Iron Deprivation in Cancer––Potential Therapeutic Implications

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
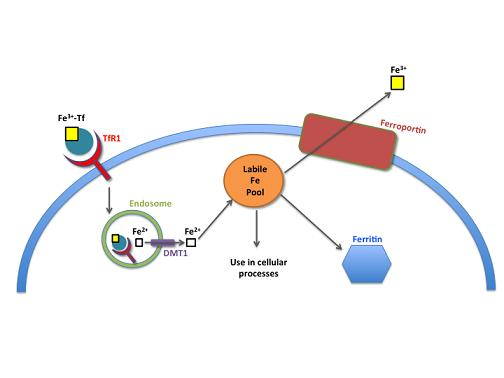
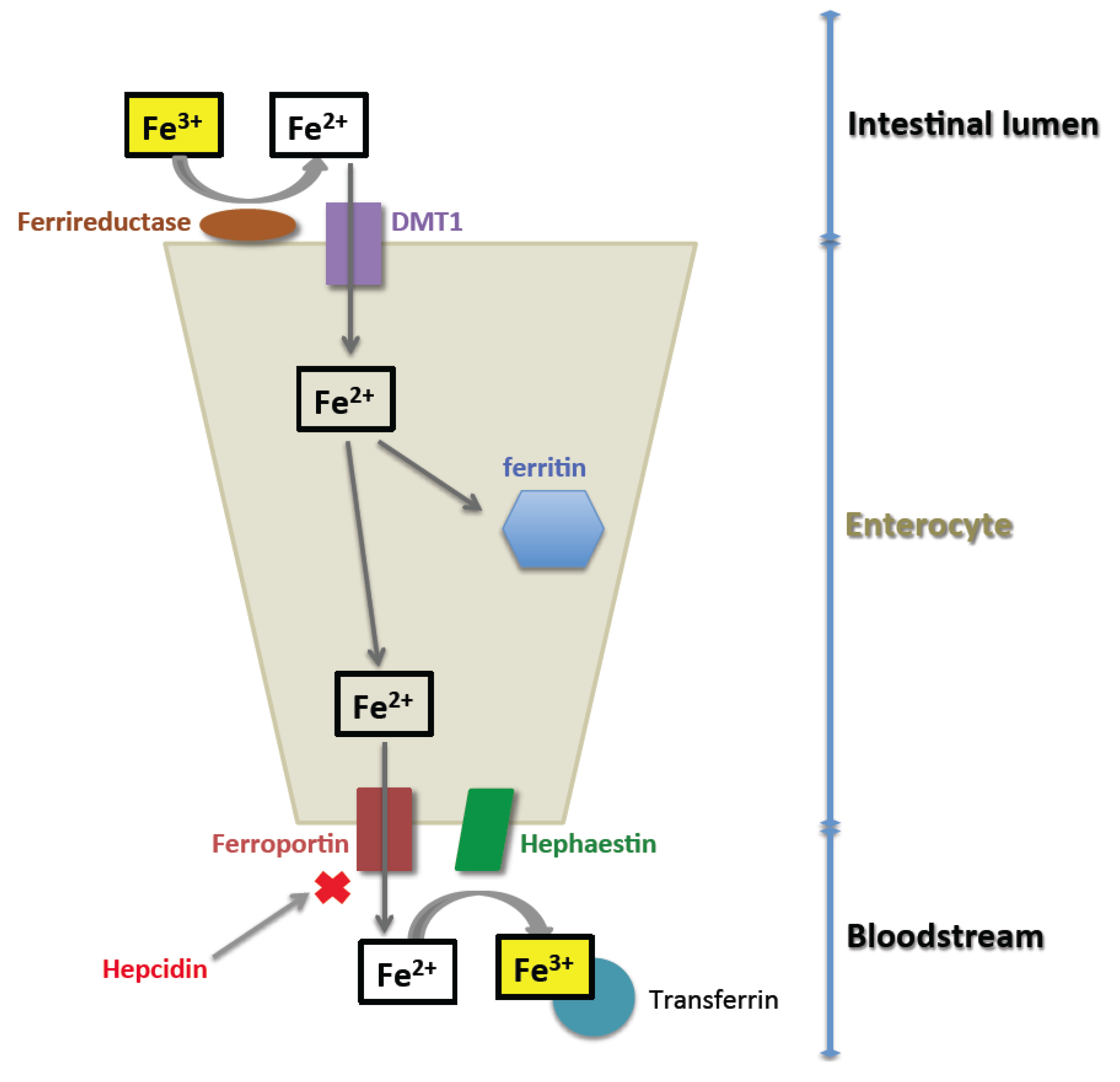
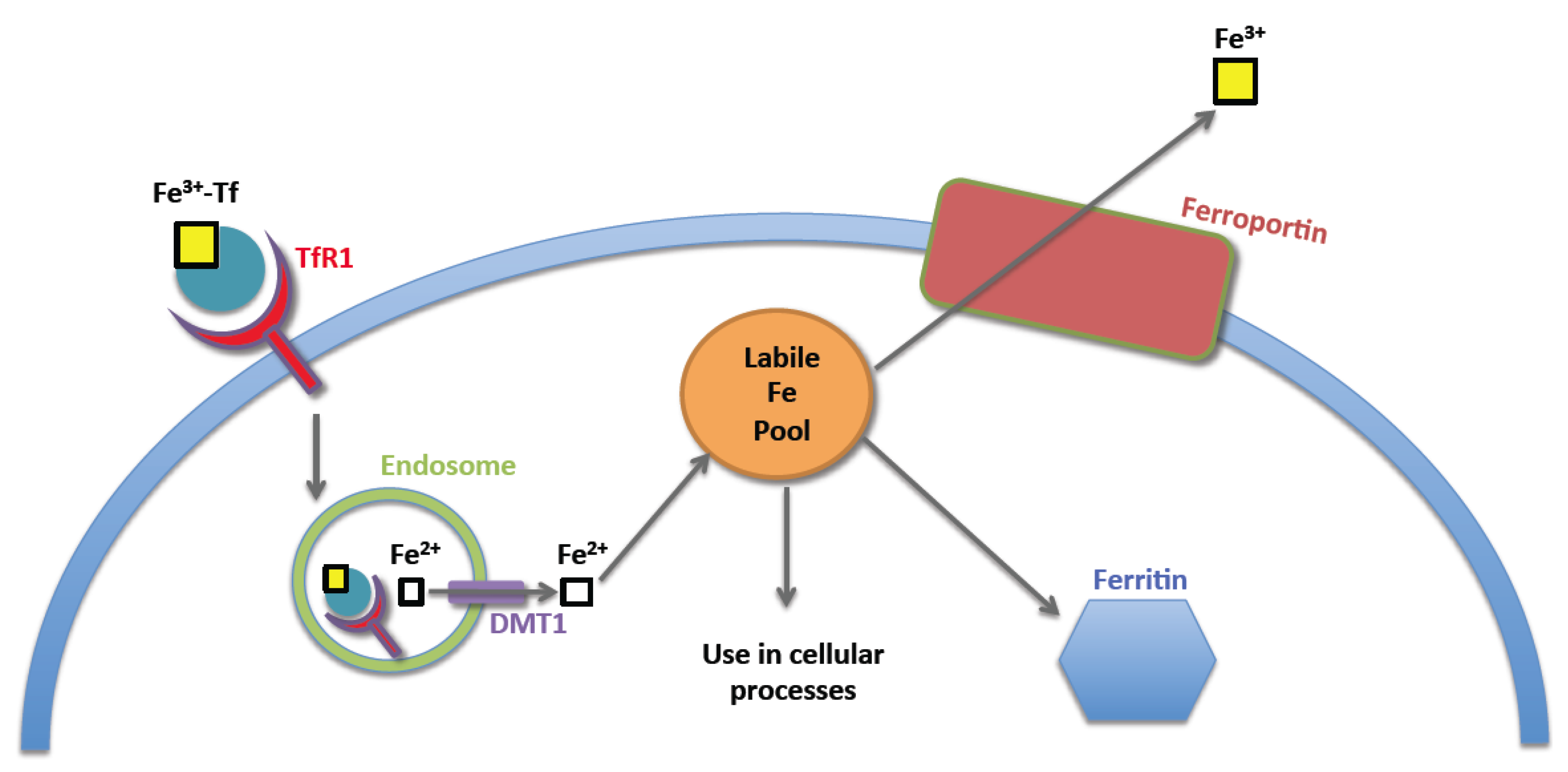
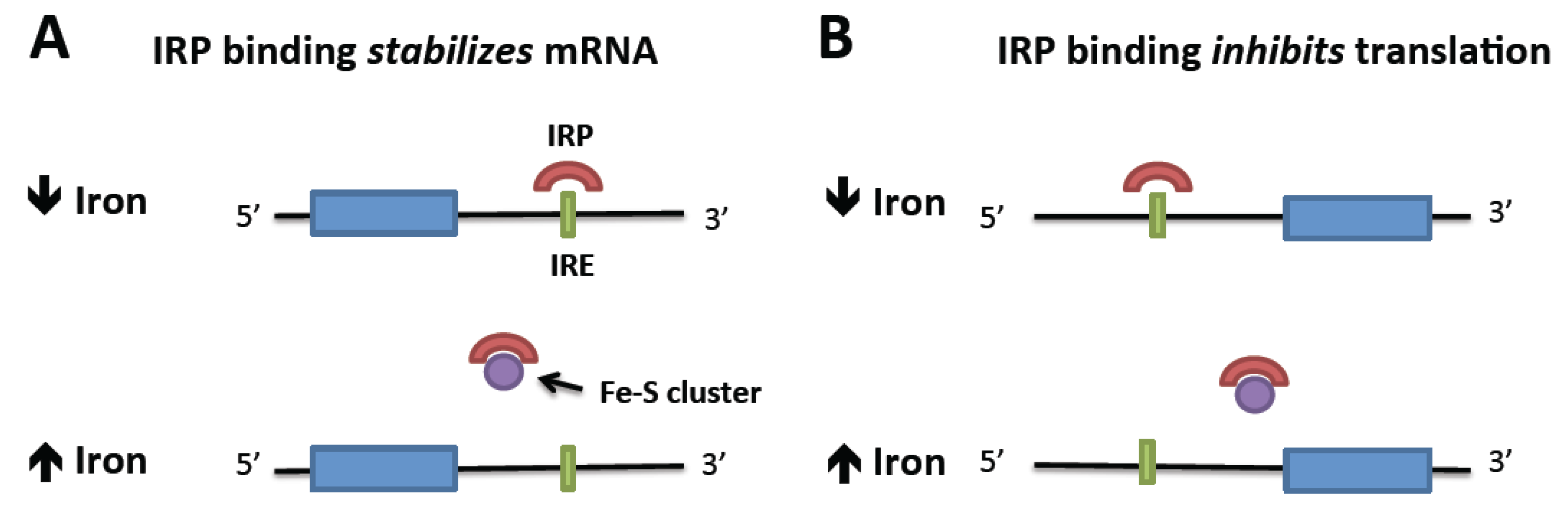
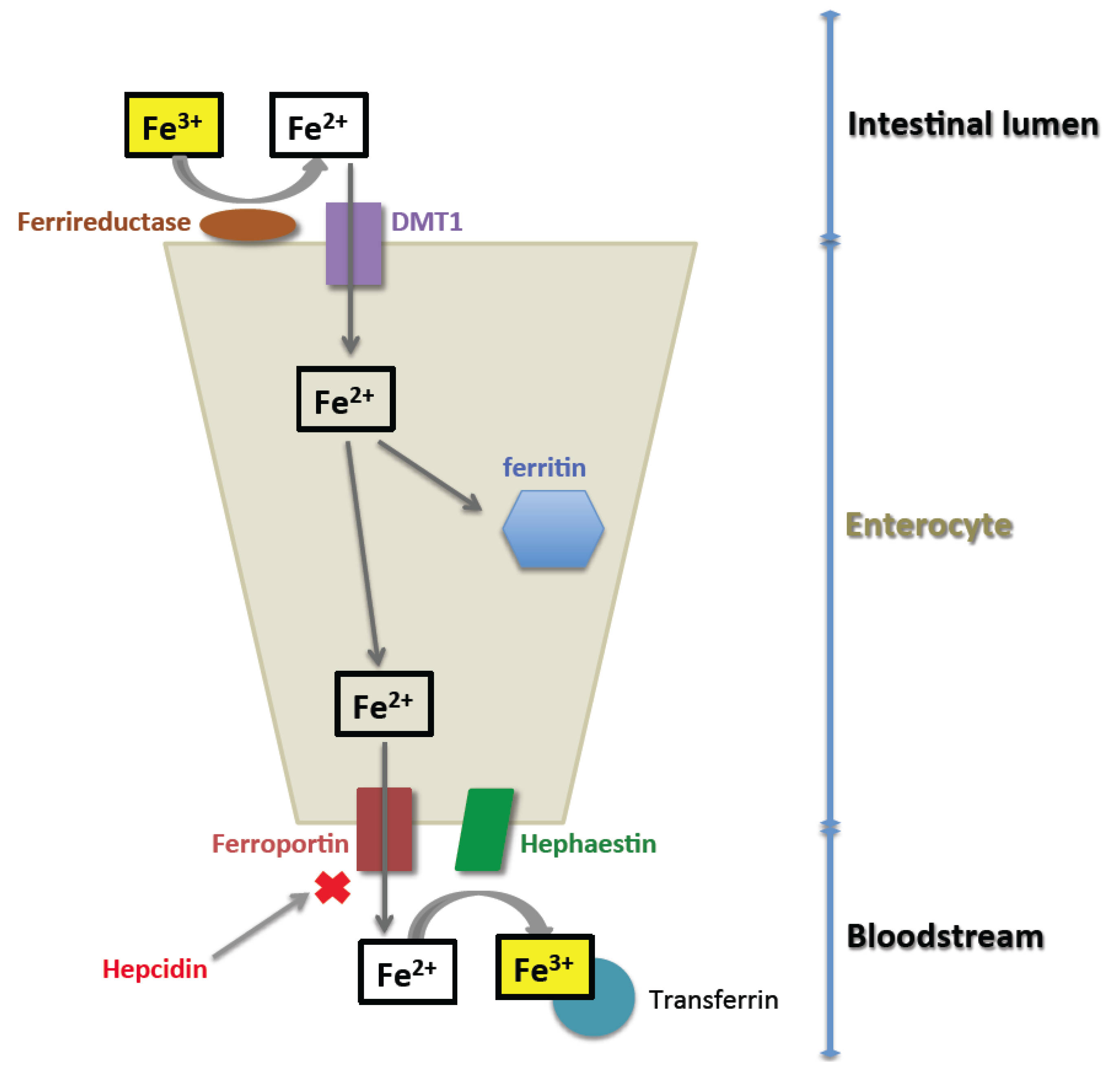
2. Normal Iron Homeostasis
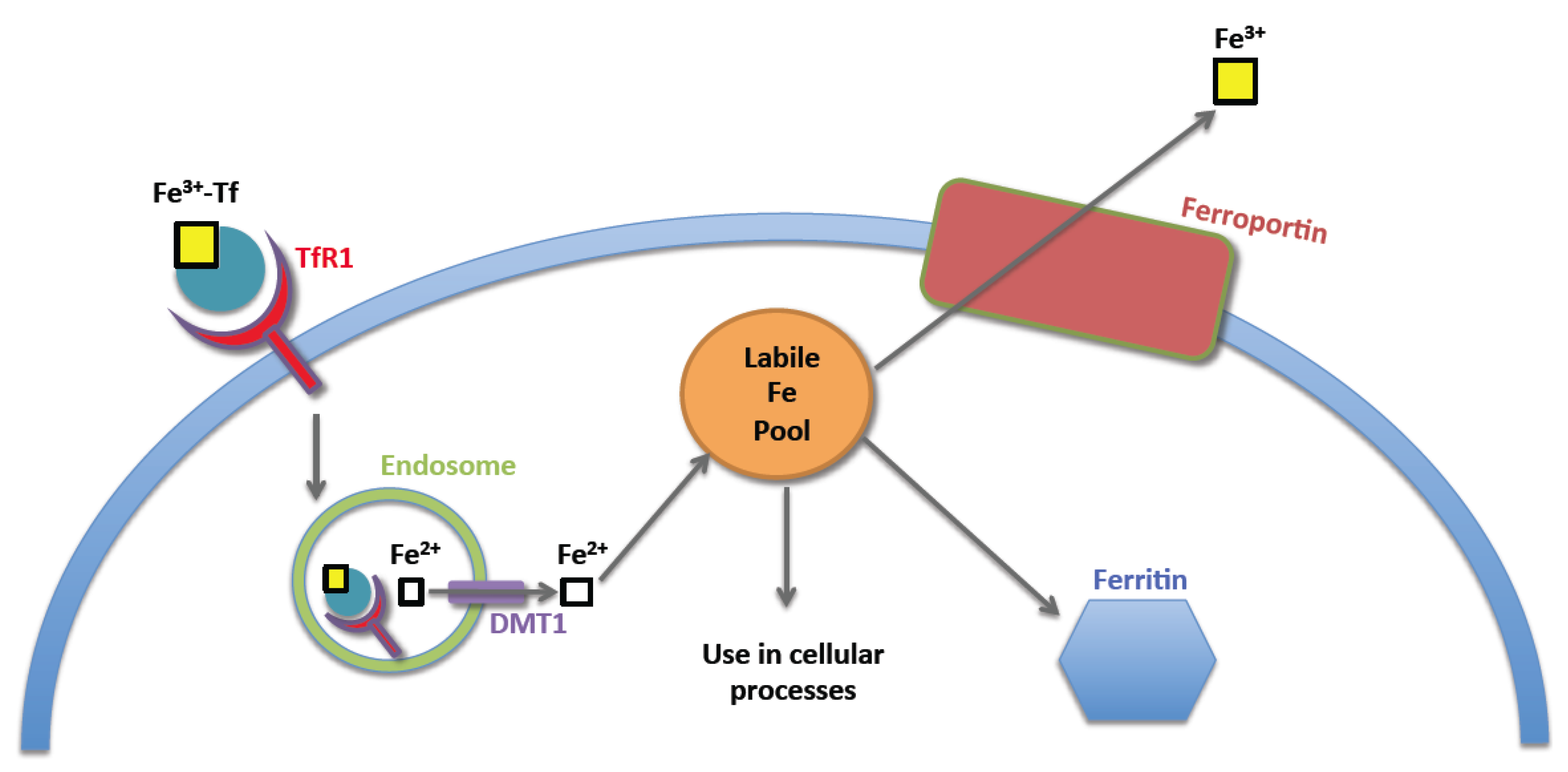
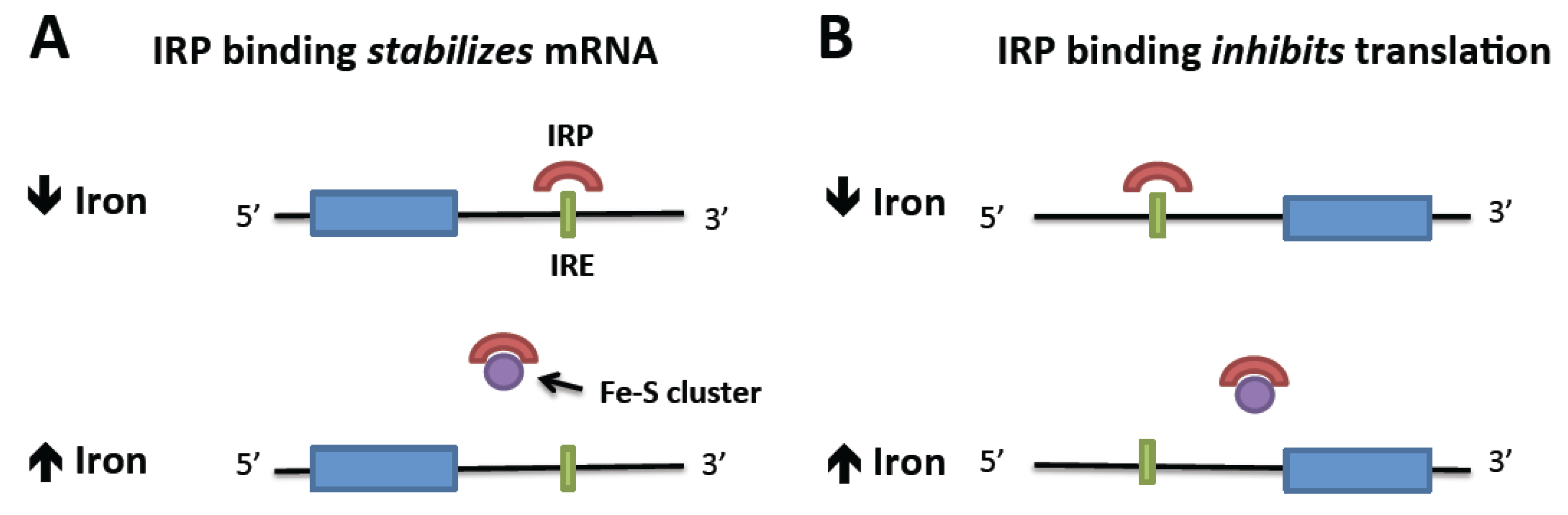
3. Iron Homeostasis in the Neoplastic Cell
4. Effects of Iron Deficiency
4.1. Cell Cycle Regulation
4.2. Cellular Metabolism
4.3. Metastatic Potential
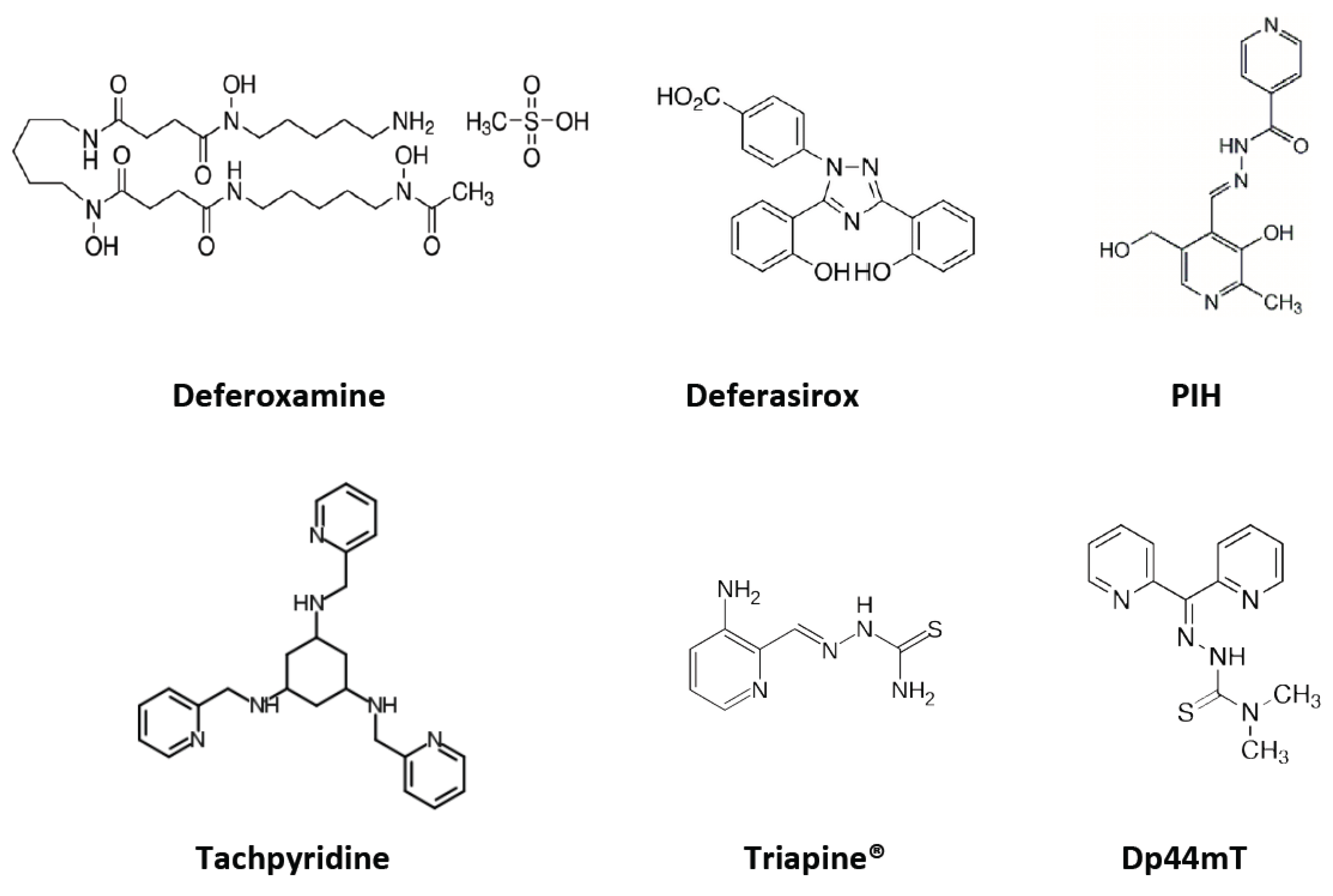
5. Iron Chelators
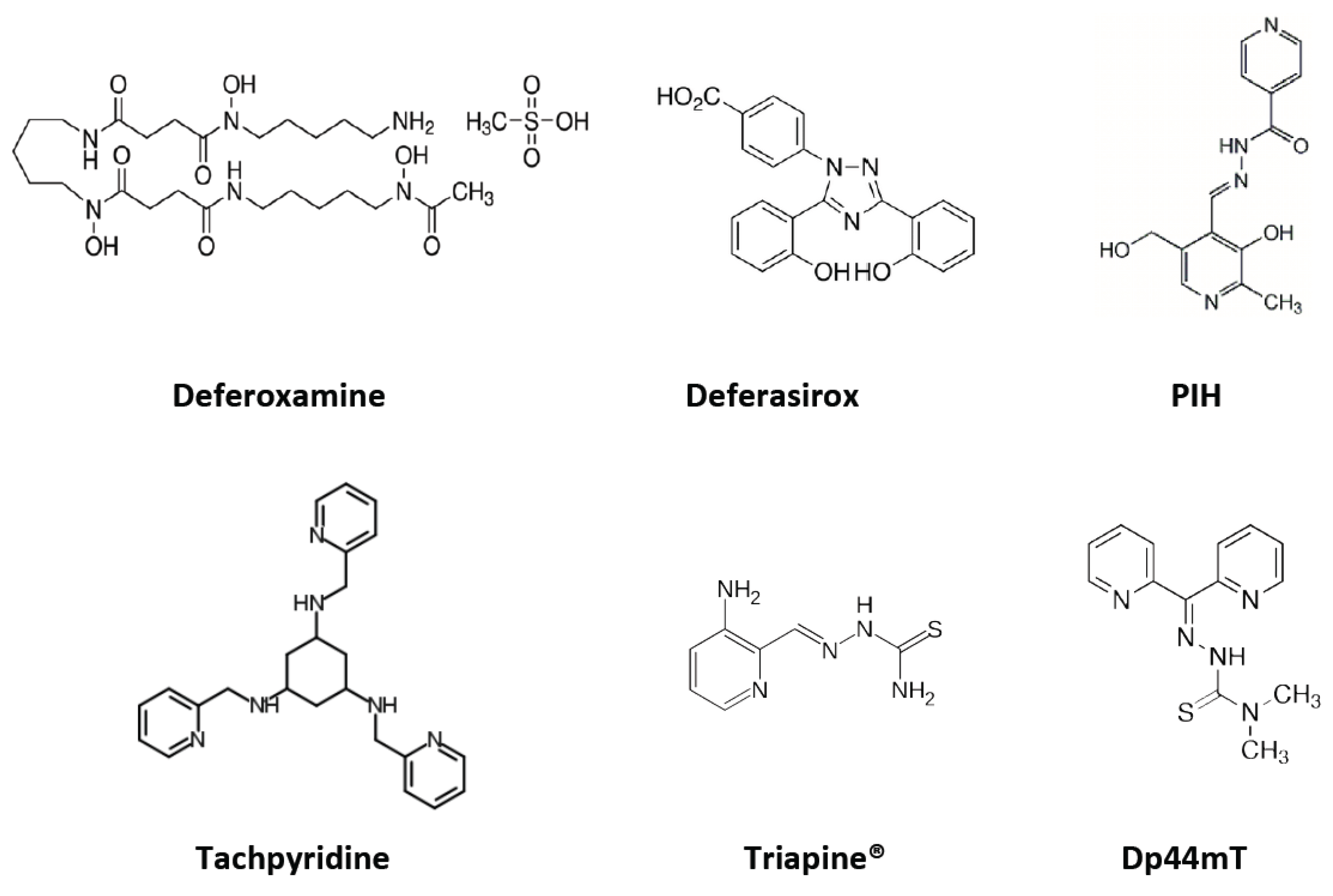
5.1. Deferoxamine
5.2. Deferasirox
5.3. Tachpyridine
5.4. Aroylhydrazones: Pyridoxal Isonicotinoyl Hydrazone (PIH) Analogs
5.5. Thiosemicarbazones
5.6. di-2-Pyridylketone Thiosemicarbazone (DpT) Series
5.7. Other Iron Chelators
5.8. Other Targeted Therapies
6. Iron Chelation as a Treatment for Cancer
6.1. Leukemia
6.1.1. In Vitro Studies
6.1.2. In Vivo Studies
6.2. Lymphoma
6.3. Neuroblastoma
6.4. Solid Tumors Other than Neuroblastoma
7. Conclusions
Conflict of Interest
References
- Fuqua, B.K.; Vulpe, C.D.; Anderson, G.J. Intestinal iron absorption. J. Trace Elem. Med. Biol. 2012, 26, 115–119. [Google Scholar] [CrossRef]
- De Domenico, I.; Ward, D.M.; Langelier, C.; Vaughn, M.B.; Nemeth, E.; Sundquist, W.I.; Ganz, T.; Musci, G.; Kaplan, J. The molecular mechanism of hepcidin-mediated ferroportin down-regulation. Mol. Biol. Cell 2007, 18, 2569–2578. [Google Scholar] [CrossRef]
- Aisen, P. Transferrin receptor 1. Int. J. Biochem. Cell Biol. 2004, 36, 2137–2143. [Google Scholar] [CrossRef]
- Dautry-Varsat, A.; Ciechanover, A.; Lodish, H.F. pH and the recycling of transferrin during receptor-mediated endocytosis. Proc. Natl. Acad. Sci. USA 1983, 80, 2258–2262. [Google Scholar] [CrossRef]
- Graf, E.; Mahoney, J.R.; Bryant, R.G.; Eaton, J.W. Iron-catalyzed hydroxyl radical formation. Stringent requirement for free iron coordination site. J. Biol. Chem. 1984, 259, 3620–3624. [Google Scholar]
- Cairo, G.; Recalcati, S. Iron-regulatory proteins: Molecular biology and pathophysiological implications. Expert Rev. Mol. Med. 2007, 9, 1–13. [Google Scholar]
- Cairo, G.; Tacchini, L.; Pogliaghi, G.; Anzon, E.; Tomasi, A.; Bernelli-Zazzera, A. Induction of ferritin synthesis by oxidative stress. Transcriptional and post-transcriptional regulation by expansion of the “free” iron pool. J. Biol. Chem. 1995, 270, 700–703. [Google Scholar]
- Meyron-Holtz, E.G.; Ghosh, M.C.; Rouault, T.A. Mammalian tissue oxygen levels modulate iron-regulatory protein activities in vivo. Science 2004, 306, 2087–2090. [Google Scholar] [CrossRef]
- Recalcati, S.; Alberghini, A.; Campanella, A.; Gianelli, U.; de Camilli, E.; Conte, D.; Cairo, G. Iron regulatory proteins 1 and 2 in human monocytes, macrophages and duodenum: Expression and regulation in hereditary hemochromatosis and iron deficiency. Haematologica 2006, 91, 303–310. [Google Scholar]
- Kennedy, M.C.; Mende-Mueller, L.; Blondin, G.A.; Beinert, H. Purification and characterization of cytosolic aconitase from beef liver and its relationship to the iron-responsive element binding protein. Proc. Natl. Acad. Sci. USA 1992, 89, 11730–11734. [Google Scholar] [CrossRef]
- Elford, H.L.; Freese, M.; Passamani, E.; Morris, H.P. Ribonucleotide reductase and cell proliferation. I. Variations of ribonucleotide reductase activity with tumor growth rate in a series of rat hepatomas. J. Biol. Chem. 1970, 245, 5228–5233. [Google Scholar]
- Trinder, D.; Zak, O.; Aisen, P. Transferrin receptor-independent uptake of differic transferrin by human hepatoma cells with antisense inhibition of receptor expression. Hepatology 1996, 23, 1512–1520. [Google Scholar] [CrossRef]
- Richardson, D.R.; Baker, E. The uptake of iron and transferrin by the human malignant melanoma cell. Biochim. Biophys. Acta 1990, 1053, 1–12. [Google Scholar] [CrossRef]
- Dang, C.V. Myc on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef]
- Adhikary, S.; Eilers, M. Transcriptional regulation and transformation by myc proteins. Nat. Rev. Mol. Cell. Biol. 2005, 6, 635–645. [Google Scholar] [CrossRef]
- Dang, C.V. C-myc target genes involved in cell growth, apoptosis, and metabolism. Mol. Cell. Biol. 1999, 19, 1–11. [Google Scholar]
- Wu, K.J.; Polack, A.; Dalla-Favera, R. Coordinated regulation of iron-controlling genes, h-ferritin and irp2, by c-myc. Science 1999, 283, 676–679. [Google Scholar] [CrossRef]
- Graslund, A.; Ehrenberg, A.; Thelander, L. Characterization of the free radical of mammalian ribonucleotide reductase. J. Biol. Chem. 1982, 257, 5711–5715. [Google Scholar]
- Green, D.A.; Antholine, W.E.; Wong, S.J.; Richardson, D.R.; Chitambar, C.R. Inhibition of malignant cell growth by 311, a novel iron chelator of the pyridoxal isonicotinoyl hydrazone class: Effect on the r2 subunit of ribonucleotide reductase. Clin. Cancer Res. 2001, 7, 3574–3579. [Google Scholar]
- Shang, H.; Li, Q.; Feng, G.; Cui, Z. Molecular analysis and functions of p53r2 in zebrafish. Gene 2011, 475, 30–38. [Google Scholar] [CrossRef]
- Wei, P.P.; Tomter, A.B.; Rohr, A.K.; Andersson, K.K.; Solomon, E.I. Circular dichroism and magnetic circular dichroism studies of the active site of p53r2 from human and mouse: Iron binding and nature of the biferrous site relative to other ribonucleotide reductases. Biochemistry 2006, 45, 14043–14051. [Google Scholar] [CrossRef]
- Chen, D.; Li, M.; Luo, J.; Gu, W. Direct interactions between hif-1 αand mdm2 modulate p53 function. J. Biol. Chem. 2003, 278, 13595–13598. [Google Scholar] [CrossRef]
- Chen, K.; Albano, A.; Ho, A.; Keaney, J.F., Jr. Activation of p53 by oxidative stress involves platelet-derived growth factor-βreceptor-mediated ataxia telangiectasia mutated (atm) kinase activation. J. Biol. Chem. 2003, 278, 39527–39533. [Google Scholar] [CrossRef]
- Fukuchi, K.; Tomoyasu, S.; Watanabe, H.; Kaetsu, S.; Tsuruoka, N.; Gomi, K. Iron deprivation results in an increase in p53 expression. Biol. Chem. Hoppe-Seyler 1995, 376, 627–630. [Google Scholar]
- Liang, S.X.; Richardson, D.R. The effect of potent iron chelators on the regulation of p53: Examination of the expression, localization and DNA-binding activity of p53 and the transactivation of waf1. Carcinogenesis 2003, 24, 1601–1614. [Google Scholar] [CrossRef]
- Saletta, F.; Suryo Rahmanto, Y.; Noulsri, E.; Richardson, D.R. Iron chelator-mediated alterations in gene expression: Identification of novel iron-regulated molecules that are molecular targets of hypoxia-inducible factor-1 alpha and p53. Mol. Pharmacol. 2010, 77, 443–458. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Fracanzani, A.L.; Cairo, G.; Megazzini, C.P.; Gatti, S.; Rametta, R.; Fargion, S.; Valenti, L. Iron-dependent regulation of mdm2 influences p53 activity and hepatic carcinogenesis. Am. J. Pathol. 2010, 176, 1006–1017. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, W.; Tsuji, Y.; Torti, S.V.; Torti, F.M. Post-transcriptional modulation of iron homeostasis during p53-dependent growth arrest. J. Biol. Chem. 2008, 283, 33911–33918. [Google Scholar] [CrossRef]
- Weizer-Stern, O.; Adamsky, K.; Margalit, O.; Ashur-Fabian, O.; Givol, D.; Amariglio, N.; Rechavi, G. Hepcidin, a key regulator of iron metabolism, is transcriptionally activated by p53. Br. J. Haematol. 2007, 138, 253–262. [Google Scholar] [CrossRef]
- Campisi, J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 2001, 11, S27–S31. [Google Scholar]
- Lagger, G.; Doetzlhofer, A.; Schuettengruber, B.; Haidweger, E.; Simboeck, E.; Tischler, J.; Chiocca, S.; Suske, G.; Rotheneder, H.; Wintersberger, E.; et al. The tumor suppressor p53 and histone deacetylase 1 are antagonistic regulators of the cyclin-dependent kinase inhibitor p21/waf1/cip1 gene. Mol. Cell. Biol. 2003, 23, 2669–2679. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. P21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Mitra, J.; Dai, C.Y.; Somasundaram, K.; El-Deiry, W.S.; Satyamoorthy, K.; Herlyn, M.; Enders, G.H. Induction of p21(waf1/cip1) and inhibition of cdk2 mediated by the tumor suppressor p16(ink4a). Mol. Cell. Biol. 1999, 19, 3916–3928. [Google Scholar]
- Cayrol, C.; Knibiehler, M.; Ducommun, B. P21 binding to pcna causes g1 and g2 cell cycle arrest in p53-deficient cells. Oncogene 1998, 16, 311–320. [Google Scholar]
- Stein, G.H.; Drullinger, L.F.; Soulard, A.; Dulic, V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol. Cell. Biol. 1999, 19, 2109–2117. [Google Scholar]
- Zhang, Y.; Fujita, N.; Tsuruo, T. Caspase-mediated cleavage of p21waf1/cip1 converts cancer cells from growth arrest to undergoing apoptosis. Oncogene 1999, 18, 1131–1138. [Google Scholar] [CrossRef]
- Kawata, S.; Ariumi, Y.; Shimotohno, K. P21(waf1/cip1/sdi1) prevents apoptosis as well as stimulates growth in cells transformed or immortalized by human t-cell leukemia virus type 1-encoded tax. J. Virol. 2003, 77, 7291–7299. [Google Scholar] [CrossRef]
- Biankin, A.V.; Kench, J.G.; Dijkman, F.P.; Biankin, S.A.; Henshall, S.M. Molecular pathogenesis of precursor lesions of pancreatic ductal adenocarcinoma. Pathology 2003, 35, 14–24. [Google Scholar]
- Weiss, R.H. P21waf1/cip1 as a therapeutic target in breast and other cancers. Cancer Cell 2003, 4, 425–429. [Google Scholar] [CrossRef]
- Gazitt, Y.; Reddy, S.V.; Alcantara, O.; Yang, J.; Boldt, D.H. A new molecular role for iron in regulation of cell cycling and differentiation of hl-60 human leukemia cells: Iron is required for transcription of p21(waf1/cip1) in cells induced by phorbol myristate acetate. J. Cell. Physiol. 2001, 187, 124–135. [Google Scholar] [CrossRef]
- Kramer, J.L.; Baltathakis, I.; Alcantara, O.S.; Boldt, D.H. Differentiation of functional dendritic cells and macrophages from human peripheral blood monocyte precursors is dependent on expression of p21 (waf1/cip1) and requires iron. Br. J. Haematol. 2002, 117, 727–734. [Google Scholar] [CrossRef]
- Fu, D.; Richardson, D.R. Iron chelation and regulation of the cell cycle: 2 Mechanisms of posttranscriptional regulation of the universal cyclin-dependent kinase inhibitor p21cip1/waf1 by iron depletion. Blood 2007, 110, 752–761. [Google Scholar] [CrossRef]
- Deshpande, A.; Sicinski, P.; Hinds, P.W. Cyclins and cdks in development and cancer: A perspective. Oncogene 2005, 24, 2909–2915. [Google Scholar] [CrossRef]
- Gao, J.; Richardson, D.R. The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents, iv: The mechanisms involved in inhibiting cell-cycle progression. Blood 2001, 98, 842–850. [Google Scholar] [CrossRef]
- Kulp, K.S.; Green, S.L.; Vulliet, P.R. Iron deprivation inhibits cyclin-dependent kinase activity and decreases cyclin d/cdk4 protein levels in asynchronous mda-mb-453 human breast cancer cells. Exp. Cell Res. 1996, 229, 60–68. [Google Scholar] [CrossRef]
- Nurtjahja-Tjendraputra, E.; Fu, D.; Phang, J.M.; Richardson, D.R. Iron chelation regulates cyclin d1 expression via the proteasome: A link to iron deficiency-mediated growth suppression. Blood 2007, 109, 4045–4054. [Google Scholar] [CrossRef]
- Masamha, C.P.; Benbrook, D.M. Cyclin d1 degradation is sufficient to induce g1 cell cycle arrest despite constitutive expression of cyclin e2 in ovarian cancer cells. Cancer Res. 2009, 69, 6565–6572. [Google Scholar] [CrossRef]
- Oexle, H.; Gnaiger, E.; Weiss, G. Iron-dependent changes in cellular energy metabolism: Influence on citric acid cycle and oxidative phosphorylation. Biochim. Biophys. Acta 1999, 1413, 99–107. [Google Scholar]
- King, A.; Selak, M.A.; Gottlieb, E. Succinate dehydrogenase and fumarate hydratase: Linking mitochondrial dysfunction and cancer. Oncogene 2006, 25, 4675–4682. [Google Scholar] [CrossRef]
- Wen, F.Q.; Jabbar, A.A.; Chen, Y.X.; Kazarian, T.; Patel, D.A.; Valentino, L.A. C-myc proto-oncogene expression in hemophilic synovitis: In vitro studies of the effects of iron and ceramide. Blood 2002, 100, 912–916. [Google Scholar] [CrossRef]
- Fan, L.; Iyer, J.; Zhu, S.; Frick, K.K.; Wada, R.K.; Eskenazi, A.E.; Berg, P.E.; Ikegaki, N.; Kennett, R.H.; Frantz, C.N. Inhibition of N-myc expression and induction of apoptosis by iron chelation in human neuroblastoma cells. Cancer Res. 2001, 61, 1073–1079. [Google Scholar]
- Shim, H.; Dolde, C.; Lewis, B.C.; Wu, C.S.; Dang, G.; Jungmann, R.A.; Dalla-Favera, R.; Dang, C.V. C-myc transactivation of ldh-a: Implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. USA 1997, 94, 6658–6663. [Google Scholar] [CrossRef]
- Keith, B.; Johnson, R.S.; Simon, M.C. Hif1α and hif2α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2012, 12, 9–22. [Google Scholar]
- Talks, K.L.; Turley, H.; Gatter, K.C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J.; Harris, A.L. The expression and distribution of the hypoxia-inducible factors hif-1α and hif-2α in normal human tissues, cancers, and tumor-associated macrophages. Am. J. Pathol. 2000, 157, 411–421. [Google Scholar] [CrossRef]
- Lidgren, A.; Hedberg, Y.; Grankvist, K.; Rasmuson, T.; Vasko, J.; Ljungberg, B. The expression of hypoxia-inducible factor 1α is a favorable independent prognostic factor in renal cell carcinoma. Clin. Cancer Res. 2005, 11, 1129–1135. [Google Scholar]
- Noguera, R.; Fredlund, E.; Piqueras, M.; Pietras, A.; Beckman, S.; Navarro, S.; Pahlman, S. Hif-1α and hif-2α are differentially regulated in vivo in neuroblastoma: High hif-1α correlates negatively to advanced clinical stage and tumor vascularization. Clin. Cancer Res. 2009, 15, 7130–7136. [Google Scholar] [CrossRef]
- Callapina, M.; Zhou, J.; Schnitzer, S.; Metzen, E.; Lohr, C.; Deitmer, J.W.; Brune, B. Nitric oxide reverses desferrioxamine- and hypoxia-evoked hif-1α accumulation—Implications for prolyl hydroxylase activity and iron. Exp. Cell Res. 2005, 306, 274–284. [Google Scholar] [CrossRef]
- Ellen, T.P.; Ke, Q.; Zhang, P.; Costa, M. Ndrg1, a growth and cancer related gene: Regulation of gene expression and function in normal and disease states. Carcinogenesis 2008, 29, 2–8. [Google Scholar] [CrossRef]
- Le, N.T.; Richardson, D.R. Iron chelators with high antiproliferative activity up-regulate the expression of a growth inhibitory and metastasis suppressor gene: A link between iron metabolism and proliferation. Blood 2004, 104, 2967–2975. [Google Scholar] [CrossRef]
- Guan, R.J.; Ford, H.L.; Fu, Y.; Li, Y.; Shaw, L.M.; Pardee, A.B. Drg-1 as a differentiation-related, putative metastatic suppressor gene in human colon cancer. Cancer Res. 2000, 60, 749–755. [Google Scholar]
- Bandyopadhyay, S.; Pai, S.K.; Gross, S.C.; Hirota, S.; Hosobe, S.; Miura, K.; Saito, K.; Commes, T.; Hayashi, S.; Watabe, M.; et al. The drg-1 gene suppresses tumor metastasis in prostate cancer. Cancer Res. 2003, 63, 1731–1736. [Google Scholar]
- Akiba, J.; Ogasawara, S.; Kawahara, A.; Nishida, N.; Sanada, S.; Moriya, F.; Kuwano, M.; Nakashima, O.; Yano, H. N-myc downstream regulated gene 1 (ndrg1)/cap43 enhances portal vein invasion and intrahepatic metastasis in human hepatocellular carcinoma. Oncol. Rep. 2008, 20, 1329–1335. [Google Scholar]
- Chua, M.S.; Sun, H.; Cheung, S.T.; Mason, V.; Higgins, J.; Ross, D.T.; Fan, S.T.; So, S. Overexpression of ndrg1 is an indicator of poor prognosis in hepatocellular carcinoma. Mod. Pathol. 2007, 20, 76–83. [Google Scholar] [CrossRef]
- Blatt, J.; Stitely, S. Antineuroblastoma activity of desferoxamine in human cell lines. Cancer Res. 1987, 47, 1749–1750. [Google Scholar]
- Foa, P.; Maiolo, A.T.; Lombardi, L.; Villa, L.; Polli, E.E. Inhibition of proliferation of human leukaemic cell populations by deferoxamine. Scand. J. Haematol. 1986, 36, 107–110. [Google Scholar]
- Reddel, R.R.; Hedley, D.W.; Sutherland, R.L. Cell cycle effects of iron depletion on t-47d human breast cancer cells. Exp. Cell Res. 1985, 161, 277–284. [Google Scholar] [CrossRef]
- Hodges, Y.K.; Antholine, W.E.; Horwitz, L.D. Effect on ribonucleotide reductase of novel lipophilic iron chelators: The desferri-exochelins. Biochem. Biophys. Res. Commun. 2004, 315, 595–598. [Google Scholar] [CrossRef]
- Kalinowski, D.S.; Richardson, D.R. The evolution of iron chelators for the treatment of iron overload disease and cancer. Pharmacol. Rev. 2005, 57, 547–583. [Google Scholar] [CrossRef]
- Keberle, H. The biochemistry of desferrioxamine and its relation to iron metabolism. Ann. N. Y. Acad. Sci. 1964, 119, 758–768. [Google Scholar] [CrossRef]
- Pullarkat, V.; Sehgal, A.; Li, L.; Meng, Z.; Lin, A.; Forman, S.; Bhatia, R. Deferasirox exposure induces reactive oxygen species and reduces growth and viability of myelodysplastic hematopoietic progenitors. Leuk. Res. 2012, 36, 966–973. [Google Scholar] [CrossRef]
- Ohyashiki, J.H.; Kobayashi, C.; Hamamura, R.; Okabe, S.; Tauchi, T.; Ohyashiki, K. The oral iron chelator deferasirox represses signaling through the mtor in myeloid leukemia cells by enhancing expression of redd1. Cancer Sci. 2009, 100, 970–977. [Google Scholar] [CrossRef]
- Kim, J.L.; Kang, H.N.; Kang, M.H.; Yoo, Y.A.; Kim, J.S.; Choi, C.W. The oral iron chelator deferasirox induces apoptosis in myeloid leukemia cells by targeting caspase. Acta Haematol. 2011, 126, 241–245. [Google Scholar] [CrossRef]
- Chantrel-Groussard, K.; Gaboriau, F.; Pasdeloup, N.; Havouis, R.; Nick, H.; Pierre, J.L.; Brissot, P.; Lescoat, G. The new orally active iron chelator icl670a exhibits a higher antiproliferative effect in human hepatocyte cultures than O-trensox. Eur. J. Pharmacol. 2006, 541, 129–137. [Google Scholar] [CrossRef]
- Torti, S.V.; Torti, F.M.; Whitman, S.P.; Brechbiel, M.W.; Park, G.; Planalp, R.P. Tumor cell cytotoxicity of a novel metal chelator. Blood 1998, 92, 1384–1389. [Google Scholar]
- Samuni, A.M.; Krishna, M.C.; DeGraff, W.; Russo, A.; Planalp, R.P.; Brechbiel, M.W.; Mitchell, J.B. Mechanisms underlying the cytotoxic effects of tachpyr—A novel metal chelator. Biochim. Biophys. Acta 2002, 1571, 211–218. [Google Scholar]
- Turner, J.; Koumenis, C.; Kute, T.E.; Planalp, R.P.; Brechbiel, M.W.; Beardsley, D.; Cody, B.; Brown, K.D.; Torti, F.M.; Torti, S.V. Tachpyridine, a metal chelator, induces g2 cell-cycle arrest, activates checkpoint kinases, and sensitizes cells to ionizing radiation. Blood 2005, 106, 3191–3199. [Google Scholar] [CrossRef]
- Baker, E.; Vitolo, M.L.; Webb, J. Iron chelation by pyridoxal isonicotinoyl hydrazone and analogues in hepatocytes in culture. Biochem. Pharmacol. 1985, 34, 3011–3017. [Google Scholar] [CrossRef]
- Richardson, D.R.; Hefter, G.T.; May, P.M.; Webb, J.; Baker, E. Iron chelators of the pyridoxal isonicotinoyl hydrazone class. III. Formation constants with calcium(II), magnesium(II) and zinc(II). Biol. Metals 1989, 2, 161–167. [Google Scholar]
- Kim, B.K.; Huebers, H.A.; Finch, C.A. Effectiveness of oral iron chelators assayed in the rat. Am. J. Hematol. 1987, 24, 277–284. [Google Scholar] [CrossRef]
- Hershko, C.; Avramovici-Grisaru, S.; Link, G.; Gelfand, L.; Sarel, S. Mechanism of in vivo iron chelation by pyridoxal isonicotinoyl hydrazone and other imino derivatives of pyridoxal. J. Lab. Clin. Med. 1981, 98, 99–108. [Google Scholar]
- Becker, E.M.; Lovejoy, D.B.; Greer, J.M.; Watts, R.; Richardson, D.R. Identification of the di-pyridyl ketone isonicotinoyl hydrazone (pkih) analogues as potent iron chelators and anti-tumour agents. Br. J. Pharmacol. 2003, 138, 819–830. [Google Scholar] [CrossRef]
- Thelander, L.; Graslund, A. Mechanism of inhibition of mammalian ribonucleotide reductase by the iron chelate of 1-formylisoquinoline thiosemicarbazone. Destruction of the tyrosine free radical of the enzyme in an oxygen-requiring reaction. J. Biol. Chem. 1983, 258, 4063–4066. [Google Scholar]
- DeConti, R.C.; Toftness, B.R.; Agrawal, K.C.; Tomchick, R.; Mead, J.A.; Bertino, J.R.; Sartorelli, A.C.; Creasey, W.A. Clinical and pharmacological studies with 5-hydroxy-2-formylpyridine thiosemicarbazone. Cancer Res. 1972, 32, 1455–1462. [Google Scholar]
- Gojo, I.; Tidwell, M.L.; Greer, J.; Takebe, N.; Seiter, K.; Pochron, M.F.; Johnson, B.; Sznol, M.; Karp, J.E. Phase I and pharmacokinetic study of triapine, a potent ribonucleotide reductase inhibitor, in adults with advanced hematologic malignancies. Leuk. Res. 2007, 31, 1165–1173. [Google Scholar] [CrossRef]
- Yuan, J.; Lovejoy, D.B.; Richardson, D.R. Novel di-2-pyridyl-derived iron chelators with marked and selective antitumor activity: In vitro and in vivo assessment. Blood 2004, 104, 1450–1458. [Google Scholar] [CrossRef]
- Whitnall, M.; Howard, J.; Ponka, P.; Richardson, D.R. A class of iron chelators with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics. Proc. Natl. Acad. Sci. USA 2006, 103, 14901–14906. [Google Scholar] [CrossRef]
- Eberhard, Y.; McDermott, S.P.; Wang, X.; Gronda, M.; Venugopal, A.; Wood, T.E.; Hurren, R.; Datti, A.; Batey, R.A.; Wrana, J.; et al. Chelation of intracellular iron with the antifungal agent ciclopirox olamine induces cell death in leukemia and myeloma cells. Blood 2009, 114, 3064–3073. [Google Scholar] [CrossRef]
- Roth, M.; Will, B.; Simkin, G.; Narayanagari, S.; Barreyro, L.; Bartholdy, B.; Tamari, R.; Mitsiades, C.S.; Verma, A.; Steidl, U. Eltrombopag inhibits the proliferation of leukemia cells via reduction of intracellular iron and induction of differentiation. Blood 2012, 120, 386–394. [Google Scholar] [CrossRef]
- Anastas, J.N.; Moon, R.T. Wnt signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef]
- Nelson, R.L. Iron and colorectal cancer risk: Human studies. Nutr. Rev. 2001, 59, 140–148. [Google Scholar] [CrossRef]
- Chua, A.C.; Klopcic, B.; Lawrance, I.C.; Olynyk, J.K.; Trinder, D. Iron: An emerging factor in colorectal carcinogenesis. World J. Gastroenterol. 2010, 16, 663–672. [Google Scholar] [CrossRef]
- Brooks, B.E.; Buchanan, S.K. Signaling mechanisms for activation of extracytoplasmic function (ecf) sigma factors. Biochim. Biophys. Acta 2008, 1778, 1930–1945. [Google Scholar]
- Song, S.; Christova, T.; Perusini, S.; Alizadeh, S.; Bao, R.Y.; Miller, B.W.; Hurren, R.; Jitkova, Y.; Gronda, M.; Isaac, M.; et al. Wnt inhibitor screen reveals iron dependence of β-catenin signaling in cancers. Cancer Res. 2011, 71, 7628–7639. [Google Scholar] [CrossRef]
- Coombs, G.S.; Schmitt, A.A.; Canning, C.A.; Alok, A.; Low, I.C.; Banerjee, N.; Kaur, S.; Utomo, V.; Jones, C.M.; Pervaiz, S.; et al. Modulation of wnt/beta-catenin signaling and proliferation by a ferrous iron chelator with therapeutic efficacy in genetically engineered mouse models of cancer. Oncogene 2012, 31, 213–225. [Google Scholar] [CrossRef]
- Daniels, T.R.; Bernabeu, E.; Rodriguez, J.A.; Patel, S.; Kozman, M.; Chiappetta, D.A.; Holler, E.; Ljubimova, J.Y.; Helguera, G.; Penichet, M.L. The transferrin receptor and the targeted delivery of therapeutic agents against cancer. Biochim. Biophys. Acta 2012, 1820, 291–317. [Google Scholar]
- Ohgami, R.S.; Campagna, D.R.; McDonald, A.; Fleming, M.D. The steap proteins are metalloreductases. Blood 2006, 108, 1388–1394. [Google Scholar] [CrossRef]
- Gomes, I.M.; Maia, C.J.; Santos, C.R. Steap proteins: From structure to applications in cancer therapy. Mol. Cancer Res. 2012, 10, 573–587. [Google Scholar] [CrossRef]
- Grunewald, T.G.; Bach, H.; Cossarizza, A.; Matsumoto, I. The steap protein family: Versatile oxidoreductases and targets for cancer immunotherapy with overlapping and distinct cellular functions. Biol. Cell 2012, 104, 641–657. [Google Scholar] [CrossRef]
- Becton, D.L.; Roberts, B. Antileukemic effects of deferoxamine on human myeloid leukemia cell lines. Cancer Res. 1989, 49, 4809–4812. [Google Scholar]
- Noulsri, E.; Richardson, D.R.; Lerdwana, S.; Fucharoen, S.; Yamagishi, T.; Kalinowski, D.S.; Pattanapanyasat, K. Antitumor activity and mechanism of action of the iron chelator, dp44mt, against leukemic cells. Am. J. Hematol. 2009, 84, 170–176. [Google Scholar] [CrossRef]
- Callens, C.; Coulon, S.; Naudin, J.; Radford-Weiss, I.; Boissel, N.; Raffoux, E.; Wang, P.H.; Agarwal, S.; Tamouza, H.; Paubelle, E.; et al. Targeting iron homeostasis induces cellular differentiation and synergizes with differentiating agents in acute myeloid leukemia. J. Exp. Med. 2010, 207, 731–750. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Luria-Perez, R.; Lopez-Valdes, H.E.; Casero, D.; Daniels, T.R.; Patel, S.; Avila, D.; Leuchter, R.; So, S.; Ortiz-Sanchez, E.; et al. Lethal iron deprivation induced by non-neutralizing antibodies targeting transferrin receptor 1 in malignant b cells. Leuk. Lymphoma 2011, 52, 2169–2178. [Google Scholar] [CrossRef]
- Fukushima, T.; Kawabata, H.; Nakamura, T.; Iwao, H.; Nakajima, A.; Miki, M.; Sakai, T.; Sawaki, T.; Fujita, Y.; Tanaka, M.; et al. Iron chelation therapy with deferasirox induced complete remission in a patient with chemotherapy-resistant acute monocytic leukemia. Anticancer Res. 2011, 31, 1741–1744. [Google Scholar]
- Estrov, Z.; Tawa, A.; Wang, X.H.; Dube, I.D.; Sulh, H.; Cohen, A.; Gelfand, E.W.; Freedman, M.H. In vitro and in vivo effects of deferoxamine in neonatal acute leukemia. Blood 1987, 69, 757–761. [Google Scholar]
- Yee, K.W.; Cortes, J.; Ferrajoli, A.; Garcia-Manero, G.; Verstovsek, S.; Wierda, W.; Thomas, D.; Faderl, S.; King, I.; O’Brien, S.M.; et al. Triapine and cytarabine is an active combination in patients with acute leukemia or myelodysplastic syndrome. Leuk. Res. 2006, 30, 813–822. [Google Scholar] [CrossRef]
- Choi, J.G.; Kim, J.L.; Park, J.; Lee, S.; Park, S.J.; Kim, J.S.; Choi, C.W. Effects of oral iron chelator deferasirox on human malignant lymphoma cells. Korean J. Hematol. 2012, 47, 194–201. [Google Scholar] [CrossRef]
- Vazana-Barad, L.; Granot, G.; Mor-Tzuntz, R.; Levi, I.; Dreyling, M.; Nathan, I.; Shpilberg, O. Mechanism of the antitumoral activity of deferasirox, an iron chelation agent, on mantle cell lymphoma. Leuk. Lymphoma 2013, 54, 851–859. [Google Scholar] [CrossRef]
- Maris, J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef]
- Blatt, J. Deferoxamine in children with recurrent neuroblastoma. Anticancer Res. 1994, 14, 2109–2112. [Google Scholar]
- Chaston, T.B.; Lovejoy, D.B.; Watts, R.N.; Richardson, D.R. Examination of the antiproliferative activity of iron chelators: Multiple cellular targets and the different mechanism of action of triapine compared with desferrioxamine and the potent pyridoxal isonicotinoyl hydrazone analogue 311. Clin. Cancer Res. 2003, 9, 402–414. [Google Scholar]
- Donfrancesco, A.; Deb, G.; Dominici, C.; Pileggi, D.; Castello, M.A.; Helson, L. Effects of a single course of deferoxamine in neuroblastoma patients. Cancer Res. 1990, 50, 4929–4930. [Google Scholar]
- Donfrancesco, A.; de Bernardi, B.; Carli, M.; Mancini, A.; Nigro, M.; de Sio, L.; Casale, F.; Bagnulo, S.; Helson, L.; Deb, G. Deferoxamine followed by cyclophosphamide, etoposide, carboplatin, thiotepa, induction regimen in advanced neuroblastoma: Preliminary results. Italian neuroblastoma cooperative group. Eur. J. Cancer 1995, 31A, 612–615. [Google Scholar]
- Lui, G.Y.; Obeidy, P.; Ford, S.J.; Tselepis, C.; Sharp, D.M.; Jansson, P.J.; Kalinowski, D.S.; Kovacevic, Z.; Lovejoy, D.B.; Richardson, D.R. The iron chelator, deferasirox, as a novel strategy for cancer treatment: Oral activity against human lung tumor xenografts and molecular mechanism of action. Mol. Pharmacol. 2013, 83, 179–190. [Google Scholar] [CrossRef]
- Ford, S.; Obeidy, P.; Lovejoy, D.; Bedford, M.; Nichols, L.; Chadwick, C.; Tucker, O.; Lui, G.; Kalinowski, D.; Jansson, P.; et al. Deferasirox (icl670a) effectively inhibits oesophageal cancer growth in vitro and in vivo. Br. J. Pharmacol. 2013, 168, 1316–1328. [Google Scholar] [CrossRef]
- Dixon, K.M.; Lui, G.Y.; Kovacevic, Z.; Zhang, D.; Yao, M.; Chen, Z.; Dong, Q.; Assinder, S.J.; Richardson, D.R. Dp44mt targets the akt, tgf-beta and erk pathways via the metastasis suppressor ndrg1 in normal prostate epithelial cells and prostate cancer cells. Br. J. Cancer 2013, 108, 409–419. [Google Scholar] [CrossRef]
- Rao, V.A.; Klein, S.R.; Agama, K.K.; Toyoda, E.; Adachi, N.; Pommier, Y.; Shacter, E.B. The iron chelator dp44mt causes DNA damage and selective inhibition of topoisomerase iiα in breast cancer cells. Cancer Res. 2009, 69, 948–957. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Heath, J.L.; Weiss, J.M.; Lavau, C.P.; Wechsler, D.S. Iron Deprivation in Cancer––Potential Therapeutic Implications. Nutrients 2013, 5, 2836-2859. https://doi.org/10.3390/nu5082836
Heath JL, Weiss JM, Lavau CP, Wechsler DS. Iron Deprivation in Cancer––Potential Therapeutic Implications. Nutrients. 2013; 5(8):2836-2859. https://doi.org/10.3390/nu5082836
Chicago/Turabian StyleHeath, Jessica L., Joshua M. Weiss, Catherine P. Lavau, and Daniel S. Wechsler. 2013. "Iron Deprivation in Cancer––Potential Therapeutic Implications" Nutrients 5, no. 8: 2836-2859. https://doi.org/10.3390/nu5082836