Effects of an Acute Exercise Bout on Serum Hepcidin Levels


 , ,
, ,
Abstract
:
1. Introduction
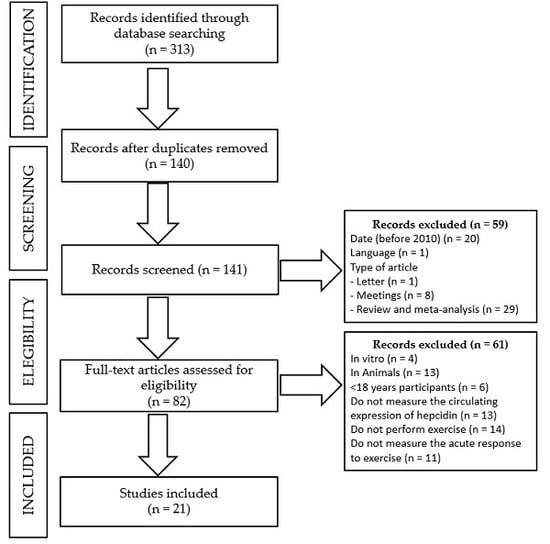
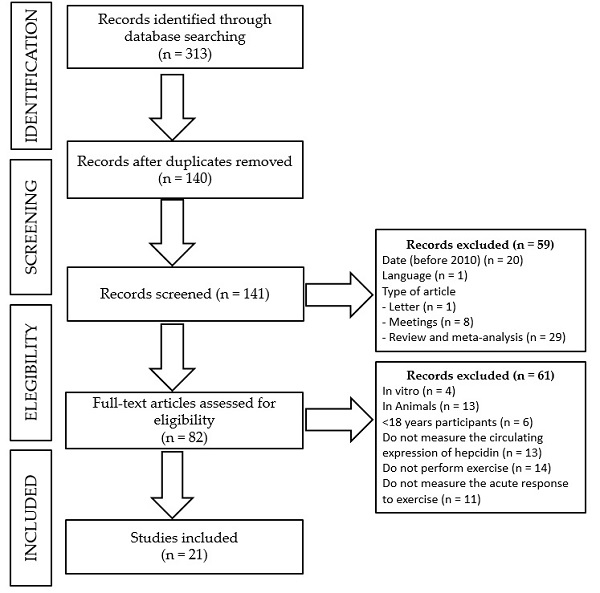
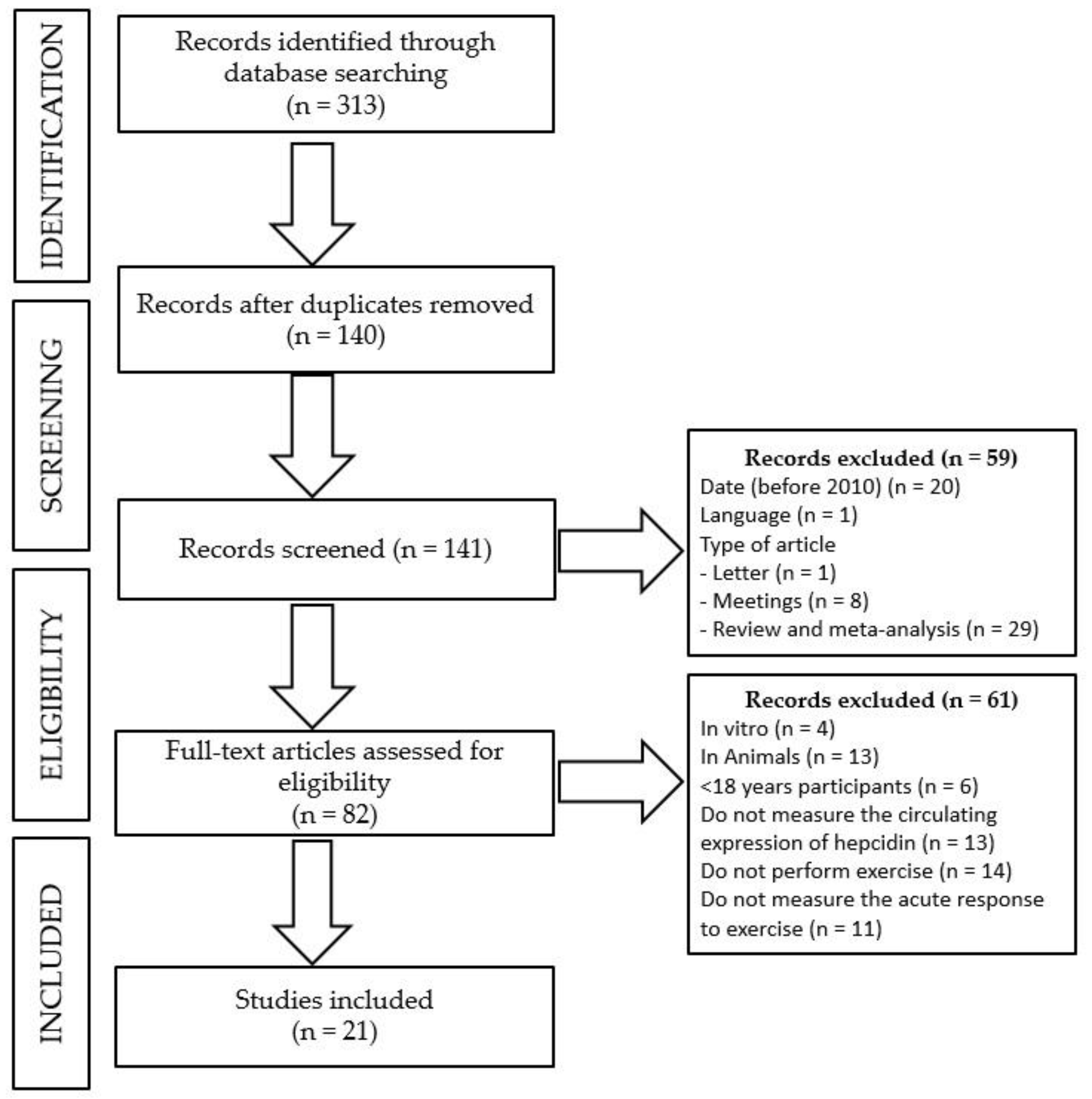
2. Materials and Methods
- -
- Date of publication: before 2010.
- -
- Language: publication in other language than English or Spanish.
- -
- Type of manuscript: others than experimental studies, such as editorials, letters to the editor, congress or meetings abstracts, reviews, or meta-analyses.
- -
- Type of study: other studies than those performed in an adult population (>18 years old) in which serum hepcidin had been analyzed in response to an acute exercise bout, such as in vitro or in vivo studies in animals, studies in children or an adolescent population, or studies in which serum hepcidin was either not measured or reported in response to an acute exercise bout.
3. Results
3.1. Population Characteristics
3.2. Measurements of Serum Hepcidin Levels
3.3. Serum Hepcidin Levels in Response to Exercise
3.3.1. Effect of Exercise Type on Serum Hepcidin Levels
3.3.2. Effect of Exercise Intensity on Serum Hepcidin Levels
3.3.3. Effect of Exercise Duration on Serum Hepcidin Levels
3.3.4. Effect of Diet and Supplementation on the Response of Serum Hepcidin Levels to Exercise
3.3.5. Effect of Hypoxia on the Response of Circulating Hepcidin to Exercise
4. Discussion
4.1. Effect of Exercise Type, Intensity, and Duration on the Circulating Expression of Hepcidin
4.2. Effect of Diet and Supplementation on the Response of Circulating Hepcidin to Exercise
4.3. A Mechanistic Approach to Exercise-Induced Hepcidin Expression
4.3.1. Iron Status
4.3.2. Inflammation
4.3.3. Hypoxia
4.3.4. Oral Contraceptives
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Umbreit, J. Iron deficiency: A concise review. Am. J. Hematol. 2005, 78, 225–331. [Google Scholar] [CrossRef] [PubMed]
- De Benois, B.; McLean, E.; Egli, I.; Cogswell, M. Worldwide Prevalence of Anaemia 1993–2005. WHO Database on Anaemia; World Health Organization: Geneve, Switzerland, 2008. [Google Scholar]
- Gropper, S.S.; Blessing, D.; Dunham, K.; Barksdale, M. Iron status of female collegiate athletes involved in different sports. Biol. Trace Elem. Res. 2006, 109, 1–14. [Google Scholar] [CrossRef]
- Woolf, K.; St Thomas, M.M.; Hahn, N.; Vaughan, L.A.; Carlson, A.G.; Hinton, P. Iron status in highly active and sedentary young women. Int. J. Sport Nutr. Exerc. Metab. 2009, 1, 519–535. [Google Scholar] [CrossRef]
- Zoller, H.; Vogel, W. Iron supplementation in athletes-first do no harm. Nutrition 2004, 20, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, L.M.; Hinton, P.S. Prevalence of iron deficiency with and without anemia in recreationally active men and women. J. Am. Diet. Assoc. 2005, 105, 975–978. [Google Scholar] [CrossRef] [PubMed]
- DellaValle, D.M.; Haas, J.D. Impact of iron depletion without anemia on performance in trained endurance athletes at the beginning of a training season: A study of female collegiate rowers. Int. J. Sport Nutr. Exerc. Metab. 2011, 21, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Lukaski, H.C. Vitamin and mineral status: Effects on physical performance. Nutrition 2004, 20, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Hinton, P.S. Iron and the endurance athlete. Appl. Physiol. Nutr. Metab. 2014, 39, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- DellaValle, D.M. Iron supplementation for female athletes: Effects on iron status and performance outcomes. Curr. Sports Med. Rep. 2013, 12, 234–239. [Google Scholar] [CrossRef] [PubMed]
- DellaValle, D.M.; Haas, J.D. Iron supplementation improves energetic efficiency in iron-depleted female rowers. Med. Sci. Sports Exerc. 2014, 46, 1204–1215. [Google Scholar] [CrossRef] [PubMed]
- Garvican, L.A.; Lobigs, L.; Telford, R.; Fallon, K.; Gore, C.J. Haemoglobin mass in an anaemic female endurance runner before and after iron supplementation. Int. J. Sports Physiol. Perform. 2011, 6, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Latunde-Dada, G.O. Iron metabolism in athletes—Achieving a gold standard. Eur. J. Haematol. 2013, 90, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Brownlie, T.T.; Utermohlen, V.; Hinton, P.S.; Giordano, C.; Haas, J.D. Marginal iron deficiency without anemia impairs aerobic adaptation among previously untrained women. Am. J. Clin. Nutr. 2002, 75, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Brownlie, T.T.; Utermohlen, V.; Hinton, P.S.; Haas, J.D. Tissue iron deficiency without anemia impairs adaptation in endurance capacity after aerobic training in previously untrained women. Am. J. Clin. Nutr. 2004, 79, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Jomova, K.; Rhodes, C.; Kuča, K.; Musílek, K. Redox- and non-redoxmetal-induced formation of free radicals and their role in human disease. Arch. Toxicol. 2016, 90, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Domínguez, R.; Garnacho-Castaño, M.V.; Maté-Muñoz, J.L. Effect of hepcidin on iron metabolism in athletes. Nutr. Hosp. 2014, 30, 1218–1231. [Google Scholar] [PubMed]
- Food and Nutrition Board. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc; National Academy Press: Washington, DC, USA, 2001; pp. 290–393. [Google Scholar]
- Peeling, P.; Dawson, B.; Goodman, C.; Landers, G.; Trinder, D. Athletic induced iron deficiency: New insights into the role of inflammation, cytokines and hormones. Eur. J. Appl. Physiol. 2008, 103, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Reeder, B.; Wilson, M. The effects of pH on the mechanism of hydrogen peroxide and lipid hydroperoxide consumption by myoglobin: A role for the protonated ferryl species. Free Radic. Biol. Med. 2001, 30, 1311–1318. [Google Scholar] [CrossRef]
- Yusof, A.; Leithauser, R.M.; Roth, H.J.; Finkernagel, H.; Wilson, M.T.; Beneke, R. Exercise-induced hemolysis is caused by protein modification and most evident during the early phase of an ultraendurance race. J. Appl. Physiol. 2007, 102, 582–586. [Google Scholar] [CrossRef] [PubMed]
- Telford, R.D.; Sly, G.J.; Hahn, A.G.; Cunningham, R.B.; Bryant, C.; Smith, J.A. Footstrike is the major cause of hemolysis during running. J. Appl. Physiol. 2003, 94, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Sim, M.; Dawson, B.; Landers, G.; Swinkels, D.W.; Tjasma, H.; Trinder, D.; Peeling, P. Effect of exercise modality and intensity on postexercise interleukin-6 and hepcidina levels. Int. J. Sports Nutr. Exerc. Metab. 2013, 23, 178–186. [Google Scholar] [CrossRef]
- Pattini, A.; Schena, F.; Guidi, G.C. Serum ferritin and serum iron after cross-country and roller sky endurance races. Eur. J. Appl. Physiol. Occpat. Phyisiol. 1990, 61, 55–60. [Google Scholar] [CrossRef]
- Babic, Z.; Papa, B.; Sikirika-Bosnjakovic, M.; Prkacin, I.; Misigoj-Durakovic, M.; Katicic, M. Occult gastrointestinal bleeding in rugby players. J. Sports Med. Phys. Fit. 2011, 41, 399–402. [Google Scholar]
- Lampre, J.W.; Slavin, J.L.; Apple, F.S. Iron status of active women and effect of running a marathon on bowel function and gastrointestinal blood-loss. Int. J. Sport Med. 1991, 12, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Lopes, T.R.; Kirsztajn, G.M. Renal analysis in 75 km ultra-marathon participants. Acta Paul. Enferm. 2009, 22, 487–489. [Google Scholar] [CrossRef]
- Roecker, L.; Meier-Buttermilch, R.; Bretchel, L.; Nemeth, E.; Ganz, T. Iron-regulatory protein hepcidin is increased in female athletes after a marathon. Eur. J. Appl. Physiol. 2005, 95, 569–571. [Google Scholar] [CrossRef] [PubMed]
- Troadec, M.B.; Lainé, F.; Daniel, V.; Rochcongar, P.; Ropert, M.; Cabillic, F.; Perrin, M.; Morcet, J.; Loréal, O.; Olbina, G.; et al. Daily regulation of serum and urinary hepcidin is not influenced by submaximal cycling exercise in humans with normal iron metabolism. Eur. J. Appl. Physiol. 2010, 106, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Le Blanc, S.; Garrick, M.D.; Arredondo, M. Heme carrier protein 1 transports heme and is involved in heme-Fe metabolism. Am. J. Physiol. Cell Physiol. 2012, 302, 1780–1785. [Google Scholar] [CrossRef] [PubMed]
- McKie, A.T.; Barrow, D.; Latunde-Dada, G.O.; Rolfs, A.; Sager, G.; Mudaly, E.; Mudaly, M.; Richardson, C.; Barlow, D.; Bomford, A.; et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 2001, 291, 1755–1759. [Google Scholar] [CrossRef] [PubMed]
- Gunshin, H.; Mackenzie, B.; Berger, U.V.; Gunshin, Y.; Romero, M.F.; Boron, W.F.; Nussberger, S.; Gollan, J.L.; Hediger, M.A. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 1997, 388, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Donker, A.E.; Raymakers, R.A.P.; Vlasveld, L.T.; van Barneveld, T.; Terink, R.; Dors, N.; Brons, P.P.; Knoers, N.V.; Swinkels, D.W. Practice guidelines for the diagnosis and management of microcytic anemias due to genetic disorders of iron metabolism or heme synthesis. Blood 2014, 123, 3873–3886. [Google Scholar] [CrossRef] [PubMed]
- Munro, H.N.; Linder, M.C. Ferritin: Structure, biosynthesis, and role in iron metabolism. Physiol. Rev. 1978, 58, 317–396. [Google Scholar] [CrossRef] [PubMed]
- Donovan, A.; Brownlie, A.; Zhou, Y.; Shepard, J.; Pratt, S.J.; Moynihan, J.; Paw, B.H.; Drejer, A.; Barut, B.; Zapata, A.; et al. Positional cloning of zebrafish ferroportin 1 identifies a conserved vertebrate iron exporter. Nature 2000, 403, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Yeh, K.Y.; Yeh, M.; Glass, J. Interactions between ferroportin and hephaestin in rat enterocytes are reduced after iron ingestion. Gastroenterology 2011, 141, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Cherukuri, S.; Potla, R.; Sarkar, J.; Nurko, S.; Harris, Z.L.; Fox, P.L. Unexpected role of ceruloplasmin in intestinal iron absorption. Cell Metab. 2005, 2, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Systemic iron homeostasis. Physiol. Rev. 2013, 93, 1721–1741. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Hepcidin and iron metabolism, 10 years later. Blood 2012, 117, 4425–4433. [Google Scholar] [CrossRef] [PubMed]
- Krause, A.; Neitz, S.; Magert, H.J.; Schulz, A.; Forssmann, W.G.; Schulz-Knappe, P.; Adermann, K. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 2000, 480, 147–150. [Google Scholar] [CrossRef]
- Park, C.H.; Valore, E.V.; Waring, A.J.; Ganz, T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J. Biol. Chem. 2001, 276, 7806–7810. [Google Scholar] [CrossRef] [PubMed]
- Brasse-Lagnel, C.; Karim, Z.; Letteron, P.; Bekri, S.; Bado, A.; Beaumont, C. Intestinal DMT1 cotransporter is down-regulated by hepcidin via proteasome internalization and degradation. Gastroenterology 2011, 140, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- Barrios, Y.; Espinoza, M.; Barón, M.A. Pro-hepcidin, its relationship with iron metabolism and inflammation indicators in hemodialyzed patients, with or without recombinant erythropoietin treatment. Nutr. Hosp. 2010, 25, 555–560. [Google Scholar] [PubMed]
- Kroot, J.C.; Tjalsma, H.; Fleming, R.; Swinkels, D.W. Hepcidin in human iron disorders: Diagnostic implications. Clin. Chem. 2011, 57, 1650–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolas, G.; Bennoun, M.; Devaux, I.; Beaumont, C.; Grandchamp, B.; Kahn, A.; Vaulont, S. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc. Natl. Acad. Sci. USA 2001, 98, 8780–8785. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Investig. 2004, 113, 1271–1276. [Google Scholar] [CrossRef] [PubMed]
- Jonker, F.A.; Calis, J.C.; Phiri, K.; Kraaijenhagen, R.J.; Brabin, B.J.; Faragher, B.; Wiegerink, E.T.; Tjalsma, H.; Swinkels, D.W.; van Hensbroek, M.B. Low hepcidin levels in severely anemic Malawian children with high incidence of infectious diseases and bone marrow iron deficiency. PLoS ONE 2013, 8, e78964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruszewski, M. Labile iron pool: The main determinant of cellular response to oxidative stress. Mutat. Res. 2003, 531, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Bresgen, N.; Eckl, P. Oxidative stress and the homeodynamics of iron metabolism. Biomolecules 2015, 5, 808–847. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Chauvet, C.; Viatte, L.; Danan, J.L.; Bigard, X.; Beaumont, C.; Kahn, A.; Vaulont, S. The gene encoding the iron regulatory peptide hepcidin is regulated by anaemia, hypoxia and inflammation. J. Clin. Investig. 2002, 110, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Antosiewicz, J.; Ziolkowski, W.; Kaczor, J.J.; Herman-Antosiewicz, A. Tumor necrosis factor-alpha-induced reactive oxygen species formation is mediated by JNK1-dependent ferritin degradation. Radic. Biol. Med. 2007, 43, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Borkowska, A.; Sielicka-Dudzin, A.; Herman-Antosiewicz, A.; Halon, M.; Wozniak, M.; Antosiewicz, J. P66Shc mediated ferritin degradation—A novel mechanism of ROS formation. Free Radic. Biol. Med. 2011, 51, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Villarino, A.V.; Huang, E.; Hunter, C.A. Understanding the pro- and anti-inflammatory properties of IL-27. J. Immunol. 2004, 173, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Wallberg, L.; Mattsson, C.M.; Enqvist, J.K.; Ekblom, B. Plasma IL-6 concentration during ultra-endurance exercise. Eur. J. Appl. Physiol. 2011, 111, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Ganz, T. The role of hepcidin in iron metabolism. Acta Haematol. 2009, 122, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Mainous, A.G.; Diaz, V.A. Relation of serum ferritin level to cardiovascular fitness among young men. Am. J. Cardiol. 2009, 103, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Peeling, P.; Dawson, B.; Goodman, C.; Landers, G.; Wiegerinck, E.T.; Swinkels, D.W.; Trinder, D. Effects of exercise on hepcidin response and iron metabolism during recovery. Int. J. Sport Nutr. Exerc. Metab. 2009, 19, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Badenhorst, C.E.; Dawson, W.; Goodman, C.; Sim, M.; Cox, G.R.; Gore, C.J.; Tjalsma, H.; Swinkels, D.W.; Peeling, P. Influence of post-exercise hypoxic exposure on hepcidina response in athletes. Eur. J. Appl. Physiol. 2014, 114, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Badenhorst, C.E.; Dawson, B.; Cox, G.R.; Laarakkers, C.M.; Swinkels, D.W.; Peeling, P. Timing of post-exercise carbohydrate ingestion: Influence on IL-6 and hepcidin responses. Eur. J. Appl. Physiol. 2015, 115, 2215–2222. [Google Scholar] [CrossRef] [PubMed]
- Badenhorst, C.E.; Dawson, B.; Cox, G.R.; Laarakkers, C.M.; Swinkels, D.W.; Peeling, P. Acute dietary carbohydrate manipulation and the subsequent inflammatory and hepcidin responses to exercise. Eur. J. Appl. Physiol. 2015, 115, 2521–2530. [Google Scholar] [CrossRef] [PubMed]
- Badenhorst, C.E.; Dawson, B.; Cox, G.R.; Sim, M.; Laarakkers, C.M.; Swinkels, D.W.; Peeling, P. Seven days of high carbohydrate ingestion does not attenuate post-exercise IL-6 and hepcidin levels. Eur. J. Appl. Physiol. 2016, 116, 1715–1724. [Google Scholar] [CrossRef] [PubMed]
- Sim, M.; Dawson, B.; Landers, G.; Swinkels, D.W.; Tjalsma, H.; Trinder, D.; Peeling, P. The effects of carbohydrate ingestion during endurance running on post-exercise inflammation and hepcidina levels. Eur. J. Appl. Physiol. 2012, 12, 1889–1898. [Google Scholar]
- Newlin, M.K.; Williams, S.; McNamara, T.; Tjalsma, H.; Swinkels, D.W.; Haymes, E.M. The effects of acute exercise bouts on hepcidina in women. Int. J. Sports Nutr. Exerc. Metab. 2012, 22, 79–89. [Google Scholar] [CrossRef]
- Peeling, P.; Sim, M.; Badenhorst, C.E.; Dawson, B.; Govus, A.D.; Abbiss, C.R.; Swinkels, D.W.; Trinder, D. Iron status and the acute post-exercise hepcidin response in athletes. PLoS ONE 2014, 9, e93002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burden, R.J.; Pollock, N.; Whyte, G.P.; Richards, T.; Moore, B.; Busbridge, M.; Srai, S.K.; Otto, J.; Pedlar, C.R. Effect of intravenous iron on aerobic capacity and iron metabolism in elite athletes. Med. Sci. Sports Exerc. 2015, 47, 1399–1407. [Google Scholar] [CrossRef] [PubMed]
- Dahlquist, D.T.; Stellingwerff, T.; Dieter, B.P.; McKenzie, D.C.; Koehle, M.S. Effects of macro- and micronutrients on exercise induced hepcidin response in highly trained endurance athletes. Appl. Physiol. Nutr. Metab. 2017, 42, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- Díaz, V.; Peinado, A.B.; Barba-Moreno, L.; Altamura, S.; Butragueño, J.; González-Gross, M.; Alteheld, B.; Stehle, P.; Zapico, A.G.; Muckenthaler, M.U.; et al. Elevated hepcidin serum level in response to inflammatory and iron signals in exercising athletes is independent of moderate supplementation with vitamin C and E. Physiol. Rep. 2015, 3, e12475. [Google Scholar] [CrossRef] [PubMed]
- Sim, M.; Dawson, B.; Landers, G.; Swinkels, D.W.; Tjasma, H.; Yeap, B.B.; Trinder, D.; Peeling, P. Oral contraception does not alter typical post-exercise interleukin-6 and hepcidin levels in females. J. Sci. Med. Sport 2015, 18, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Peeling, P.; McKay, A.K.A.; Pyne, D.B.; Guelfi, K.J.; McCormick, R.H.; Laarakkers, C.M.; Swinkels, D.W.; Garvican-Lewis, L.A.; Ross, M.L.R.; Sharma, A.P.; et al. Factors influencing the post-exercise hepcidin-25 response in elite Athletes. Eur. J. Appl. Physiol. 2017, 117, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Govus, A.D.; Abbiss, C.R.; Garvican-rvica, L.A.; Swinkels, D.W.; Laarakkers, C.M.; Gore, C.J.; Peeling, P. Acute hypoxic exercise does not alter post-exercise iron metabolism in moderately trained endurance athletes. Eur. J. Appl. Physiol. 2014, 114, 2183–2191. [Google Scholar] [CrossRef] [PubMed]
- Govus, A.D.; Peeling, P.; Abbiss, C.R.; Lawler, N.G.; Swinkels, D.W.; Laarakkers, C.M.; Thompson, K.G.; Peiffer, J.J.; Gore, C.J.; Garvican-Lewis, L.A. Live high, train low-influence on resting and post-exercise hepcidin levels. Scand. J. Med. Sci. Sports 2017, 27, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Antosiewicz, J.; Kaczor, J.J.; Kasprowicz, K.; Laskowski, R.; Kujach, S.; Luszczyk, M.; Radziminski, L.; Ziemann, E. Repeated “all out” interval exercise causes an increase in serum hepcidin concentration in both trained and untrained men. Cell Immunol. 2013, 283, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Tomczyk, M.; Kortas, J.; Flis, D.; Skrobot, W.; Camilleri, R.; Antosiewicz, J. Simple sugar supplementation abrogates exercise-induced increase in hepcidin in young men. J. Int. Soc. Sports Nutr. 2017, 14, 10. [Google Scholar] [CrossRef] [PubMed]
- Kasprovicz, K.; Ziemann, E.; Ratkowski, W.; Laskowski, R.; Kaczor, J.J.; Dadci, R.; Antosiewicz, J. Running a 100-km-ultra-marathon induces an inflammatory response but does not raise the level of the plasma iron-regulatory protein hepcidina. J. Sports Med. Phys. Fit. 2013, 53, 533–537. [Google Scholar]
- Skarpanska-Stejnborn, A.; Basta, P.; Trzeciak, J.; Szczesniak-Pilaczynska, L. Effect of intense physical exercise on hepcidin levels and selected parameters of iron metabolism in rowing athletes. Eur. J. Appl. Physiol. 2015, 115, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Skarpańska-Stejnborn, A.; Basta, P.; Trzeciak, J.; Michalska, A.; Kafkas, M.E.; Woitas–Ślubowska, D. Effects of cranberry (Vaccinum macrocarpon) supplementation on iron status and inflammatory markers in rowers. J. Int. Soc. Sports Nutr. 2017, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Robson-Ansley, P.; Walsh, Q.; Sala, D. The effect of carbohydrate ingestion on plasma interleukin-6, hepcidina and iron concentrations following prolonged exercise. Cytokine 2011, 53, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, W.; Prommer, N. Impact of alterations in total hemoglobin mass on VO2max. Exerc. Sport Sci. Rev. 2010, 38, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Asai, T.; Matsuo, T.; Okamura, K. Effect of resistance exercise on iron status in moderately iron-deficient rats. Biol. Trace Elem. Res. 2011, 144, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T.; Suzuki, H.; Suzuki, M. Resistance exercise increses the capacity of heme biosynthesis more than aerobic exercise in rats. J. Clin. Biohem. Nutr. 2000, 29, 19–27. [Google Scholar] [CrossRef]
- Fujii, T.; Matsuo, T.; Okamura, K. Effects of resistance exercise on iron absorption and balance in iron-deficient rats. Biol. Trace Elem. Res. 2014, 161, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Deruisseau, K.C.; Roberts, L.M.; Kushnick, R.M.; Evans, A.M.; Austin, K.; Haymes, E.M. Iron status of young males and females performing weight-training exercise. Med. Sci. Sports Exerc. 2004, 36, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Helgerud, J.; Høydal, K.; Wang, E.; Karlsen, T.; Berg, P.; Bjerkaas, M.; Simonsen, T.; Helgesen, C.; Hjorth, N.; Bach, R.; et al. Aerobic high-intensity intervals improve VO2max more than moderate training. Med. Sci. Sports Exerc. 2007, 39, 665–671. [Google Scholar] [CrossRef] [PubMed]
- Domínguez, R.; Garnacho-Castaño, M.V.; Maté-Muñoz, J.L. Methodology to determine the aerobic-anaerobic transition in functional evaluation. Arch. Med. Deporte 2015, 32, 387–392. [Google Scholar]
- Amine, E.K.; Hegsed, D.M. Effect of diet on iron absortion in rion-deficient rats. J. Nutr. 1971, 101, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Bacchetta, J.; Zaritsky, J.J.; Sea, J.L.; Chun, R.F.; Lisse, T.S.; Zavala, K.; Nayak, A.; Wesseling-Perry, K.; Westerman, M.; Hollis, B.W.; et al. Suppression of iron-regulatory hepcidin by vitamin D. J. Am. Soc. Nephrol. 2014, 25, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Zughaier, S.M.; Alvarez, J.A.; Sloan, J.H.; Konrad, R.J.; Tangpricha, V. The role of vitamin D in regulating the iron-hepcidin-ferroportin axis in monocytes. J. Clin. Transl. Endocrinol. 2014, 1, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Ohsaki, Y.; Shirakawa, H.; Hiwatashi, K.; Furukawa, Y.; Mizutani, T.; Komai, M. Vitamin K suppresses lipopolysaccharide-induced inflammation in the rat. Biosci. Biotechnol. Biochem. 2006, 70, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Anhê, F.F.; Roy, D.; Pilon, G.; Dudonné, S.; Matamoros, S.; Varin, T.V.; Garofalo, C.; Moine, Q.; Desjardins, Y.; Levy, E.; et al. A polyphenol-rich cranberry extract protects from diet-induced obesity, insulin resistance and intestinal inflammation in association with increased Akkermansia spp. population in the gut microbiota of mice. Gut 2015, 64, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Pappas, E.; Schaich, K.M. Phytochemicals of cranberries and cranberry products: Characterization, potential health effects, and processing stability. Crit. Rev. Food Sci. Nutr. 2009, 49, 741–781. [Google Scholar] [CrossRef] [PubMed]
- Denis, M.C.; Desjardins, Y.; Furtos, A.; Marcil, V.; Dudonné, S.; Montoudis, A.; Garofalo, C.; Delvin, E.; Marette, A.; Levy, E. Prevention of oxidative stress, inflammation and mitochondrial dysfunction in the intestine by different cranberry phenolic fractions. Clin. Sci. 2015, 128, 197–212. [Google Scholar] [CrossRef] [PubMed]
- Hinton, P.S.; Sinclair, L.M. Iron supplementation maintains ventilatory threshold and improves energetic efficiency in iron-deficient nonanaemic athletes. Eur. J. Clin. Nutr. 2007, 61, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Garvican, L.A.; Saunders, P.U.; Cardoso, T.; Macdougall, I.C.; Lobigs, L.M.; Fazakerley, R.; Fallon, K.E.; Anderson, B.; Anson, J.M.; Thompson, K.G.; et al. Intravenous iron supplementation in distance runners with low or suboptimal ferritin. Med. Sci. Sports Exerc. 2014, 46, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Blee, T.; Goodman, C.; Dawson, B.; Stapff, A. The effect of intramuscular iron injections of serum ferritin levels and physical performance in elite netballers. J. Sci. Med. Sport 1999, 2, 311–321. [Google Scholar] [CrossRef]
- Pedlar, C.R.; Whyte, G.P.; Burden, R.J.; Moore, B.; Horgan, G.; Pollock, N. A case study of an iron-deficient female Olympic 15,000 m runner. Int. J. Sports Physiol. Perform. 2013, 8, 696–698. [Google Scholar] [CrossRef]
- Ishibashi, A.; Maeda, N.; Kamei, A.; Goto, K. Iron supplementation during three consecutive days of endurance training augmented hepcidin levels. Nutrients 2017, 9, 809. [Google Scholar] [CrossRef] [PubMed]
- Darshan, D.; Frazer, D.M.; Wilkins, S.J.; Anderson, G.J. Severe iron deficiency blunts the response of the iron regulatory gene Hamp and pro-inflammatory cytokines to lipopolysaccharide. Haematologica 2010, 95, 1660–1667. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Valore, E.V.; Territo, M.; Schiller, G.; Lichtenstein, A.; Ganz, T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acutephase protein. Blood 2003, 101, 2461–2463. [Google Scholar] [CrossRef] [PubMed]
- Van Santen, S.; van Dongen-Lases, E.C.; de Vegt, F.; van Riel, P.L.; van Ede, A.E.; Swinkels, D.W. Hepcidin and hemoglobin content parameters in the diagnosis of iron deficiency in rheumatoid arthritis patients with anemia. Arthritis Rheum. 2011, 63, 3672–3680. [Google Scholar] [CrossRef] [PubMed]
- Jaeggi, T.; Moretti, D.; Kvalsvig, J.; Holding, P.A.; Tjalsma, H.; Kortman, G.A.; Jooste, I.; Mwangi, A.; Zimmermann, M.B. Iron status and systemic inflammation, but not gut inflammation, strongly predict gender specific concentrations of serum hepcidin in infants in rural Kenya. PLoS ONE 2013, 8, e57513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganz, T.; Nemeth, E. Hepcidin and iron homeostasis. Biochim. Biophys. Acta 2012, 1823, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Banzet, S.; Sanchez, H.; Chapot, R.; Bigard, X.; Vaulont, S.; Koulmann, N. Interleukin-6 contributes to hepcidin mRNA increase in response to exercise. Cytokine 2012, 58, 158–161. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.E.; López, A.M.; de Cabo, M.R.; Rodríguez, F.M.; Losada, J.P.; Sarmiento, R.G.; López, A.J.; Arellano, J.L. Cyclosporin A decreases human machrophage interleukin-6 synthesis at post-transcriptional level. Mediat. Inflamm. 1999, 8, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Williamson, M.S.; Miller, E.K.; Plemons, J.; Rees, T.; Iacopino, A.M. Cyclosporine A upregulates interleukin-6 gene expression in human gingiva: Possible mechanism for gingival overgrowth. J. Periodontol. 1994, 65, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Keller, C.; Hellsten, Y.; Steensberg, A.; Pedersen, B.K. Differential regulation of IL-6 and TNF-alpha via calcineurin in human skeletal muscle cells. Cytokine 2006, 36, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Keller, C.; Steensberg, A.; Pilegaard, H.; Osada, T.; Saltin, B.; Pedersen, B.K.; Neufer, P.D. Transcriptional activation of the IL-6 gene in human contracting skeletal muscle: Influence of muscle glycogen content. FASEB J. 2001, 15, 2748–2750. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Febbraio, M.A. Muscle as an endocrine organ: Focus on muscle-derived interleukin-6. Physiol. Rev. 2008, 88, 1379–1406. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K. The diseasome of physical inactivity—And the role of myokines in muscle-fat cross talk. J. Physiol. 2009, 587, 5559–5568. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, L.; Idorn, M.; Olofsson, G.H.; Lauenborg, B.; Nookaew, I.; Hansen, R.H.; Johannesen, H.H.; Becker, J.C.; Pedersen, K.S.; Dethlefsen, C.; et al. Voluntary running suppresses tumor growth through epinephrine- and IL-6-Dependent NK cell mobilization and redistribution. Cell Metab. 2016, 23, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Starkie, R.; Ostrowski, S.R.; Jauffred, S.; Febbraio, M.; Pedersen, B.K. Exercise and IL-6 infusion inhibit endotoxin-induced TNF-alpha production in humans. FASEB J. 2003, 17, 884–886. [Google Scholar] [CrossRef] [PubMed]
- Steensberg, A.; Fischer, C.P.; Keller, C.; Moller, K.; Pedersen, B.K. IL-6 enhances plasma IL-1ra, IL-10, and cortisol in humans. Am. J. Physiol. Endocrinol. Metab. 2003, 285, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P.; Aljada, A.; Bandyopadhyay, A. Inflammation: The link between insulin resistance, obesity and diabetes. Trends Immunol. 2004, 25, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Duncan, B.B.; Schmidt, M.I.; Pankow, J.S.; Ballantyne, C.M.; Couper, D.; Vigo, A.; Hoogeveen, R.; Folsom, A.R.; Heiss, G.; Atherosclerosis Risk in Communities Study. Low-grade systemic inflammation and the development of type 2 diabetes: The atherosclerosis risk in communities study. Diabetes 2003, 52, 1799–1805. [Google Scholar] [CrossRef] [PubMed]
- Budagian, V.; Bulanova, E.; Paus, R.; Bulfone-Paus, S. IL-1/IL-15 receptor biology: A guided tour through an expanding universe. Cytokine Growth Factor Rev. 2006, 17, 259–280. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.; Watanabe, K.; Kantani, T.; Hayashi, J.; Ishida, N.; Kaneki, M. Upregulation of circulating IL-15 by treadmill running in healthy individuals: Is IL-15 an endocrine mediator of the beneficial effects of endurance exercise. Endocrin. J. 2011, 58, 211–215. [Google Scholar] [CrossRef]
- Pérez-López, A.; McKendry, J.; Martin-Rincón, M.; Morales-Alamo, D.; Pérez-Köhler, B.; Valadés, D.; Buján, J.; Calbet, J.A.L.; Breen, L. Skeletal muscle IL-15/IL-15Rα and myofibrillar protein synthesis after resistance exercise. Scand. J. Med. Sci. Sports 2017, 28, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Quinn, L.S.; Strait-Bodey, L.; Anderson, B.G.; Argiles, J.M.; Havel, P.J. Interleukin-15 stimulates adiponectin secretion by 3T3-L1 adipocytes: Evidence for a skeletal muscle-to-fat signaling pathway. Cell Biol. Int. 2005, 29, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Pérez-López, A.; Valadés, D.; Vázquez Martínez, C.; de Cos Blanco, A.I.; Buján, J.; Garcia-Honduvilla, N. Serum IL-15 and IL-15Rα levels are decreased in lean and obese physically active humans. Scand. J. Med. Sci. Sports 2017. [Google Scholar] [CrossRef] [PubMed]
- Kemna, E.; Pickkers, P.; Nemeth, E.; van der Hoeven, H.; Swinkels, D. Time-course analysis of hepcidin, serum iron, and plasma cytokine levels in humans injected with LPS. Blood 2005, 106, 1864–1866. [Google Scholar] [CrossRef] [PubMed]
- Christoulas, K.; Karamouzis, M.; Mandroukas, K. “Living high-training low” vs. “living high-training high”: Erythropoietic responses and performance of adolescent cross-country skiers. J. Sport Med. Phys. Fit. 2011, 51, 74–81. [Google Scholar]
- Son, H.J.; Kim, H.J.; Kim, J.H.; Ohno, H.; Kim, C.K. Erythropoietin, 2,3 DPG, oxygen transport capacity, and altitude training in adolescent Alpine skiers. Aviat. Space Envirn. Med. 2012, 83, 50–53. [Google Scholar] [CrossRef]
- Peyssonnaux, C.; Zinkernagel, A.S.; Schuepbach, R.A.; Rankin, E.; Vaulont, S.; Haase, V.H.; Nizet, V.; Johnson, R.S. Regulation of iron homeostasis by the hypoxia-inducible transcription factors(HIFs). J. Clin. Investig. 2007, 117, 1926–1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kautz, L.; Jung, G.; Valore, E.V.; Rivella, S.; Nemeth, E.; Ganz, T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet. 2014, 46, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Talbot, N.P.; Lakhal, S.; Smith, T.G.; Privat, C.; Nickol, A.H.; Rivera-Ch, M.; León-Velarde, F.; Dorrington, K.L.; Mole, D.R.; Robbins, P.A. Regulation of hepcidin expression at high altitude. Blood 2012, 119, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Goetze, O.; Schmitt, J.; Spliethoff, K.; Theurl, I.; Weiss, G.; Swinkels, D.W.; Tjalsma, H.; Maggiorini, M.; Krayenbuhl, P.; Rau, M.; et al. Adaptation of iron transport and metabolism to acute highaltitude hypoxia in mountaineers. Hepatology 2013, 58, 2153–2162. [Google Scholar] [CrossRef] [PubMed]
- Lui, Q.; Davidoff, O.; Niss, K.; Haase, V.H. Hypoxia-inducible factor regulates hepcidin via erythropoietin-induced erythropoiesis. J. Clin. Investig. 2012, 122, 4635–4644. [Google Scholar]
- Robach, P.; Cairo, G.; Gelfi, C.; Bernuzzi, F.; Pilegaard, H.; Viganò, A.; Santambrogio, P.; Cerretelli, P.; Calbet, J.A.; Moutereau, S.; et al. Strong iron demand during hypoxia-induced erythropoiesis is associated with down-regulation of iron-related proteins and myoglobin in human skeletal muscle. Blood 2007, 109, 4724–4731. [Google Scholar] [CrossRef] [PubMed]
- Robach, P.; Recalcati, S.; Girelli, D.; Gelfi, C.; Aachmann-Andersen, N.J.; Thomsen, J.J.; Norgaard, A.M.; Alberghini, A.; Campostrini, N.; Castagna, A.; et al. Alterations of systemic and muscle iron metabolism in human subjects treated with low dose recombinant erythropoietin. Blood 2009, 113, 6707–6715. [Google Scholar] [CrossRef] [PubMed]
- Volke, M.; Gale, D.P.; Maegdefrau, U.; Schley, G.; Klanke, B.; Bosserhoff, A.K.; Maxwell, P.H.; Eckardt, K.U.; Warnecke, C. Evidence for a lack of a direct transcriptional suppression of the iron regulatory peptide hepcidin by hypoxia-inducible factors. PLoS ONE 2009, 4, e7875. [Google Scholar] [CrossRef] [PubMed]
- Choudhry, H.; Harris, A.L. Advances in Hypoxia-inducible factor biology. Cell Metab. 2017, 27, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Piperno, A.; Galimberti, S.; Mariani, R.; Pelucchi, S.; Ravasi, G. Modulation of hepcidin production during hypoxia-induced erythropoiesis in humans in vivo: Data from the HIGHCARE project. Blood 2011, 117, 2953–2959. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.; Troesch, S.; Saugy, J.J.; Schmitt, L.; Cejuela-Anta, R.; Faiss, R.; Steiner, T.; Robinson, N.; Millet, G.P.; Wehrlin, J.P. Individual hemoglobin mass response to normobaric and hypobaric “live high-train low”: A one-year crossover study. J. Appl. Physiol. 2017, 123, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Harvey, L.J.; Armah, C.N.; Dainty, J.R.; Foxall, R.J.; John Lewis, D.; Langford, N.J.; Fairweather-Tait, S.J. Impact of menstrual blood loss and diet on iron deficiency among women in the UK. Br. J. Nutr. 2005, 94, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Tajima, S.; Izawa-Ishizawa, Y.; Kihira, Y.; Ishizawa, K.; Tomita, S.; Tsuchiya, K.; Tamaki, T. Estrogen regulates hepcidin expression via GPR30-BMP6-dependent signaling in hepatocytes. PLoS ONE 2012, 7, e40465. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Bachman, E.; Li, M.; Roy, C.N.; Blusztajn, J.; Wong, S.; Chan, S.Y.; Serra, C.; Jasuja, R.; Travison, T.G.; et al. Testosterone administration inhibits hepcidin transcription and is associated with increased iron incorporation into red blood cells. Aging Cell 2013, 12, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Díaz, P.; Domínguez, R. Effects of testosterone supplementation on endurance performance. Rev. Andal. Med. Deporte 2016, 9, 131–137. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Author | Population | n | Exercise Protocol | Experimental Conditions | TP | Main Outcomes | |
|---|---|---|---|---|---|---|---|
| Pre vs. Post Comparison | EC Differences | ||||||
| Sim et al. [23] | Trained males (66 ± 2 mL·kg−1·min−1 VO2peak) | 10 | Endurance exercise (Running and Cycling) EP1: 60 min at 65% VO2peak EP2: 8 × (3 min at 85% VO2peak & 1.5 min at 60% VO2peak) | EC1: EP1 running EC2: EP1 cycling EC3: EP2 running EC4: EP2 cycling | Pre & 3 h PE | * EC1: ~1.6 vs. ~2.4 nmol·L−1 * EC2: ~1.1 vs. ~2.0 nmol·L−1 * EC3: ~1.5 vs. ~2.5 nmol·L−1 * EC4: ~1.2 vs. ~2.6 nmol·L−1 | ANOVA time but no EC or interaction effect |
| Badenhorst et al. [58] | Male endurance athletes (63 ± 6 mL·kg−1·min−1 VO2peak) | 10 | Endurance exercise (Running) 8 × (3 min at 85% VO2peak & 1.5 min at 60% VO2peak) | EC1: Recovery in hypoxia (FIO2 ~0.1513) EC1: Recovery in normoxia (FIO2 ~0.2093) | Pre, 3 h & 24 h PE | Pre vs. 3 h PE * EC1: 3.2 ± 1.9 vs. 5.4 ± 3.2 nM * EC2: 3.2 ± 1.2 vs. 7.4 ± 4.0 nM | ANOVA time and interaction effect. EC1 > EC2 at 3 h PE |
| Badenhorst et al. [59] | Male endurance athletes (63 ± 4 mL·kg−1·min−1 VO2peak) | 11 | Endurance exercise (Running) 8 × (3 min at 85% VO2peak & 1.5 min at 60% VO2peak) | EC1: Early recovery (0.5 & 2 h) CHO (1.2 g·kg−1) intake EC2: Late recovery (2 & 4 h PE) CHO (1.2 g·kg−1) intake | Pre, 3 h, 5 h PE. | Pre vs. 3 h PE * EC1: 6.5 ± 9.6 vs. 9.7 ± 3.5 nM * EC2: 4.9 ± 2.4 vs. 7.5 ± 3.6 nM Pre vs. 5 h PE * EC1: 6.5 ± 9.6 vs. 9.7 ± 3.8 nM * EC2: 4.9 ± 2.4 vs. 7.1 ± 3.5 nM | ANOVA time, but no EC or interaction effect |
| Sim et al. [60] | Male endurance athletes (63 ± 4 mL·kg−1·min−1 VO2peak) | 11 | Endurance exercise (Running) 8 × 3 min at 85% VO2peak & 1.5 min at 60% VO2peak | EC1: 24 h LCHO (3 g·kg·day−1) EC2: 24 h HCHO (10 g·kg·day−1) | Pre & 3 h PE | * EC1: (Pre vs. 3 h PE): 4.2 ± 3.6 vs. 6.4 ± 5.1 nM * EC2 (Pre vs. 3 h PE): 2.2 ± 1.1 vs. 4.1 ± 3.2 nM | ANOVA time and EC, but no interaction effect. * EC1 > EC2 at pre-exercise NS, EC1 vs. EC2 at 3 h PE |
| Badenhorst et al. [61] | Male endurance athletes (64 ± 5 mL·kg−1·min−1 VO2peak) | 12 | Endurance exercise (Running) Two sessions of 45 min at 65% VO2peak (day 1 -D1- and day 7 -D7-) | EC1: LCHO diet (3 g·kg·day−1) EC2: HCHO diet (8 g·kg·day−1) | Pre & 3 h PE | EC1 (Pre vs. 3 h PE): * D1: 2.0 ± 1.9 vs. 7.6 ± 6.0 nM * D7: 1.8 ± 1.2 vs. 6.5 ± 4.7 nM EC2 (Pre vs. 3 h PE): * D1: 1.9 ± 1.2 vs. 6.4 ± 3.9 nM * D7: 1.8 ± 0.7 vs. 5.4 ± 3.4 nM | ANOVA time, but no EC or interaction effect |
| Sim et al. [62] | Male endurance athletes (60 ± 1 mL·kg−1·min−1 VO2peak) | 11 | Endurance exercise (Running) 90 min at 75% VO2peak | EC1: CHO drink (6%) during exercise EC2: H2O during exercise | Pre, 3 h, 24 h PE | Pre vs. 3 h PE: * EC1: ~3.0 vs. ~7.5 nm·l−1 * EC2: ~3.0 vs. ~9.0 nm·l−1 | ANOVA time but no EC or interaction effect |
| Newlin et al. [63] | PA females (52 ± 4 mL·kg−1·min−1 VO2peak) | 11 | Endurance exercise (Running) 65% VO2peak | EC1: 60 min EC2: 120 min | Pre, 0 h, 3 h, 6 h, 9 h & 24 h PE | * EC1 (Pre vs. 3 h PE): ~0.7 vs. ~1.9 nmol·L−1 * EC2 (Pre vs. 3 h PE): ~1.1 vs. ~4.5 nmol·L−1 | ANOVA time and EC, but no interaction effect * EC2 > EC1 at 3 h PE |
| Peeling et al. [64] | Endurance athletes (60 ± 7 mL·kg−1·min−1 VO2peak). | ♂ 38 ♀ 54 | Endurance exercise (5 Running sessions) S1: 8 × 3 min at 85% VO2peak S2: 5 × 4 min at 90% VO2peak S3: 90 min at 75% VO2peak S4: 40 min at 75% VO2peak S5: 40 min at 65% VO2peak | Baseline SF: SF1 (n = 12): SF ≤ 30 μg·L−1 SF2 (n = 8): SF = 30–50 μg·L−1 SF3 (n = 14): SF = 50–100 μg·L−1 SF4 (n = 20): SF ≥ 100 μg·L−1 | Pre & 3 h PE | SF1: ~0.8 vs. ~1.2 nM * SF2: ~2.1 vs. ~4.5 nM * SF3: ~2.2 vs. ~5.3 nM * SF4: ~3.5 vs. ~8.0 nM | ANOVA effect (Pre and 3 h PE) particularly SF1 compared with SF2, SF3, and SF4. Baseline SF and 3 h PE hepcidin correlation (r = 0.52). |
| Burden et al. [65] | ID endurance athletes without anemia (64 ± 6 mL·kg−1·min−1 VO2peak) | ♂ 6 ♀ 9 | Endurance exercise (Running) Incremental test at day 1 (D1), day 2 (D2) and week 4 (W4) | EC1: Iron (500 mg) EC2: Placebo | Pre, 0 h, and 3 h PE | EC1 (Pre vs. 3 h PE) * D2: ~110 vs. ~210 ng·mL−1 * W4: ~70 vs. ~210 ng·mL−1 NS increase in EC2. | D1: ANOVA time effect D2: ANOVA time and EC effect (EC1 > EC2) W4: ANOVA time and EC effect (EC1 > EC2 at 3 h post-exercise). |
| Dahlquist et al. [66] | Male trained cyclists (67 ± 4 mL·kg−1·min−1 VO2peak) | 10 | Endurance exercise (Running) 8 × 3 min at 85% & 1.5 min at 60% VO2peak | EC1: PE CHO (75 g), Pro (25 g), vit.D (5000 IU) & vit.K(100 mcg). EC2: PE CHO (75 g), Pro (25 g) EC3: Placebo PE | Pre, 0 h, and 3 h PE | Pre vs. 0 h PE * EC1: 14.2 ± 14.9 vs. 17.8 ± 19.8 nmol·L−1 * EC2: 9.9 ± 8.9 vs. 11.8 ± 10.2 nmol·L−1 * EC3: 10.4 ± 14.6 vs. 10.1 ± 7.7 nmol·L−1 Pre vs. 3 h PE * EC1: 14.2 ± 14.9 vs. 25.4 ± 11.9 nmol·L−1 * EC2: 9.9 ± 8.9 vs. 22.3 ± 13.4 nmol·L−1 * EC3: 10.4 ± 14.6 vs. 22.6 ± 15.6 nmol·L−1 | ANOVA time (in EC1 & EC2), but no EC effect or interaction |
| Díaz et al. [67] | Trained males (70 ± 6 mL·kg−1·min−1 VO2peak) | 10 | Endurance exercise (Running) 90 min at 75% VO2peak in before (D1) & after the 4 W intervention (W4). | EC1: Vit.C (500 mg) & vit.E (400 IU). EC2: Placebo | Pre, 0 h, 3 h, 6 h, and 10 h PE | Pre vs. 3 h PE (D1 & W4) * EC1: ~11 vs. ~26 ng·mL−1 EC2: NR Pre vs. 6 h PE (D1 & W4) * EC1: ~11 vs. ~21 ng·mL−1 EC2: NR | ANOVA time but no EC effect. |
| Sim et al. [68] | PA females who ingested oral contraceptives (53 ± 2 mL·kg−1·min−1 VO2peak) | 10 | Endurance exercise (Running) 40 min at 75% VO2peak | EC1: D2 to D4 of the menstrual cycle EC2: D12 to D14 of the menstrual cycle | Pre and 3 h PE | * EC1: ~1.9 vs. ~4.4 ng·mL−1 * EC1: ~3.6 vs. ~4.5 ng·mL−1 | ANOVA time, but no EC or interaction effect. |
| Peeling et al. [69] | Male race-walker athletes (64.9 ± 5.9 mL·kg−1·min−1 VO2peak) | 24 | Endurance exercise (Running) 25 km race-walk at 75% VO2peak | EC1: All walkers EC2: lower 50th percentile EC3: higher 50th percentile | Pre and 3 h PE | * EC1: 1.1 ± 1.0 vs. 8.6 ± 5.3 nM * EC2: 0.8 ± 0.5 vs. 6.0 ± 3.6 nM * EC3: 1.5 ± 1.2 vs. 11.3 ± 5.4 nM | EC differences at baseline. Correlation of hepcidin at 3 h with SF (r = 0.69) and serum iron (r = 0.62). |
| Govus et al. [70] | Endurance athletes (males 61 ± 6.3 and females 55.0 ± 5.9 mL·kg−1·min−1 VO2max) | ♂ 7 ♀ 6 | Endurance exercise (Running) 5 × 4 min at 90% VO2peak & 1.5 min of passive recovery | EC1: hypoxia (FIO2 ~0.1450) EC2: normoxia (FIO2 ~0.2093) | Pre, 0 h, and 3 h PE | Pre vs. 3 h PE * EC1: 3.32 vs. 4.17 nmol·L−1 * EC2: 2.85 vs. 4.44 nmol·L−1 | ANOVA time, but no EC or interaction effect. |
| Govus et al. [71] | Endurance athletes (65.6 ± 8.1 mL·kg−1·min−1 VO2max) | ♂ 6 ♀ 4 | Endurance exercise (Running) 6 × 1000 m at 90% VO2peak & 1.5 min of passive recovery | EC1: hypoxia (FIO2 ~0.155) EC2: normoxia (600 m) EC3: 11 days of LHTL EC4: Iron (105 mg) plus Vit.C (1000 mg) during 1 week before the trials in participants with baseline SF < 100 μg·L−1 (EG1, n = 5), no placebo was provided for those with SF ≥ 100 μg·L−1 (EG2, n = 5). | Pre and 3 h PE | * EC1: aumento (NR) * EC2: aumento (NR) * EC3: Pre 4.0 vs. 2.0 nmol·L−1 | Baseline differences between EG1 and EG2 were observed. ANOVA time but not EC1, EC2, EC4 or interaction effect. ANOVA time and EC3 effect |
| Antosiewicz et al. [72] | Trained males (judokas) A and sedentary males B (NR VO2peak) | 11 A 10 B | Endurance exercise (Cycling) 3 × 30 s all-out sprint. (4.5 min recovery) | Population comparison: Trained (A) vs. Sedentary population (B). | Pre, 1 h, 24 h, and 5 D | Pre vs. 1 h PE * A: 64.7 ± 14.5 vs. 83.3 ± 23.3 ng·L−1 * B: 32.0 ± 5.5 vs. 43.7 ± 9.9 ng·L−1 | NR ANOVA differences A > B at baseline and 1 h PE |
| Tomczyk et al. [73] | PA males (50.1 ± 8.9 mL·kg−1·min−1 VO2peak) | 17 | Endurance exercise (Cycling) Incremental test before (D1) & after 3 days (D3) intervention | EC1: Glucose (4 g·kg−1) EC2: Fructose (4 g·kg−1) EC3: Placebo | Pre & 1 h PE | EC1: ~61.3 vs. ~60.0 ng·mL−1 EC2: ~61.5 vs. ~57.5 ng·mL−1 * EC3: ~56.0 vs. ~63.5 ng·mL−1 | NR ANOVA EC effect |
| Kasprovicz et al. [74] | Trained males (NR VO2peak not specified) | 6 | Endurance exercise (Running) 100 km ultramarathon | Pre, 25 km, 50 km, 75 km, 0 h, and 14 h PE | Pre: ~43 ng·L−1 25 km: ~45 ng·L−1 50 km: ~45 ng·L−1 75 km: ~43 ng·L−1 0 h PE or 100 km: ~44.5 ng·L−1 14 h PE: ~48 ng·L−1 | ||
| Skarpanska-Stejnborn et al. [75] | Male rowing athletes (NS VO2peak) | 20 | Endurance exercise (Rowing) 2000 m maximum test | Pre, 0 h, and 1D PE | Pre: ~0.25 ng·mL−1 * 0 h PE: ~1.7 ng·mL−1 # 1D PE: ~0.25 ng·mL−1 | ||
| Skarpanska-Stejnborn et al. [76] | Male rowing athletes (NS VO2peak) | 16 | Endurance exercise (Rowing) 2000 m maximum test before (D1) and after 6 weeks (W6) | EC1: Cranberry extract (648 mg·day−1) (n = 9) EC2: Placebo (n = 7) | Pre, 0 h, and 1D PE | D1: NS W6 (Pre vs. 0 h Post): * EC1: ~0.12 vs. ~0.32 ng·dL−1 EC2: ~0.11 vs. ~0.15 ng·dL−1 | No ANOVA time or EC effect EC1: ANOVA time effect |
| Robson-Ansley et al. [77] | Trained males (58 ± 4 mL·kg−1·min−1 VO2max) | 9 | Endurance exercise (Running) 120 min at 60% VO2peak & 5 km time trial | EC1: CHO drink (6%) during exercise EC2: H2O during exercise | Pre, 0 h, and 24 h PE | Pre vs. 0 h PE * EC1: ~20 vs. ~34 pg·mL−1 * EC2: ~15 vs. ~30 pg·mL−1 | ANOVA time but no EC or interaction effect Plasma hepcidin and IL-6 correlation at 0 h PE: EC1 (R2 = 0.13), EC2 (R2 = 0.65). |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domínguez, R.; Sánchez-Oliver, A.J.; Mata-Ordoñez, F.; Feria-Madueño, A.; Grimaldi-Puyana, M.; López-Samanes, Á.; Pérez-López, A. Effects of an Acute Exercise Bout on Serum Hepcidin Levels. Nutrients 2018, 10, 209. https://doi.org/10.3390/nu10020209
Domínguez R, Sánchez-Oliver AJ, Mata-Ordoñez F, Feria-Madueño A, Grimaldi-Puyana M, López-Samanes Á, Pérez-López A. Effects of an Acute Exercise Bout on Serum Hepcidin Levels. Nutrients. 2018; 10(2):209. https://doi.org/10.3390/nu10020209
Chicago/Turabian StyleDomínguez, Raúl, Antonio Jesús Sánchez-Oliver, Fernando Mata-Ordoñez, Adrián Feria-Madueño, Moisés Grimaldi-Puyana, Álvaro López-Samanes, and Alberto Pérez-López. 2018. "Effects of an Acute Exercise Bout on Serum Hepcidin Levels" Nutrients 10, no. 2: 209. https://doi.org/10.3390/nu10020209