Simple Sugar Intake and Hepatocellular Carcinoma: Epidemiological and Mechanistic Insight


Abstract
:1. Introduction
2. Insulin Resistance, Hyperinsulinemia and HCC
3. Obesity and HCC
4. NAFLD and HCC
5. Sugar Intake and Cancer Incidence
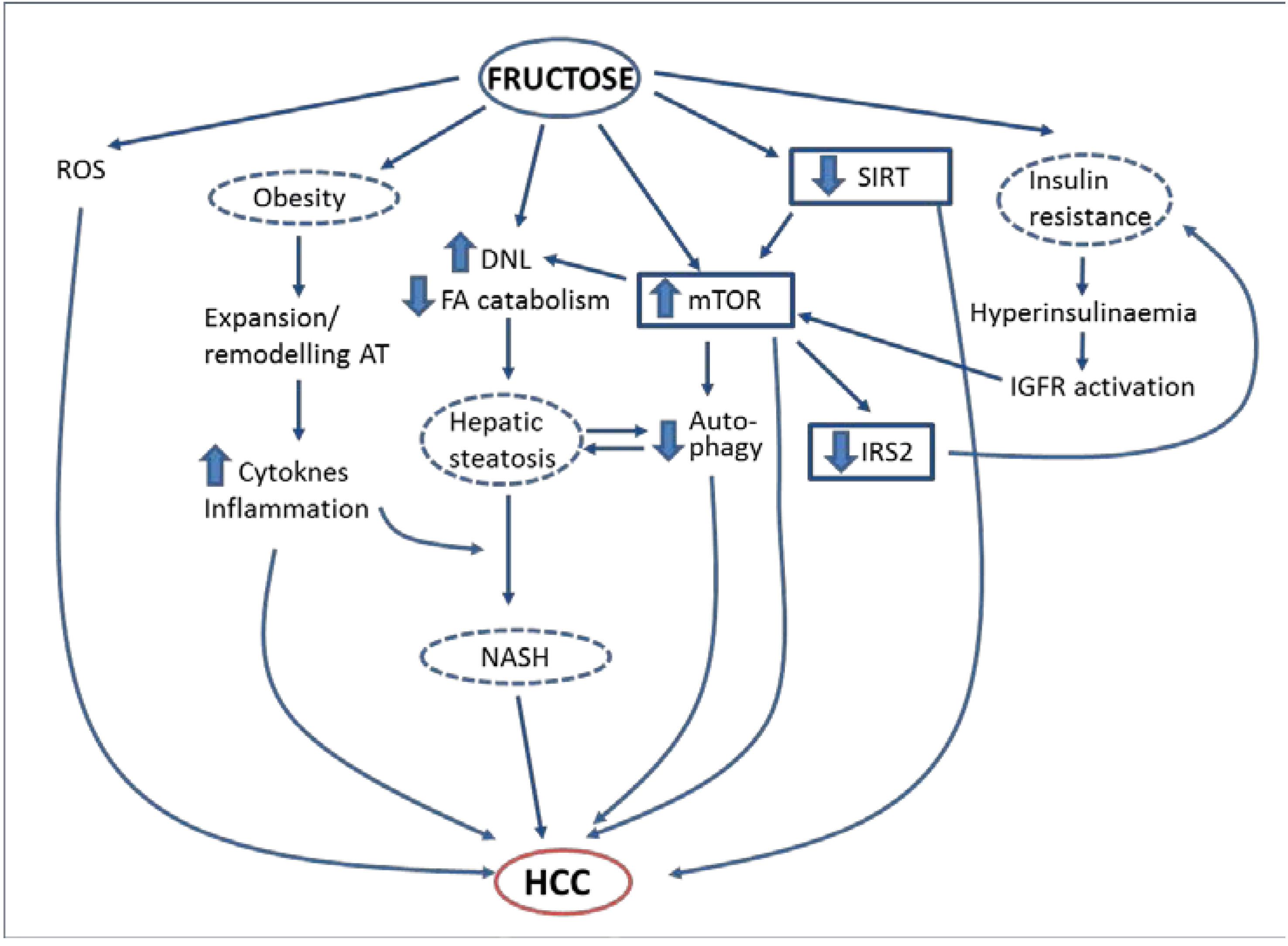
6. Simple Sugars, Insulin Resistance, Obesity, NAFLD and HCC: What Mechanisms Are Involved?

{kind=link}
| Authors | Type of Cancer | Study Characteristics | Sugar | Main Results |
|---|---|---|---|---|
| Aune et al. [39] | Pancreatic | Meta-analysis of ten cohort studies | GI, GL, TCH, S, F | Positive association with F intake (RR 1.22 per 25 g/day) |
| Bao et al. [43] | Pancreatic | Prospective study in 487,922 participants (NIH-AARP Diet and Health Study), 7.2 years follow-up | S, SSB | No association |
| Boyle et al. [44] | Cancer in general | Meta-analysis and review | SSB | No association |
| Chan et al. [45] | Pancreatic | Population based case (532) control (1701) study | SSB, F, L, SU | Positive association only with L (OR 2.0 comparing extreme quartiles) |
| Drake et al. [46] | Prostate | Prospective study in 8128 men | TCH, SSB | Positive association with SSB (HR 1.38 between highest and lowest consumption) |
| Fedirko et al. [41] | Liver and biliary tract | Prospective study in 477,206 participants | GI, GL, TCH | Positive association for TCH and HCC (HR: 1.45 per 50 g/day) |
| Gallus et al. [47] | Pancreatic | Combined data of a case (326) control (659) Italian study and other studies appearing before June, 2010 | SSB | No association |
| Genkinger et al. [48] | Pancreatic | Pooled analysis of 14 prospective cohort studies (853,894 participants) | SSB | Positive small association (RR 1.06) with SSB when modeled as a continuous variable |
| Jackson et al. [49] | Prostate | Case (243) control (273) study | RCH (including SSB) | Positive association (OR 2.02) between highest and lowest tertiles of consumption |
| Jiao et al. [38] | Pancreatic | Prospective study in 482,362 participants (NIH-AARP Diet and Health Study), 11 years follow-up | GI, GL, TCH, ST, F, G | Positive association with F (RR 1.35) and G (RR 1.29) between highest and lowest quintiles of consumption |
| Lagiou et al. [36] | HCC | Case (333) control (360) study | GL | Positive association (OR 1.92) between highest and lowest quintiles in patients with chronic HBV and/or HCV infection |
| Larsson et al. [50] | Pancreatic | Prospective study in 77,797 participants, 8 years follow-up | S, SSB | Positive association, highest with SSB (HR 1.93 between highest and lowest consumption) |
| Mueller et al. [51] | Pancreatic | Prospective study in 60,524 participants (Singapore Chinese Health Study), 14 years follow-up | SSB | Positive association (HR 2.0 between highest and lowest consumption) |
| Rossi et al. [37] | HCC | Case (185) control (412) study | GL | Positive association (OR 3.02) between highest and lowest quintiles |
| Schernhammer et al. [52] | Pancreatic | Prospective study in 138,158 participants, 20 years follow-up | SSB | Positive association in women with an underlying degree of insulin resistance |
| Schernhammer et al. [53] | Lymphoma and leukemia | Prospective study in 138,158 participants, 22 years follow-up | SSB | Positive association (RR 1.66) with higher consumption of SSB in men |
| Tasevska et al. [40] | Cancer in general | Prospective study in 435,674 participants (NIH-AARP Diet and Health Study), 7.2 years follow-up | S, F, SU | Positive association of F with small intestine cancer (HR 2.20 between highest and lowest quintiles of consumption); Negative association of all sugars with ovarian cancer |
| Tasevska et al. [42] | Cancer-related mortality | Prospective study in 353,751 participants (NIH-AARP Diet and Health Study), 13 years follow-up | S,F,SU | No association between intake of individual or added sugars with cancer mortality |
| Theodoratou et al. [54] | Colon | Case (2062) control (2778) study | SSB | Positive association |
| Witte et al. [55] | Breast | Familial matched case (140) control study | TCH, SSB | Positive association with SSB |
| Zhang et al. [56] | Colon | Pooled data from 13 cohort studies (731,441 participants), 6–20 years follow-up | SSB | No association |
6.1. Fructose and NAFLD
6.2. Fructose and Insulin Resistance
6.3. Fructose and Obesity
6.4. Other Mechanisms

7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ezzati, M.; Riboli, E. Behavioral and dietary risk factors for noncommunicable diseases. N. Engl. J. Med. 2013, 369, 954–964. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Zhang, C.; Yan, J.; Ruan, Y.; Zhao, X.; San, X.; Mao, Y.; Sun, Q.; Zhang, K.; Fan, Z. Diabetes mellitus is associated with hepatocellular carcinoma: a retrospective case-control study in hepatitis endemic area. PLoS One 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Jinjuvadia, R.; Patel, S.; Liangpunsakul, S. The association between metabolic syndrome and hepatocellular carcinoma: systemic review and meta-analysis. J. Clin. Gastroenterol. 2014, 48, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Atchison, E.; Gridley, G.; Carreon, J.; Leitzmann, M.; McGlynn, K. Risk of cancer in a large cohort of U.S. veterans with diabetes. Int. J. Cancer 2011, 128, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Turati, F.; Talamini, R.; Pelucchi, C.; Polesel, J.; Franceschi, S.; Crispo, A; Izzo, F.; La Vecchia, C.; Boffetta, P.; Montella, M. Metabolic syndrome and hepatocellular carcinoma risk. Br. J. Cancer 2013, 108, 222–228. [Google Scholar] [CrossRef]
- Hosokawa, T.; Kurosaki, M.; Tsuchiya, K.; Matsuda, S.; Muraoka, M.; Suzuki, Y.; Tamaki, N.; Yasui, Y.; Nakata, T.; Nishimura, T.; et al. Hyperglycemia is a significant prognostic factor of hepatocellular carcinoma after curative therapy. World J. Gastroenterol. 2013, 19, 249–257. [Google Scholar] [CrossRef]
- Miao Jonasson, J.; Cederholm, J.; Eliasson, B.; Zethelius, B.; Eeg-Olofsson, K.; Gudbjörnsdottir, S. HbA1C and cancer risk in patients with type 2 diabetes—A nationwide population-based prospective cohort study in Sweden. PLoS One 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Seshasai, S.R.K.; Kaptoge, S.; Thompson, A.; Di Angelantonio, E.; Gao, P.; Sarwar, N.; Whincup, P.H.; Mukamal, K.J.; Gillum, R.F.; Holme, I.; et al. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N. Engl. J. Med. 2011, 364, 829–841. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Giovannucci, E.; Pollak, M.; Leavitt, A.; Tao, Y.; Gaziano, J.M.; Stampfer, M.J. A prospective study of plasma C-peptide and colorectal cancer risk in men. J. Natl. Cancer Inst. 2004, 96, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Li, H.; Giovannucci, E.; Mucci, L.; Qiu, W.; Nguyen, P.L.; Gaziano, J.M.; Pollak, M.; Stampfer, M.J. Prediagnostic body-mass index, plasma C-peptide concentration, and prostate cancer-specific mortality in men with prostate cancer: A long-term survival analysis. Lancet Oncol. 2008, 9, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, A.; Bell, J.R.; Chen, E.Y.; Herrera, R.; Petruzzelli, L.M.; Dull, T.J.; Gray, A.; Coussens, L.; Liao, Y.C.; Tsubokawa, M. Human insulin receptor and its relationship to the tyrosine kinase family of oncogenes. Nature 1985, 313, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat. Rev. Cancer 2012, 12, 159–169. [Google Scholar] [PubMed]
- Thissen, J.P.; Ketelslegers, J.M.; Underwood, L.E. Nutritional regulation of the insulin-like growth factors. Endocr. Rev. 1994, 15, 80–101. [Google Scholar] [PubMed]
- Djiogue, S.; Nwabo Kamdje, A.H.; Vecchio, L.; Kipanyula, M.J.; Farahna, M.; Aldebasi, Y.; Seke Etet, P.F. Insulin resistance and cancer: The role of insulin and IGFs. Endocr. Relat. Cancer 2013, 20, 1–17. [Google Scholar] [CrossRef] [PubMed]
- McCarty, M.F. mTORC1 activity as a determinant of cancer risk—Rationalizing the cancer-preventive effects of adiponectin, metformin, rapamycin, and low-protein vegan diets. Med. Hypotheses 2011, 77, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Jalving, M.; Gietema, J.A.; Lefrandt, J.D.; de Jong, S.; Reyners, A.K.L.; Gans, R.O.B.; de Vries, E.G.E. Metformin: Taking away the candy for cancer? Eur. J. Cancer 2010, 46, 2369–2380. [Google Scholar] [CrossRef] [PubMed]
- Ben Sahra, I.; Le Marchand-Brustel, Y.; Tanti, J.-F.; Bost, F. Metformin in cancer therapy: A new perspective for an old antidiabetic drug? Mol. Cancer Ther. 2010, 9, 1092–1099. [Google Scholar] [CrossRef] [PubMed]
- Basen-Engquist, K.; Chang, M. Obesity and cancer risk: Recent review and evidence. Curr. Oncol. Rep. 2011, 13, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Borena, W.; Strohmaier, S.; Lukanova, A.; Bjørge, T.; Lindkvist, B.; Hallmans, G.; Edlinger, M.; Stocks, T.; Nagel, G.; Manjer, J.; et al. Metabolic risk factors and primary liver cancer in a prospective study of 578,700 adults. Int. J. Cancer 2012, 131, 193–200. [Google Scholar] [CrossRef]
- Schlesinger, S.; Aleksandrova, K.; Pischon, T.; Jenab, M.; Fedirko, V.; Trepo, E.; Overvad, K.; Roswall, N.; Tjønneland, A.; Boutron-Ruault, M.C.; et al. Diabetes mellitus, insulin treatment, diabetes duration, and risk of biliary tract cancer and hepatocellular carcinoma in a European cohort. Ann. Oncol. 2013, 24, 2449–2455. [Google Scholar] [CrossRef]
- Karagozian, R.; Derdák, Z.; Baffy, G. Obesity-associated mechanisms of hepatocarcinogenesis. Metabolism 2014, 63, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Calle, E.E.; Rodriguez, C.; Walker-thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Wolk, A. Overweight, obesity and risk of liver cancer: A meta-analysis of cohort studies. Br. J. Cancer 2007, 97, 1005–1008. [Google Scholar] [PubMed]
- Bhaskaran, K.; Douglas, I.; Forbes, H.; dos-Santos-Silva, I.; Leon, D.A.; Smeeth, L. Body-mass index and risk of 22 specific cancers: A population-based cohort study of 5.24 million UK adults. Lancet 2014, 384, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Liu, J.; Yang, H.I.; Lee, M.H.; Lu, S.N.; Wang, L.Y.; Iloeje, U.H.; You, S.L.; Brenner, D.; Chen, C.-J. Synergistic effects of family history of hepatocellular carcinoma and hepatitis B virus infection on risk for incident hepatocellular carcinoma. Clin. Gastroenterol Hepatol. 2013, 11, 1636–1645. [Google Scholar] [CrossRef]
- Nomura, K.; Yamanouchi, T. The role of fructose-enriched diets in mechanisms of nonalcoholic fatty liver disease. J. Nutr. Biochem. 2012, 23, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Rosmorduc, O.; Fartoux, L. HCC and NASH: How strong is the clinical demonstration? Clin. Res. Hepatol. Gastroenterol. 2012, 36, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.R.; Mohanty, S.R. Nonalcoholic fatty liver disease: A review and update. Dig. Dis. Sci. 2010, 55, 560–578. [Google Scholar] [CrossRef] [PubMed]
- Tokushige, K.; Hashimoto, E.; Horie, Y.; Taniai, M.; Higuchi, S. Hepatocellular carcinoma in Japanese patients with nonalcoholic fatty liver disease, alcoholic liver disease, and chronic liver disease of unknown etiology: report of the nationwide survey. J. Gastroenterol. 2011, 46, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-L.; Yang, H.-I.; Yang, W.-S.; Liu, C.-J.; Chen, P.-J.; You, S.-L.; Wang, L.-Y.; Sun, C.-A.; Lu, S.-N.; Chen, D.-S.; et al. Metabolic factors and risk of hepatocellular carcinoma by chronic hepatitis B/C infection: A follow-up study in Taiwan. Gastroenterology 2008, 135, 111–121. [Google Scholar] [CrossRef]
- Baffy, G.; Brunt, E.M.; Caldwell, S.H. Hepatocellular carcinoma in non-alcoholic fatty liver disease: An emerging menace. J. Hepatol. 2012, 56, 1384–1391. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Heaney, A.P. Refined fructose and cancer. Expert Opin. Ther. Targets 2011, 15, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Port, A.M.; Ruth, M.R.; Istfan, N.W. Fructose consumption and cancer: Is there a connection? Curr. Opin. Endocrinol. Diabetes Obes. 2012, 19, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Lagiou, P.; Rossi, M.; Tzonou, A; Georgila, C.; Trichopoulos, D.; La Vecchia, C. Glycemic load in relation to hepatocellular carcinoma among patients with chronic hepatitis infection. Ann. Oncol. 2009, 20, 1741–1745. [Google Scholar] [CrossRef]
- Rossi, M.; Lipworth, L.; Maso, L.D.; Talamini, R.; Montella, M.; Polesel, J.; McLaughlin, J.K.; Parpinel, M.; Franceschi, S.; Lagiou, P.; et al. Dietary glycemic load and hepatocellular carcinoma with or without chronic hepatitis infection. Ann. Oncol. 2009, 20, 1736–1740. [Google Scholar] [CrossRef]
- Jiao, L.; Flood, A.; Subar, A.F.; Hollenbeck, A.R.; Schatzkin, A.; Stolzenberg-Solomon, R. Glycemic index, carbohydrates, glycemic load, and the risk of pancreatic cancer in a prospective cohort study. Cancer Epidemiol. Biomark. Prev. 2009, 18, 1144–1151. [Google Scholar] [CrossRef]
- Aune, D.; Chan, D.S.M.; Vieira, A.R.; Navarro Rosenblatt, D.A.; Vieira, R.; Greenwood, D.C.; Cade, J.E.; Burley, V.J.; Norat, T. Dietary fructose, carbohydrates, glycemic indices and pancreatic cancer risk: A systematic review and meta-analysis of cohort studies. Ann. Oncol. 2012, 23, 2536–2546. [Google Scholar] [CrossRef] [PubMed]
- Tasevska, N.; Jiao, L.; Cross, A.J.; Kipnis, V.; Subar, A.F.; Hollenbeck, A.; Schatzkin, A.; Potischman, N. Sugars in diet and risk of cancer in the NIH-AARP Diet and Health Study. Int. J. Cancer 2012, 130, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Fedirko, V.; Lukanova, A; Bamia, C.; Trichopolou, A.; Trepo, E.; Nöthlings, U.; Schlesinger, S.; Aleksandrova, K.; Boffetta, P.; Tjønneland, A.; et al. Glycemic index, glycemic load, dietary carbohydrate, and dietary fiber intake and risk of liver and biliary tract cancers in Western Europeans. Ann. Oncol. 2013, 24, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Tasevska, N.; Park, Y.; Jiao, L.; Hollenbeck, A.; Subar, A.F.; Potischman, N. Sugars and risk of mortality in the NIH-AARP Diet and Health Study. Am. J. Clin. Nutr. 2014, 99, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Stolzenberg-solomon, R.; Jiao, L.; Silverman, D.T.; Subar, A.F.; Park, Y.; Michael, F. Added sugar and sugar-sweetened foods and beverages and the risk of pancreatic cancer in the National Institutes of Health—AARP. Am. J. Clin. Nutr. 2008, 88, 431–440. [Google Scholar] [PubMed]
- Boyle, P.; Koechlin, A.; Autier, P. Sweetened carbonated beverage consumption and cancer risk: Meta-analysis and review. Eur. J. Cancer Prev. 2014, 23, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.M.; Wang, F.; Holly, E.A. Sweets, sweetened beverages, and risk of pancreatic cancer in a large population-based case-control study. Cancer Causes Control 2009, 20, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Drake, I.; Sonestedt, E.; Gullberg, B.; Bjartell, A.; Wallstro, P.; Wirfa, E. Dietary intakes of carbohydrates in relation to prostate cancer risk : A prospective study in the Malmo. Am. J. Clin. Nutr. 2012, 96, 1409–1418. [Google Scholar] [CrossRef] [PubMed]
- Gallus, S.; Turati, F.; Tavani, A.; Polesel, J.; Talamini, R.; Franceschi, S.; La Vecchia, C. Soft drinks, sweetened beverages and risk of pancreatic cancer. Cancer Causes Control 2011, 22, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Genkinger, J.M.; Li, R.; Spiegelman, D.; Anderson, K.E.; Albanes, D.; Bergkvist, L.; Bernstein, L.; Black, A.; van den Brandt, P.A.; English, D.R.; et al. A Coffee, tea, and sugar-sweetened carbonated soft drink intake and pancreatic cancer risk: A pooled analysis of 14 cohort studies. Cancer Epidemiol. Biomark. Prev. 2012, 21, 305–318. [Google Scholar] [CrossRef]
- Jackson, M.; Tulloch-Reid, M.; Walker, S.; McFarlane-Anderson, N.; Bennett, F.; Francis, D.; Coard, K. Dietary patterns as predictors of prostate cancer in Jamaican men. Nutr. Cancer 2013, 65, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Bergkvist, L.; Wolk, A. Consumption of sugar and sugar-sweetened foods and the risk of pancreatic cancer in a prospective study. Am. J. Clin. Nutr. 2006, 84, 1171–1176. [Google Scholar] [PubMed]
- Mueller, N.T.; Odegaard, A.; Anderson, K.; Yuan, J.-M.; Gross, M.; Koh, W.-P.; Pereira, M.A. Soft drink and juice consumption and risk of pancreatic cancer: The Singapore Chinese health study. Cancer Epidemiol. Biomark. Prev. 2010, 19, 447–455. [Google Scholar] [CrossRef]
- Schernhammer, E.S.; Hu, F.B.; Giovannucci, E.; Michaud, D.S.; Colditz, G.A.; Stampfer, M.J.; Fuchs, C.S. Sugar-sweetened soft drink consumption and risk of pancreatic cancer in two prospective cohorts. Cancer Epidemiol. Biomark. Prev. 2005, 14, 2098–2105. [Google Scholar] [CrossRef]
- Schernhammer, E.S.; Bertrand, K.A.; Birmann, B.M.; Sampson, L.; Willett, W.C.; Feskanich, D. Consumption of artificial sweetener—And sugar-containing soda and risk of lymphoma and leukemia in men and women. Am. J. Clin. Nutr. 2012, 96, 1419–1428. [Google Scholar] [CrossRef] [PubMed]
- Theodoratou, E.; Farrington, S.M.; Tenesa, A.; McNeill, G.; Cetnarskyj, R.; Korakakis, E.; Din, F.V.N.; Porteous, M.E.; Dunlop, M.G.; Campbell, H. Associations between dietary and lifestyle risk factors and colorectal cancer in the Scottish population. Eur. J. Cancer Prev. 2014, 23, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Witte, J.; Ursin, G.; Siemiatycki, J.; Thompson, W.; Paganini-Hill, A.; Haile, R. Diet and premenopausal bilateral breast cancer: A case-control study. Breast Cancer Res. Treat. 1997, 42, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Albanes, D.; Beeson, W.L.; van den Brandt, P.A.; Buring, J.E.; Flood, A.; Freudenheim, J.L.; Giovannucci, E.L.; Goldbohm, R.A.; Jaceldo-Siegl, K.; et al. Risk of colon cancer and coffee, tea, and sugar-sweetened soft drink intake: Pooled analysis of prospective cohort studies. J. Natl. Cancer Inst. 2010, 102, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Roglans, N.; Vilà, L.; Farré, M.; Alegret, M.; Sánchez, R.M.; Vázquez-Carrera, M.; Laguna, J.C. Impairment of hepatic Stat-3 activation and reduction of PPARalpha activity in fructose-fed rats. Hepatology 2007, 45, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Vilà, L.; Roglans, N.; Alegret, M.; Sánchez, R.M.; Vázquez-Carrera, M.; Laguna, J.C. Suppressor of cytokine signaling-3 (SOCS-3) and a deficit of serine/threonine (Ser/Thr) phosphoproteins involved in leptin transduction mediate the effect of fructose on rat liver lipid metabolism. Hepatology 2008, 48, 1506–1516. [Google Scholar] [CrossRef] [PubMed]
- Vilà, L.; Roglans, N.; Perna, V.; Sánchez, R.M.; Vázquez-Carrera, M.; Alegret, M.; Laguna, J.C. Liver AMP/ATP ratio and fructokinase expression are related to gender differences in AMPK activity and glucose intolerance in rats ingesting liquid fructose. J. Nutr. Biochem. 2011, 22, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Igarashi, K.; Koeda, T.; Sugimoto, K.; Nakagawa, K.; Hayashi, S.; Yamaji, R.; Inui, H.; Fukusato, T.; Yamanouchi, T. Rats fed fructose-enriched diets have characteristics of nonalcoholic hepatic steatosis. J. Nutr. 2009, 139, 2067–2071. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-M.; Li, Y.-C.; Kong, L.-D.; Hu, Q.-H. Curcumin inhibits hepatic protein-tyrosine phosphatase 1B and prevents hypertriglyceridemia and hepatic steatosis in fructose-fed rats. Hepatology 2010, 51, 1555–1566. [Google Scholar] [CrossRef] [PubMed]
- Rebollo, A.; Roglans, N.; Baena, M.; Sánchez, R.M.; Merlos, M.; Alegret, M.; Laguna, J.C. Liquid fructose downregulates Sirt1 expression and activity and impairs the oxidation of fatty acids in rat and human liver cells. Biochim. Biophys. Acta 2014, 1841, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Le, K.-A.; Ith, M.; Kreis, R.; Faeh, D.; Bortolotti, M.; Tran, C.; Boesch, C.; Tappy, L. Fructose overconsumption causes dyslipidemia and ectopic lipid deposition in healthy subjects with and without a family history of. Am. J. Clin. Nutr. 2009, 89, 1760–1765. [Google Scholar] [CrossRef] [PubMed]
- Sobrecases, H.; Lê, K.; Bortolotti, M.; Schneiter, P.; Ith, M.; Kreis, R.; Boesch, C.; Tappy, L. Effects of short-term overfeeding with fructose, fat and fructose plus fat on plasma and hepatic lipids in healthy men. Diabetes Metab. 2010, 36, 244–246. [Google Scholar] [CrossRef] [PubMed]
- Maersk, M.; Belza, A.; Stødkilde-jørgensen, H.; Ringgaard, S.; Chabanova, E.; Thomsen, H.; Pedersen, S.B.; Astrup, A.; Richelsen, B. Sucrose-sweetened beverages increase fat storage in the liver, muscle, and visceral fat depot: A 6-mo randomized intervention study. Am. J. Clin. Nutr. 2012, 95, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Havel, P.J. Dietary Fructose: Implications for dysregulation of energy homeostasis and lipid/carbohydrate metabolism. Nutr. Rev. 2005, 63, 133–157. [Google Scholar] [CrossRef] [PubMed]
- Baena, M.; Sangüesa, G.; Hutter, N.; Sánchez, R.M.; Roglans, N.; Laguna, J.C.; Alegret, M. Fructose supplementation impairs rat liver autophagy through mTORC activation without inducing endoplasmic reticulum stress. Biochim. Biophys. Acta 2014, 1841, 107–116. [Google Scholar]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Lv, G.; Sheng, J.; Yang, Y. Effect of miRNA-10b in regulating cellular steatosis level by targeting PPAR-alpha expression, a novel mechanism for the pathogenesis of NAFLD. J. Gastroenterol. Hepatol. 2010, 25, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Frayn, K.N.; Kingman, M. Dietary sugars and lipid metabolsim in humans. Am. J. Clin. Nutr. 1995, 62, 250–263. [Google Scholar]
- Hirano, T.; Mamo, J.C.; Poapst, M.E.; Kuksis, A.; Steiner, G. Impaired very low-density lipoprotein-triglyceride catabolism in acute and chronic fructose-fed rats. Am. J. Physiol. Endocrinol. Metab. 1989, 256, 559–565. [Google Scholar]
- Mamo, J.C.; Hirano, T.; James, L.; Szeto, L.; Steiner, G. Partial characterization of the fructose-induced defect in very-low-density lipoprotein triglyceride metabolism. Metabolism 1991, 40, 888–893. [Google Scholar] [CrossRef] [PubMed]
- Bergheim, I.; Weber, S.; Vos, M.; Krämer, S.; Volynets, V.; Kaserouni, S.; McClain, C.J.; Bischoff, S.C. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: Role of endotoxin. J. Hepatol. 2008, 48, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Spruss, A.; Kanuri, G.; Wagnerberger, S.; Haub, S.; Bischoff, S.C.; Bergheim, I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology 2009, 50, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Spruss, A.; Kanuri, G.; Uebel, K.; Bischoff, S.C.; Bergheim, I. Role of the inducible nitric oxide synthase in the onset of fructose-induced steatosis in mice. Antioxid. Redox Signal. 2011, 14, 2121–2235. [Google Scholar] [CrossRef] [PubMed]
- Spruss, A.; Henkel, J.; Kanuri, G.; Blank, D.; Püschel, G.P.; Bischoff, S.C.; Bergheim, I. Female mice are more susceptible to nonalcoholic fatty liver disease: Sex-specific regulation of the hepatic AMP-activated protein kinase-plasminogen activator inhibitor 1 cascade, but not the hepatic endotoxin response. Mol. Med. 2012, 18, 1346–1355. [Google Scholar] [CrossRef] [PubMed]
- Kanuri, G.; Spruss, A.; Wagnerberger, S.; Bischoff, S.C.; Bergheim, I. Fructose-induced steatosis in mice: Role of plasminogen activator inhibitor-1, microsomal triglyceride transfer protein and NKT cells. Lab. Investig. 2011, 91, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Wagnerberger, S.; Spruss, A.; Kanuri, G.; Volynets, V.; Stahl, C.; Bischoff, S.C.; Bergheim, I. Toll-like receptors 1-9 are elevated in livers with fructose-induced hepatic steatosis. Br. J. Nutr. 2012, 107, 1727–1738. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalek, M.F.; Suzuki, A.; Guy, C.; Unalp-Arida, A.; Colvin, R.; Johnson, R.J.; Diehl, A.M. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Alegret, M.; Roglans, N.; Laguna, J.C. Fructose consumption and leptin resistance: What have we learnt from animal studies? 400. In Leptin: Hormonal Fuctions, Dysfunctions and Clinical Uses; Nova Science: Hauppaugue, NY, USA, 2011; pp. 209–230. [Google Scholar]
- Wang, Y.; Ausman, L.M.; Greenberg, A.S.; Russell, R.M.; Wang, X.-D. Nonalcoholic steatohepatitis induced by a high-fat diet promotes diethylnitrosamine-initiated early hepatocarcinogenesis in rats. Int. J. Cancer 2009, 124, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, F.T.; Luedde, T.; Singer, S.; Schmidt-supprian, M.; Baumgartl, J.; Schirmacher, P.; Pasparakis, M.; Bru, J.C. Hepatic NF-B essential modulator deficiency prevents obesity-induced insulin resistance but synergizes with high-fat feeding in tumorigenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 1297–1302. [Google Scholar] [CrossRef] [PubMed]
- Kumamoto, R.; Uto, H.; Oda, K.; Ibusuki, R.; Tanoue, S.; Arima, S.; Mawatari, S.; Kumagai, K.; Numata, M.; Tamai, T.; et al. Dietary fructose enhances the incidence of precancerous hepatocytes induced by administration of diethylnitrosamine in rat. Eur. J. Med. Res. 2013, 18. [Google Scholar] [CrossRef]
- Park, E.J.; Lee, J.H.; Yu, G.-Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Enzmann, H.; Ohlhauser, D.; Dettler, T.; Bannasch, P. Enhancement of hepatocarcinogenesis in rats by dietary fructose. Carcinogenesis 1989, 10, 1242–1252. [Google Scholar] [CrossRef]
- Dowman, J.K.; Hopkins, L.J.; Reynolds, G.M.; Nikolaou, N.; Armstrong, M.J.; Shaw, J.C.; Houlihan, D.D.; Lalor, P.F.; Tomlinson, J.W.; Hübscher, S.G.; et al. Development of hepatocellular carcinoma in a murine model of nonalcoholic steatohepatitis induced by use of a high-fat/fructose diet and sedentary lifestyle. Am. J. Pathol. 2014, 184, 1550–1561. [Google Scholar] [CrossRef] [PubMed]
- Rebollo, A.; Roglans, N.; Baena, M.; Padrosa, A.; Sánchez, R.M.; Merlos, M.; Alegret, M.; Laguna, J.C. Liquid fructose down-regulates liver insulin receptor substrate 2 and gluconeogenic enzymes by modifying nutrient sensing factors in rats. J. Nutr. Biochem. 2014, 25, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Boura-Halfon, S.; Zick, Y. Phosphorylation of IRS proteins , insulin action , and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2009, 296, 581–591. [Google Scholar] [CrossRef]
- Cota, D. Mammalian target of rapamycin complex 1 (mTORC1) signaling in energy balance and obesity. Physiol. Behav. 2009, 97, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Howell, J.J.; Manning, B.D. mTOR couples cellular nutrient sensing to organismal metabolic homeostasis. Trends Endocrinol. Metab. 2011, 22, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Sahin, F.; Kannangai, R.; Adegbola, O.; Wang, J.; Su, G.; Torbenson, M. mTOR and P70 S6 kinase expression in primary liver neoplasms mTOR and P70 S6 kinase expression in primary liver neoplasms. Clin. Cancer Res. 2004, 10, 8421–8425. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Huang, Y.; Li, J.; Wang, Z. The mTOR pathway is associated with the poor prognosis of human hepatocellular carcinoma. Med. Oncol. 2010, 27, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jin, W.; Jin, H.; Wang, X. mTOR in viral hepatitis and hepatocellular carcinoma: Function and treatment. Biomed Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Wang, C.; Ho, C.; Ladu, S.; Lee, S.A.; Mattu, S.; Destefanis, G.; Delogu, S.; Zimmermann, A.; Ericsson, J.; et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011, 140, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Kanzawa, T.; Sawaya, R.; Kondo, S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 2005, 5, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.-M.; Wang, G.-L.; Chen, L.; Xu, Y.-Y.; He, S.; Cao, X.-L.; Qin, J.; Zhou, J.-M.; Zhang, Y.-X.; Qun, E. The expression of beclin-1, an autophagic gene, in hepatocellular carcinoma associated with clinical pathological and prognostic significance. BMC Cancer 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Rebollo, A.; Roglans, N.; Alegret, M.; Laguna, J.C. Way back for fructose and liver metabolism: Bench side to molecular insights. World J. Gastroenterol. 2012, 18, 6552–6559. [Google Scholar] [CrossRef] [PubMed]
- Vilà, L.; Rebollo, A.; Ađalsteisson, G.S.; Alegret, M.; Merlos, M.; Roglans, N.; Laguna, J.C. Reduction of liver fructokinase expression and improved hepatic inflammation and metabolism in liquid fructose-fed rats after atorvastatin treatment. Toxicol. Appl. Pharmacol. 2011, 251, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Alzamendi, A.; Giovambattista, A.; García, M.E.; Rebolledo, O.R.; Gagliardino, J.J.; Spinedi, E. Effect of pioglitazone on the fructose-induced abdominal adipose tissue dysfunction. PPAR Res. 2012, 2012. [Google Scholar] [CrossRef]
- Crescenzo, R.; Bianco, F.; Coppola, P.; Mazzoli, A.; Valiante, S.; Liverini, G.; Iossa, S. Adipose tissue remodeling in rats exhibiting fructose-induced obesity. Eur. J. Nutr. 2014, 53, 413–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, E.; Tokushige, K. Hepatocellular carcinoma in non-alcoholic steatohepatitis: Growing evidence of an epidemic? Hepatol. Res. 2012, 42, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Naugler, W.E.; Sakurai, T.; Kim, S.; Maeda, S.; Kim, K.; Elsharkawy, A.M.; Karin, M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 2007, 317, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Veličković, N.; Djordjevic, A.; Vasiljević, A.; Bursać, B.; Milutinović, D.V.; Matić, G. Tissue-specific regulation of inflammation by macrophage migration inhibitory factor and glucocorticoids in fructose-fed Wistar rats. Br. J. Nutr. 2013, 110, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Baena, M.; Sangüesa, G.; Hutter, N.; Beltrán, J.M.; Roglans, N.; Alegret, M.; Laguna, J.C. Fructose induces mouse liver insulin resistance independently of the caloric intake. In Proceedings of the XIIth International Symposium on Insulin Receptors and Insulin Action, Barcelona, Spain, 7–9 November 2013.
- Feige, J.N.; Lagouge, M.; Canto, C.; Strehle, A.; Houten, S.M.; Milne, J.C.; Lambert, P.D.; Mataki, C.; Elliott, P.J.; Auwerx, J. Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 2008, 8, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Y.; Wang, D.H.; Yen, R.C.; Luo, J.; Gu, W.; Baylin, S.B. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell 2005, 123, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y.; Hong, D.; Nam, S.H.; Kim, J.M.; Paik, Y.H.; Joh, J.W.; Kwon, C.H.D.; Park, J.B.; Choi, G.-S.; Jang, K.Y.; et al. SIRT1 regulates oncogenesis via a mutant p53-dependent pathway in hepatocellular carcinoma. J. Hepatol. 2014, in press. [Google Scholar]
- Herranz, D.; Muñoz-Martin, M.; Cañamero, M.; Mulero, F.; Martinez-Pastor, B.; Fernandez-Capetillo, O.; Serrano, M. Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat. Commun. 2010, 1. [Google Scholar] [CrossRef]
- Ghosh, H.S.; McBurney, M.; Robbins, P.D. SIRT1 negatively regulates the mammalian target of rapamycin. PLoS One 2010, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upadhyay, M.; Samal, J.; Kandpal, M.; Singh, O.V.; Vivekanandan, P. The Warburg effect: Insights from the past decade. Pharmacol. Ther. 2013, 137, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Urtasun, R.; Conde de la Rosa, L.; Nieto, N. Oxidative and nitrosative stress and fibrogenic response. Clin. Liver Dis. 2008, 12, 769–790. [Google Scholar] [CrossRef] [PubMed]
- López-Lázaro, M. Excessive superoxide anion generation plays a key role in carcinogenesis. Int. J. Cancer 2007, 120, 1378–1380. [Google Scholar] [CrossRef] [PubMed]
- Lee, O.; Bruce, W.R.; Dong, Q.; Bruce, J.; Mehta, R.; O’Brien, P.J. Fructose and carbonyl metabolites as endogenous toxins. Chem. Biol. Interact. 2009, 178, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Kohli, R.; Kirby, M.; Xanthakos, S.A.; Softic, S.; Feldstein, A.E.; Tang, P.H.; Miles, L.; Miles, M.V.; Balistreri, W.F.; Stephen, C.; et al. High-fructose medium-chain-trans-fat diet induces liver fibrosis & elevates plasma coenzyme Q9 in a novel murine model of obesity and NASH. Hepatology 2011, 52, 934–944. [Google Scholar] [CrossRef]
- Bagul, P.K.; Middela, H.; Matapally, S.; Padiya, R.; Bastia, T.; Madhusudana, K.; Reddy, B.R.; Chakravarty, S.; Banerjee, S.K. Attenuation of insulin resistance, metabolic syndrome and hepatic oxidative stress by resveratrol in fructose-fed rats. Pharmacol. Res. 2012, 66, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Levi, B.; Werman, M.J. Fructose triggers DNA modification and damage in an Escherichia coli plasmid. J. Nutr. Biochem. 2001, 12, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Levi, B.; Werman, M.J. Fructose and related phosphate derivatives impose DNA damage and apoptosis in L5178Y mouse lymphoma cells. J. Nutr. Biochem. 2003, 14, 49–60. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laguna, J.C.; Alegret, M.; Roglans, N. Simple Sugar Intake and Hepatocellular Carcinoma: Epidemiological and Mechanistic Insight. Nutrients 2014, 6, 5933-5954. https://doi.org/10.3390/nu6125933
Laguna JC, Alegret M, Roglans N. Simple Sugar Intake and Hepatocellular Carcinoma: Epidemiological and Mechanistic Insight. Nutrients. 2014; 6(12):5933-5954. https://doi.org/10.3390/nu6125933
Chicago/Turabian StyleLaguna, Juan Carlos, Marta Alegret, and Núria Roglans. 2014. "Simple Sugar Intake and Hepatocellular Carcinoma: Epidemiological and Mechanistic Insight" Nutrients 6, no. 12: 5933-5954. https://doi.org/10.3390/nu6125933