Phylum Level Change in the Cecal and Fecal Gut Communities of Rats Fed Diets Containing Different Fermentable Substrates Supports a Role for Nitrogen as a Factor Contributing to Community Structure

Abstract
:1. Introduction
2. Experimental Section
2.1. Feeding Trial
2.2. SCFA and BCFA Analysis
2.3. Isolation of Community DNA
2.4. Gut Community 16S rRNA Gene Analysis
2.5. Metagenomic Analysis
2.6. Statistical Analysis
3. Results
3.1. Fecal and Cecal Community Metabolic Activity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fatty Acid | Cecal (%) | Fecal (%) | Fecal (μmol·gdw−1·y−1) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| C | WB | RS | C | WB | RS | C | WB | RS | |
| Acetic | 59.4 ± 0.6 | 53.7 ± 1.1 | 59.9 ± 0.8 | 73.0 ± 1.0 a | 72.6 ± 1.1 b | 57.5 ± 0.9 b | 48.0 ± 3.4 a | 103.5 ± 4.5 b | 79.7 ± 2.4 b |
| Propionic | 19.1 ± 0.2 a | 14.6 ± 0.3 b | 20.3 ± 0.6 a | 10.7 ± 0.2 a | 11.7 ± 0.6 a | 21.6 ± 0.9 b | 6.9 ± 0.5 a | 16.4 ± 0.8 b | 30.3 ± 1.7 b |
| Butyric | 12.8 ± 0.2 a | 25.3 ± 0.8 b | 11.2 ± 0.4 a | 8.2 ± 1.2 | 8.2 ± 0.8 | 10.8 ± 0.4 | 5.2 ± 1.0 a | 13.1 ± 1.8 b | 14.8 ± 0.6 b |
| Isobutyric | 2.3 ± 0.1 a | 1.7 ± 0.02 b | 1.8 ± 0.04 ab | 1.4 ± 0.2 a | 1.7 ± 0.1 b | 1.6 ± 0.0 ab | 1.0 ± 0.2 | 2.3 ± 0.2 | 2.2 ± 0.1 |
| Isovaleric | 3.0 ± 0.1 a | 2.0 ± 0.02 b | 1.8 ± 0.06 b | 3.4 ± 0.3 | 2.7 ± 0.2 | 2.8 ± 0.1 | 2.3 ± 0.2 | 3.9 ± 0.3 | 3.8 ± 0.1 |
| Valeric | 3.4 ± 0.1 | 2.6 ± 0.04 | 2.9 ± 0.1 | 2.5 ± 0.3 a | 2.5 ± 0.1 a | 3.9 ± 0.1 b | 1.6 ± 0.2 | 3.5 ± 0.3 | 5.4 ± 0.2 |
| Caproic | 0.03 ± 0.01 | 0.13 ± 0.05 | 1.4 ± 0.2 | 0.7 ± 0.2 a | 0.6 ± 0.1 a | 1.3 ± 0.1 b | 0.4 ± 0.1 | 1.0 ± 0.2 | 1.9 ± 0.2 |
| Heptanoic | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.6 ± 0.10 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.5 ± 0.1 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.7 ± 0.1 |
| Total | - | - | - | - | - | - | 65.5 ± 4.6 a | 143.7 ± 6.9 b | 138.9 ± 3.8 b |
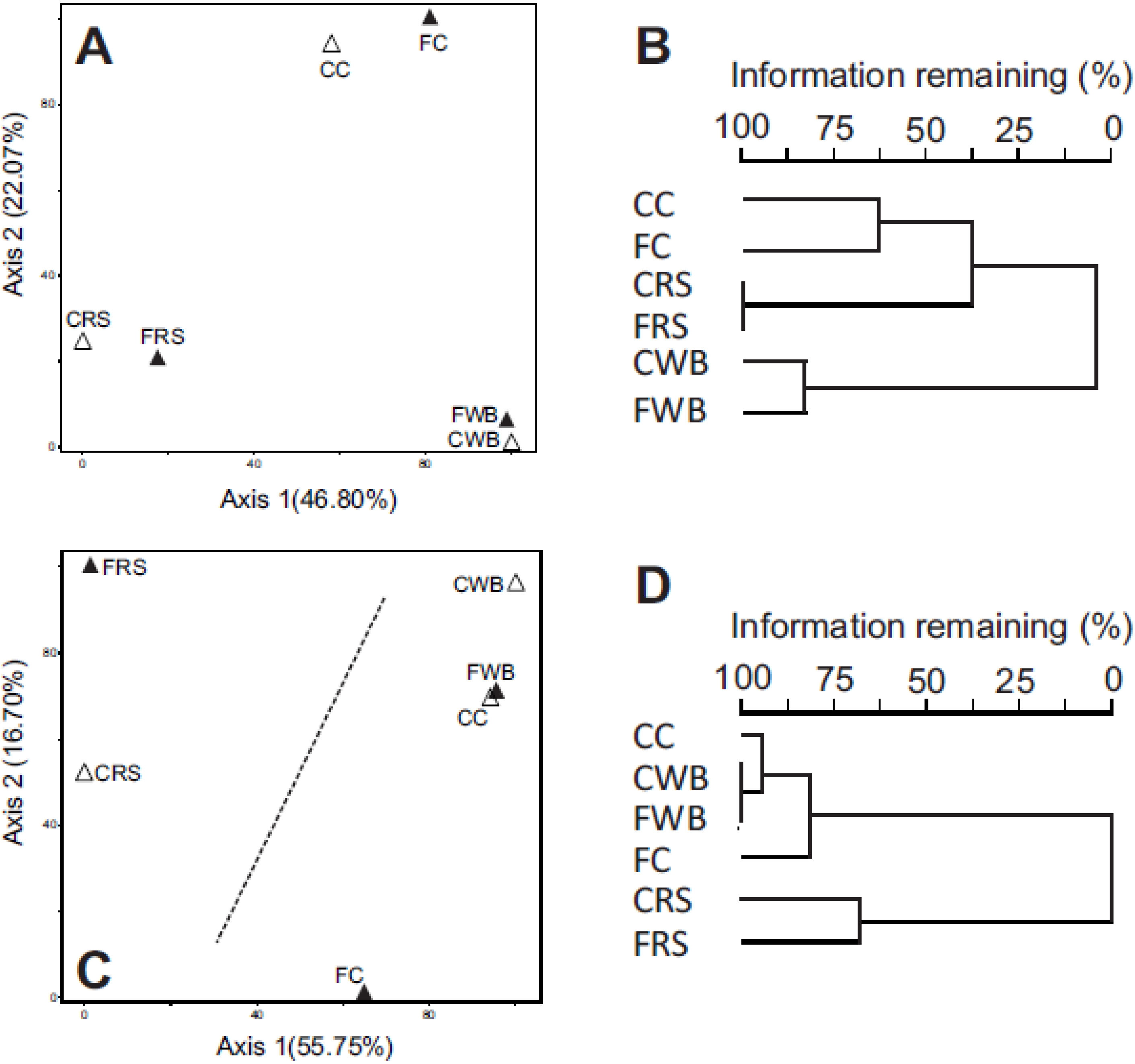
3.2. 16S rRNA Profiling of Fecal and Cecal Communities
| Index | Cecal | Fecal | ||||
|---|---|---|---|---|---|---|
| Community | C | WB | RS | C | WB | RS |
| Chao1 | 122 | 131 | 83 | 111 | 153 | 93 |
| Shannon index | 3.6 | 3.8 | 2.5 | 3.5 | 3.8 | 2.5 |

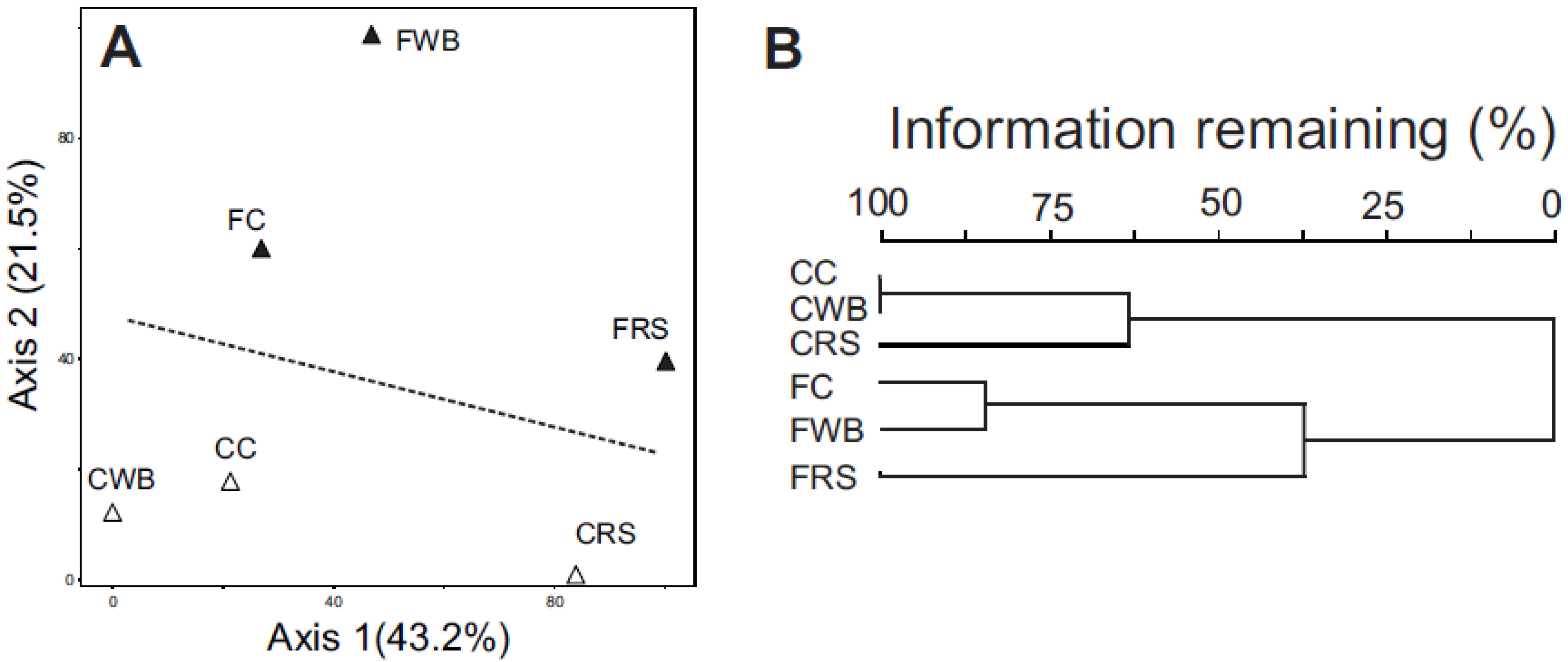
3.3. Taxonomic Profiling by Analysis of Community Shot Gun DNA Sequences

| Estimator | Cecal Contents | Feces | ||||
|---|---|---|---|---|---|---|
| C | WB | RS | C | WB | RS | |
| 16S rRNA 1 | 29 | 24 | 31 | 22 | 37 | 64 |
| MEGAN | 36 | 31 | 46 | 43 | 56 | 64 |
| MG-RAST | 28 | 24 | 39 | 34 | 41 | 56 |
| CAZymes | 33 | 27 | 39 | 40 | 53 | 53 |
| Average 2 | 32 | 27 | 41 | 39 | 50 | 58 |
3.4. Metagenome Profiling by Analysis of Community DNA Sequences


3.5. Potential Community Glycoside Hydrolase Capacity
4. Discussion
| CAZyme Family | Function | % Abundance | |||||
|---|---|---|---|---|---|---|---|
| CC | CWB | CRS | FC | FWB | FRS | ||
| GH3 | β-glucosidase, xylan 1,4-β-xylosidase, glucan 1,3-β-glucosidase, etc. | 6.0 | 4.5 | 3.9 | 7.8 | 6.0 | 5.9 |
| GH2 | β-galactosidase, β-glucuronidase, β-mannosidase, etc. | 5.2 | 4.6 | 3.6 | 5.8 | 4.7 | 3.2 |
| GH13 | α-amylase, pullulanase, etc. | 4.4 | 3.9 | 5.4 | 2.0 | 3.4 | 5.4 |
| GH23 | lysozyme type G, peptidoglycan lyase, chitinase. | 3.9 | 2.9 | 3.6 | 5.0 | 4.5 | 2.7 |
| GH43 | β-xylosidase, β-1,3-xylosidase, α-l-arabinofuranosidase, arabinanase, xylanase, etc. | 3.5 | 2.5 | 2.7 | 1.8 | 3.5 | 2.0 |
| GH31 | α-glucosidase, α-1,3-glucosidase, α-xylosidase, etc. | 2.3 | 1.9 | 2.9 | 1.3 | 1.7 | 1.2 |
| GH13|CBM26 | GH13 + starch binding module. | 1.1 | 0.4 | 3.1 | 0.3 | 1.3 | 3.7 |
| GH92 | α-mannosidase, α-1,2-Mannosidase, etc. | 1.8 | 0.4 | 1.0 | 1.4 | 1.3 | 1.7 |
| GH36 | α-galactosidase, α-N-acetylgalactosaminidase, stachyose synthase, raffinose synthase | 0.4 | 1.8 | 1.2 | 1.1 | 1.9 | 1.0 |
| GH73 | peptidoglycan hydrolase with endo-β-N-acetylglucosaminidase specificity. | 0.5 | 1.1 | 0.7 | 2.0 | 1.7 | 1.0 |
| GH97 | α-glucosidase, α-galactosidase. | 0.4 | 0.6 | 1.7 | 0.8 | 2.0 | 1.6 |
| GH94 | cellobiose phosphorylase, cellodextrin phosphorylase, chitobiose phosphorylase, etc. | 1.0 | 1.1 | 1.1 | 1.8 | 0.9 | 0.7 |
| GH1 | β-glucosidase , β-galactosidase, 6-P-β-glucosidase, β-glucuronidase, β-d-fucosidase, etc. | 1.2 | 0.8 | 0.5 | 2.5 | 0.6 | 0.2 |
| GH32 | invertase, endo-inulinase, endo-levanase, exo-inulinase, etc. | 1.2 | 1.4 | 0.0 | 1.3 | 0.8 | 0.8 |
| GH77 | amylomaltase or 4-α-glucanotransferase. | 1.0 | 1.3 | 1.2 | 0.6 | 0.2 | 0.5 |
| GH51 | α-l-arabinofuranosidase, endoglucanase | 0.9 | 0.9 | 1.0 | 0.5 | 0.5 | 0.9 |
| GH95 | α-1,2-l-fucosidase, α-l-fucosidase | 0.4 | 0.1 | 0.5 | 1.6 | 0.7 | 1.0 |
| GH20 | β-hexosaminidase, lacto-N-biosidase, β-1,6-N-acetylglucosaminidase, etc. | 0.7 | 0.6 | 0.2 | 1.2 | 1.1 | 0.5 |
| GH29 | α-l-fucosidase, α-1,3/1,4-l-fucosidase | 0.7 | 1.0 | 0.6 | 0.6 | 0.5 | 0.7 |
| GH25 | lysozyme | 1.1 | 1.0 | 0.4 | 0.4 | 0.5 | 0.8 |
| GH39 | α-l-iduronidase, β-xylosidase | 0.5 | 0.8 | 0.6 | 0.8 | 0.5 | 0.6 |
| GH18 | chitinase, lysozyme, endo-β-N-acetylglucosaminidase, peptidoglycan hydrolase, etc. | 0.9 | 0.4 | 0.5 | 0.6 | 0.6 | 0.8 |
| GH105 | unsaturated rhamnogalacturonyl hydrolase. | 0.8 | 0.1 | 0.6 | 0.8 | 0.5 | 0.8 |
| GH127 | β-l-arabinofuranosidase. | 1.5 | 0.0 | 0.4 | 0.6 | 0.8 | 0.2 |
| GH2|CBM32 | GH2 + binding to galactose, lactose, polygalacturonic acid, etc. | 0.0 | 0.1 | 0.1 | 0.9 | 1.4 | 0.8 |
| GH77|CBM20 | GH77 + The granular starch-binding function. | 0.1 | 0.6 | 0.7 | 0.5 | 0.6 | 0.7 |
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Walker, A.W.; Ince, J.; Duncan, S.H.; Webster, L.M.; Holtrop, G.; Ze, X.; Brown, D.; Stares, M.D.; Scott, P.; Bergerat, A.; et al. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 2011, 5, 220–230. [Google Scholar] [CrossRef]
- Abnous, K.; Brooks, S.P.; Kwan, J.; Matias, F.; Green-Johnson, J.; Selinger, L.B.; Thomas, M.; Kalmokoff, M. Diets enriched in oat bran or wheat bran temporally and differentially alter the composition of the fecal community of rats. J. Nutr. 2009, 139, 2024–2031. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.S.; Dowd, S.E.; Suchodolski, J.S.; Middelbos, I.S.; Vester, B.M.; Barry, K.A.; Nelson, K.E.; Torralba, M.; Henrissat, B.; Coutinho, P.M.; et al. Phylogenetic and gene-centric metagenomics of the canine intestinal microbiome reveals similarities with humans and mice. ISME J. 2011, 5, 639–649. [Google Scholar] [CrossRef]
- Hildebrandt, M.A.; Hoffmann, C.; Sherrill-Mix, S.A.; Keilbaugh, S.A.; Hamady, M.; Chen, Y.Y.; Knight, R.; Ahima, R.S.; Bushman, F.; Wu, G.D. High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology 2009, 137, 1716–1724.e2. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Hooda, S.; Boler, B.M.; Serao, M.C.; Brulc, J.M.; Staeger, M.A.; Boileau, T.W.; Dowd, S.E.; Fahey, G.C., Jr.; Swanson, K.S. 454 pyrosequencing reveals a shift in fecal microbiota of healthy adult men consuming polydextrose or soluble corn fiber. J. Nutr. 2012, 142, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Martinez, I.; Walter, J.; Keshavarzian, A.; Rose, D.J. In vitro characterization of the impact of selected dietary fibers on fecal microbiota composition and short chain fatty acid production. Anaerobe 2013, 23, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Martinez, I.; Kim, J.; Duffy, P.R.; Schlegel, V.L.; Walter, J. Resistant starches types 2 and 4 have differential effects on the composition of the fecal microbiota in human subjects. PLoS ONE 2010, 5, e15046. [Google Scholar] [CrossRef] [PubMed]
- Abell, G.C.; Cooke, C.M.; Bennett, C.N.; Conlon, M.A.; McOrist, A.L. Phylotypes related to Ruminococcus bromii are abundant in the large bowel of humans and increase in response to a diet high in resistant starch. FEMS Microbiol. Ecol. 2008, 66, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Abell, G.C.; Christophersen, C.T.; McOrist, A.L.; Clarke, J.M. Dietary resistant and butyrylated starches have different effects on the faecal bacterial flora of azoxymethane-treated rats. Br. J. Nutr. 2011, 105, 1480–1485. [Google Scholar] [CrossRef] [PubMed]
- Kalmokoff, M.; Zwicker, B.; O’Hara, M.; Matias, F.; Green, J.; Shastri, P.; Green-Johnson, J.; Brooks, S.P. Temporal change in the gut community of rats fed high amylose cornstarch is driven by endogenous urea rather than strictly on carbohydrate availability. J. Appl. Microbiol. 2013, 114, 1516–1528. [Google Scholar] [CrossRef]
- Klieve, A.V.; O’Leary, M.N.; McMillen, L.; Ouwerkerk, D. Ruminococcus bromii, identification and isolation as a dominant community member in the rumen of cattle fed a barley diet. J. Appl. Microbiol. 2007, 103, 2065–2073. [Google Scholar] [CrossRef]
- Bouhnik, Y.; Vahedi, K.; Achour, L.; Attar, A.; Salfati, J.; Pochart, P.; Marteau, P.; Flourie, B.; Bornet, F.; Rambaud, J.C. Short-chain fructo-oligosaccharide administration dose-dependently increases fecal bifidobacteria in healthy humans. J. Nutr. 1999, 129, 113–116. [Google Scholar] [PubMed]
- Depeint, F.; Tzortzis, G.; Vulevic, J.; I’Anson, K.; Gibson, G.R. Prebiotic evaluation of a novel galactooligosaccharide mixture produced by the enzymatic activity of Bifidobacterium bifidum NCIMB 41171, in healthy humans: A randomized, double-blind, crossover, placebo-controlled intervention study. Am. J. Clin. Nutr. 2008, 87, 785–791. [Google Scholar] [PubMed]
- Gostner, A.; Blaut, M.; Schaffer, V.; Kozianowski, G.; Theis, S.; Klingeberg, M.; Dombrowski, Y.; Martin, D.; Ehrhardt, S.; Taras, D.; et al. Effect of isomalt consumption on faecal microflora and colonic metabolism in healthy volunteers. Br. J. Nutr. 2006, 95, 40–50. [Google Scholar] [CrossRef]
- Finney, M.; Smullen, J.; Foster, H.A.; Brokx, S.; Storey, D.M. Effects of low doses of lactitol on faecal microflora, pH, short chain fatty acids and gastrointestinal symptomology. Eur. J. Nutr. 2007, 46, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Grieshop, C.M.; Flickinger, E.A.; Fahey, G.C., Jr. Oral administration of arabinogalactan affects immune status and fecal microbial populations in dogs. J. Nutr. 2002, 132, 478–482. [Google Scholar] [PubMed]
- Macfarlane, S.; Macfarlane, G.T.; Cummings, J.H. Review article: Prebiotics in the gastrointestinal tract. Aliment. Pharmacol. Ther. 2006, 24, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Everard, A.; Lazarevic, V.; Derrien, M.; Girard, M.; Muccioli, G.G.; Neyrinck, A.M.; Possemiers, S.; Van Holle, A.; Francois, P.; de Vos, W.M.; et al. Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes 2011, 60, 2775–2786. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- Duncan, S.H.; Lobley, G.E.; Holtrop, G.; Ince, J.; Johnstone, A.M.; Louis, P.; Flint, H.J. Human colonic microbiota associated with diet, obesity and weight loss. Int. J. Obes. (Lond.) 2008, 32, 1720–1724. [Google Scholar] [CrossRef]
- Roberfroid, M. Prebiotics: The concept revisited. J. Nutr. 2007, 137, 830S–837S. [Google Scholar] [PubMed]
- Herbeck, J.L.; Bryant, M.P. Nutritional features of the intestinal anaerobe Ruminococcus bromii. Appl. Microbiol. 1974, 28, 1018–1022. [Google Scholar] [PubMed]
- Faith, J.J.; McNulty, N.P.; Rey, F.E.; Gordon, J.I. Predicting a human gut microbiota’s response to diet in gnotobiotic mice. Science 2011, 333, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.H.; Macfarlane, G.T. The control and consequences of bacterial fermentation in the human colon. J. Appl. Bacteriol. 1991, 70, 443–459. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, G.T.; Cummings, J.H.; Allison, C. Protein degradation by human intestinal bacteria. J. Gen. Microbiol. 1986, 132, 1647–1656. [Google Scholar] [PubMed]
- Pridmore, R.D.; Berger, B.; Desiere, F.; Vilanova, D.; Barretto, C.; Pittet, A.C.; Zwahlen, M.C.; Rouvet, M.; Altermann, E.; Barrangou, R.; et al. The genome sequence of the probiotic intestinal bacterium Lactobacillus johnsonii NCC 533. Proc. Natl. Acad. Sci. USA 2004, 101, 2512–2517. [Google Scholar] [CrossRef]
- Bryant, M.P.; Robinson, I.M. Some nutritional characteristics of predominant culturable ruminal bacteria. J. Bacteriol. 1962, 84, 605–614. [Google Scholar] [PubMed]
- Dehority, B.A. Pectin-fermenting bacteria isolated from the bovine rumen. J. Bacteriol. 1969, 99, 189–196. [Google Scholar] [PubMed]
- Maczulak, A.E.; Dawson, K.A.; Baker, J.P. Nitrogen utilization in bacterial isolates from the equine cecum. Appl. Environ. Microbiol. 1985, 50, 1439–1443. [Google Scholar] [PubMed]
- Macy, J.M.; Probst, I. The biology of gastrointestinal Bacteroides. Annu. Rev. Microbiol. 1979, 33, 561–594. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, G.T.; Gibson, G.R.; Cummings, J.H. Comparison of fermentation reactions in different regions of the human colon. J. Appl. Bacteriol. 1992, 72, 57–64. [Google Scholar] [PubMed]
- Louis, P.; Flint, H.J. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 2009, 294, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Birkett, A.; Muir, J.; Phillips, J.; Jones, G.; O’Dea, K. Resistant starch lowers fecal concentrations of ammonia and phenols in humans. Am. J. Clin. Nutr. 1996, 63, 766–772. [Google Scholar]
- Wen, Z.; Morrison, M. The NAD(P)H-dependent glutamate dehydrogenase activities of Prevotella ruminicola b(1)4 can be attributed to one enzyme (GDHA), and gdhA expression is regulated in response to the nitrogen source available for growth. Appl. Environ. Microbiol. 1996, 62, 3826–3833. [Google Scholar]
- Reeves, P.G.; Nielsen, F.H.; Fahey, G.C., Jr. Ain-93 purified diets for laboratory rodents: Final report of the american institute of nutrition ad hoc writing committee on the reformulation of the AIN-76A rodent diet. J. Nutr. 1993, 123, 1939–1951. [Google Scholar] [PubMed]
- Scheppach, W.M.; Fabian, C.E.; Kasper, H.W. Fecal short-chain fatty acid (SCFA) analysis by capillary gas-liquid chromatography. Am. J. Clin. Nutr. 1987, 46, 641–646. [Google Scholar] [PubMed]
- Gouy, M.; Guindon, S.; Gascuel, O. Seaview version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Cai, Y.; Liu, L.; Yu, F.; Farrell, M.L.; McKendree, W.; Farmerie, W. Esprit: Estimating species richness using large collections of 16s rna pyrosequences. Nucleic Acids Res. 2009, 37, e76. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glockner, F.O. The silva and “all-species living tree project (ltp)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.R.; Chai, B.; Marsh, T.L.; Farris, R.J.; Wang, Q.; Kulam, S.A.; Chandra, S.; McGarrell, D.M.; Schmidt, T.M.; Garrity, G.M.; et al. The ribosomal database project (rdp-ii): Previewing a new autoaligner that allows regular updates and the new prokaryotic taxonomy. Nucleic Acids Res. 2003, 31, 442–443. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Mitra, S.; Ruscheweyh, H.J.; Weber, N.; Schuster, S.C. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011, 21, 1552–1560. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Begley, T.; Butler, R.M.; Choudhuri, J.V.; Chuang, H.Y.; de Cohoon, M.; Crécy-Lagard, V.; Diaz, N.; Disz, T.; Edwards, R.; et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005, 33, 5691–5702. [Google Scholar] [CrossRef] [PubMed]
- Glasman, S.; Seeman, T. Velvetoptimiser, v2.2.5. Victorian Bioinformatics Consortium: Melbourne, Australia, 2012. [Google Scholar]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZY) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed]
- McCune, B.; Mefford, M.J. PC-ORD: Multivariate Analysis of Ecological Data; MjM Software Design: Gleneden Beach, OR, USA, 1999. [Google Scholar]
- Beals, E.W. Bray-curtis ordination: An effective strategy for analysis of multivariate ecological data. Adv. Ecol. Res. 1984, 14, 1–55. [Google Scholar]
- Yu, Y.; Breitbart, M.; McNairnie, P.; Rohwer, F. Fastgroupii: A web-based bioinformatics platform for analyses of large 16s rdna libraries. BMC Bioinform. 2006, 7, 57. [Google Scholar]
- R Development Core Team R: A Language and Environment for Statistical Computing. Available online: http://www.R-project.org/ (accessed on 07 May 2014).
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A. The metagenomics rast server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Xu, W.; Yang, F.; Li, J.; Yang, Y.; Tang, X.; Mu, Y.; Zhou, J.; Huang, Z. Metagenomic analysis of the pygmy loris fecal microbiome reveals unique functional capacity related to metabolism of aromatic compounds. PLoS ONE 2013, 8, e56565. [Google Scholar] [CrossRef] [PubMed]
- Lamendella, R.; Domingo, J.W.; Ghosh, S.; Martinson, J.; Oerther, D.B. Comparative fecal metagenomics unveils unique functional capacity of the swine gut. BMC Microbiol. 2011, 11, 103. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, K.; Itoh, T.; Kuwahara, T.; Oshima, K.; Toh, H.; Toyoda, A.; Takami, H.; Morita, H.; Sharma, V.K.; Srivastava, T.P.; et al. Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res. 2007, 14, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Kalmokoff, M.; Allard, S.; Austin, J.; Whitford, M.; Hefford, M.; Teather, R. Biochemical and genetic characterization of the flagellar filaments from the rumen anaerobe Butyrivibrio fibrisolvens OR77. Anaerobe 2000, 6, 93–109. [Google Scholar] [CrossRef]
- Martin, J.H.; Savage, D.C. Cloning, nucleotide sequence, and taxonomic implications of the flagellin gene of Roseburia cecicola. J. Bacteriol. 1988, 170, 2612–2617. [Google Scholar] [PubMed]
- Mongeau, R.; Brooks, S.P.J. Dietary fibre: Properties and sources. In Encyclopaedia of Food Sciences and Nutrition; Academic Press: New York, NY, USA, 2003; pp. 1813–1823. [Google Scholar]
- Mongeau, R.; Sarwar, G.; Peace, R.W.; Brassard, R. Relationship between dietary fiber levels and protein digestibility in selected foods as determined in rats. Plant Foods Hum. Nutr. 1989, 39, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Inglis, G.D.; Thomas, M.C.; Thomas, D.K.; Kalmokoff, M.L.; Brooks, S.P.; Selinger, L.B. Molecular methods to measure intestinal bacteria: A review. J. AOAC Int. 2012, 95, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Stoddard, S.F.; Smith, B.J.; Hein, R.; Roller, B.R.; Schmidt, T.M. RNAdb: Improved tools for interpreting rRNA gene abundance in bacteria and archaea and a new foundation for future development. Nucleic Acids Res. 2015, 43, D593–D598. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.E.; Seipp, R.P.; Betts, M.; Hawkins, L.; Van Kessel, A.G.; Crosby, W.L.; Hemmingsen, S.M. Extensive profiling of a complex microbial community by high-throughput sequencing. Appl. Environ. Microbiol. 2002, 68, 3055–3066. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.; Zakrzewski, M.; Wibberg, D.; Eikmeyer, F.; Schluter, A.; Madamwar, D. Taxonomic profiling and metagenome analysis of a microbial community from a habitat contaminated with industrial discharges. Microb. Ecol. 2013, 66, 533–550. [Google Scholar] [CrossRef] [PubMed]
- Hanreich, A.; Schimpf, U.; Zakrzewski, M.; Schluter, A.; Benndorf, D.; Heyer, R.; Rapp, E.; Puhler, A.; Reichl, U.; Klocke, M. Metagenome and metaproteome analyses of microbial communities in mesophilic biogas-producing anaerobic batch fermentations indicate concerted plant carbohydrate degradation. Syst. Appl. Microbiol. 2013, 36, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.W.; Duncan, S.H.; Leitch, E.C.M.; Child, M.W.; Flint, H.J. pH and peptide supply can radically alter bacterial populations and short-chain fatty acid ratios within microbial communities from the human colon. Appl. Environ. Microbiol. 2005, 71, 3692–3700. [Google Scholar] [CrossRef] [PubMed]
- Teather, R.M.; Erfle, J.D.; Boila, R.J.; Sauer, F.D. Effect of dietary nitrogen on the rumen microbial population in lactating dairy cattle. J. Appl. Bacteriol. 1980, 49, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Hespell, R.; Smith, C. Utilization of nitrogen sources by gastrointestinal tract bacteria. In Human Intestinal Microflora in Health and Disease; Hentges, D.J., Ed.; Academic Press: New York, NY, USA, 1983; pp. 167–187. [Google Scholar]
- Pittman, K.A.; Bryant, M.P. Peptides and other nitrogen sources for growth of Bacteroides ruminicola. J. Bacteriol. 1964, 88, 401–410. [Google Scholar] [PubMed]
- Moore, W.E.; Holdeman, L.V. Human fecal flora: The normal flora of 20 japanese-hawaiians. Appl. Microbiol. 1974, 27, 961–979. [Google Scholar] [PubMed]
- Turnbaugh, P.J.; Gordon, J.I. The core gut microbiome, energy balance and obesity. J. Physiol. 2009, 587, 4153–4158. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Young, P.; Holtrop, G.; Flint, H.J. Diversity of human colonic butyrate-producing bacteria revealed by analysis of the butyryl-CoA:Acetate CoA-transferase gene. Environ. Microbiol. 2010, 12, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, G.T.; Macfarlane, S. Bacteria, colonic fermentation, and gastrointestinal health. J. AOAC Int. 2012, 95, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Islam, K.B.; Fukiya, S.; Hagio, M.; Fujii, N.; Ishizuka, S.; Ooka, T.; Ogura, Y.; Hayashi, T.; Yokota, A. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology 2011, 141, 1773–1781. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- De La Serre, C.B.; Ellis, C.L.; Lee, J.; Hartman, A.L.; Rutledge, J.C.; Raybould, H.E. Propensity to high-fat diet-induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G440–G448. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalmokoff, M.; Franklin, J.; Petronella, N.; Green, J.; Brooks, S.P.J. Phylum Level Change in the Cecal and Fecal Gut Communities of Rats Fed Diets Containing Different Fermentable Substrates Supports a Role for Nitrogen as a Factor Contributing to Community Structure. Nutrients 2015, 7, 3279-3299. https://doi.org/10.3390/nu7053279
Kalmokoff M, Franklin J, Petronella N, Green J, Brooks SPJ. Phylum Level Change in the Cecal and Fecal Gut Communities of Rats Fed Diets Containing Different Fermentable Substrates Supports a Role for Nitrogen as a Factor Contributing to Community Structure. Nutrients. 2015; 7(5):3279-3299. https://doi.org/10.3390/nu7053279
Chicago/Turabian StyleKalmokoff, Martin, Jeff Franklin, Nicholas Petronella, Judy Green, and Stephen P.J. Brooks. 2015. "Phylum Level Change in the Cecal and Fecal Gut Communities of Rats Fed Diets Containing Different Fermentable Substrates Supports a Role for Nitrogen as a Factor Contributing to Community Structure" Nutrients 7, no. 5: 3279-3299. https://doi.org/10.3390/nu7053279