Antioxidant Effect of Barley Sprout Extract via Enhancement of Nuclear Factor-Erythroid 2 Related Factor 2 Activity and Glutathione Synthesis

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of BSE
2.2. Animal Experiments
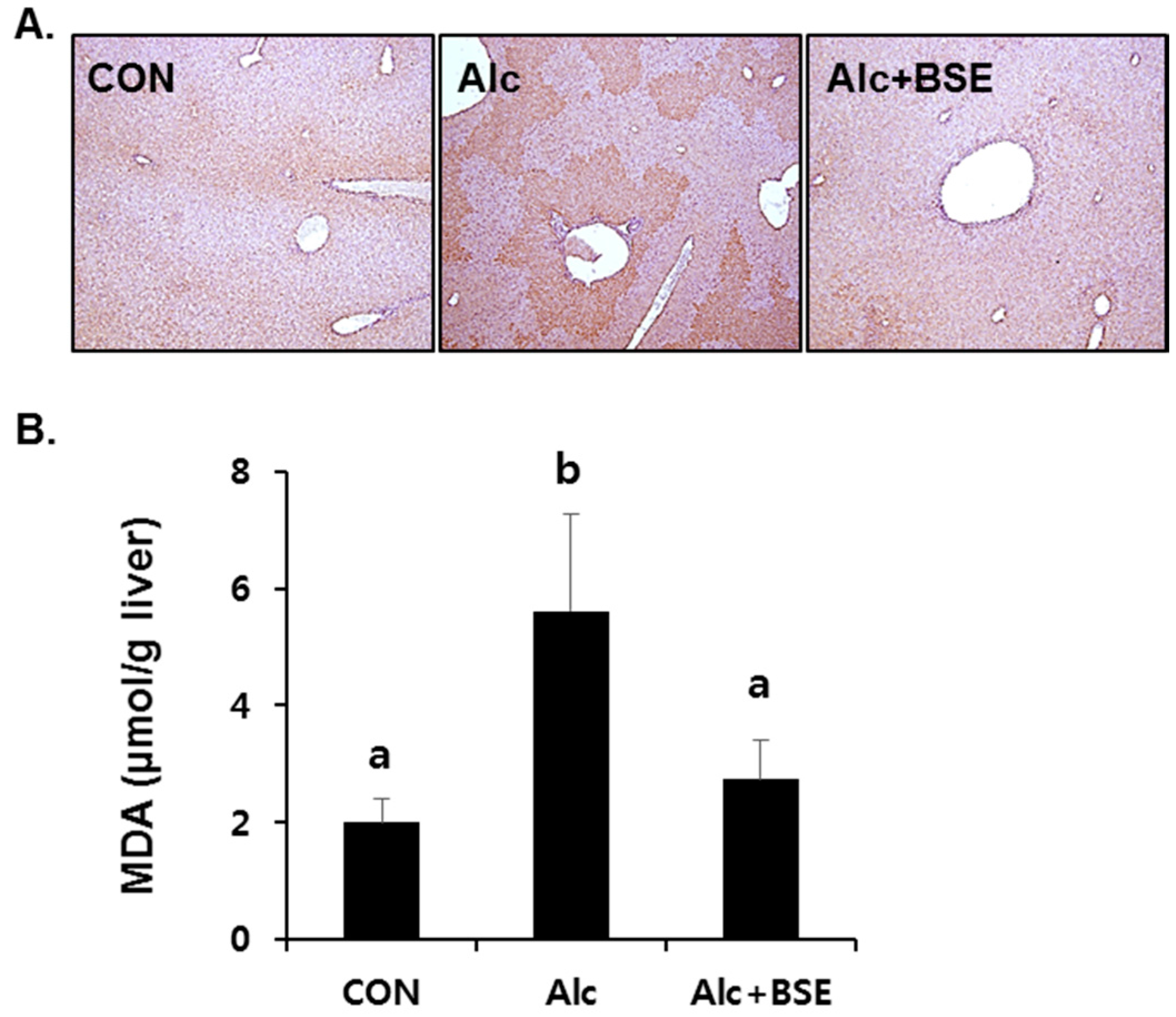
2.3. Determination of Lipid Peroxidation in the Liver Tissue
2.4. Immunohistochemical Staining of 4-Hydroxynonenal (4-HNE)
2.5. Cell Culture
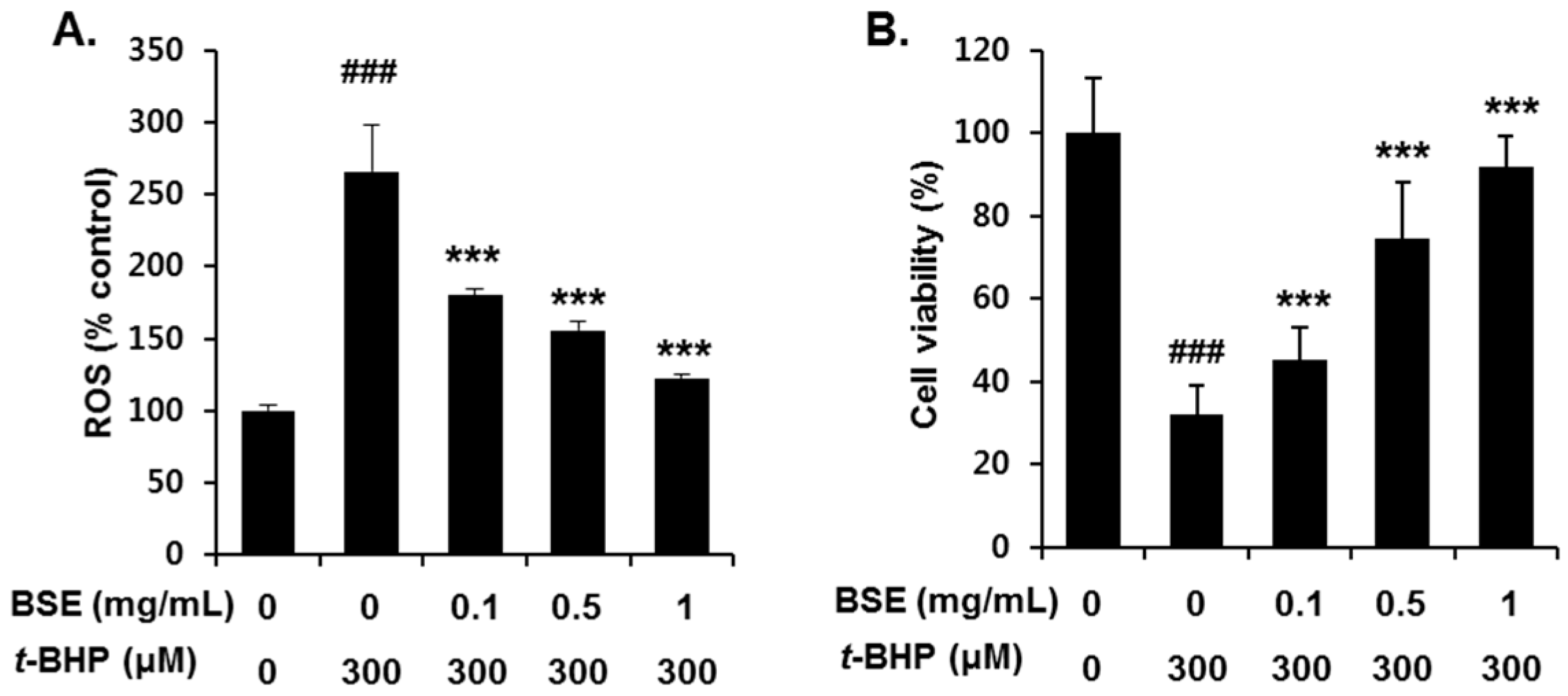
2.6. Measurement of ROS Generation
2.7. Determination of Cell Viability
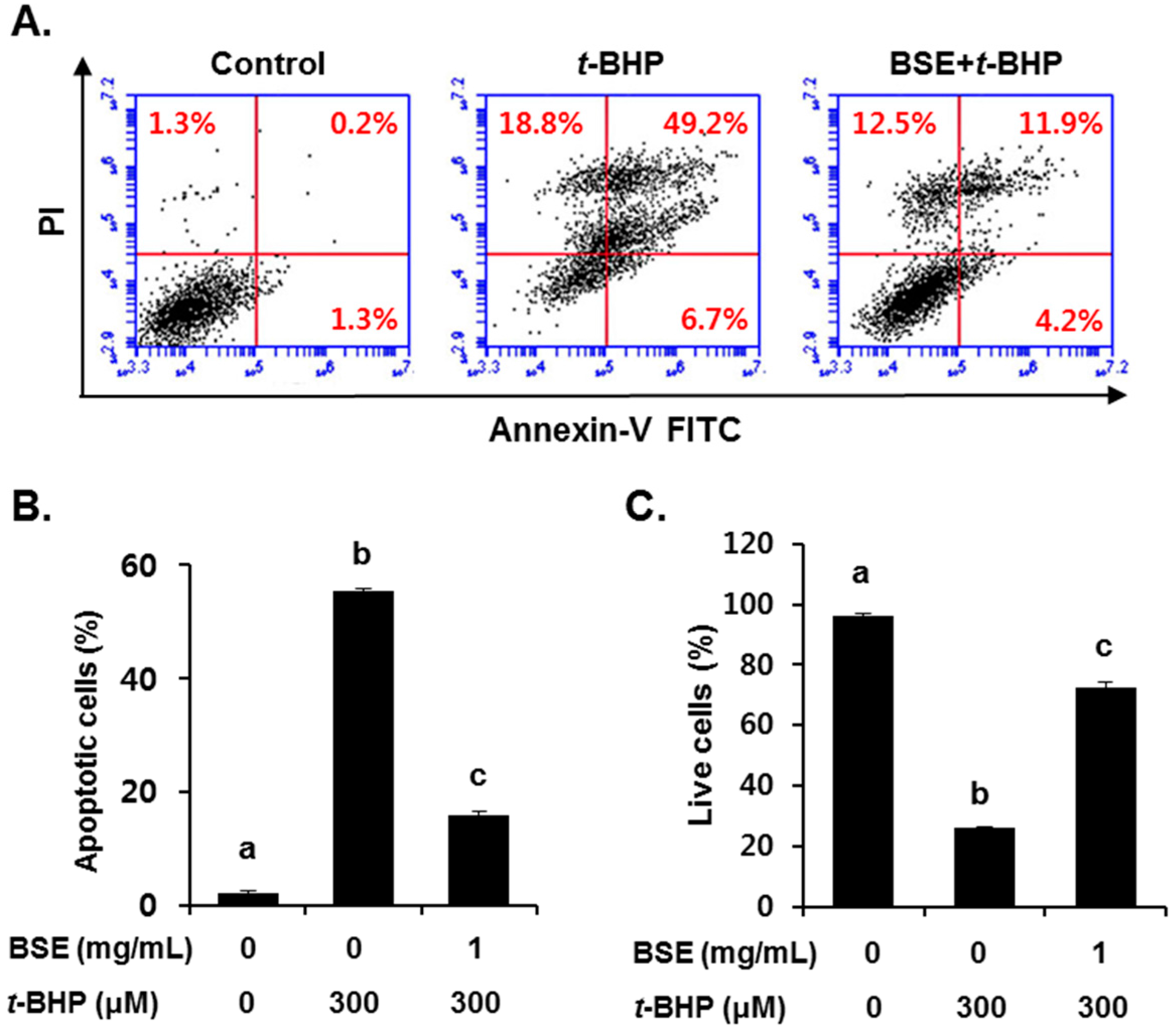
2.8. Fluorescence-Activated Cell Sorting (FACS) Analysis of Apoptosis
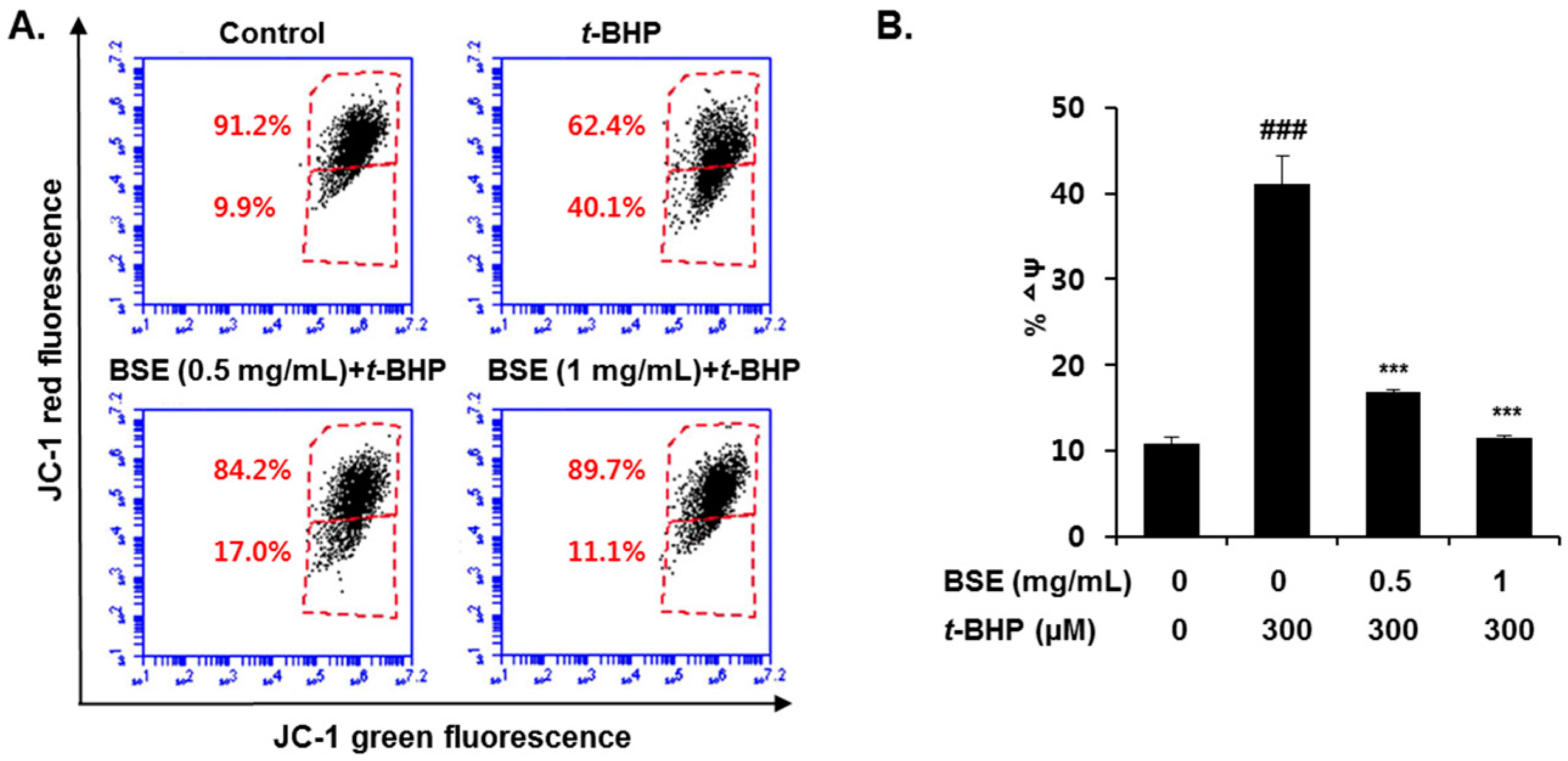
2.9. FACS Analysis of Mitochondrial Membrane Potential (∆ψ)
2.10. Antioxidant Response Element (ARE) Luciferase Assay
2.11. Real-Time Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
2.12. Western Blotting
2.13. Determination of Cysteine, Taurine, and GSH Levels
2.14. Statistical Analysis
3. Results
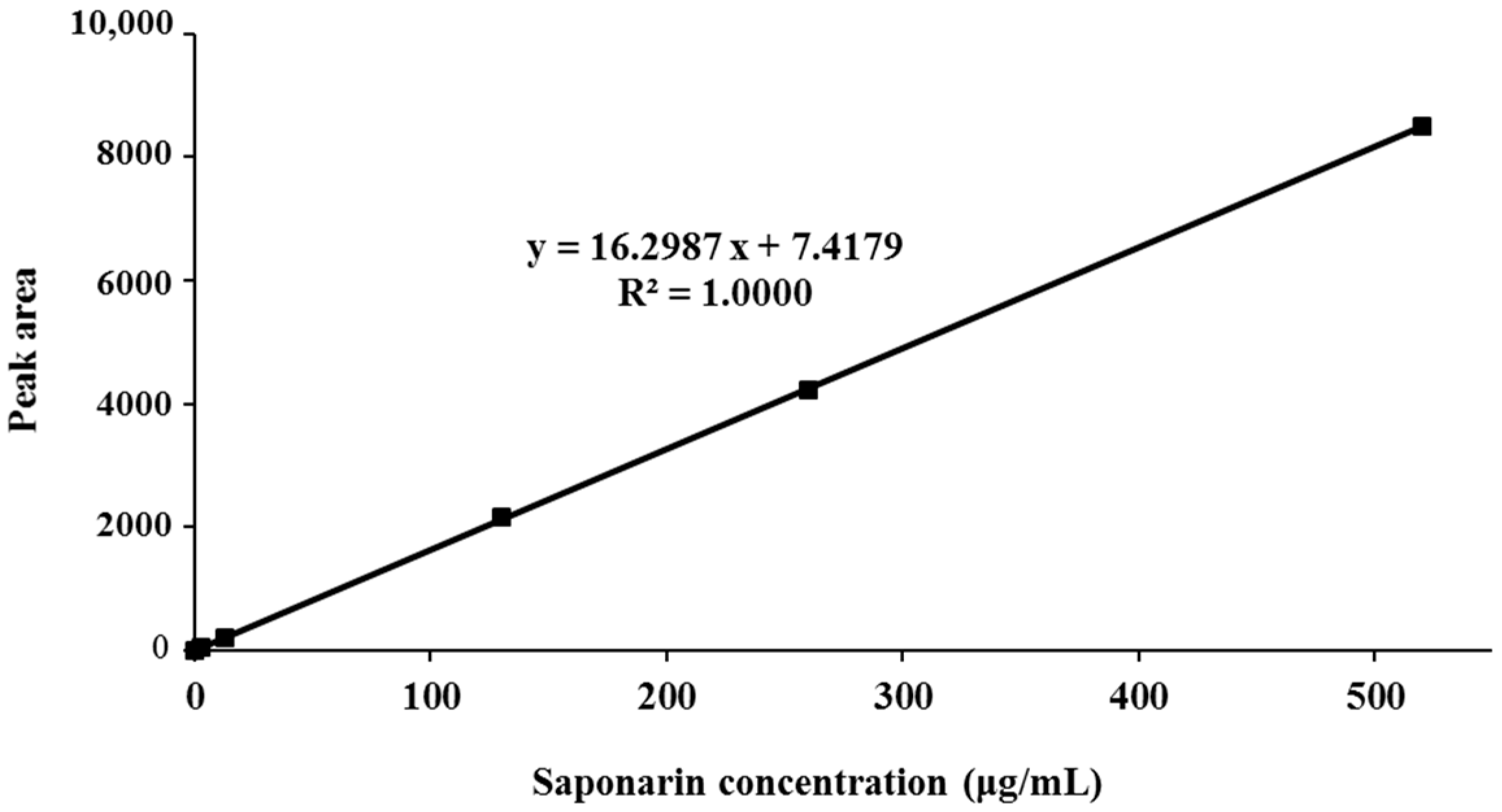
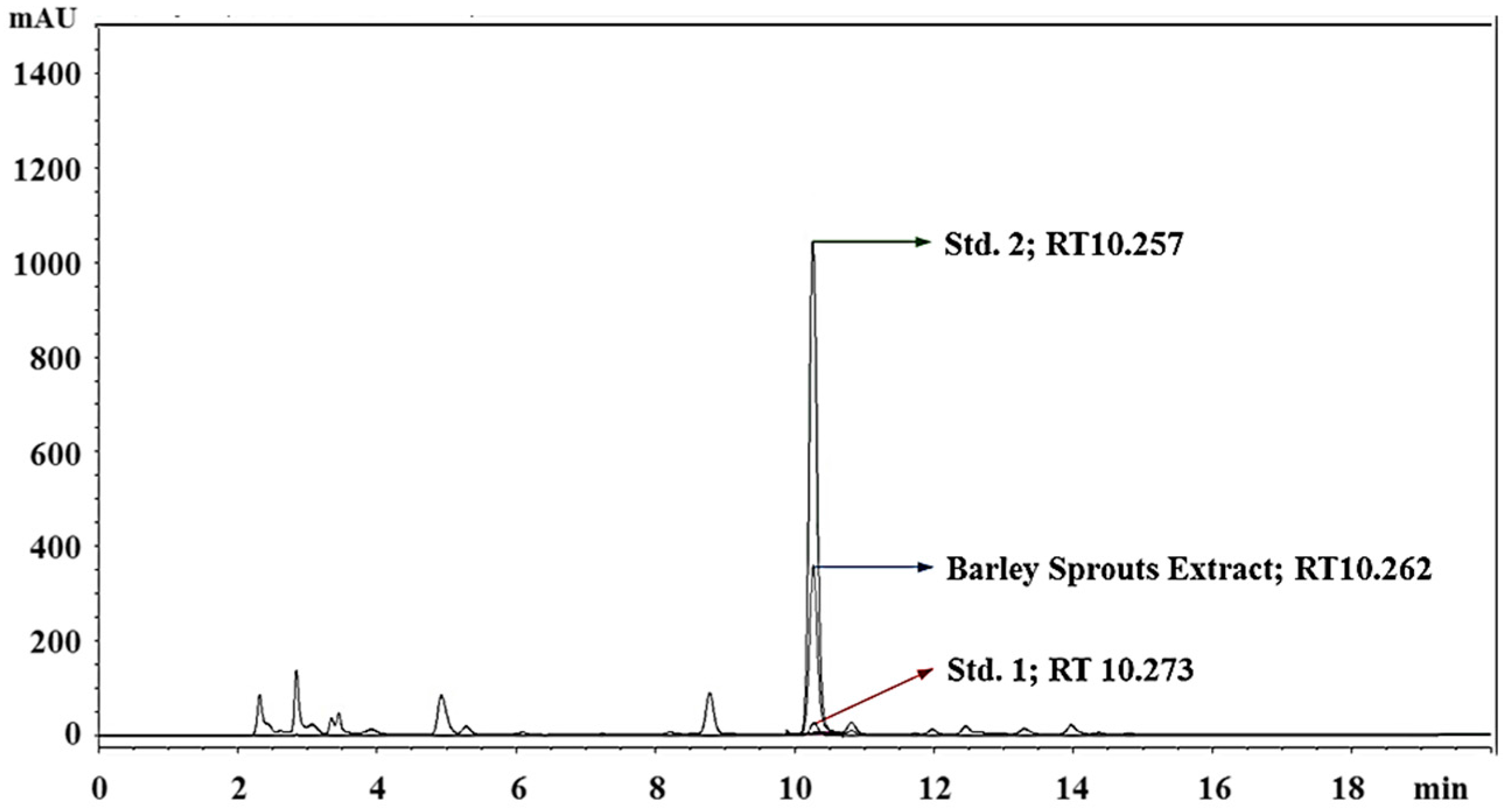
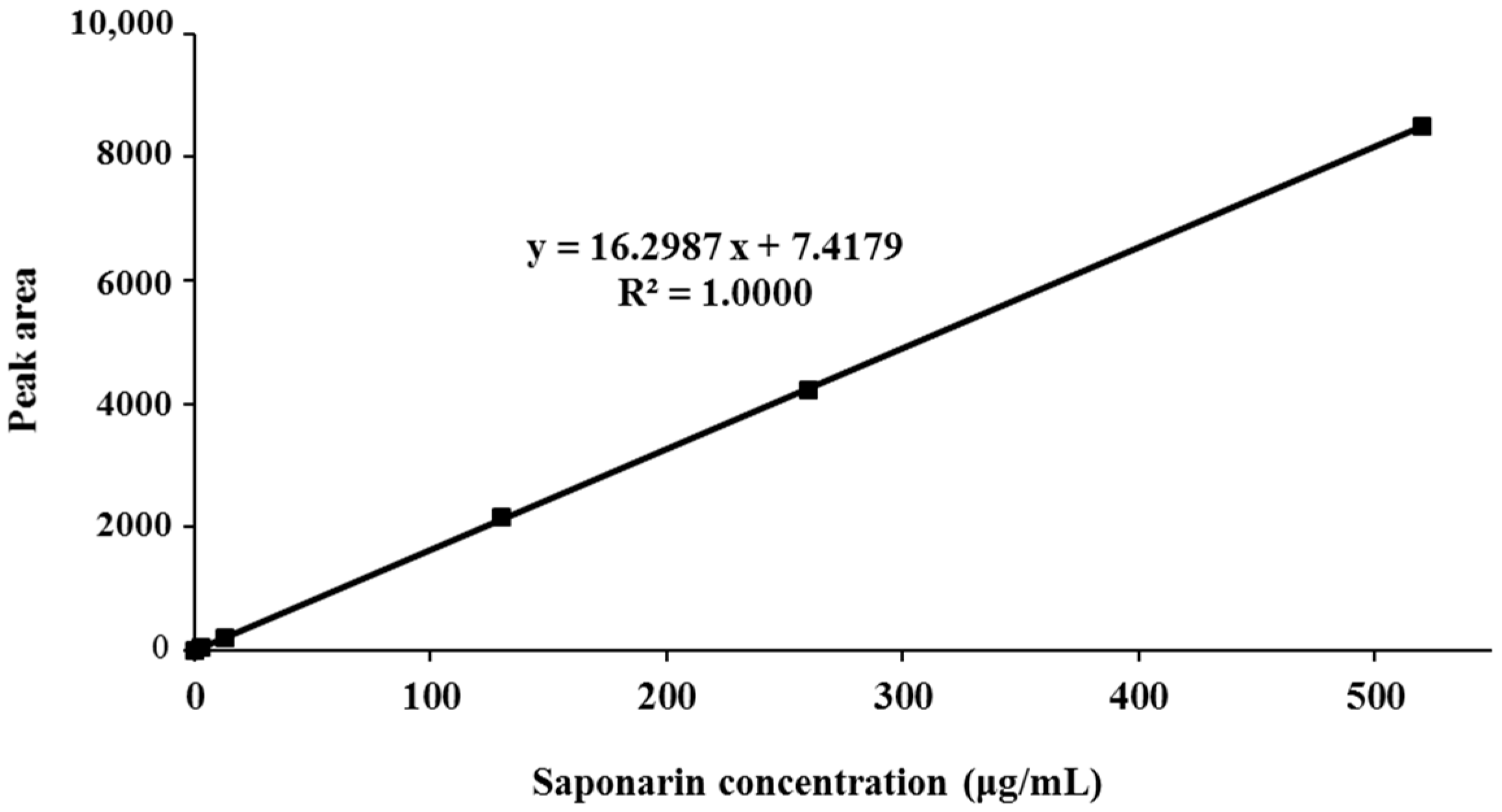
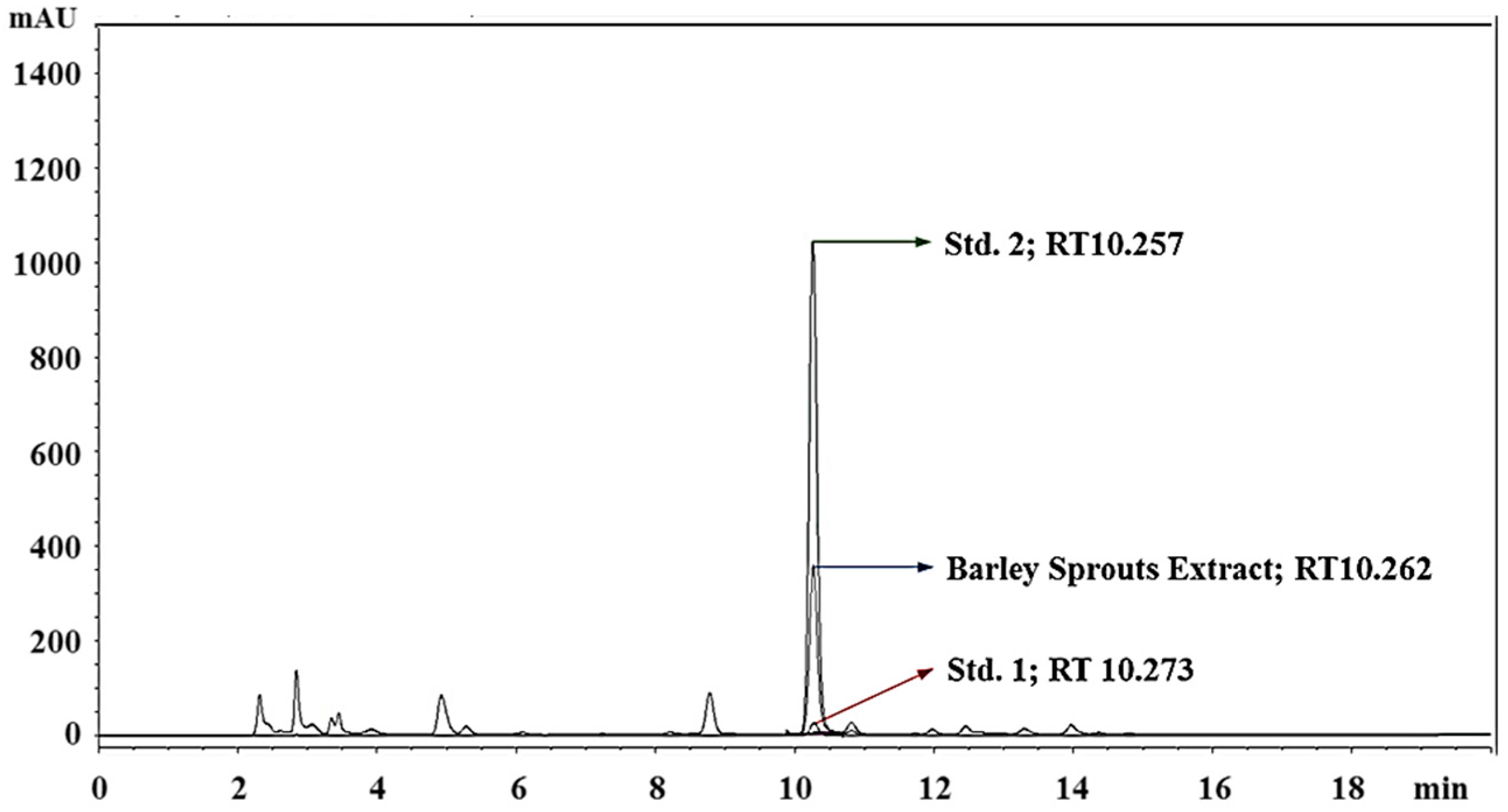
3.1. Standardization of BSE
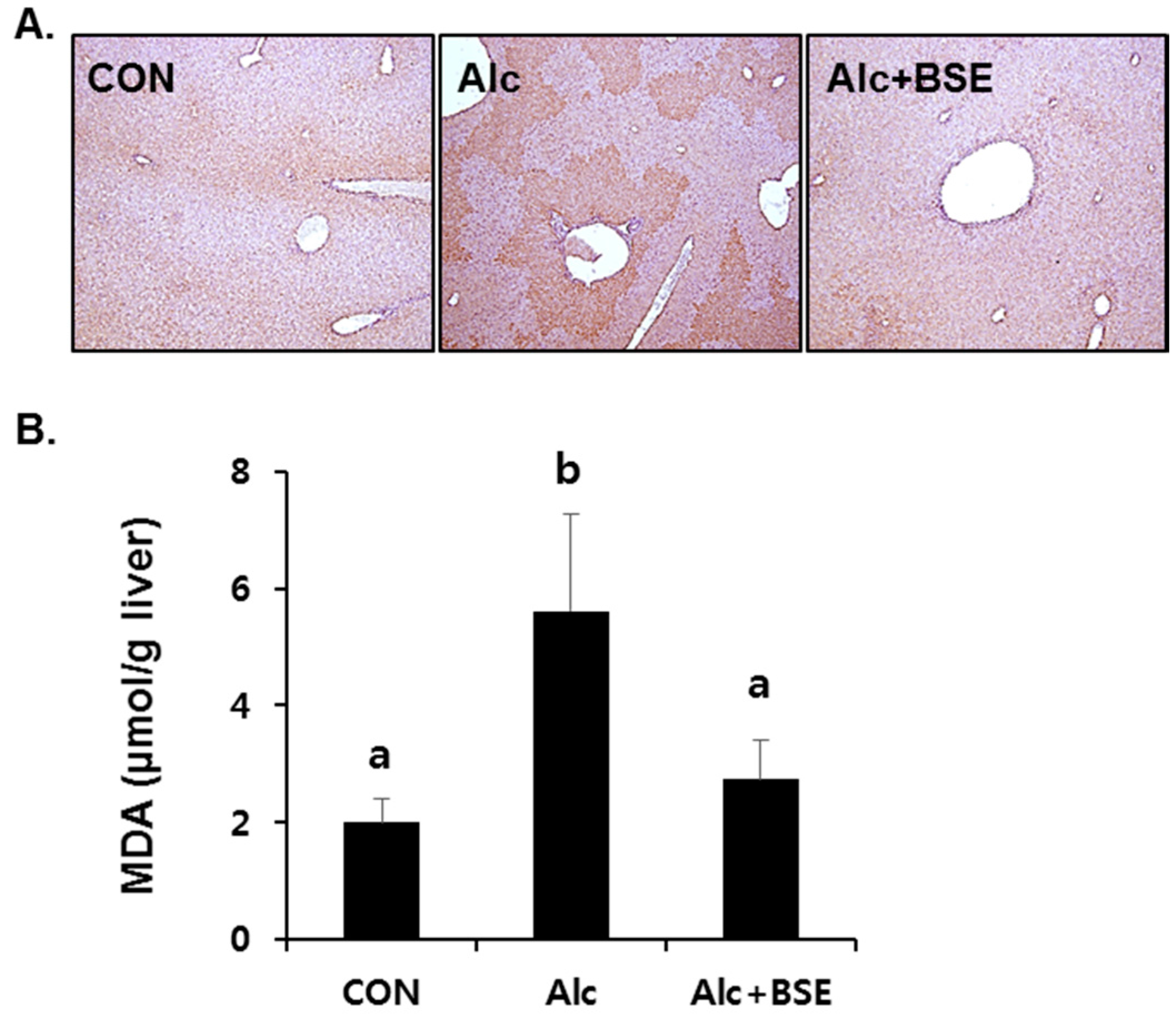
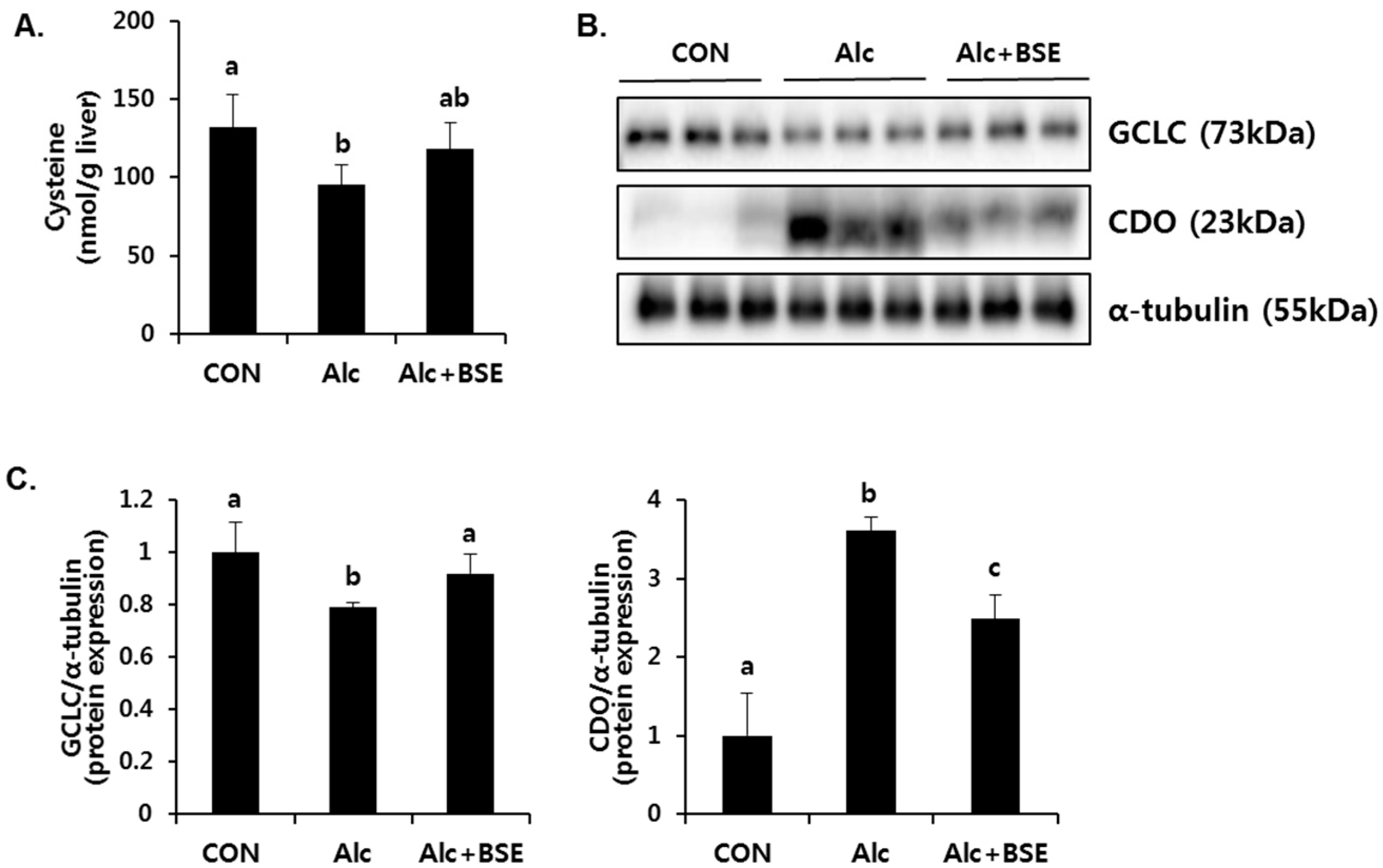
3.2. Prevention of Alcohol-Induced Oxidative Liver Injury by BSE
3.3. Inhibition of t-BHP-Induced Oxidative Cell Death by BSE in HepG2 Cells
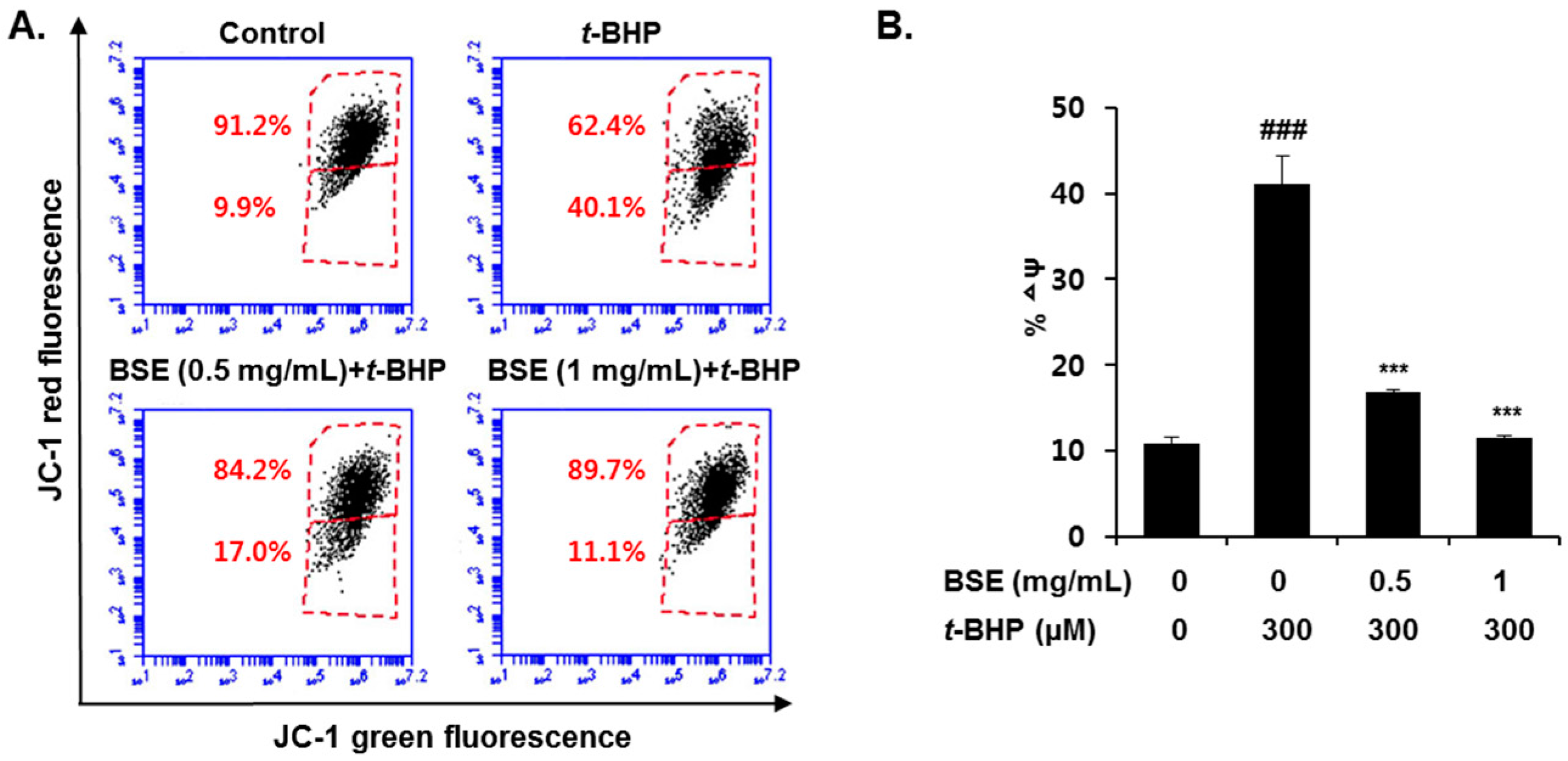
3.4. Prevention of t-BHP-Induced Mitochondrial Dysfunction by BSE in HepG2 Cells
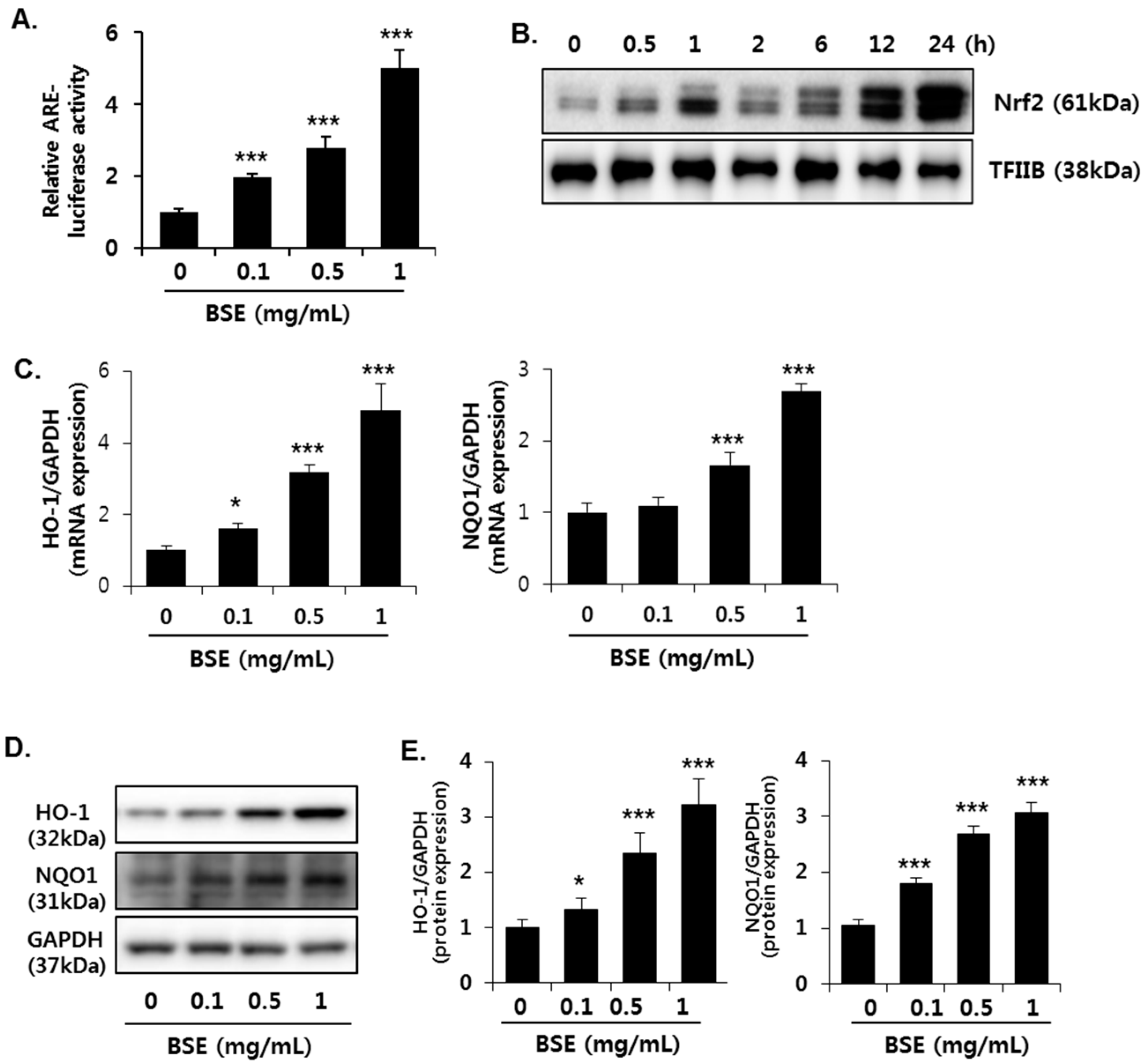
3.5. Induction of Nrf2 Activity in HepG2 Cells by BSE
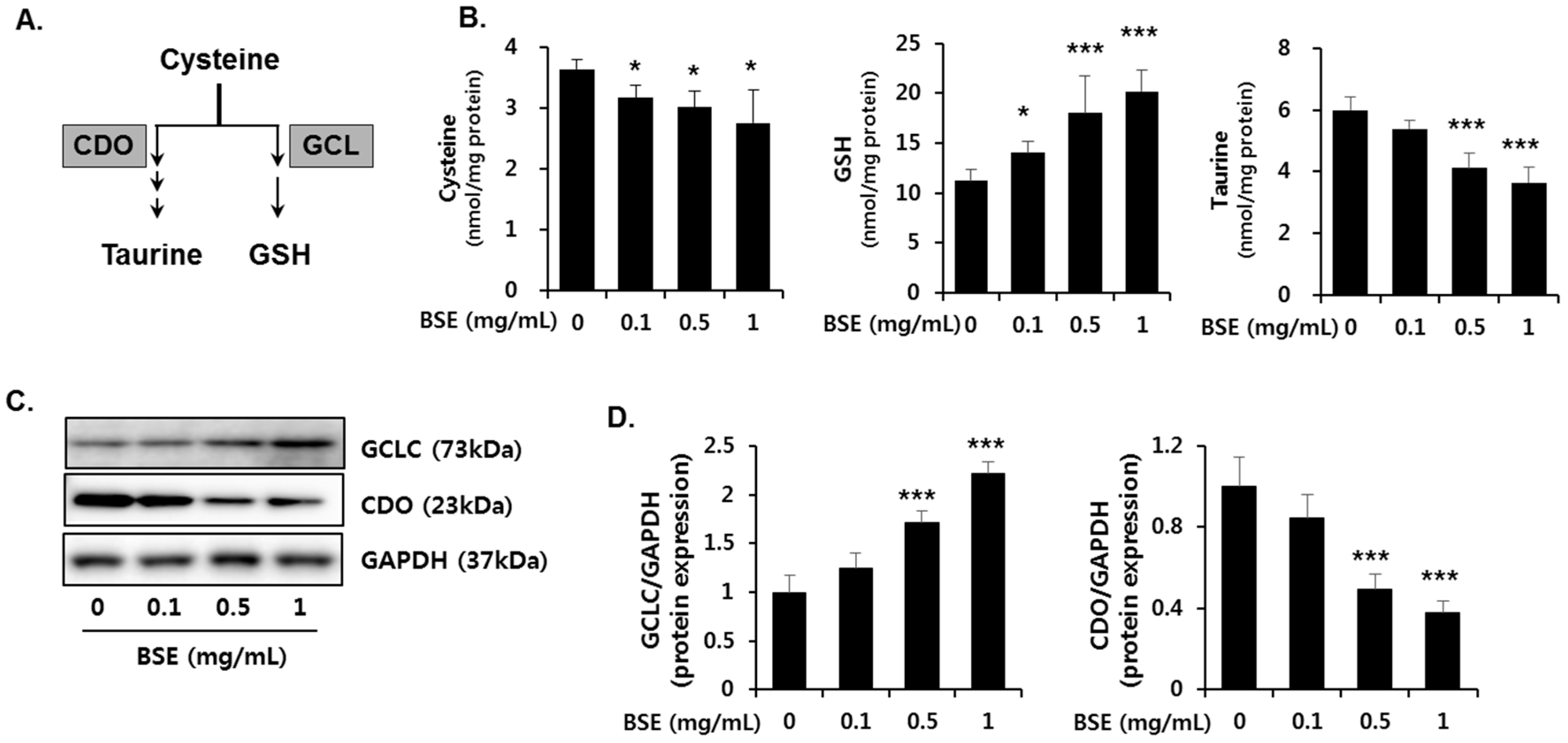
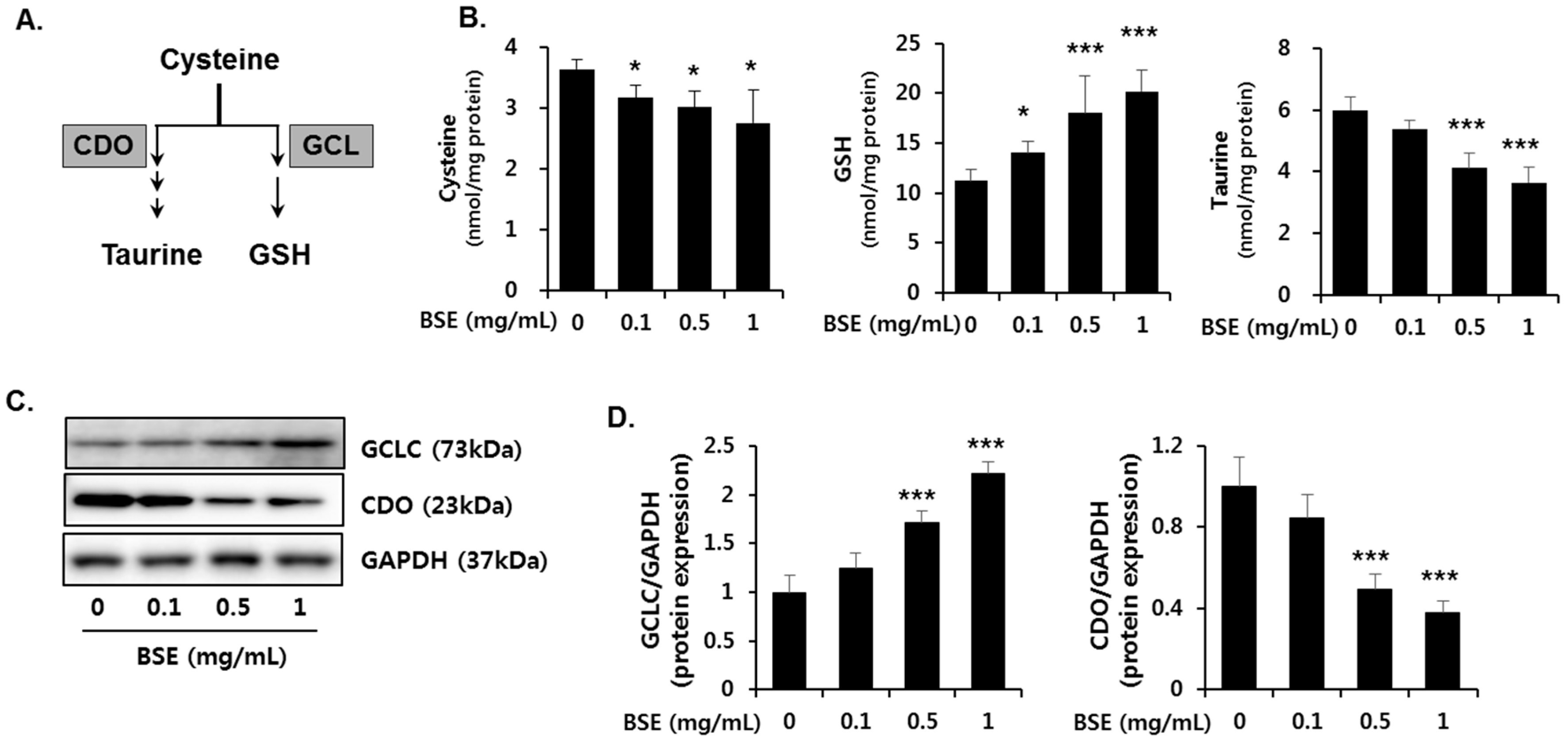
3.6. Effect of BSE on the De Novo Synthesis of GSH
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yip, W.W.; Burt, A.D. Alcoholic liver disease. Semin. Diagn. Pathol. 2006, 23, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P. Genes or environment to determine alcoholic liver disease and non-alcoholic fatty liver disease. Liver Int. 2006, 26, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Cederbaum, A.I. Oxidative stress and alcoholic liver disease. Semin. Liver Dis. 2009, 29, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.; Suzuki, H.; Sakaguchi, S. Common pathogenic mechanism in development progression of liver injury caused by non-alcoholic or alcoholic steatohepatitis. J. Toxicol. Sci. 2007, 32, 453–468. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.M.; Mantena, S.K.; Millender-Swain, T.; Cakir, Y.; Jhala, N.C.; Chhieng, D.; Pinkerton, K.E.; Ballinger, S.W. Ethanol and tobacco smoke increase hepatic steatosis and hypoxia in the hypercholesterolemic apoe(-/-) mouse: Implications for a “multihit” hypothesis of fatty liver disease. Free Radic. Biol. Med. 2009, 46, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Albano, E.; Clot, P.; Morimoto, M.; Tomasi, A.; Ingelman-Sundberg, M.; French, S.W. Role of cytochrome p4502e1-dependent formation of hydroxyethyl free radical in the development of liver damage in rats intragastrically fed with ethanol. Hepatology 1996, 23, 155–163. [Google Scholar] [CrossRef] [PubMed]
- French, S.W.; Wong, K.; Jui, L.; Albano, E.; Hagbjork, A.L.; Ingelman-Sundberg, M. Effect of ethanol on cytochrome p450 2e1 (cyp2e1), lipid peroxidation, and serum protein adduct formation in relation to liver pathology pathogenesis. Exp. Mol. Pathol. 1993, 58, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Butura, A.; Nilsson, K.; Morgan, K.; Morgan, T.R.; French, S.W.; Johansson, I.; Schuppe-Koistinen, I.; Ingelman-Sundberg, M. The impact of cyp2e1 on the development of alcoholic liver disease as studied in a transgenic mouse model. J. Hepatol. 2009, 50, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Banerjee, A.; Jang, S.; Yoo, S.H.; Yun, J.W.; Gonzalez, F.J.; Keshavarzian, A.; Song, B.J. Cyp2e1 potentiates binge alcohol-induced gut leakiness, steatohepatitis, and apoptosis. Free Radic. Biol. Med. 2013, 65, 1238–1245. [Google Scholar] [CrossRef] [PubMed]
- Mantena, S.K.; King, A.L.; Andringa, K.K.; Landar, A.; Darley-Usmar, V.; Bailey, S.M. Novel interactions of mitochondria and reactive oxygen/nitrogen species in alcohol mediated liver disease. World J. Gastroenterol. 2007, 13, 4967–4973. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S.; Casini, A.; DeCarli, L.M.; Kim, C.I.; Lowe, N.; Sasaki, R.; Leo, M.A. S-adenosyl-l-methionine attenuates alcohol-induced liver injury in the baboon. Hepatology 1990, 11, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Zhou, Z.; Chen, T.; Hill, D.; Kang, J.; Barve, S.; McClain, C. S-adenosylmethionine (same) protects against acute alcohol induced hepatotoxicity in mice. J. Nutr. Biochem. 2003, 14, 591–597. [Google Scholar] [CrossRef]
- Ronis, M.J.; Butura, A.; Sampey, B.P.; Shankar, K.; Prior, R.L.; Korourian, S.; Albano, E.; Ingelman-Sundberg, M.; Petersen, D.R.; Badger, T.M. Effects of n-acetylcysteine on ethanol-induced hepatotoxicity in rats fed via total enteral nutrition. Free Radic. Biol. Med. 2005, 39, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Ozaras, R.; Tahan, V.; Aydin, S.; Uzun, H.; Kaya, S.; Senturk, H. N-acetylcysteine attenuates alcohol-induced oxidative stress in the rat. World J. Gastroenterol. 2003, 9, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Effect of graded nrf2 activation on phase-i and -ii drug metabolizing enzymes and transporters in mouse liver. PLoS ONE 2012, 7, e39006. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the keap1-nrf2-are pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Han, X.D.; Kan, Y.W. An important function of nrf2 in combating oxidative stress: Detoxification of acetaminophen. Proc. Natl. Acad. Sci. USA 2001, 98, 4611–4616. [Google Scholar] [CrossRef] [PubMed]
- Chowdhry, S.; Nazmy, M.H.; Meakin, P.J.; Dinkova-Kostova, A.T.; Walsh, S.V.; Tsujita, T.; Dillon, J.F.; Ashford, M.L.; Hayes, J.D. Loss of nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2010, 48, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Xu, W.; Zhang, F.; Shao, J.; Zheng, S. Nrf2 knockdown disrupts the protective effect of curcumin on alcohol-induced hepatocyte necroptosis. Mol. Pharm. 2016, 13, 4043–4053. [Google Scholar] [CrossRef] [PubMed]
- Reisman, S.A.; Buckley, D.B.; Tanaka, Y.; Klaassen, C.D. Cddo-im protects from acetaminophen hepatotoxicity through induction of nrf2-dependent genes. Toxicol. Appl. Pharmacol. 2009, 236, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Okawa, H.; Motohashi, H.; Kobayashi, A.; Aburatani, H.; Kensler, T.W.; Yamamoto, M. Hepatocyte-specific deletion of the keap1 gene activates nrf2 and confers potent resistance against acute drug toxicity. Biochem. Biophys. Res. Commun. 2006, 339, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Kim, J.H.; Kim, S.H.; Oh, J.Y.; Seo, W.D.; Kim, K.M.; Jung, J.C.; Jung, Y.S. Barley sprouts extract attenuates alcoholic fatty liver injury in mice by reducing inflammatory response. Nutrients 2016, 8, 440. [Google Scholar] [CrossRef] [PubMed]
- Volpi, N.; Tarugi, P. Improvement in the high-performance liquid chromatography malondialdehyde level determination in normal human plasma. J. Chromatogr. B Biomed. Sci. Appl. 1998, 713, 433–437. [Google Scholar] [CrossRef]
- Jung, Y.S.; Kim, S.J.; Kwon, D.Y.; Kim, Y.C. Comparison of the effects of buthioninesulfoximine and phorone on the metabolism of sulfur-containing amino acids in rat liver. Biochem. Biophys. Res. Commun. 2008, 368, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Ide, T. Simple high-performance liquid chromatographic method for assaying cysteinesulfinic acid decarboxylase activity in rat tissue. J. Chromatogr. B Biomed. Sci. Appl. 1997, 694, 325–332. [Google Scholar] [CrossRef]
- Nolin, T.D.; McMenamin, M.E.; Himmelfarb, J. Simultaneous determination of total homocysteine, cysteine, cysteinylglycine, and glutathione in human plasma by high-performance liquid chromatography: Application to studies of oxidative stress. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 852, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Castillo, T.; Koop, D.R.; Kamimura, S.; Triadafilopoulos, G.; Tsukamoto, H. Role of cytochrome P-450 2E1 in ethanol-, carbon tetrachloride- and iron-dependent microsomal lipid peroxidation. Hepatology 1992, 16, 992–996. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.; Sies, H. Role of alcohol dehydrogenase activity and the acetaldehyde in ethanol- induced ethane and pentane production by isolated perfused rat liver. Biochem. J. 1982, 206, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Kera, Y.; Komura, S.; Ohbora, Y.; Kiriyama, T.; Inoue, K. Ethanol induced changes in lipid peroxidation and nonprotein sulfhydryl content. Different sensitivities in rat liver and kidney. Res. Commun. Chem. Pathol. Pharmacol. 1985, 47, 203–209. [Google Scholar] [PubMed]
- Videla, L.A.; Fernandez, V.; Ugarte, G.; Valenzuela, A. Effect of acute ethanol intoxication on the content of reduced glutathione of the liver in relation to its lipoperoxidative capacity in the rat. FEBS Lett. 1980, 111, 6–10. [Google Scholar] [CrossRef]
- Vina, J.; Estrela, J.M.; Guerri, C.; Romero, F.J. Effect of ethanol on glutathione concentration in isolated hepatocytes. Biochem. J. 1980, 188, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Kono, H.; Rusyn, I.; Bradford, B.U.; Connor, H.D.; Mason, R.P.; Thurman, R.G. Allopurinol prevents early alcohol-induced liver injury in rats. J. Pharmacol. Exp. Ther. 2000, 293, 296–303. [Google Scholar] [PubMed]
- Kono, H.; Arteel, G.E.; Rusyn, I.; Sies, H.; Thurman, R.G. Ebselen prevents early alcohol-induced liver injury in rats. Free Radic. Biol. Med. 2001, 30, 403–411. [Google Scholar] [CrossRef]
- Kono, H.; Rusyn, I.; Uesugi, T.; Yamashina, S.; Connor, H.D.; Dikalova, A.; Mason, R.P.; Thurman, R.G. Diphenyleneiodonium sulfate, an nadph oxidase inhibitor, prevents early alcohol-induced liver injury in the rat. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 280, G1005–G1012. [Google Scholar] [PubMed]
- McKim, S.E.; Konno, A.; Gabele, E.; Uesugi, T.; Froh, M.; Sies, H.; Thurman, R.G.; Arteel, G.E. Cocoa extract protects against early alcohol-induced liver injury in the rat. Arch. Biochem. Biophys. 2002, 406, 40–46. [Google Scholar] [CrossRef]
- Lamle, J.; Marhenke, S.; Borlak, J.; von Wasielewski, R.; Eriksson, C.J.; Geffers, R.; Manns, M.P.; Yamamoto, M.; Vogel, A. Nuclear factor-eythroid 2-related factor 2 prevents alcohol-induced fulminant liver injury. Gastroenterology 2008, 134, 1159–1168. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.C.; Liu, J.; Klaassen, C.D. Role of nrf2 in preventing ethanol-induced oxidative stress and lipid accumulation. Toxicol. Appl. Pharmacol. 2012, 262, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Zhang, F.; Xu, W.; Wu, X.; Lian, N.; Jin, H.; Chen, Q.; Chen, L.; Shao, J.; Wu, L.; et al. Curcumin attenuates ethanol-induced hepatic steatosis through modulating nrf2/fxr signaling in hepatocytes. IUBMB Life 2015, 67, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.H.; Stipanuk, M.H. Cysteine regulates expression of cysteine dioxygenase and gamma-glutamylcysteine synthetase in cultured rat hepatocytes. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E804–E815. [Google Scholar] [PubMed]
- Stipanuk, M.H.; Dominy, J.E.; Lee, J.I.; Coloso, R.M. Mammalian cysteine metabolism: New insights into regulation of cysteine metabolism. J. Nutr. 2006, 136, 1652S–1659S. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Primer Sequences | |
|---|---|---|
| HO-1 | F: CGGGCCAGCAACAAAGTG | R: AGTGTAAGGACCCATCGGAGAA |
| NQO1 | F: AGGCTGGTTTGAGCGAGT | R: ATTGAATTCGGGCGTCTGCTG |
| GAPDH | F: ATCACCATCTTCCAGGAGCGA | R: GCCAGTGAGCTTCCCGTTCA |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.-H.; Kim, S.H.; Lee, S.; Kim, K.-M.; Jung, J.-C.; Son, T.G.; Ki, S.H.; Seo, W.-D.; Kwak, J.-H.; Hong, J.T.; et al. Antioxidant Effect of Barley Sprout Extract via Enhancement of Nuclear Factor-Erythroid 2 Related Factor 2 Activity and Glutathione Synthesis. Nutrients 2017, 9, 1252. https://doi.org/10.3390/nu9111252
Lee Y-H, Kim SH, Lee S, Kim K-M, Jung J-C, Son TG, Ki SH, Seo W-D, Kwak J-H, Hong JT, et al. Antioxidant Effect of Barley Sprout Extract via Enhancement of Nuclear Factor-Erythroid 2 Related Factor 2 Activity and Glutathione Synthesis. Nutrients. 2017; 9(11):1252. https://doi.org/10.3390/nu9111252
Chicago/Turabian StyleLee, Yun-Hee, Sou Hyun Kim, Seunghyun Lee, Kyung-Mi Kim, Jae-Chul Jung, Tae Gen Son, Sung Hwan Ki, Woo-Duck Seo, Jae-Hwan Kwak, Jin Tae Hong, and et al. 2017. "Antioxidant Effect of Barley Sprout Extract via Enhancement of Nuclear Factor-Erythroid 2 Related Factor 2 Activity and Glutathione Synthesis" Nutrients 9, no. 11: 1252. https://doi.org/10.3390/nu9111252