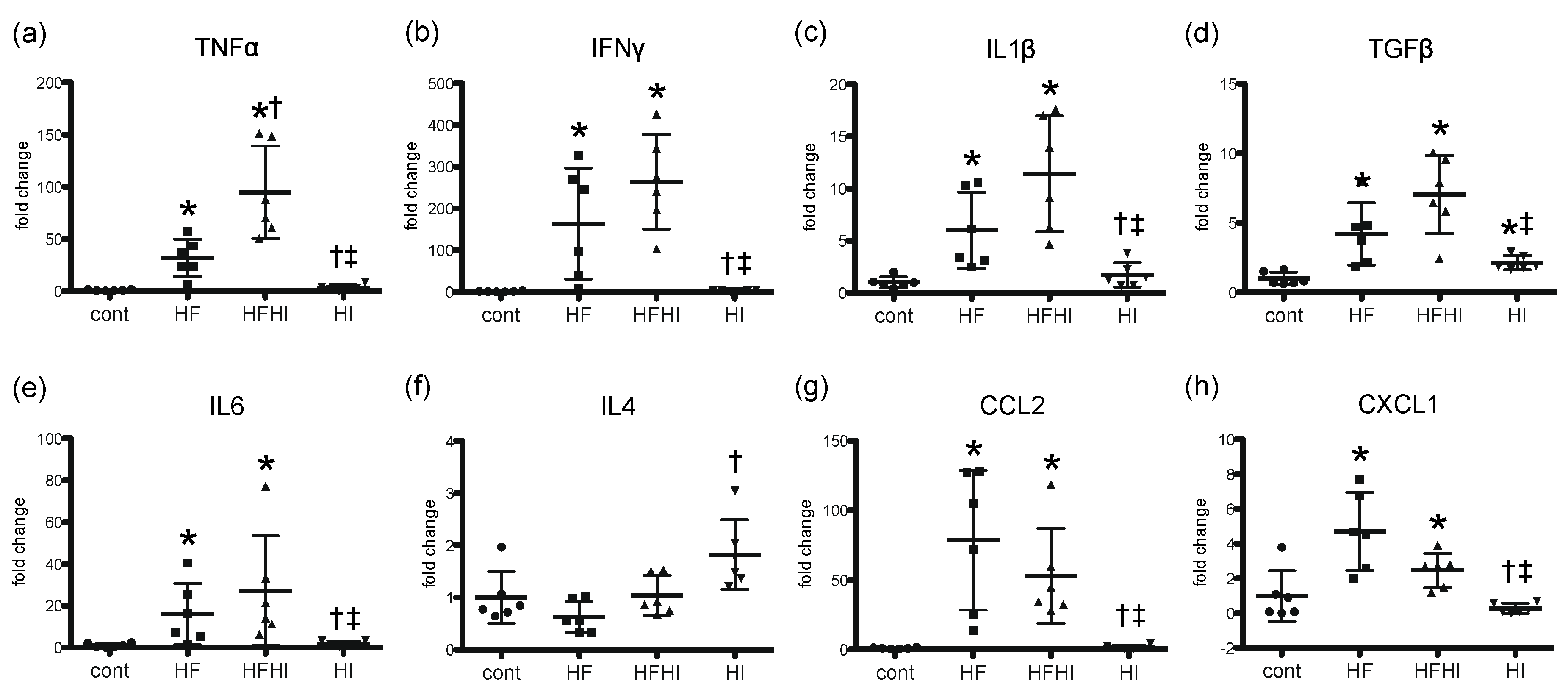
2.5. Real-Time Reverse Transcriptase Polymerase Chain Reaction (RT-PCR)
2.6. Gut Microbiota Analysis
2.7. Statistical Analysis
3. Results
3.1. Dietary Iron Burden Enhances High-Fat Diet-Induced Hepatic Inflammation
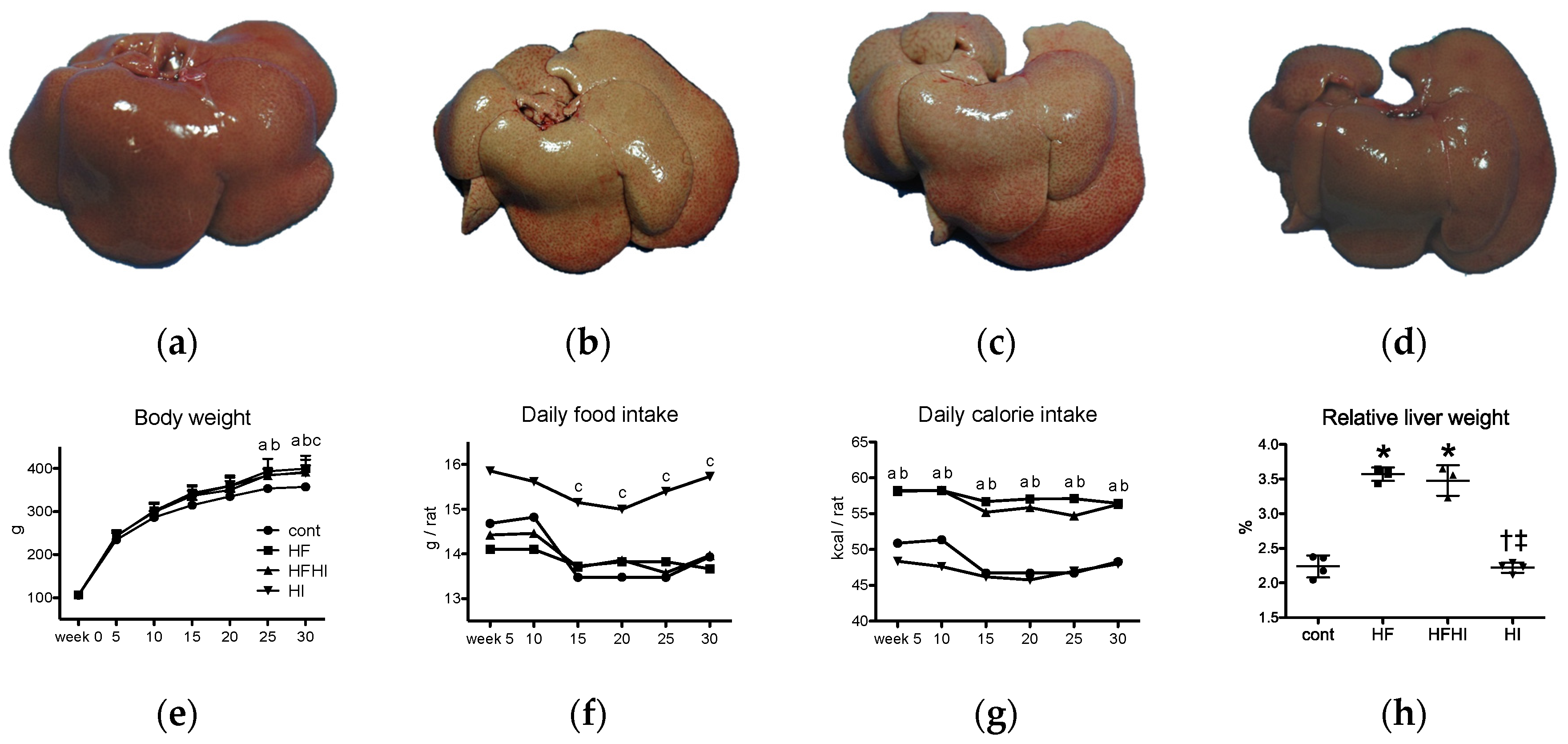
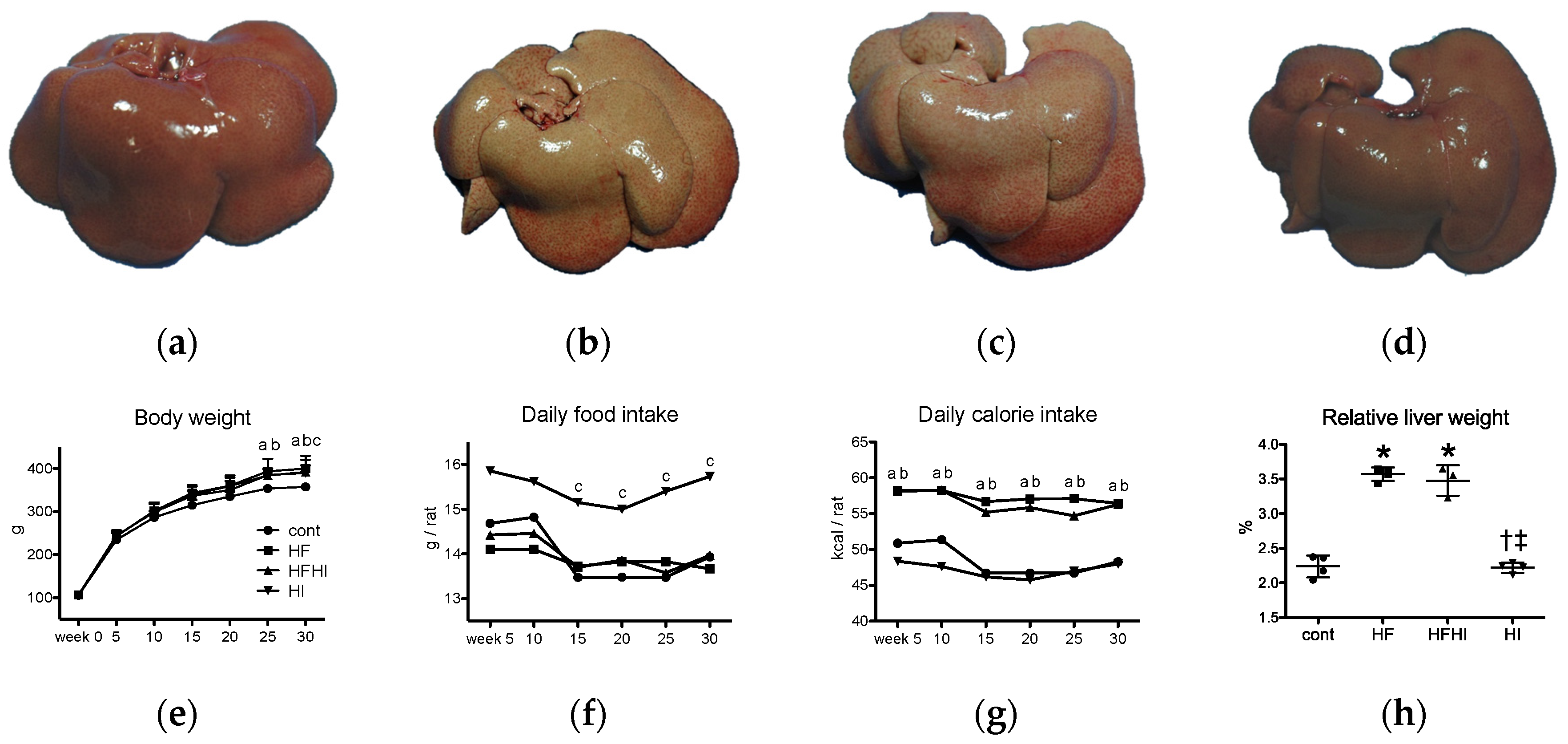
3.1.1. Clinical and Gross Findings
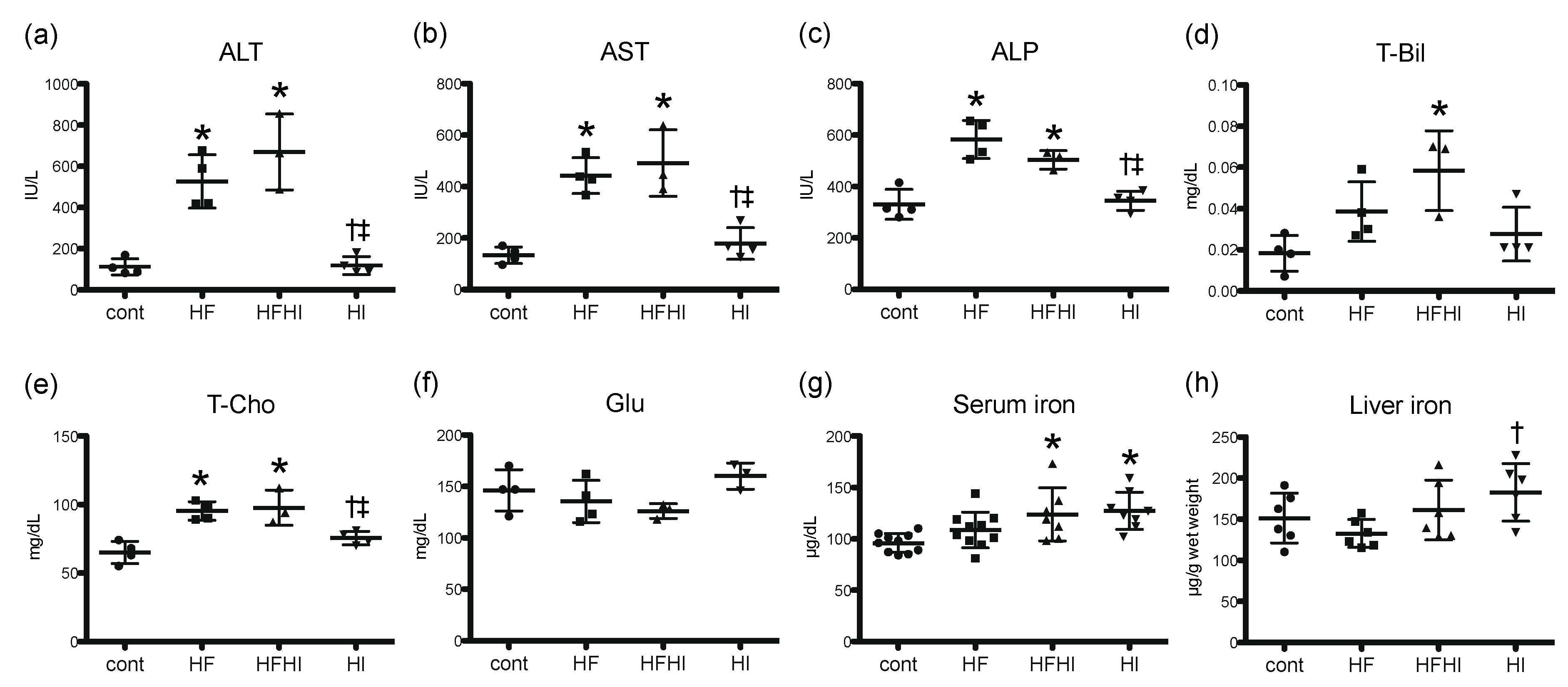
3.1.2. Blood Biochemical Findings
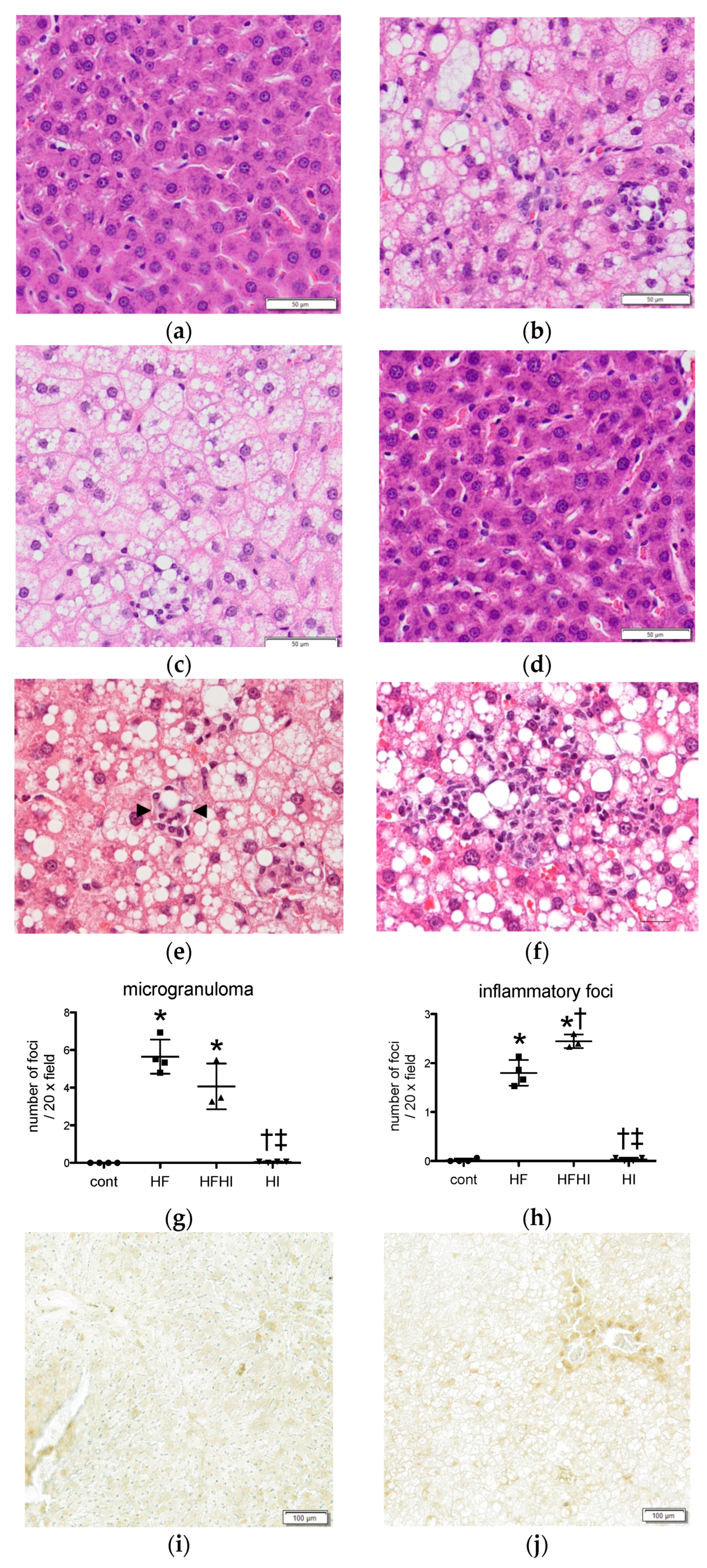
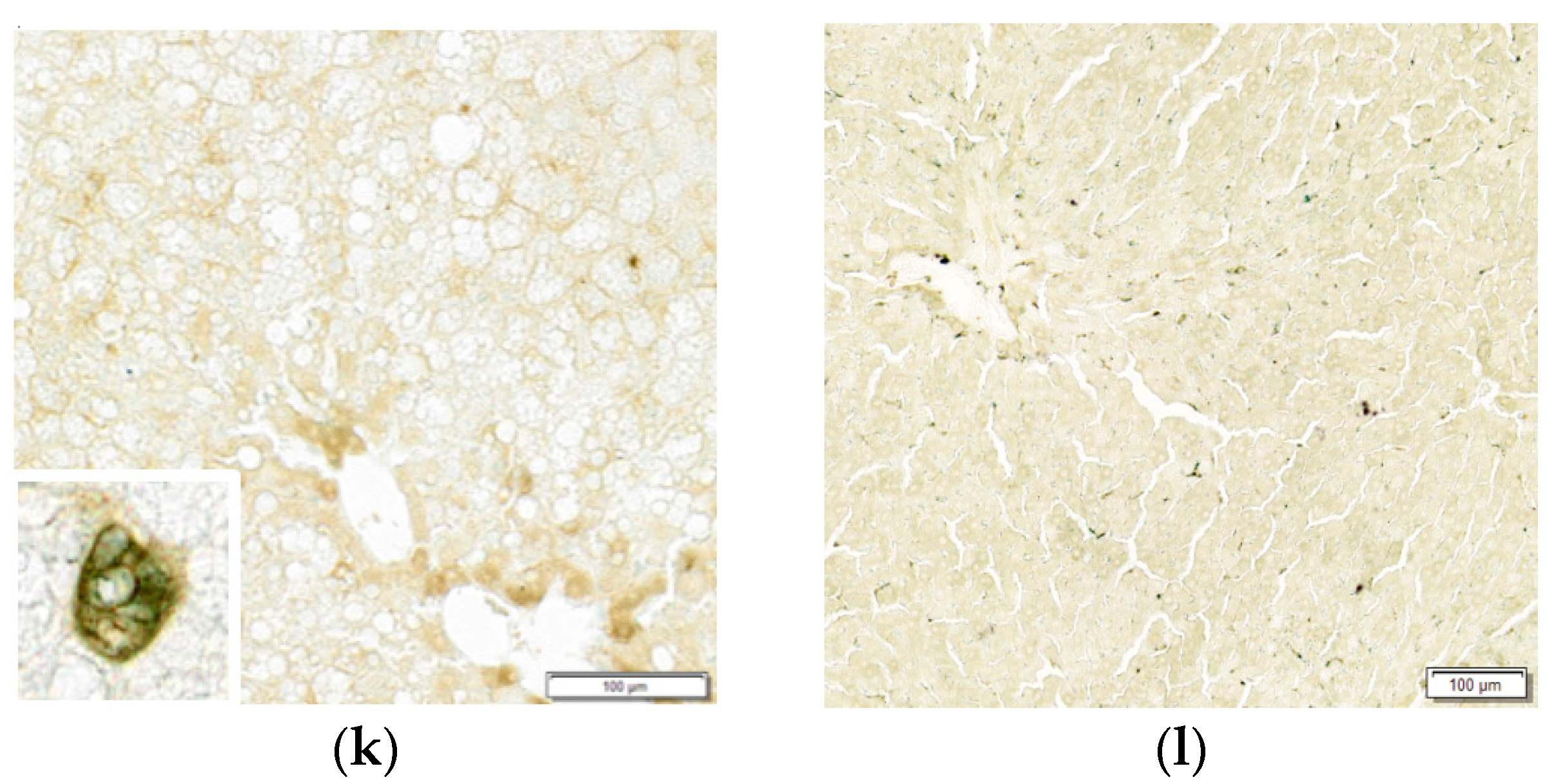
3.1.3. Histopathological Findings
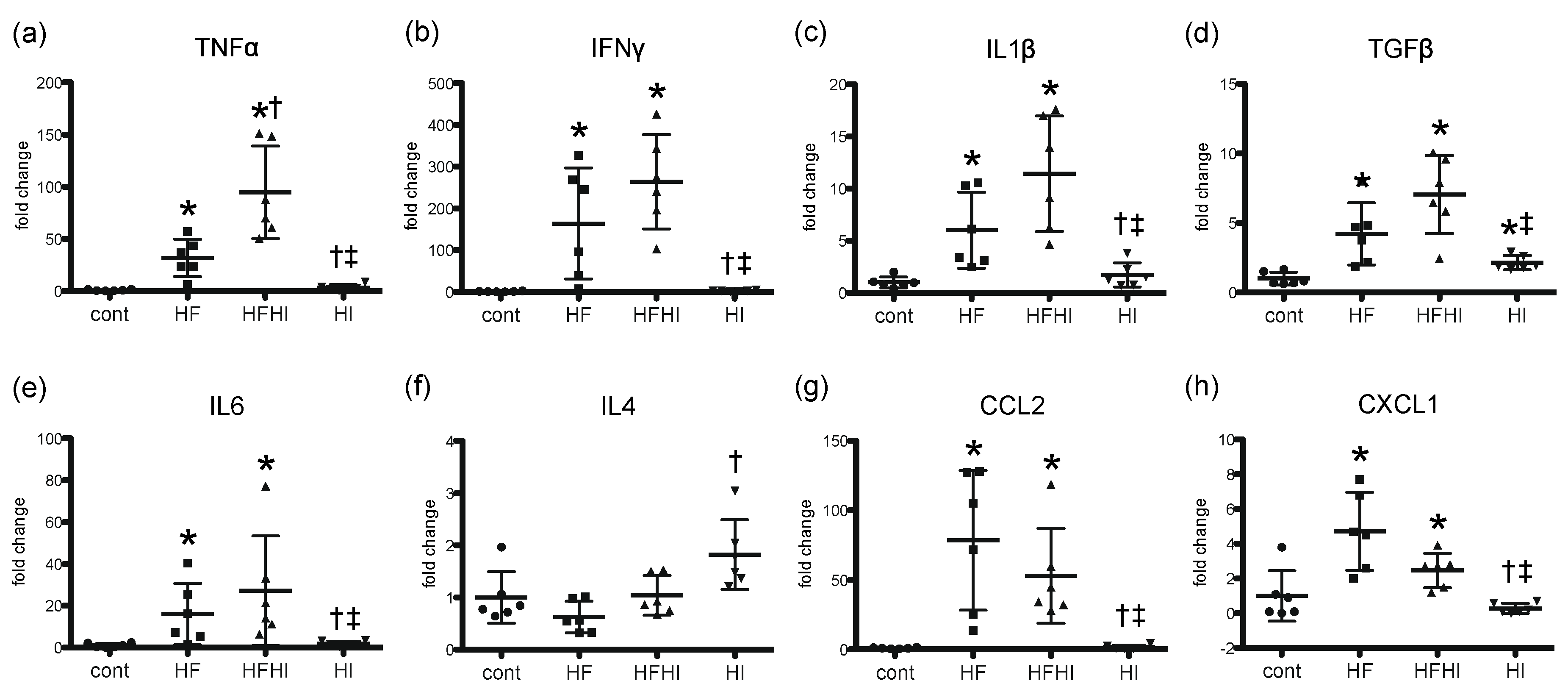
3.1.4. Expression Patterns of Inflammatory Cytokines
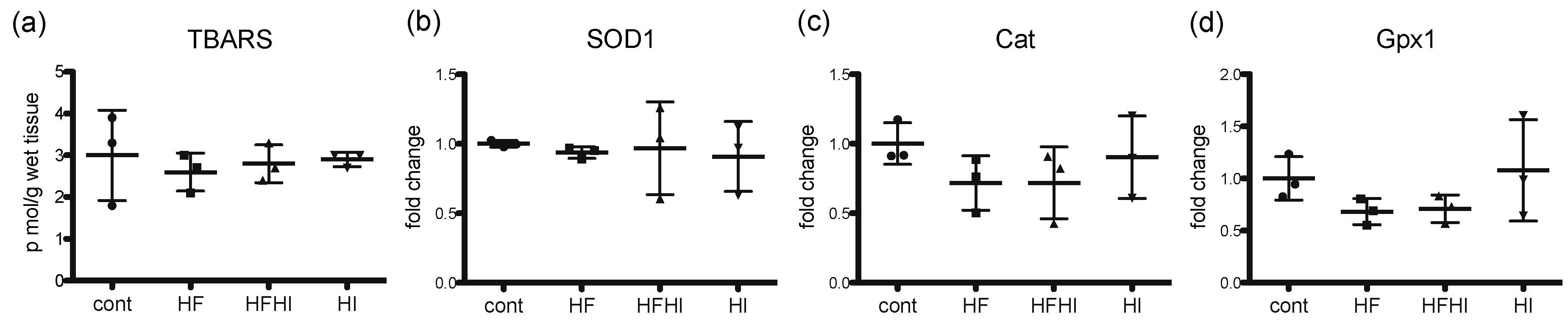
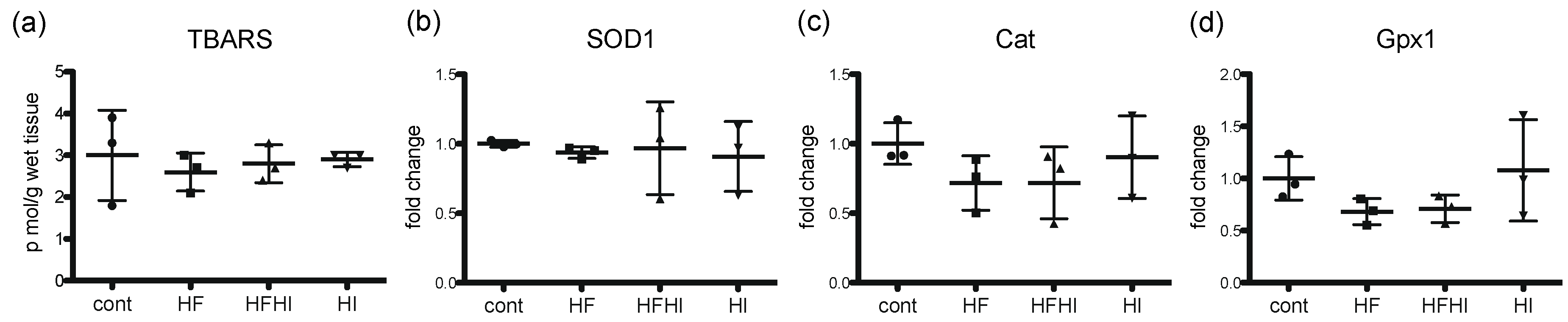
3.2. Changes in Oxidative Stress-Related Molecules
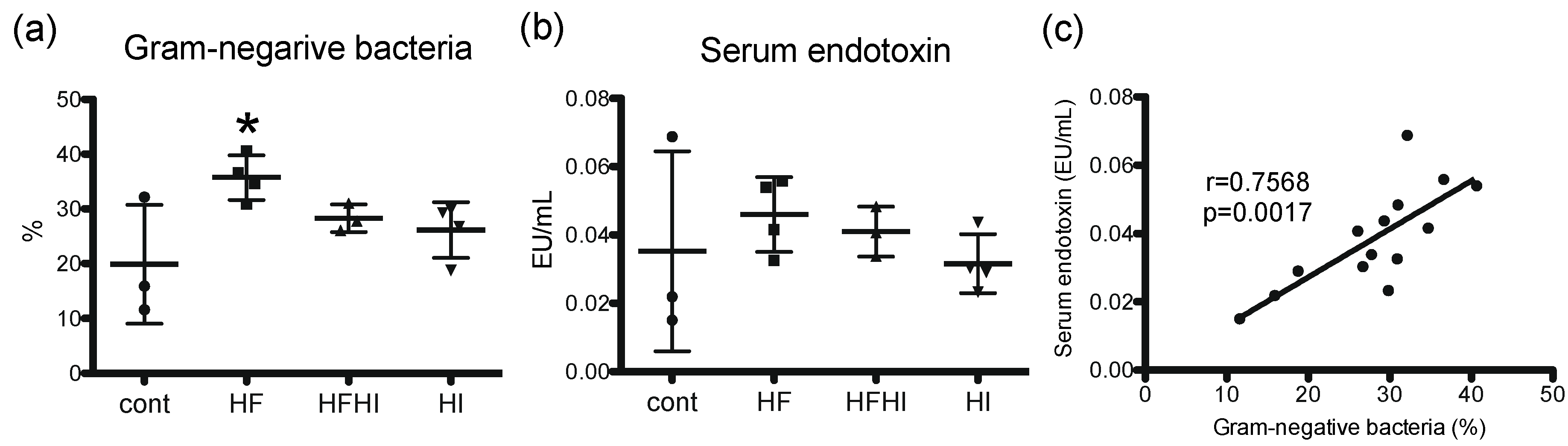
3.3. Changes in Gut Microbiota
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CLD | chronic liver disease |
| HCC | hepatocellular carcinoma |
| NAFLD | nonalcoholic fatty liver disease |
| ROS | reactive oxygen species |
| NASH | nonalcoholic steatohepatitis |
| HF | high-fat |
| HFHI | high-fat and high-iron |
| HI | high-iron |
| TBARS | thiobarbituric acid reactive substances |
| LPS | lipopolysaccharide |
| HE | hematoxylin and eosin |
| DAB | 3,3′-diaminobenzidine |
| RT-PCR | real-time reverse transcriptase polymerase chain reaction |
| ALT | alanine aminotransferase |
| AST | aspartate aminotransferase |
| ALP | alkaline phosphatase |
| T-Bil | total bilirubin |
| NTBI | non-transferrin-bound iron |
References
- Younossi, Z.M.; Otgonsuren, M.; Henry, L.; Venkatesan, C.; Mishra, A.; Erario, M.; Hunt, S. Association of nonalcoholic fatty liver disease (NAFLD) with hepatocellular carcinoma (HCC) in the United States from 2004 to 2009. Hepatology 2015, 62, 1723–1730. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Stepanova, M.; Afendy, M.; Fang, Y.; Younossi, Y.; Mir, H.; Srishord, M. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin. Gastroenterol. Hepatol. 2011, 9, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Piscaglia, F.; Svegliati-Baroni, G.; Barchetti, A.; Pecorelli, A.; Marinelli, S.; Tiribelli, C.; Bellentani, S.; Group, H.-N.I.S. Clinical patterns of hepatocellular carcinoma in nonalcoholic fatty liver disease: A multicenter prospective study. Hepatology 2016, 63, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Brissot, P.; Loreal, O. Iron metabolism and related genetic diseases: A cleared land, keeping mysteries. J. Hepatol. 2016, 64, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Munoz, M.; Garcia-Erce, J.A.; Remacha, A.F. Disorders of iron metabolism. Part 1: Molecular basis of iron homoeostasis. J. Clin. Pathol. 2011, 64, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Maras, J.S.; Maiwall, R.; Harsha, H.C.; Das, S.; Hussain, M.S.; Kumar, C.; Bihari, C.; Rastogi, A.; Kumar, M.; Trehanpati, N.; et al. Dysregulated iron homeostasis is strongly associated with multiorgan failure and early mortality in acute-on-chronic liver failure. Hepatology 2015, 61, 1306–1320. [Google Scholar] [CrossRef] [PubMed]
- Kessler, S.M.; Barghash, A.; Laggai, S.; Helms, V.; Kiemer, A.K. Hepatic hepcidin expression is decreased in cirrhosis and HCC. J. Hepatol. 2015, 62, 977–979. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.E.; Wilson, L.; Brunt, E.M.; Yeh, M.M.; Kleiner, D.E.; Unalp-Arida, A.; Kowdley, K.V.; Nonalcoholic Steatohepatitis Clinical Research, N. Relationship between the pattern of hepatic iron deposition and histological severity in nonalcoholic fatty liver disease. Hepatology 2011, 53, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Philippe, M.A.; Ruddell, R.G.; Ramm, G.A. Role of iron in hepatic fibrosis: One piece in the puzzle. World J. Gastroenterol. 2007, 13, 4746–4754. [Google Scholar] [CrossRef] [PubMed]
- Guyader, D.; Thirouard, A.S.; Erdtmann, L.; Rakba, N.; Jacquelinet, S.; Danielou, H.; Perrin, M.; Jouanolle, A.M.; Brissot, P.; Deugnier, Y. Liver iron is a surrogate marker of severe fibrosis in chronic hepatitis C. J. Hepatol. 2007, 46, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Sebastiani, G.; Tempesta, D.; Alberti, A. Hepatic iron overload is common in chronic hepatitis B and is more severe in patients coinfected with hepatitis D virus. J. Viral Hepat. 2012, 19, e170–e176. [Google Scholar] [CrossRef] [PubMed]
- Erhardt, A.; Maschner-Olberg, A.; Mellenthin, C.; Kappert, G.; Adams, O.; Donner, A.; Willers, R.; Niederau, C.; Haussinger, D. HFE mutations and chronic hepatitis C: H63D and C282Y heterozygosity are independent risk factors for liver fibrosis and cirrhosis. J. Hepatol. 2003, 38, 335–342. [Google Scholar] [CrossRef]
- Nelson, J.E.; Bhattacharya, R.; Lindor, K.D.; Chalasani, N.; Raaka, S.; Heathcote, E.J.; Miskovsky, E.; Shaffer, E.; Rulyak, S.J.; Kowdley, K.V. HFE C282Y mutations are associated with advanced hepatic fibrosis in Caucasians with nonalcoholic steatohepatitis. Hepatology 2007, 46, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Tung, B.Y.; Emond, M.J.; Bronner, M.P.; Raaka, S.D.; Cotler, S.J.; Kowdley, K.V. Hepatitis C, iron status, and disease severity: Relationship with HFE mutations. Gastroenterology 2003, 124, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Lambrecht, R.W.; Sterling, R.K.; Naishadham, D.; Stoddard, A.M.; Rogers, T.; Morishima, C.; Morgan, T.R.; Bonkovsky, H.L.; Group, H.-C.T. Iron levels in hepatocytes and portal tract cells predict progression and outcomes of patients with advanced chronic hepatitis C. Gastroenterology 2011, 140, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Nahon, P.; Sutton, A.; Rufat, P.; Ziol, M.; Thabut, G.; Schischmanoff, P.O.; Vidaud, D.; Charnaux, N.; Couvert, P.; Ganne-Carrie, N.; et al. Liver iron, HFE gene mutations, and hepatocellular carcinoma occurrence in patients with cirrhosis. Gastroenterology 2008, 134, 102–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, J.E.; Brunt, E.M.; Kowdley, K.V.; Nonalcoholic Steatohepatitis Clinical Research, N. Lower serum hepcidin and greater parenchymal iron in nonalcoholic fatty liver disease patients with C282Y HFE mutations. Hepatology 2012, 56, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Valenti, L.; Fracanzani, A.L.; Bugianesi, E.; Dongiovanni, P.; Galmozzi, E.; Vanni, E.; Canavesi, E.; Lattuada, E.; Roviaro, G.; Marchesini, G.; et al. HFE genotype, parenchymal iron accumulation, and liver fibrosis in patients with nonalcoholic fatty liver disease. Gastroenterology 2010, 138, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Maliken, B.D.; Nelson, J.E.; Klintworth, H.M.; Beauchamp, M.; Yeh, M.M.; Kowdley, K.V. Hepatic reticuloendothelial system cell iron deposition is associated with increased apoptosis in nonalcoholic fatty liver disease. Hepatology 2013, 57, 1806–1813. [Google Scholar] [CrossRef] [PubMed]
- Bugianesi, E.; Manzini, P.; D’Antico, S.; Vanni, E.; Longo, F.; Leone, N.; Massarenti, P.; Piga, A.; Marchesini, G.; Rizzetto, M. Relative contribution of iron burden, HFE mutations, and insulin resistance to fibrosis in nonalcoholic fatty liver. Hepatology 2004, 39, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Bugianesi, E.; Marzocchi, R.; Villanova, N.; Marchesini, G. Non-alcoholic fatty liver disease/non-alcoholic steatohepatitis (NAFLD/NASH): Treatment. Best Pract. Res. Clin. Gastroenterol. 2004, 18, 1105–1116. [Google Scholar] [CrossRef]
- Adams, L.A.; Crawford, D.H.; Stuart, K.; House, M.J.; St Pierre, T.G.; Webb, M.; Ching, H.L.; Kava, J.; Bynevelt, M.; MacQuillan, G.C.; et al. The impact of phlebotomy in nonalcoholic fatty liver disease: A prospective, randomized, controlled trial. Hepatology 2015, 61, 1555–1564. [Google Scholar] [CrossRef] [PubMed]
- Valenti, L.; Fracanzani, A.L.; Dongiovanni, P.; Rovida, S.; Rametta, R.; Fatta, E.; Pulixi, E.A.; Maggioni, M.; Fargion, S. A randomized trial of iron depletion in patients with nonalcoholic fatty liver disease and hyperferritinemia. World J. Gastroenterol. 2014, 20, 3002–3010. [Google Scholar] [CrossRef] [PubMed]
- Savard, C.; Tartaglione, E.V.; Kuver, R.; Haigh, W.G.; Farrell, G.C.; Subramanian, S.; Chait, A.; Yeh, M.M.; Quinn, L.S.; Ioannou, G.N. Synergistic interaction of dietary cholesterol and dietary fat in inducing experimental steatohepatitis. Hepatology 2013, 57, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Izawa, T.; Murakami, H.; Wijesundera, K.K.; Golbar, H.M.; Kuwamura, M.; Yamate, J. Inflammatory regulation of iron metabolism during thioacetamide-induced acute liver injury in rats. Exp. Toxicol. Pathol. 2014, 66, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Bondoc, A.; Golbar, H.M.; Pervin, M.; Katou-Ichikawa, C.; Tanaka, M.; Izawa, T.; Kuwamura, M.; Yamate, J. Participation of Tumor-Associated Myeloid Cells in Progression of Amelanotic Melanoma (RMM Tumor Line) in F344 Rats, with Particular Reference to MHC Class II- and CD163-Expressing Cells. Cancer Microenviron. 2017, 10, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Pervin, M.; Golbar, H.M.; Bondoc, A.; Izawa, T.; Kuwamura, M.; Yamate, J. Immunophenotypical characterization and influence on liver homeostasis of depleting and repopulating hepatic macrophages in rats injected with clodronate. Exp. Toxicol. Pathol. 2016, 68, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.E.; Klintworth, H.; Kowdley, K.V. Iron metabolism in Nonalcoholic Fatty Liver Disease. Curr. Gastroenterol. Rep. 2012, 14, 8–16. [Google Scholar] [CrossRef] [PubMed]
- George, D.K.; Goldwurm, S.; MacDonald, G.A.; Cowley, L.L.; Walker, N.I.; Ward, P.J.; Jazwinska, E.C.; Powell, L.W. Increased hepatic iron concentration in nonalcoholic steatohepatitis is associated with increased fibrosis. Gastroenterology 1998, 114, 311–318. [Google Scholar] [CrossRef]
- Malaguarnera, L.; Madeddu, R.; Palio, E.; Arena, N.; Malaguarnera, M. Heme oxygenase-1 levels and oxidative stress-related parameters in non-alcoholic fatty liver disease patients. J. Hepatol. 2005, 42, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, A. Mechanisms of iron hepatotoxicity. J. Hepatol. 2016, 65, 226–227. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Miyachi, H.; Tanaka, H.; Takeo, M.; Nakagawa, N.; Kobayashi, Y.; Iwasa, M.; Watanabe, S.; Takei, Y. Iron overload is associated with hepatic oxidative damage to DNA in nonalcoholic steatohepatitis. Cancer Epidemiol. Prev. Biomark. 2009, 18, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- She, H.; Xiong, S.; Lin, M.; Zandi, E.; Giulivi, C.; Tsukamoto, H. Iron activates NF-kappaB in Kupffer cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G719–G726. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H. Iron regulation of hepatic macrophage TNFalpha expression. Free Radic. Biol. Med. 2002, 32, 309–313. [Google Scholar] [CrossRef]
- Xiong, S.; She, H.; Sung, C.K.; Tsukamoto, H. Iron-dependent activation of NF-kappaB in Kupffer cells: A priming mechanism for alcoholic liver disease. Alcohol 2003, 30, 107–113. [Google Scholar] [CrossRef]
- Del Chierico, F.; Nobili, V.; Vernocchi, P.; Russo, A.; Stefanis, C.; Gnani, D.; Furlanello, C.; Zandona, A.; Paci, P.; Capuani, G.; et al. Gut microbiota profiling of pediatric nonalcoholic fatty liver disease and obese patients unveiled by an integrated meta-omics-based approach. Hepatology 2017, 65, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Hui, S.T.; Parks, B.W.; Org, E.; Norheim, F.; Che, N.; Pan, C.; Castellani, L.W.; Charugundla, S.; Dirks, D.L.; Psychogios, N.; et al. The genetic architecture of NAFLD among inbred strains of mice. Elife 2015, 4, e05607. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Wang, H.; Yuan, F.; Li, N.; Huang, Q.; He, L.; Wang, L.; Liu, Z. Perilla Oil Has Similar Protective Effects of Fish Oil on High-Fat Diet-Induced Nonalcoholic Fatty Liver Disease and Gut Dysbiosis. BioMed Res. Int. 2016, 2016, 9462571. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Sanduzzi Zamparelli, M.; Compare, D.; Coccoli, P.; Rocco, A.; Nardone, O.M.; Marrone, G.; Gasbarrini, A.; Grieco, A.; Nardone, G.; Miele, L. The Metabolic Role of Gut Microbiota in the Development of Nonalcoholic Fatty Liver Disease and Cardiovascular Disease. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Axling, U.; Olsson, C.; Xu, J.; Fernandez, C.; Larsson, S.; Strom, K.; Ahrne, S.; Holm, C.; Molin, G.; Berger, K. Green tea powder and Lactobacillus plantarum affect gut microbiota, lipid metabolism and inflammation in high-fat fed C57BL/6J mice. Nutr. Metab. 2012, 9, 105. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Wang, X.; Liu, P.; Wei, R.; Chen, W.; Rajani, C.; Hernandez, B.Y.; Alegado, R.; Dong, B.; Li, D.; et al. Distinctly altered gut microbiota in the progression of liver disease. Oncotarget 2016, 7, 19355–19366. [Google Scholar] [CrossRef] [PubMed]
- Hofmeister, A.E.; Berger, S.; Buckel, W. The iron-sulfur-cluster-containing l-serine dehydratase from Peptostreptococcus asaccharolyticus. Stereochemistry of the deamination of l-threonine. Eur. J. Biochem. 1992, 205, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Tessitore, A.; Mastroiaco, V.; Vetuschi, A.; Sferra, R.; Pompili, S.; Cicciarelli, G.; Barnabei, R.; Capece, D.; Zazzeroni, F.; Capalbo, C.; et al. Development of hepatocellular cancer induced by long term low fat-high carbohydrate diet in a NAFLD/NASH mouse model. Oncotarget 2017, 8, 53482–53494. [Google Scholar] [CrossRef] [PubMed]
- Asgharpour, A.; Cazanave, S.C.; Pacana, T.; Seneshaw, M.; Vincent, R.; Banini, B.A.; Kumar, D.P.; Daita, K.; Min, H.K.; Mirshahi, F.; et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J. Hepatol. 2016, 65, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Dietary Supplement Fact Sheet: Iron—Health Professional Fact Sheet. Available online: https://ods.od.nih.gov/factsheets/Iron-HealthProfessional/.

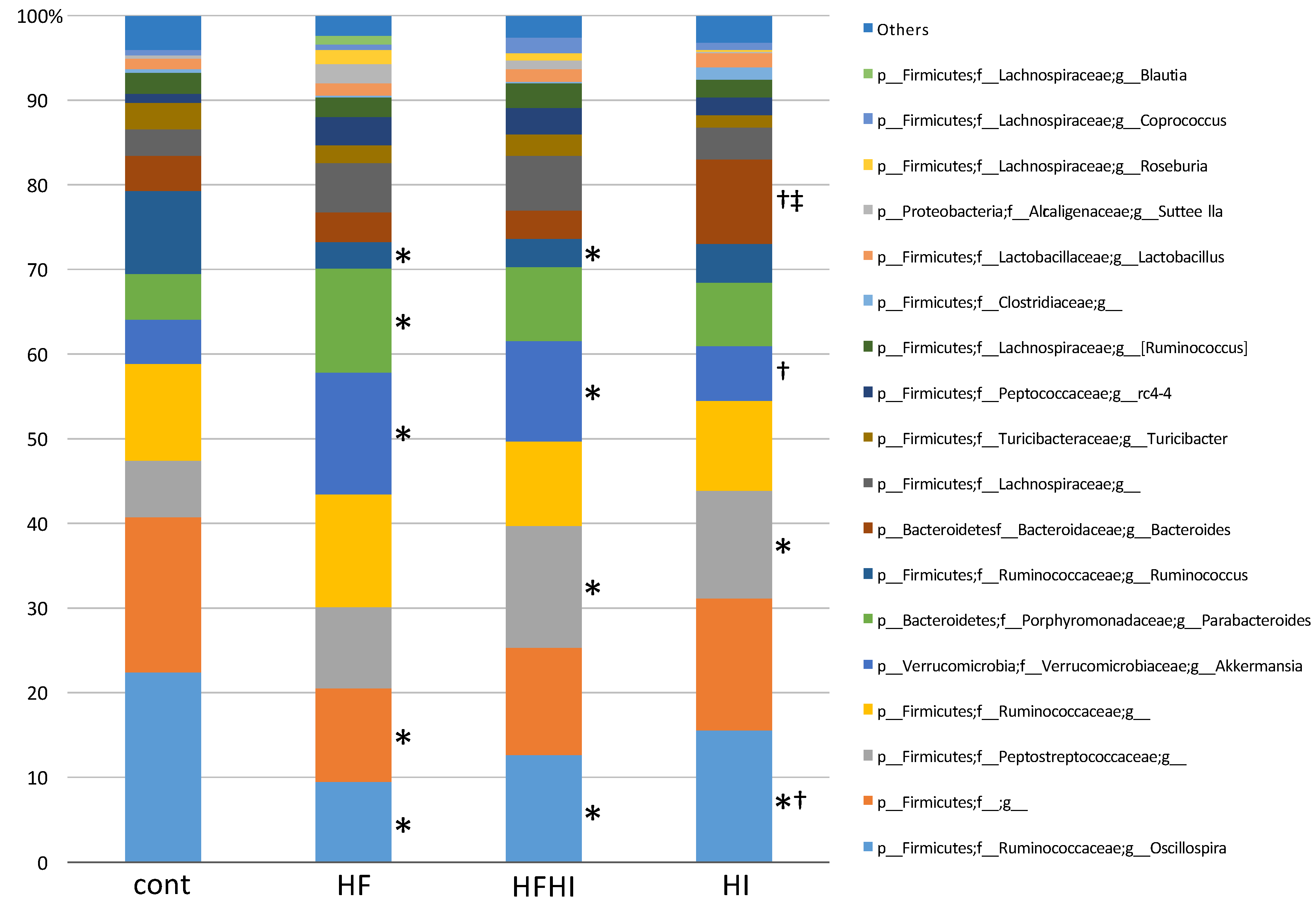

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
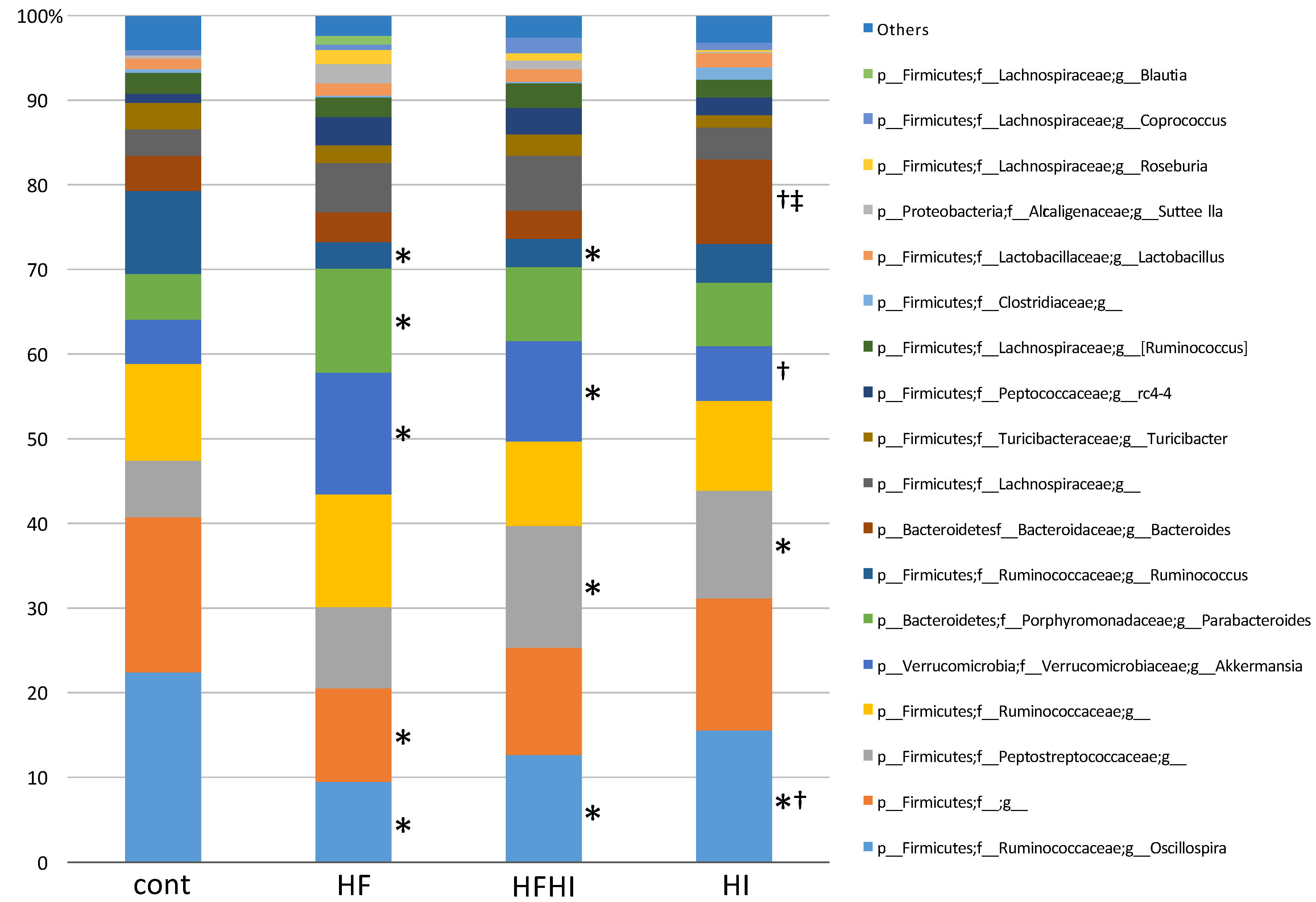{kind=link}
| Diet | Cont | HF | HFHI | HI |
|---|---|---|---|---|
| CE-2 * (g %) | 100 | 88 | 85 | 97 |
| cocoa butter (g %) | 0 | 11 | 11 | 0 |
| cholesterol (g %) | 0 | 1 | 1 | 0 |
| iron (III) citrate (g %) | 0 | 0 | 2.9 | 2.9 |
| iron content (g %) | 0.03 | 0.03 | 0.50 | 0.51 |
| protein (g %) | 24.7 | 21.7 | 21.0 | 21.7 |
| fat (g %) | 4.8 | 16.2 | 16.0 | 4.2 |
| carbohydrate (g %) | 51.3 | 45.1 | 43.6 | 45.1 |
| protein (kcal %) | 28.5 | 21.0 | 20.9 | 28.5 |
| fat (kcal %) | 12.3 | 35.3 | 35.8 | 12.3 |
| carbohydrate (kcal %) | 59.2 | 43.7 | 43.3 | 59.2 |
| total calories (kcal/100 g diet) | 346.5 | 412.9 | 402.9 | 309.4 |
| Gene Name | Abbreviation | Probe Assay ID |
|---|---|---|
| Tumor necrosis factor-α | TNFα | Rn01525859_g1 |
| Interferon-γ | IFNγ | Rn00594078_m1 |
| Interleukin-1β | IL1β | Rn00580432_m1 |
| Transforming growth factor-β | TGFβ | Rn00572010_m1 |
| Interleukin-6 | IL6 | Rn01410330_m1 |
| Interleukin-4 | IL4 | Rn01456866_m1 |
| Chemokine (C-X-C motif) ligand 1 | CXCL1 | Rn00578225_m1 |
| Chemokine (C-C motif) ligand 2 | CCL2 | Rn00580555_m1 |
| Superoxide dismutase 1, soluble | SOD1 | Rn00566938_m1 |
| Catalase | Cat | Rn00560930_m1 |
| Glutathione peroxidase 1 | GPx1 | Rn00577994_g1 |
| 18S ribosomal RNA | 18S rRNA | Hs99999901_s1 |
| Bacteria | Cont | HF | HFHI | HI |
|---|---|---|---|---|
| Bacteroidetes | 9.7 | 15.9 | 12.3 | 17.4 |
| Bacteroidaceae | 4.1 | 3.5 | 3.4 | 9.9 |
| Bacteroides | 4.1 | 3.5 | 3.4 | 9.9 †,‡ |
| Porphyromonadaceae | 5.6 | 12.3 | 8.9 | 7.5 |
| Parabacteroides | 5.6 | 12.3 * | 8.9 | 7.5 |
| Firmicutes | 84.3 | 67.3 | 74.5 | 75.5 |
| Clostridiaceae | 0.9 | 0.6 | 0.5 | 1.6 |
| Lachnospiraceae | 6.9 | 12.0 | 12.4 | 7.3 |
| Blautia | 0.0 | 1.0 | 0.0 | 0.0 |
| Coprococcus | 0.5 | 0.7 | 1.8 | 0.8 |
| Roseburia | 0.1 | 1.7 | 0.9 | 0.2 |
| Lactobacillaceae | 1.2 | 1.4 | 1.6 | 1.7 |
| Lactobacillus | 1.2 | 1.4 | 1.6 | 1.7 |
| Peptococcaceae | 1.2 | 3.3 | 3.3 | 2.3 |
| Peptostreptococcaceae | 6.5 | 9.8 | 14.3 * | 12.7 * |
| Ruminococcaceae | 44.1 | 26.2 | 26.2 | 30.9 |
| Oscillospira | 22.5 | 9.6 * | 12.7 * | 15.5 *,† |
| Ruminococcus | 9.8 | 3.2 * | 3.3 * | 4.6 |
| Turicibacteraceae | 3.1 | 2.2 | 2.5 | 1.6 |
| Turicibacter | 3.1 | 2.2 | 2.5 | 1.6 |
| Proteobacteria | 0.4 | 2.3 | 1.0 | 0.2 |
| Alcaligenaceae | 0.3 | 2.3 | 1.0 | 0.1 |
| Sutterella | 0.3 | 2.3 | 1.0 | 0.1 |
| Verrucomicrobia | 5.1 | 14.2 | 11.7 | 6.6 |
| Verrucomicrobiaceae | 5.1 | 14.2 | 11.7 | 6.6 |
| Akkermansia | 5.1 | 14.2 * | 11.7 * | 6.6 † |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atarashi, M.; Izawa, T.; Miyagi, R.; Ohji, S.; Hashimoto, A.; Kuwamura, M.; Yamate, J. Dietary Iron Supplementation Alters Hepatic Inflammation in a Rat Model of Nonalcoholic Steatohepatitis. Nutrients 2018, 10, 175. https://doi.org/10.3390/nu10020175
Atarashi M, Izawa T, Miyagi R, Ohji S, Hashimoto A, Kuwamura M, Yamate J. Dietary Iron Supplementation Alters Hepatic Inflammation in a Rat Model of Nonalcoholic Steatohepatitis. Nutrients. 2018; 10(2):175. https://doi.org/10.3390/nu10020175
Chicago/Turabian StyleAtarashi, Machi, Takeshi Izawa, Rena Miyagi, Shoko Ohji, Ai Hashimoto, Mitsuru Kuwamura, and Jyoji Yamate. 2018. "Dietary Iron Supplementation Alters Hepatic Inflammation in a Rat Model of Nonalcoholic Steatohepatitis" Nutrients 10, no. 2: 175. https://doi.org/10.3390/nu10020175