Low-Density Lipoprotein Receptor-Related Protein 6 (LRP6) Is a Novel Nutritional Therapeutic Target for Hyperlipidemia, Non-Alcoholic Fatty Liver Disease, and Atherosclerosis

Abstract
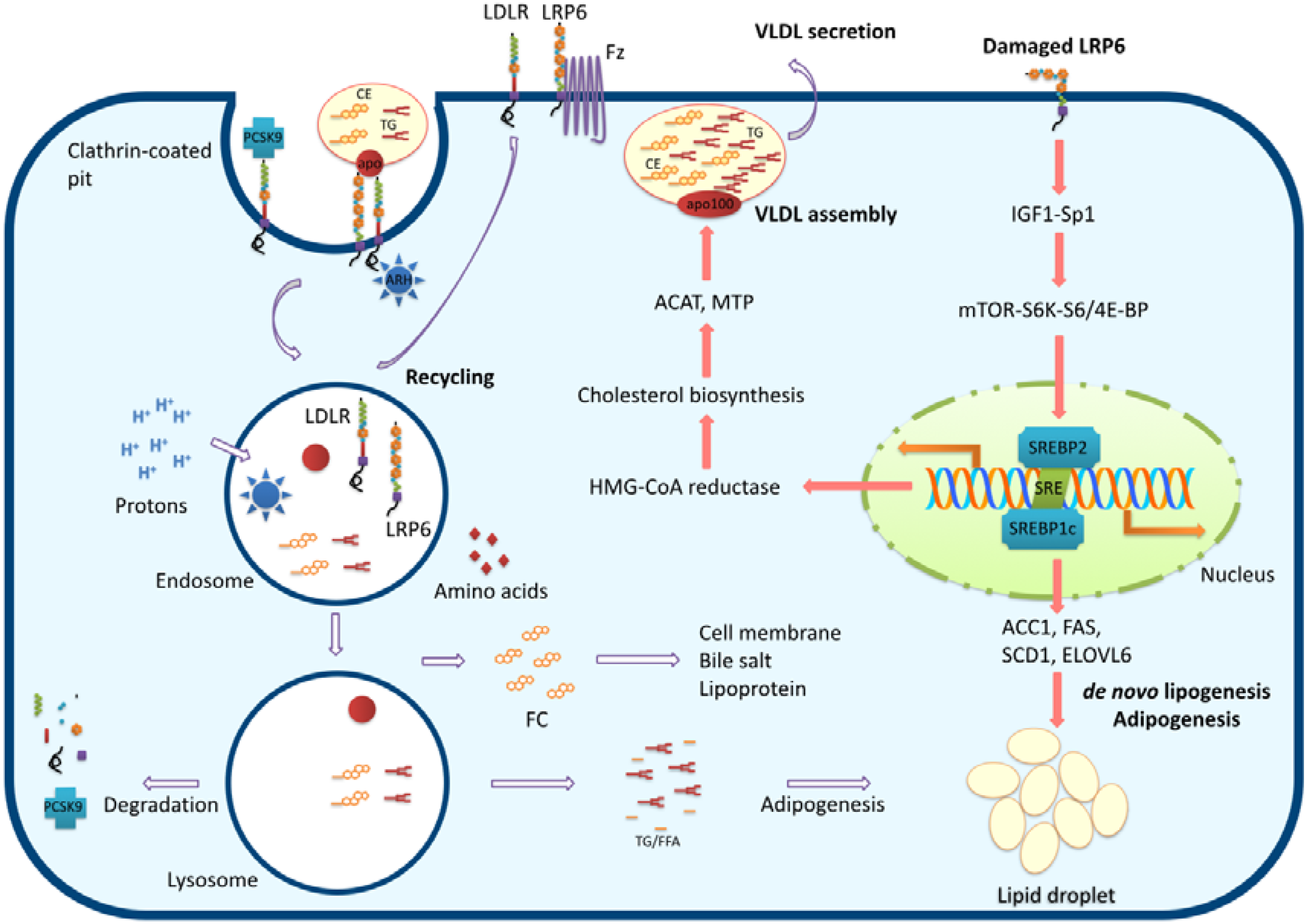
:1. Introduction to LRP6

2. Impaired Function of Mutant LRP6

{kind=link}
{kind=link}
| Disorders/Diseases | Phenotypes | Pathway | Reference |
|---|---|---|---|
| Hypertriglyceridemia | Increased serum TG levels Increased TG synthesis and VLDL secretion | IGF1-AKT-mTOR-SREBP1 | [3,20] |
| Hypercholesterolemia | Decreased LDL clearance in liver and peripheral tissues Increased hepatic synthesis of cholesterol and serum LDL levels | Insig1-SREBP2-HMGCR | [17,21] |
| Non-alcoholic fatty liver disease | Increased lipid/TG accumulation in liver Increased hepatic de novo lipogenesis and TG synthesis | MTP-apoB | [20] |
| Atherosclerosis | Early onset of atherosclerosis Increase of lesions and plagues in aorta | PDGF TCF4 | [3,16] |

3. LRP6 and Hyperlipidemia
3.1. LRP6 Conducts Cholesterol Homeostasis
3.2. Malfunctioning LRP6 Enhances Hepatic Lipogenesis via the Nutrient-Sensing Pathway
4. LRP6 and the Onset of Atherosclerosis
5. Conclusions
Acknowledgments
Abbreviations
| TG | triglyceride |
| VLDL | very low-density lipoprotein |
| LDL | low-density lipoprotein |
| IGF1 | insulin-like growth factor 1 |
| mTOR | mechanistic target of rapamycin |
| Insig1 | insulin induced gene 1 |
| SREBP, | sterol regulatory element-binding protein |
| MTP | microsomal triglyceride transfer protein |
| PDGF | Platelet-derived growth factor |
| TCF4 | transcription factor 4 |
Conflict of Interest
References
- Bilic, J.; Huang, Y.L.; Davidson, G.; Zimmermann, T.; Cruciat, C.M.; Bienz, M.; Niehrs, C. Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science 2007, 316, 1619–1622. [Google Scholar] [CrossRef] [PubMed]
- Cselenyi, C.S.; Jernigan, K.K.; Tahinci, E.; Thorne, C.A.; Lee, L.A.; Lee, E. LRP6 transduces a canonical Wnt signal independently of axin degradation by inhibiting GSK3’s phosphorylation of beta-catenin. Proc. Natl. Acad. Sci. USA 2008, 105, 8032–8037. [Google Scholar] [CrossRef] [PubMed]
- Mani, A.; Radhakrishnan, J.; Wang, H.; Mani, A.; Mani, M.-A.; Nelson-Williams, C.; Carew, K.S.; Mane, S.; Najmabadi, H.; Wu, D.; et al. LRP6 mutation in a family with early coronary disease and metabolic risk factors. Science 2007, 315, 1278–1282. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Liu, C.-C.; Thottassery, J.V.; Bu, G.; Li, Y. MESD is a universal inhibitor of Wnt coreceptors LRP5 and LRP6 and blocks Wnt/beta-catenin signaling in cancer cells. Biochemistry 2010, 49, 4635–4643. [Google Scholar] [CrossRef] [PubMed]
- Sladek, R.; Rocheleau, G.; Rung, J.; Dina, C.; Shen, L.; Serre, D.; Boutin, P.; Vincent, D.; Belisle, A.; Hadjadj, S.; et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 2007, 445, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Perez-Martinez, P.; Perez-Caballero, A.I.; Garcia-Rios, A.; Yubero-Serrano, E.M.; Camargo, A.; Gomez-Luna, M.J.; Marin, C.; Gomez-Luna, P.; Dembinska-Kiec, A.; Rodriguez-Cantalejo, F.; et al. Effects of rs7903146 variation in the TCF7L2 gene in the lipid metabolism of three different populations. PLoS ONE 2012, 7, e43390. [Google Scholar] [CrossRef] [PubMed]
- Muendlein, A.; Saely, C.H.; Geller-Rhomberg, S.; Sonderegger, G.; Rein, P.; Winder, T.; Beer, S.; Vonbank, A.; Drexel, H. Single nucleotide polymorphisms of TCF7L2 are linked to diabetic coronary atherosclerosis. PLoS ONE 2011, 6, e17978. [Google Scholar] [CrossRef] [PubMed]
- Tomaszewski, M.; Charchar, F.J.; Barnes, T.; Gawron-Kiszka, M.; Sedkowska, A.; Podolecka, E.; Kowalczyk, J.; Rathbone, W.; Kalarus, Z.; Grzeszczak, W.; et al. A common variant in low-density lipoprotein receptor-related protein 6 gene (LRP6) is associated with LDL-cholesterol. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1316–1321. [Google Scholar] [CrossRef] [PubMed]
- Huertas-Vazquez, A.; Plaisier, C.; Weissglas-Volkov, D.; Sinsheimer, J.; Canizales-Quinteros, S.; Cruz-Bautista, I.; Nikkola, E.; Herrera-Hernandez, M.; Davila-Cervantes, A.; Tusie-Luna, T.; et al. TCF7L2 is associated with high serum triacylglycerol and differentially expressed in adipose tissue in families with familial combined hyperlipidaemia. Diabetologia 2008, 51, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Sarzani, R.; Salvi, F.; Bordicchia, M.; Guerra, F.; Battistoni, I.; Pagliariccio, G.; Carbonari, L.; Dessì-Fulgheri, P.; Rappelli, A. Carotid artery atherosclerosis in hypertensive patients with a functional LDL receptor-related protein 6 gene variant. Nutr. Metab. Cardiovasc. Dis. 2011, 21, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.H.; Chang, Y.C.; Jiang, Y.D.; Chen, W.J.; Chang, T.J.; Kuo, S.S.; Lee, K.C.; Hsiao, P.C.; Chiu, K.C.; Chuang, L.M. Genetic variants of TCF7L2 are associated with insulin resistance and related metabolic phenotypes in taiwanese adolescents and caucasian young adults. J. Clin. Endocrinol. MeTable 2009, 94, 3575–3582. [Google Scholar] [CrossRef] [PubMed]
- Saxena, R.; Gianniny, L.; Burtt, N.P.; Lyssenko, V.; Giuducci, C.; Sjögren, M.; Florez, J.C.; Almgren, P.; Isomaa, B.; Orho-Melander, M.; et al. Common single nucleotide polymorphisms in TCF7L2 are reproducibly associated with type 2 diabetes and reduce the insulin response to glucose in nondiabetic individuals. Diabetes 2006, 55, 2890–2895. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kuusisto, J.; Vänttinen, M.; Kuulasmaa, T.; Lindström, J.; Tuomilehto, J.; Uusitupa, M.; Laakso, M. Variants of transcription factor 7-like 2 (TCF7L2) gene predict conversion to type 2 diabetes in the finnish diabetes prevention study and are associated with impaired glucose regulation and impaired insulin secretion. Diabetologia 2007, 50, 1192–1200. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Lista, J.; Perez-Martinez, P.; García-Rios, A.; Phillips, C.M.; Williams, C.M.; Gulseth, H.L.; Helal, O.; Blaak, E.E.; Kiec-Wilk, B.; Basu, S.; et al. Pleiotropic effects of TCF7L2 gene variants and its modulation in the metabolic syndrome: From the lipgene study. Atherosclerosis 2011, 214, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; de Aguiar, R.B.; Naik, S.; Mani, S.; Ostadsharif, K.; Wencker, D.; Sotoudeh, M.; Malekzadeh, R.; Sherwin, R.S.; Mani, A. LRP6 enhances glucose metabolism by promoting TCF7L2-dependent insulin receptor expression and IGF receptor stabilization in humans. Cell MeTable 2013, 17, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Keramati, A.R.; Singh, R.; Lin, A.; Faramarzi, S.; Ye, Z.-J.; Mane, S.; Tellides, G.; Lifton, R.P.; Mani, A. Wild-type LRP6 inhibits, whereas atherosclerosis-linked LRP6R611C increases PDGF-dependent vascular smooth muscle cell proliferation. Proc. Natl. Acad. Sci. USA 2011, 108, 1914–1918. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.J.; Go, G.W.; Singh, R.; Liu, W.Z.; Keramati, A.R.; Mani, A. LRP6 protein regulates low density lipoprotein (LDL) receptor-mediated LDL uptake. J. Biol. Chem. 2012, 287, 1335–1344. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Biechele, T.; Wei, Z.; Morrone, S.; Moon, R.T.; Wang, L.; Xu, W. Crystal structures of the extracellular domain of LRP6 and its complex with DKK1. Nat. Struct. Mol. Biol. 2011, 18, 1204–1210. [Google Scholar] [CrossRef] [PubMed]
- Bryja, V.; Andersson, E.R.; Schambony, A.; Esner, M.; Bryjová, L.; Biris, K.K.; Hall, A.C.; Kraft, B.; Cajanek, L.; Yamaguchi, T.P.; et al. The extracellular domain of Lrp5/6 inhibits noncanonical Wnt signaling in vivo. Mol. Biol. Cell 2009, 20, 924–936. [Google Scholar]
- Go, G.W.; Srivastava, R.; Hernandez-Ono, A.; Gang, G.; Smith, S.B.; Booth, C.J.; Ginsberg, H.N.; Mani, A. The combined hyperlipidemia caused by impaired Wnt-LRP6 signaling is reversed by Wnt3a rescue. Cell MeTable 2014, 19, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Mani, S.; Davis, N.R.; Sarrafzadegan, N.; Kavathas, P.B.; Mani, A. Mutation in EGFP domain of LDL receptor-related protein 6 impairs cellular LDL clearance. Circ. Res. 2008, 103, 1280–1288. [Google Scholar] [CrossRef] [PubMed]
- Register, T.C.; Hruska, K.A.; Divers, J.; Bowden, D.W.; Palmer, N.D.; Carr, J.J.; Wagenknecht, L.E.; Hightower, R.C.; Xu, J.; Smith, S.C.; et al. Plasma dickkopf1 (DKK1) concentrations negatively associate with atherosclerotic calcified plaque in african-americans with type 2 diabetes. J. Clin. Endocrinol. MeTable 2013, 98, E60–E65. [Google Scholar] [CrossRef] [PubMed]
- Goliasch, G.; Wiesbauer, F.; Kastl, S.P.; Katsaros, K.M.; Blessberger, H.; Maurer, G.; Schillinger, M.; Huber, K.; Wojta, J.; Speidl, W.S. Premature myocardial infarction is associated with low serum levels of Wnt-1. Atherosclerosis 2012, 222, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Morales-Santana, S.; García-Fontana, B.; García-Martín, A.; Rozas-Moreno, P.; García-Salcedo, J.A.; Reyes-García, R.; Muñoz-Torres, M. Atherosclerotic disease in type 2 diabetes is associated with an increase in sclerostin levels. Diabetes Care 2013, 36, 1667–1674. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Singh, R.; Choi, C.S.; Lee, H.Y.; Keramati, A.R.; Samuel, V.T.; Lifton, R.P.; Shulman, G.I.; Mani, A. Low density lipoprotein (LDL) receptor-related protein 6 (LRP6) regulates body fat and glucose homeostasis by modulating nutrient sensing pathways and mitochondrial energy expenditure. J. Biol. Chem. 2012, 287, 7213–7223. [Google Scholar] [CrossRef] [PubMed]
- Ballantyne, C.; Arroll, B.; Shepherd, J. Lipids and CVD management: Towards a global consensus. Eur. Heart J. 2005, 26, 2224–2231. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Salinas, C.; Gómez-Díaz, R.; Tusié-Luna, M.T. Fifty years studying hiperlipidemias: The case of familial combined hyperlipidemia. Invest. Clin. 2010, 51, 145–158. [Google Scholar] [PubMed]
- Brown, M.S.; Goldstein, J.L. Familial hypercholesterolemia: Defective binding of lipoproteins to cultured fibroblasts associated with impaired regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity. Proc. Natl. Acad. Sci. USA 1974, 71, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S.; Stone, N.J. Genetics of the LDL receptor: Evidence that the mutations affecting binding and internalization are allelic. Cell 1977, 12, 629–641. [Google Scholar] [CrossRef]
- Hobbs, H.H.; Brown, M.S.; Goldstein, J.L. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum. Mutat. 1992, 1, 445–466. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, S.; Brown, M.S.; Goldstein, J.L.; Gerard, R.D.; Hammer, R.E.; Herz, J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J. Clin. Invest. 1993, 92, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.K.; Wilund, K.; Arca, M.; Zuliani, G.; Fellin, R.; Maioli, M.; Calandra, S.; Bertolini, S.; Cossu, F.; Grishin, N.; Barnes, R.; Cohen, J.C.; Hobbs, H.H. Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science 2001, 292, 1394–1398. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.C.; Kiss, R.S.; Pertsemlidis, A.; Marcel, Y.L.; McPherson, R.; Hobbs, H.H. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science 2004, 305, 869–872. [Google Scholar] [CrossRef] [PubMed]
- Lifton, R.P.; Gharavi, A.G.; Geller, D.S. Molecular mechanisms of human hypertension. Cell 2001, 104, 545–556. [Google Scholar] [CrossRef]
- Gwack, Y.; Sharma, S.; Nardone, J.; Tanasa, B.; Iuga, A.; Srikanth, S.; Okamura, H.; Bolton, D.; Feske, S.; Hogan, P.G.; et al. A genome-wide drosophila RNAi screen identifies DYRK-family kinases as regulators of NFAT. Nature 2006, 441, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Bartz, F.; Kern, L.; Erz, D.; Zhu, M.; Gilbert, D.; Meinhof, T.; Wirkner, U.; Erfle, H.; Muckenthaler, M.; Pepperkok, R.; et al. Identification of cholesterol-regulating genes by targeted RNAi screening. Cell MeTable 2009, 10, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, M.; Fu, H.; Xu, H.; Wei, J.; Wang, T.; Wang, X. Mammalian target of the rapamycin pathway is involved in non-alcoholic fatty liver disease. Mol. Med. Rep. 2010, 3, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Mauvoisin, D.; Rocque, G.; Arfa, O.; Radenne, A.; Boissier, P.; Mounier, C. Role of the PI3-kinase/mtor pathway in the regulation of the stearoyl coa desaturase (SCD1) gene expression by insulin in liver. J. Cell Commun. Signal 2007, 1, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; Flowers, M.T.; Sampath, H.; Chu, K.; Otzelberger, C.; Liu, X.; Ntambi, J.M. Hepatic stearoyl-CoA desaturase-1 deficiency protects mice from carbohydrate-induced adiposity and hepatic steatosis. Cell MeTable 2007, 6, 484–496. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by AKT and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Lamming, D.W.; Sabatini, D.M. A central role for mTOR in lipid homeostasis. Cell MeTable 2013, 18, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, H.; Summers, B.D.; Nicholson, A.C.; Gotto, A.M., Jr.; Hajjar, D.P.; Han, J. Atherosclerosis in Ldlr-knockout mice is inhibited, but not reversed, by the PPARγ ligand pioglitazone. Am. J. Pathol. 2009, 174, 2007–2014. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.; Garuti, R.; Michaely, P.; Li, W.-P.; Maeda, N.; Cohen, J.C.; Herz, J.; Hobbs, H.H. Disruption of LDL but not VLDL clearance in autosomal recessive hypercholesterolemia. J. Clin. Invest. 2007, 117, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Plump, A.S.; Breslow, J.L. Apolipoprotein e and the apolipoprotein E-deficient mouse. Ann. Rev. Nutr. 1995, 15, 495–518. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Smith, E.; Fathzadeh, M.; Liu, W.; Go, G.W.; Subrahmanyan, L.; Faramarzi, S.; McKenna, W.; Mani, A. Rare nonconservative LRP6 mutations are associated with metabolic syndrome. Hum. Mutat. 2013, 34, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Keramati, A.R.; Fathzadeh, M.; Go, G.W.; Singh, R.; Choi, M.; Faramarzi, S.; Mane, S.; Kasaei, M.; Sarajzadeh-Fard, K.; Hwa, J.; et al. A form of the metabolic syndrome associated with mutations in DYRK1B. N. Engl. J. Med. 2014, 370, 1909–1919. [Google Scholar] [CrossRef] [PubMed]
- Zenibayashi, M.; Miyake, K.; Horikawa, Y.; Hirota, Y.; Teranishi, T.; Kouyama, K.; Sakaguchi, K.; Takeda, J.; Kasuga, M. Lack of association of LRP5 and LRP6 polymorphisms with type 2 diabetes mellitus in the japanese population. Endocr. J. 2008, 55, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.; Go, G.W.; Zhang, J.; Mani, A. Loss of LRP6/TCF7L2 regulation of vascular smooth muscle cell phenotype contributes to coronary artery disease. Circulation 2014, 130, A19224. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Go, G.-w. Low-Density Lipoprotein Receptor-Related Protein 6 (LRP6) Is a Novel Nutritional Therapeutic Target for Hyperlipidemia, Non-Alcoholic Fatty Liver Disease, and Atherosclerosis. Nutrients 2015, 7, 4453-4464. https://doi.org/10.3390/nu7064453
Go G-w. Low-Density Lipoprotein Receptor-Related Protein 6 (LRP6) Is a Novel Nutritional Therapeutic Target for Hyperlipidemia, Non-Alcoholic Fatty Liver Disease, and Atherosclerosis. Nutrients. 2015; 7(6):4453-4464. https://doi.org/10.3390/nu7064453
Chicago/Turabian StyleGo, Gwang-woong. 2015. "Low-Density Lipoprotein Receptor-Related Protein 6 (LRP6) Is a Novel Nutritional Therapeutic Target for Hyperlipidemia, Non-Alcoholic Fatty Liver Disease, and Atherosclerosis" Nutrients 7, no. 6: 4453-4464. https://doi.org/10.3390/nu7064453