Nutrients in Energy and One-Carbon Metabolism: Learning from Metformin Users

 , , , , , and
, , , , , and 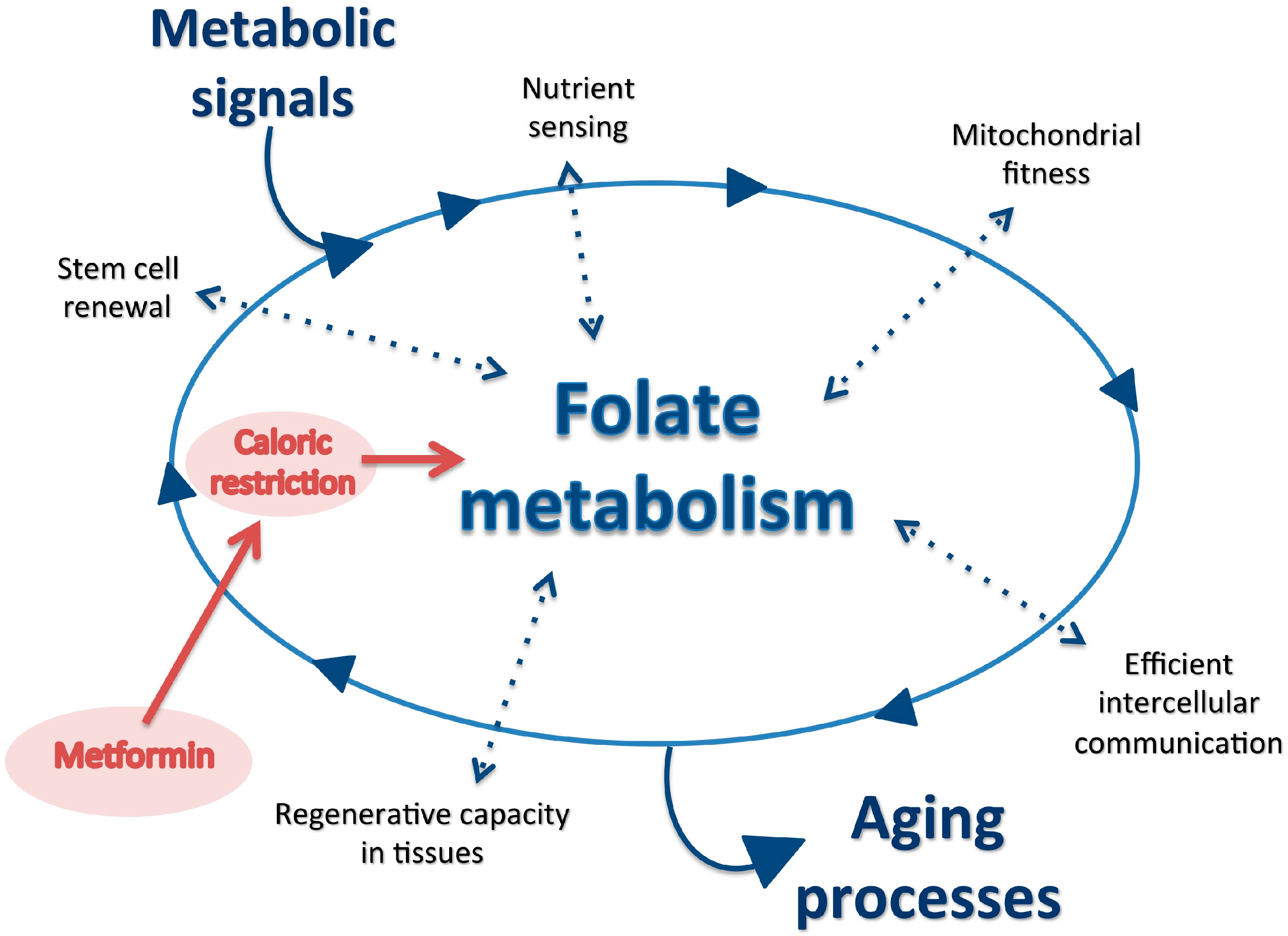{kind=link}
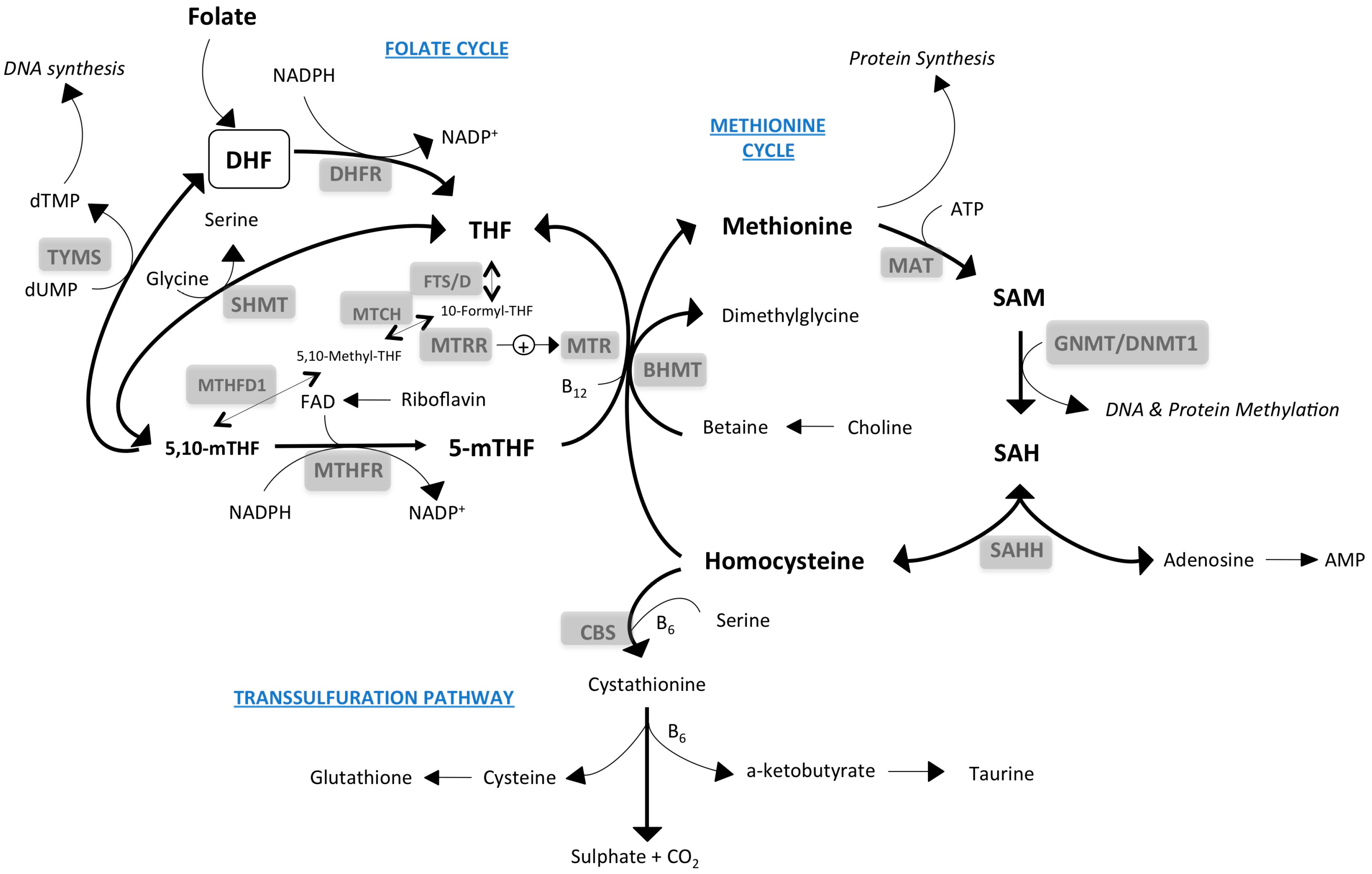{kind=link}
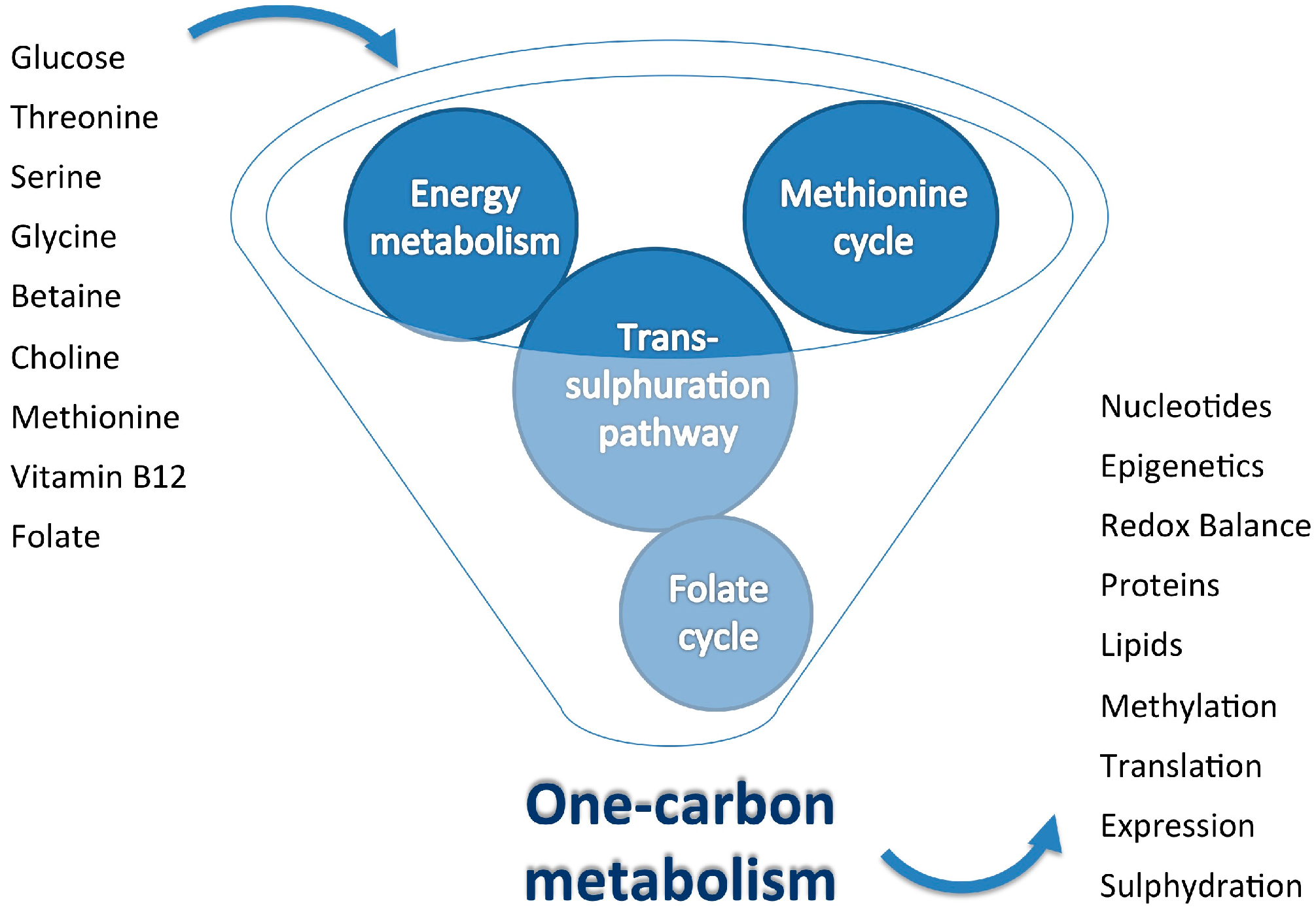{kind=link}
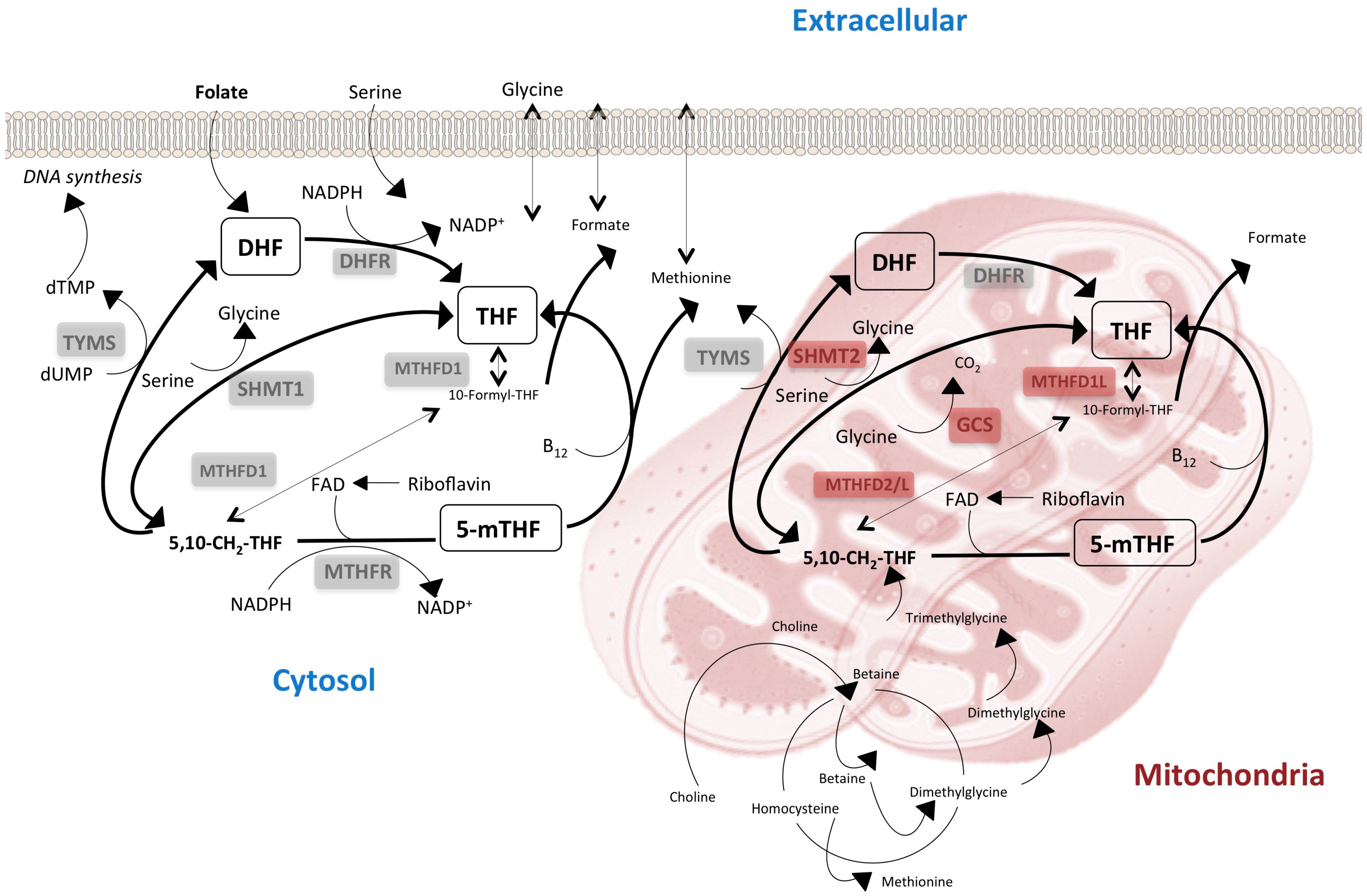{kind=link}
Abstract
:1. Introduction
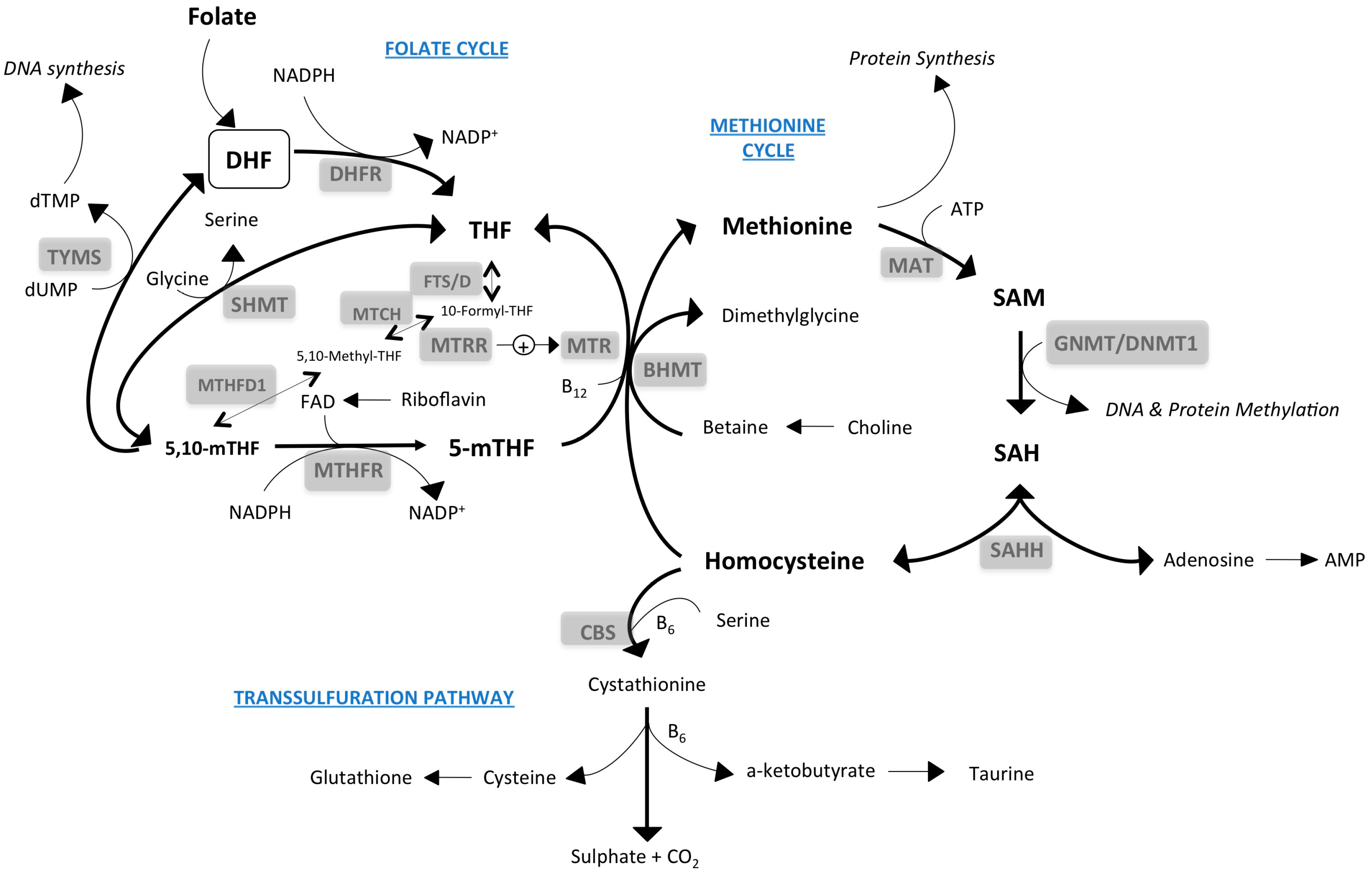
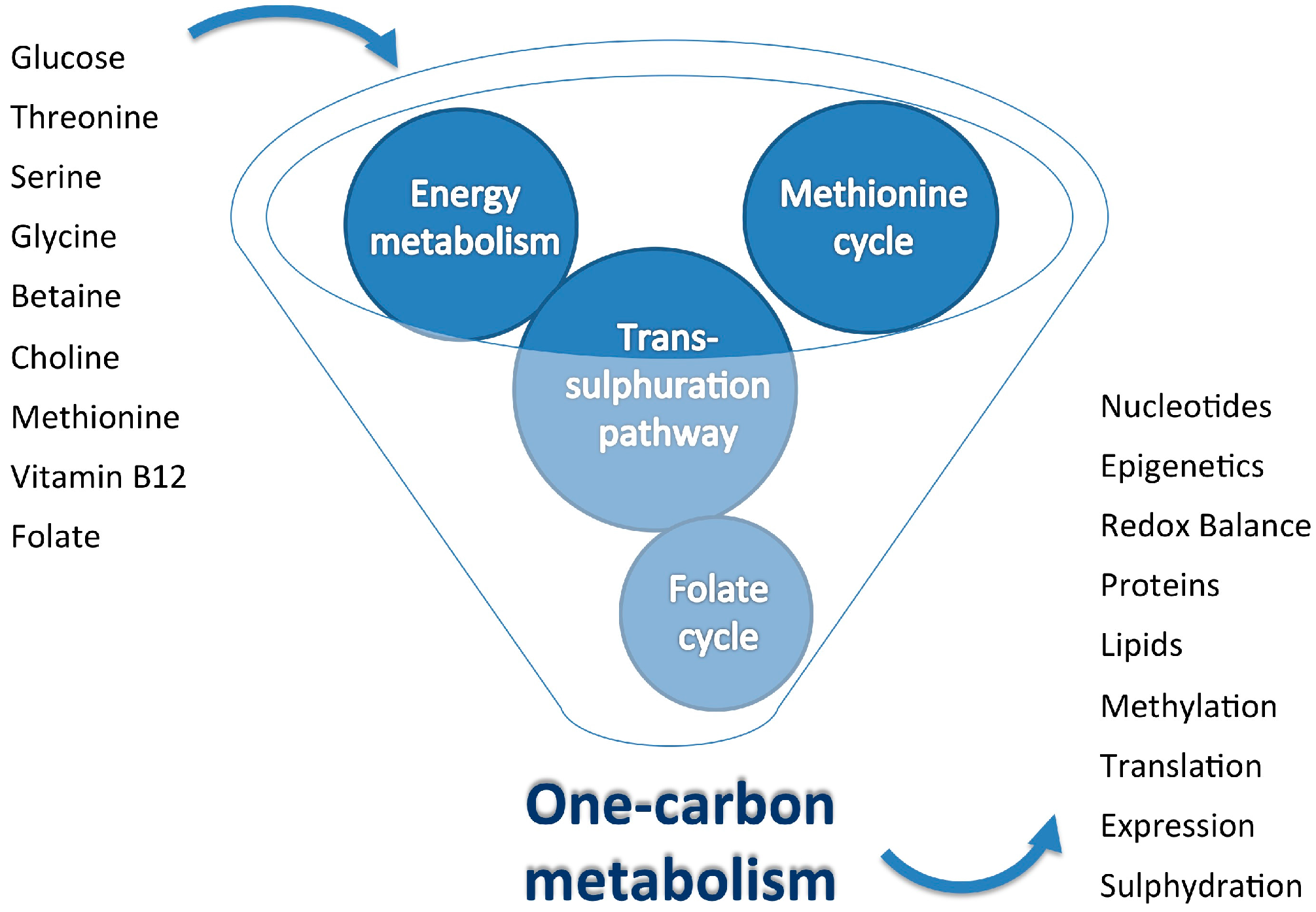
2. One-Carbon Metabolism: Inputs and Outputs
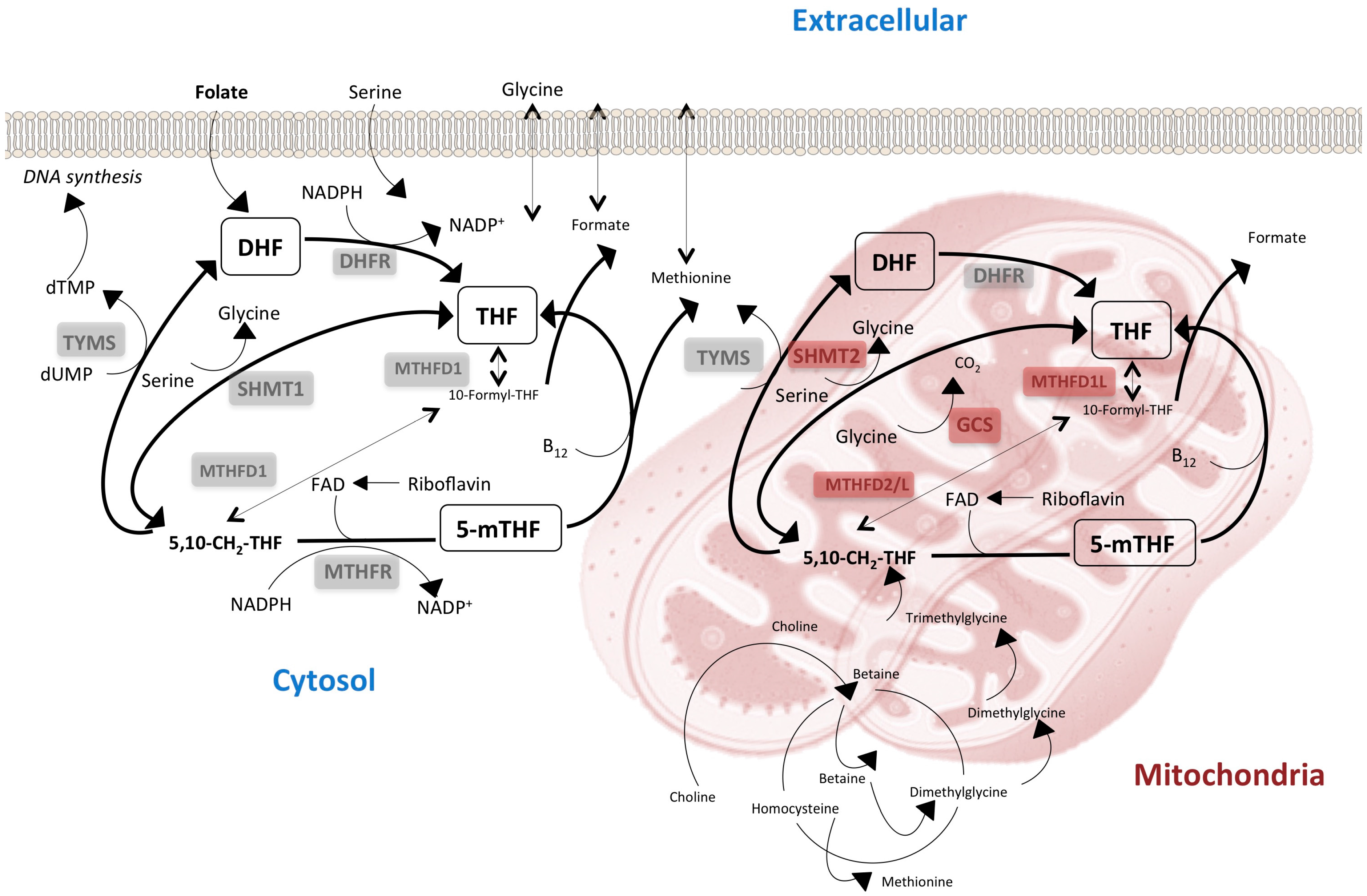
3. The Lack of Redundancy between Cytoplasmatic and Mitochondrial One-Carbon Metabolism Affects Tissue-Specific Responses to Nutrients
4. The Importance of Drugs and Diets That Modulate Mitochondrial Activity
5. Is Gene Expression Reprogrammed in Response to Metabolic and Dietary Stimuli Affecting One-Carbon Metabolism?
6. The Ability of Metformin to Target One-Carbon Metabolism: Perspectives in Clinical Practice Outcomes
7. The One-Carbon Cycle in Metformin Users and Potential Adverse Effects
8. Measuring the Impact of Folate-Related Deficiencies in the Clinical Setting: Potential for Targeted Metabolomics
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mirzaei, H.; Di Biase, S.; Longo, V.D. Dietary Interventions, Cardiovascular Aging, and Disease: Animal Models and Human Studies. Circ. Res. 2016, 118, 1612–1625. [Google Scholar] [CrossRef] [PubMed]
- Hall, K.D.; Bemis, T.; Brychta, R.; Chen, K.Y.; Courville, A.; Crayner, E.J.; Goodwin, S.; Guo, J.; Howard, L.; Knuth, N.D.; et al. Calorie for Calorie, Dietary Fat Restriction Results in More Body Fat Loss than Carbohydrate Restriction in People with Obesity. Cell Metab. 2015, 22, 427–436. [Google Scholar] [CrossRef]
- Kim, W.; Woo, H.D.; Lee, J.; Choi, I.J.; Kim, Y.W.; Sung, J.; Kim, J. Dietary folate, one-carbon metabolism-related genes, and gastric cancer risk in Korea. Mol. Nutr. Food Res. 2016, 60, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial one-carbon metabolism maintains redox balance during hypoxia. Cancer Discov. 2014, 4, 1371–1373. [Google Scholar] [CrossRef] [PubMed]
- Duncan, T.M.; Reed, M.C.; Nijhout, H.F. A population model of folate-mediated one-carbon metabolism. Nutrients 2013, 5, 2457–2474. [Google Scholar] [CrossRef] [PubMed]
- Bailey, L.B.; Stover, P.J.; McNulty, H.; Fenech, M.F.; Gregory, J.F.; Mills, J.L.; Pfeiffer, C.M.; Fazili, Z.; Zhang, M.; Ueland, P.M.; et al. Biomarkers of Nutrition for Development. J. Nutr. 2015, 145, 1636S–1680S. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Galluzzi, L.; Freije, J.M.; Madeo, F.; Kroemer, G. Metabolic Control of Longevity. Cell 2016, 166, 802–821. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Quirantes-Piné, R.; Rodríguez-Gallego, E.; Cufí, S.; Corominas-Faja, B.; Cuyàs, E.; Bosch-Barrera, J.; Martin-Castillo, B.; Segura-Carretero, A.; Joven, J. Oncobiguanides: Paracelsus’ law and nonconventional routes for administering diabetobiguanides for cancer treatment. Oncotarget 2014, 5, 2344–2348. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M. Potential applications for biguanides in oncology. J. Clin. Investig. 2013, 123, 3693–3700. [Google Scholar] [CrossRef]
- Coperchini, F.; Leporati, P.; Rotondi, M.; Chiovato, L. Expanding the therapeutic spectrum of metformin: From diabetes to cancer. J. Endocrinol. Investig. 2015, 38, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Pryor, R.; Cabreiro, F. Repurposing metformin: An old drug with new tricks in its binding pockets. Biochem. J. 2015, 471, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Corominas-Faja, B.; Quirantes-Pine, R.; Oliveras-Ferraros, C.; Vazquez-Martin, A.; Cufi, S.; Martin-Castillo, B.; Micol, V.; Joven, J.; Segura-Carretero, A.; Menendez, J.A. Metabolomic fingerprint reveals that metformin impairs one-carbon metabolism in a manner similar to the antifolate class of chemotherapy drugs. Aging 2012, 4, 480–498. [Google Scholar] [CrossRef] [PubMed]
- Novelle, M.G.; Ali, A.; Diéguez, C.; Bernier, M.; de Cabo, R. Metformin: A Hopeful Promise in Aging Research. Cold Spring Harb. Perspect. Med. 2016, 6, a025932. [Google Scholar] [CrossRef] [PubMed]
- Carmona, J.J.; Michan, S. Biology of Healthy Aging and Longevity. Rev. Investig. Clin. 2016, 68, 7–16. [Google Scholar]
- Cabreiro, F.; Au, C.; Leung, K.Y.; Vergara-Irigaray, N.; Cochemé, H.M.; Noori, T.; Weinkove, D.; Schuster, E.; Greene, N.D.; Gems, D. Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell 2013, 153, 228–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domecq, J.P.; Prutsky, G.; Leppin, A.; Sonbol, M.B.; Altayar, O.; Undavalli, C.; Wang, Z.; Elraiyah, T.; Brito, J.P.; Mauck, K.F.; et al. Clinical review: Drugs commonly associated with weight change: A systematic review and meta-analysis. J. Clin. Endocrinol. Metab. 2015, 100, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Syngelaki, A.; Nicolaides, K.H.; Balani, J.; Hyer, S.; Akolekar, R.; Kotecha, R.; Pastides, A.; Shehata, H. Metformin versus Placebo in Obese Pregnant Women without Diabetes Mellitus. N. Engl. J. Med. 2016, 374, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Cassina, M.; Donà, M.; Di Gianantonio, E.; Litta, P.; Clementi, M. First-trimester exposure to metformin and risk of birth defects: A systematic review and meta-analysis. Hum. Reprod. Update 2014, 20, 656–669. [Google Scholar] [CrossRef] [PubMed]
- Legro, R.S.; Arslanian, S.A.; Ehrmann, D.A.; Hoeger, K.M.; Murad, M.H.; Pasquali, R.; Welt, C.K. Diagnosis and treatment of polycystic ovary syndrome: An Endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 2013, 98, 4565–4592. [Google Scholar] [CrossRef] [PubMed]
- Knowler, W.C.; Fowler, S.E.; Hamman, R.F.; Christophi, C.A.; Hoffman, H.J.; Brenneman, A.T.; Brenneman, A.T.; Brown-Friday, J.O.; Goldberg, R.; Venditti, E.; et al. 10-year follow-up of diabetes incidence and weight loss in the Diabetes Prevention Program Outcomes Study. Lancet 2009, 374, 1677–1686. [Google Scholar] [PubMed]
- Hostalek, U.; Gwilt, M.; Hildemann, S. Therapeutic Use of Metformin in Prediabetes and Diabetes Prevention. Drugs 2015, 75, 1071–1094. [Google Scholar] [CrossRef] [PubMed]
- Igel, L.I.; Sinha, A.; Saunders, K.H.; Apovian, C.M.; Vojta, D.; Aronne, L.J. Metformin: An Old Therapy that Deserves a New Indication for the Treatment of Obesity. Curr. Atheroscler. Rep. 2016, 18, 16. [Google Scholar] [CrossRef] [PubMed]
- Lamanna, C.; Monami, M.; Marchionni, N.; Mannucci, E. Effect of metformin on cardiovascular events and mortality: A meta-analysis of randomized clinical trials. Diabetes Obes. Metab. 2011, 13, 221–228. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Goodman, A.M. Efficacy of metformin in patients with non-insulin-dependent diabetes mellitus. The Multicenter Metformin Study Group. N. Engl. J. Med. 1995, 333, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.R.; Morrison, V.L.; Levin, D.; Mohan, M.; Forteath, C.; Beall, C.; McNeilly, A.D.; Balfour, D.J.; Savinko, T.; Wong, A.K.; et al. Anti-Inflammatory Effects of Metformin Irrespective of Diabetes Status. Circ. Res. 2016, 119, 652–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, P.J. Obesity and Breast Cancer Outcomes: How Much Evidence Is Needed to Change Practice? J. Clin. Oncol. 2016, 34, 646–648. [Google Scholar] [CrossRef] [PubMed]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a Tool to Target Aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.H. Current Information Gaps in Micronutrient Research, Programs and Policy: How Can We Fill Them? World Rev. Nutr. Diet 2016, 115, 109–117. [Google Scholar] [PubMed]
- Bruins, M.J.; Kupka, R.; Zimmermann, M.B.; Lietz, G.; Engle-Stone, R.; Kraemer, K. Maximizing the benefits and minimizing the risks of intervention programs to address micronutrient malnutrition: Symposium report. Matern. Child Nutr. 2016, 12, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.R.; Sobczyńska-Malefora, A. The adverse effects of an excessive folic acid intake. Eur. J. Clin. Nutr. 2016. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Mattaini, K.R.; Sullivan, M.R.; Vander Heiden, M.G. The importance of serine metabolism in cancer. J. Cell Biol. 2016, 214, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Kopp, M.; Morisset, R.; Koehler, P.; Rychlik, M. Stable Isotope Dilution Assays for Clinical Analyses of Folates and Other One-Carbon Metabolites: Application to Folate-Deficiency Studies. PLoS ONE 2016, 11, E0156610. [Google Scholar] [CrossRef] [PubMed]
- Mönch, S.; Netzel, M.; Netzel, G.; Ott, U.; Frank, T.; Rychlik, M. Folate bioavailability from foods rich in folates assessed in a short term human study using stable isotope dilution assays. Food Funct. 2015, 6, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Mato, J.M.; Martinez-Chantar, M.L.; Lu, S.C. Methionine metabolism and liver disease. Annu. Rev. Nutr. 2008, 28, 273–293. [Google Scholar] [CrossRef] [PubMed]
- Pacana, T.; Cazanave, S.; Verdianelli, A.; Patel, V.; Min, H.K.; Mirshahi, F.; Quinlivan, E.; Sanyal, A.J. Dysregulated Hepatic Methionine Metabolism Drives Homocysteine Elevation in Diet-Induced Nonalcoholic Fatty Liver Disease. PLoS ONE 2015, 10, E0136822. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.C.; Kaya, A.; Gladyshev, V.N. Methionine restriction and life-span control. Ann. N. Y. Acad. Sci. 2016, 1363, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Tibbetts, A.S.; Appling, D.R. Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 2010, 30, 57–81. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.F.; Huang, R.F.; Shane, B. Regulation of folate and one-carbon metabolism in mammalian cells. III. Role of mitochondrial folylpoly-gamma-glutamate synthetase. J. Biol. Chem. 1993, 268, 21674–21679. [Google Scholar] [PubMed]
- Lawrence, S.A.; Titus, S.A.; Ferguson, J.; Heineman, A.L.; Taylor, S.M.; Moran, R.G. Mammalian mitochondrial and cytosolic folylpolyglutamate synthetase maintain the subcellular compartmentalization of folates. J. Biol. Chem. 2014, 289, 29386–29396. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, E.A.; Titus, S.A.; Taylor, S.M.; Jackson-Cook, C.; Moran, R.G. A mutation inactivating the mitochondrial inner membrane folate transporter creates a glycine requirement for survival of Chinese hamster cells. J. Biol. Chem. 2004, 279, 33829–33836. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.E.; Patel, H.; Kuzmanov, U.; Mejia, N.R.; MacKenzie, R.E. Disruption of the mthfd1 gene reveals a monofunctional 10-formyltetrahydrofolate synthetase in mammalian mitochondria. J. Biol. Chem. 2005, 280, 7597–7602. [Google Scholar] [CrossRef] [PubMed]
- Field, M.S.; Kamynina, E.; Stover, P.J. MTHFD1 regulates nuclear de novo thymidylate biosynthesis and genome stability. Biochimie 2016, 126, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Giardina, G.; Brunotti, P.; Fiascarelli, A.; Cicalini, A.; Costa, M.G.; Buckle, A.M.; di Salvo, M.L.; Giorgi, A.; Marani, M.; Paone, A.; et al. How pyridoxal 5′-phosphate differentially regulates human cytosolic and mitochondrial serine hydroxymethyltransferase oligomeric state. FEBS J. 2015, 282, 1225–1241. [Google Scholar] [CrossRef] [PubMed]
- Hampson, R.K.; Barron, L.L.; Olson, M.S. Regulation of the glycine cleavage system in isolated rat liver mitochondria. J. Biol. Chem. 1983, 258, 2993–2999. [Google Scholar] [PubMed]
- Horne, D.W.; Holloway, R.S.; Wagner, C. Transport of S-adenosylmethionine in isolated rat liver mitochondria. Arch. Biochem. Biophys. 1997, 343, 201–206. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, N.; Sweeney, T.; Donagh, R.; Clarke, K.J.; Porter, R.K. Control of choline oxidation in rat kidney mitochondria. Biochim. Biophys. Acta 2009, 1787, 1135–1139. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Dreumont, N.; Coelho, D.; Guéant, J.L.; Arnold, C. Genetic animal models to decipher the pathogenic effects of vitamin B12 and folate deficiency. Biochimie 2016, 126, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.Y.; Makar, K.W.; Neuhouser, M.L.; Miller, J.W.; Song, X.; Brown, E.C.; Beresford, S.A.; Zheng, Y.; Poole, E.M.; Galbraith, R.L.; et al. Folate-mediated one-carbon metabolism genes and interactions with nutritional factors on colorectal cancer risk: Women’s Health Initiative Observational Study. Cancer 2015, 121, 3684–3691. [Google Scholar] [CrossRef] [PubMed]
- Pangilinan, F.; Molloy, A.M.; Mills, J.L.; Troendle, J.F.; Parle-McDermott, A.; Kay, D.M.; Browne, M.L.; McGrath, E.C.; Abaan, H.O.; Sutton, M.; et al. Replication and exploratory analysis of 24 candidate risk polymorphisms for neural tube defects. BMC Med. Genet. 2014, 15, 102. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.M.; Dear, A.; Craig, J.M.; Norman, P.E.; Golledge, J. The potential role of homocysteine mediated DNA methylation and associated epigenetic changes in abdominal aortic aneurysm formation. Atherosclerosis 2013, 228, 295–305. [Google Scholar] [CrossRef] [PubMed]
- De Vilbiss, E.A.; Gardner, R.M.; Newschaffer, C.J.; Lee, B.K. Maternal folate status as a risk factor for autism spectrum disorders: A review of existing evidence. Br. J. Nutr. 2015, 114, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Martorell, L.; Segués, T.; Folch, G.; Valero, J.; Joven, J.; Labad, A.; Vilella, E. New variants in the mitochondrial genomes of schizophrenic patients. Eur. J. Hum. Genet. 2006, 14, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Vilella, E.; Virgos, C.; Murphy, M.; Martorell, L.; Valero, J.; Simó, J.M.; Joven, J.; Fernández-Ballart, J.; Labad, A. Further evidence that hyperhomocysteinemia and methylenetetrahydrofolate reductase C677T and A1289C polymorphisms are not risk factors for schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2005, 29, 1169–1174. [Google Scholar] [CrossRef] [PubMed]
- Ron-Harel, N.; Santos, D.; Ghergurovich, J.M.; Sage, P.T.; Reddy, A.; Lovitch, S.B.; Dephoure, N.; Satterstrom, F.K.; Sheffer, M.; Spinelli, J.B.; et al. Mitochondrial Biogenesis and Proteome Remodeling Promote One-Carbon Metabolism for T Cell Activation. Cell Metab. 2016, 24, 104–117. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Vander Heiden, M.G. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 2015, 162, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fan, J.; Venneti, S.; Wan, Y.W.; Pawel, B.R.; Zhang, J.; Finley, L.W.; Lu, C.; Lindsten, T.; Cross, J.R. Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov. 2014, 4, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.L.; Kafri, R.; Kirschner, M.W.; Clish, C.B.; Mootha, V.K. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Maddocks, O.D.; Berkers, C.R.; Mason, S.M.; Zheng, L.; Blyth, K.; Gottlieb, E.; Vousden, K.H. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 2013, 493, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. The metabolic regulation of aging. Nat. Med. 2015, 21, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Suliman, H.B.; Piantadosi, C.A. Mitochondrial Quality Control as a Therapeutic Target. Pharmacol. Rev. 2016, 68, 20–48. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Zhai, D.; Cabezas, E.; Welsh, K.; Nouraini, S.; Satterthwait, A.C.; Reed, J.C. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature 2003, 423, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Zeng, J.; Drew, B.G.; Sallam, T.; Martin-Montalvo, A.; Wan, J.; Kim, S.J.; Mehta, H.; Hevener, A.L.; de Cabo, R.; et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 2015, 21, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Quirós, P.M.; Mottis, A.; Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 2016, 17, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Morton, N.M.; Beltram, J.; Carter, R.N.; Michailidou, Z.; Gorjanc, G.; McFadden, C.; Barrios-Llerena, M.E.; Rodriguez-Cuenca, S.; Gibbins, M.T.; Aird, R.E.; et al. Genetic identification of thiosulfate sulfurtransferase as an adipocyte-expressed antidiabetic target in mice selected for leanness. Nat. Med. 2016, 22, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Fitzgibbons, T.P.; Czech, M.P. Emerging evidence for beneficial macrophage functions in atherosclerosis and obesity-induced insulin resistance. J. Mol. Med. 2016, 94, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 2016, 26, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Beltrán-Debón, R.; Rodríguez-Gallego, E.; Fernández-Arroyo, S.; Senan-Campos, O.; Massucci, F.A.; Hernández-Aguilera, A.; Sales-Pardo, M.; Guimerà, R.; Camps, J.; Menendez, J.A.; et al. The acute impact of polyphenols from Hibiscus sabdariffa in metabolic homeostasis: An approach combining metabolomics and gene-expression analyses. Food Funct. 2015, 6, 2957–2966. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wu, J.; Zhu, J.; Kuei, C.; Yu, J.; Shelton, J.; Sutton, S.W.; Li, X.; Yun, S.J.; Mirzadegan, T.; et al. Lactate inhibits lipolysis in fat cells through activation of an orphan G-protein-coupled receptor, GPR81. J. Biol. Chem. 2009, 284, 2811–2822. [Google Scholar] [CrossRef] [PubMed]
- Haas, R.; Cucchi, D.; Smith, J.; Pucino, V.; Macdougall, C.E.; Mauro, C. Intermediates of Metabolism: From Bystanders to Signalling Molecules. Trends Biochem. Sci. 2016, 41, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.R.; Ong, S.E.; Goldberger, O.; Peng, J.; Sharma, R.; Thompson, D.A.; Vafai, S.B.; Cox, A.G.; Marutani, E.; Ichinose, F.; et al. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. eLife 2016, 5, E10575. [Google Scholar] [CrossRef] [PubMed]
- Cuyàs, E.; Fernández-Arroyo, S.; Corominas-Faja, B.; Rodríguez-Gallego, E.; Bosch-Barrera, J.; Martin-Castillo, B.; de Llorens, R.; Joven, J.; Menendez, J.A. Oncometabolic mutation IDH1 R132H confers a metformin-hypersensitive phenotype. Oncotarget 2015, 6, 12279–12296. [Google Scholar] [CrossRef] [PubMed]
- Katada, S.; Imhof, A.; Sassone-Corsi, P. Connecting threads: Epigenetics and metabolism. Cell 2012, 148, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Huypens, P.; Sass, S.; Wu, M.; Dyckhoff, D.; Tschöp, M.; Theis, F.; Marschall, S.; Hrabe de Angelis, M.; Beckers, J. Epigenetic germline inheritance of diet-induced obesity and insulin resistance. Nat. Genet. 2016, 48, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Öst, A.; Lempradl, A.; Casas, E.; Weigert, M.; Tiko, T.; Deniz, M.; Pantano, L.; Boenisch, U.; Itskow, P.M.; Stoeckius, M.; et al. Paternal diet defines offspring chromatin state and intergenerational obesity. Cell 2014, 159, 1352–1364. [Google Scholar] [CrossRef] [PubMed]
- Foster, G.D.; Wyatt, H.R.; Hill, J.O.; Makris, A.P.; Rosenbaum, D.L.; Brill, C.; Stein, R.I.; Mohammed, B.S.; Miller, B.; Rader, D.J.; et al. Weight and metabolic outcomes after 2 years on a low-carbohydrate versus low-fat diet: A randomized trial. Ann. Intern. Med. 2010, 153, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Painter, R.C.; Roseboom, T.J.; Bleker, O.P. Prenatal exposure to the Dutch famine and disease in later life: An overview. Reprod. Toxicol. 2005, 20, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; He, Y.; Qi, L.; Jaddoe, V.W.; Feskens, E.J.; Yang, X.; Ma, G.; Hu, F.N. Exposure to the Chinese famine in early life and the risk of hyperglycemia and type 2 diabetes in adulthood. Diabetes 2010, 59, 2400–2406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanner, S.A.; Bulmer, K.; Andrès, C.; Lantseva, O.E.; Borodina, V.; Poteen, V.V.; Yudkin, J.S. Does malnutrition in utero determine diabetes and coronary heart disease in adulthood? Results from the Leningrad siege study, a cross sectional study. BMJ 1997, 315, 1342–1348. [Google Scholar] [CrossRef] [PubMed]
- Finer, S.; Saravanan, P.; Hitman, G.; Yajnik, C. The role of the one-carbon cycle in the developmental origins of Type 2 diabetes and obesity. Diabet. Med. 2014, 31, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Tobi, E.W.; Goeman, J.J.; Monajemi, R.; Gu, H.; Putter, H.; Zhang, Y.; Slieker, R.C.; Stok, A.P.; Thijssen, P.E.; Müller, F.; et al. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat. Commun. 2014, 5, 5592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Aguilera, A.; Fernández-Arroyo, S.; Cuyàs, E.; Luciano-Mateo, F.; Cabre, N.; Camps, J.; Lopez-Miranda, J.; Menendez, J.A.; Joven, J. Epigenetics and nutrition-related epidemics of metabolic diseases: Current perspectives and challenges. Food. Chem. Toxicol. 2016, 96, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Klosin, A.; Lehner, B. Mechanisms, timescales and principles of trans-generational epigenetic inheritance in animals. Curr. Opin. Genet. Dev. 2016, 36, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Partridge, L. Promoting health and longevity through diet: From model organisms to humans. Cell 2015, 161, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Peiris-Pagés, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Maddocks, O.D.; Labuschagne, C.F.; Adams, P.D.; Vousden, K.H. Serine metabolism supports the methionine cycle and DNA/RNA methylation through de novo ATP Synthesis in Cancer Cells. Mol. Cell 2016, 61, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Mentch, S.J.; Locasale, J.W. One-carbon metabolism and epigenetics: Understanding the specificity. Ann. N. Y. Acad. Sci. 2016, 1363, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; Gan, H.; Lee, J.H.; Han, J.; Wang, Z.; Riester, S.M.; Jin, L.; Chen, J.; Zhou, H.; Wang, J.; et al. The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science 2016, 352, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Orgeron, M.L.; Stone, K.P.; Wanders, D.; Cortez, C.C.; Van, N.T.; Gettys, T.W. The impact of dietary methionine restriction on biomarkers of metabolic health. Prog. Mol. Biol. Transl. Sci. 2014, 121, 351–376. [Google Scholar] [PubMed]
- Cuyàs, E.; Fernández-Arroyo, S.; Joven, J.; Menendez, J.A. Metformin targets histone acetylation in cancer-prone epithelial cells. Cell Cycle 2016, 15, 3413–3416. [Google Scholar] [CrossRef] [PubMed]
- Farber, S.; Diamond, L.K. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N. Engl. J. Med. 1948, 238, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Tannenbaum, A. The dependence of tumor formation on the composition of the calorie- restricted diet as well as on the degree of restriction. Cancer Res. 1945, 5, 616–625. [Google Scholar]
- Heckman-Stoddard, B.M.; Gandini, S.; Puntoni, M.; Dunn, B.K.; De Censi, A.; Szabo, E. Repurposing old drugs to chemoprevention: The case of metformin. Semin. Oncol. 2016, 43, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Marini, C.; Bianchi, G.; Buschiazzo, A.; Ravera, S.; Martella, R.; Bottoni, G.; Petretto, A.; Emionite, L.; Monteverde, E.; Capitanio, S.; et al. Divergent targets of glycolysis and oxidative phosphorylation result in additive effects of metformin and starvation in colon and breast cancer. Sci. Rep. 2016, 6, 19569. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Oliveras-Ferraros, C.; Cufí, S.; Corominas-Faja, B.; Joven, J.; Martin-Castillo, B. Vazquez-Martin, A. Metformin is synthetically lethal with glucose withdrawal in cancer cells. Cell Cycle 2012, 11, 2782–2792. [Google Scholar] [CrossRef] [PubMed]
- Van Wijk, J.P.; de Koning, E.J.; Cabezas, M.C.; op’t Roodt, J.; Joven, J.; Rabelink, T.J.; Hoepelman, A.I. Comparison of rosiglitazone and metformin for treating HIV lipodystrophy: A randomized trial. Ann. Intern. Med. 2005, 143, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Ross, F.A.; Chevtzoff, C.; Green, K.A.; Evans, A.; Fogarty, S.; Towler, M.C.; Brown, L.J.; Ogunbayo, O.A.; Evans, A.M.; et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 2010, 11, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014, 510, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Cufí, S.; Oliveras-Ferraros, C.; Martin-Castillo, B.; Joven, J.; Vellon, L.; Vazquez-Martín, A. Metformin and the ATM DNA damage response (DDR): Accelerating the onset of stress-induced senescence to boost protection against cancer. Aging 2011, 3, 1063–1077. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Joven, J. One-carbon metabolism: An aging-cancer crossroad for the gerosuppressant metformin. Aging 2012, 4, 894–898. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Martin-Castillo, B.; Joven, J. Metformin and cancer: Quo vadis et cui bono? Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Municio, C.; Soler-Palacios, B.; Estrada-Capetillo, L.; Benguria, A.; Dopazo, A.; García-Lorenzo, E.; Fernández-Arroyo, S.; Joven, J.; Miranda-Carús, M.E.; González-Álvaro, I.; et al. Methotrexate selectively targets human proinflammatory macrophages through a thymidylate synthase/p53 axis. Ann. Rheum. Dis. 2016, 75, 2157–2165. [Google Scholar] [CrossRef] [PubMed]
- Rena, G.; Pearson, E.R.; Sakamoto, K. Molecular mechanism of action of metformin: Old or new insights? Diabetologia 2013, 56, 1898–1906. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Sheardown, S.A.; Brown, C.; Owen, R.P.; Zhang, S.; Castro, R.A.; Ianculescu, A.G.; Yue, L.; Lo, J.C.; Burchard, E.G.; et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J. Clin. Investig. 2007, 117, 1422–1431. [Google Scholar] [CrossRef] [PubMed]
- Dujic, T.; Zhou, K.; Donnelly, L.A.; Tavendale, R.; Palmer, C.N.; Pearson, E.R. Association of organic cation transporter 1 with intolerance to metformin in type 2 diabetes: A GoDARTS study. Diabetes 2015, 64, 1786–1793. [Google Scholar] [CrossRef] [PubMed]
- Dujic, T.; Causevic, A.; Bego, T.; Malenica, M.; Velija-Asimi, Z.; Pearson, E.R.; Semiz, S. Organic cation transporter 1 variants and gastrointestinal side effects of metformin in patients with type 2 diabetes. Diabet. Med. 2016, 33, 511–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, K.; Bellenguez, C.; Spencer, C.C.; Bennett, A.J.; Coleman, R.L.; Tavendale, R.; Hawley, S.A.; Donnelly, L.A.; Schofield, C.; Groves, C.J.; et al. Common variants near ATM are associated with glycemic response to metformin in type 2 diabetes. Nat. Genet. 2011, 43, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A.J. Personalising metformin therapy: A clinician’s perspective. Lancet Diabetes Endocrinol. 2014, 2, 442–444. [Google Scholar] [CrossRef]
- Ter Borg, S.; de Groot, L.C.; Mijnarends, D.M.; de Vries, J.H.; Verlaan, S.; Meijboom, S.; Luiking, Y.C.; Schols, J.M. Differences in Nutrient Intake and Biochemical Nutrient Status Between Sarcopenic and Nonsarcopenic Older Adults-Results From the Maastricht Sarcopenia Study. J. Am. Med. Dir. Assoc. 2016, 17, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, G.; Gagliano, C.; Salomone, S.; Giordano, M.; Bucolo, C.; Pappalardo, A.; Drago, F.; Caraci, F.; Avitabile, T.; Motta, M. Folate status in type 2 diabetic patients with and without retinopathy. Clin. Ophthalmol. 2015, 9, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Mudryj, A.N.; de Groh, M.; Aukema, H.M.; Yu, N. Folate intakes from diet and supplements may place certain Canadians at risk for folic acid toxicity. Br. J. Nutr. 2016, 116, 1236–1245. [Google Scholar] [CrossRef] [PubMed]
- Danenberg, P.V.; Gustavsson, B.; Johnston, P.; Lindberg, P.; Moser, R.; Odin, E.; Peters, G.J.; Petrelli, N. Folates as adjuvants to anticancer agents: Chemical rationale and mechanism of action. Crit. Rev. Oncol. Hematol. 2016, 106, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Yajnik, C.S.; Deshpande, S.S.; Jackson, A.A.; Refsum, H.; Rao, S.; Fisher, D.J.; Bhat, D.S.; Naik, S.S.; Coyaki, K.J.; Joglekar, C.V.; et al. Vitamin B12 and folate concentrations during pregnancy and insulin resistance in the offspring: The Pune Maternal Nutrition Study. Diabetologia 2008, 51, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.M.; Weir, D.G. The methyl folate trap. A physiological response in man to prevent methyl group deficiency in kwashiorkor (methionine deficiency) and an explanation for folic-acid induced exacerbation of subacute combined degeneration in pernicious anaemia. Lancet 1981, 2, 337–340. [Google Scholar] [CrossRef]
- Stowers, J.M.; Smith, O.A. Vitamin B12 and metformin. BMJ 1971, 3, 246–247. [Google Scholar] [CrossRef] [PubMed]
- Ting, R.Z.; Szeto, C.C.; Chan, M.H.; Ma, K.K.; Chow, K.M. Risk factors of vitamin B12 deficiency in patients receiving metformin. Arch. Intern. Med. 2006, 166, 1975–1979. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, S.; Quan, H.; Li, J. Vitamin B12 status in metformin treated patients: Systematic review. PLoS ONE 2014, 9, E100379. [Google Scholar] [CrossRef] [PubMed]
- Obeid, R.; Jung, J.; Falk, J.; Herrmann, W.; Geisel, J.; Friesenhahn-Ochs, B.; Lammerts, F.; Fassbender, K.; Kostopoulos, P. Serum vitamin B12 not reflecting vitamin B12 status in patients with type 2 diabetes. Biochimie 2013, 95, 1056–1061. [Google Scholar] [CrossRef] [PubMed]
- Greibe, E.; Trolle, B.; Bor, M.V.; Lauszus, F.F.; Nexo, E. Metformin lowers serum cobalamin without changing other markers of cobalamin status: A study on women with polycystic ovary syndrome. Nutrients 2013, 5, 2475–2482. [Google Scholar] [CrossRef] [PubMed]
- Leung, S.; Mattman, A.; Snyder, F.; Kassam, R.; Meneilly, G.; Nexo, E. Metformin induces reductions in plasma cobalamin and haptocorrin bound cobalamin levels in elderly diabetic patients. Clin. Biochem. 2010, 43, 759–760. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.A. Metformin and Vitamin B12 Deficiency: Where Do We Stand? J. Pharm. Pharm. Sci. 2016, 19, 382–398. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.; Fleming, G.A.; Chen, K.; Bicsak, T.A. Metformin-associated lactic acidosis: Current perspectives on causes and risk. Metabolism 2016, 65, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Pernicova, I.; Korbonits, M. Metformin—Mode of action and clinical implications for diabetes and cancer. Nat. Rev. Endocrinol. 2014, 10, 143–156. [Google Scholar] [CrossRef] [PubMed]
- De Jager, J.; Kooy, A.; Lehert, P.; Wulffelé, M.G.; van der Kolk, J.; Bets, D.; Verburg, J.; Donker, A.J.; Stehouwer, C.D. Long term treatment with metformin in patients with type 2 diabetes and risk of vitamin B-12 deficiency: Randomised placebo controlled trial. BMJ 2010, 340, c2181. [Google Scholar] [CrossRef] [PubMed]
- McCreight, L.J.; Bailey, C.J.; Pearson, E.R. Metformin and the gastrointestinal tract. Diabetologia 2016, 59, 426–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.; Thazhath, S.S.; Bound, M.J.; Jones, K.L.; Horowitz, M.; Rayner, C.K. Mechanism of increase in plasma intact GLP-1 by metformin in type 2 diabetes: Stimulation of GLP-1 secretion or reduction in plasma DPP-4 activity? Diabetes Res. Clin. Pract. 2014, 106, e3–e6. [Google Scholar] [CrossRef] [PubMed]
- Yee, S.W.; Lin, L.; Merski, M.; Keiser, M.J.; Gupta, A.; Zhang, Y.; Chien, H.C.; Shoichet, B.K.; Giacomini, K.M. Prediction and validation of enzyme and transporter off-targets for metformin. J. Pharmacokinet. Pharmacodyn. 2015, 42, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Scarpello, J.H.; Hodgson, E.; Howlett, H.C. Effect of metformin on bile salt circulation and intestinal motility in type 2 diabetes mellitus. Diabet. Med. 1998, 15, 651–656. [Google Scholar] [CrossRef]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Krogh Pedersen, H.; et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, J.H.; Johnson, M.; Johnson, J.; Hsia, D.S.; Greenway, F.L.; Heiman, M.L. Addition of a gastrointestinal microbiome modulator to metformin improves metformin tolerance and fasting glucose levels. J. Diabetes Sci. Technol. 2015, 9, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Boren, J.; Smith, U. Confounding Effects of Metformin on the Human Gut Microbiome in Type 2 Diabetes. Cell Metab. 2016, 23, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Bhalerao, K.D.; Lee, S.C.; Soboyejo, W.O.; Soboyejo, A.B. A folic acid-based functionalized surface for biosensor systems. J. Mater. Sci. Mater. Med. 2007, 18, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Armbruster, D.A.; Alexander, D.B. Sample to sample carryover: A source of analytical laboratory error and its relevance to integrated clinical chemistry/immunoassay systems. Clin. Chim. Acta 2006, 373, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Bertran, N.; Camps, J.; Fernández-Ballart, J.; Murphy, M.M.; Arija, V.; Ferré, N.; Tous, M.; Joven, J. Evaluation of a high-sensitivity turbidimetric immunoassay for serum C-reactive protein: Application to the study of longitudinal changes throughout normal pregnancy. Clin. Chem. Lab. Med. 2005, 43, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, P.; Narayanan, S.; Cook, P. False-normal vitamin B12 results in a patient with pernicious anaemia. Clin. Biochem. 2015, 48, 1366–1377. [Google Scholar] [CrossRef] [PubMed]
- Harrington, D.J. Holotranscobalamin: In the middle of difficultly lies opportunity. Clin. Chem. Lab. Med. 2016, 54, 1407–1409. [Google Scholar] [CrossRef] [PubMed]
- Kancherla, V.; Garn, J.V.; Zakai, N.A.; Williamson, R.S.; Cashion, W.T.; Odewole, O.; Judd, S.E.; Oakley, G.P., Jr. Multivitamin Use and Serum Vitamin B12 Concentrations in Older-Adult Metformin Users in REGARDS, 2003–2007. PLoS ONE 2016, 11, E0160802. [Google Scholar] [CrossRef] [PubMed]
- Russo, G.T.; Giandalia, A.; Romeo, E.L.; Scarcella, C.; Gambadoro, N.; Zingale, R.; Forte, F.; Perdichizzi, G.; Alibrandi, A.; Cucinotta, D. Diabetic neuropathy is not associated with homocysteine, folate, vitamin B12 levels, and MTHFR C677T mutation in type 2 diabetic outpatients taking metformin. J. Endocrinol. Investig. 2016, 39, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Bird, J.K.; Ronnenberg, A.G.; Choi, S.W.; Du, F.; Mason, J.B.; Liu, Z. Obesity is associated with increased red blood cell folate despite lower dietary intakes and serum concentrations. J. Nutr. 2015, 145, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Denimal, D.; Brindisi, M.C.; Lemaire, S.; Duvillard, L. Assessment of Folate Status in Obese Patients: Should We Measure Folate in Serum or in Red Blood Cells? Obes. Surg. 2016. [Google Scholar] [CrossRef] [PubMed]
- Yetley, E.A.; Johnson, C.L. Folate and vitamin B-12 biomarkers in NHANES: History of their measurement and use. Am. J. Clin. Nutr. 2011, 94, 322S–331S. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, S.; Li, L.; Li, Q.; Ren, K.; Sun, X.; Li, J. Metformin treatment and homocysteine: A systematic review and meta-analysis of randomized controlled trials. Nutrients 2016, 8, 798. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, C.M.; Sternberg, M.R.; Fazili, Z.; Lacher, D.A.; Zhang, M.; Johnson, C.L.; Hamner, H.C.; Bailey, R.L.; Rader, J.I.; Yamini, S. Folate status and concentrations of serum folate forms in the US population: National Health and Nutrition Examination Survey 2011–2012. Br. J. Nutr. 2015, 113, 1965–1977. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Salas, P.; Moore, S.E.; Cole, D.; da Costa, K.A.; Cox, S.E.; Dyer, R.A.; Fuldord, A.J.; Innis, S.M.; Waterland, R.A.; Zeisel,, S.H.; et al. DNA methylation potential: Dietary intake and blood concentrations of one-carbon metabolites and cofactors in rural African women. Am. J. Clin. Nutr. 2013, 97, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Kühnen, P.; Handke, D.; Waterland, R.A.; Hennig, B.J.; Silver, M.; Fulford, A.J.; Dominguez-Salas, P.; Moore, S.E.; Prentice, A.M.; Spranger, J.; et al. Interindividual Variation in DNA Methylation at a Putative POMC Metastable Epiallele Is Associated with Obesity. Cell Metab. 2016, 24, 502–509. [Google Scholar] [CrossRef]
- Eicher-Miller, H.A.; Fulgoni, V.L.; Keast, D.R. Processed Food Contributions to Energy and Nutrient Intake Differ among US Children by Race/Ethnicity. Nutrients 2015, 7, 10076–10088. [Google Scholar] [CrossRef] [PubMed]
- Colditz, G.A. Overview of the epidemiology methods and applications: Strengths and limitations of observational study designs. Crit. Rev. Food. Sci. Nutr. 2010, 50 (Suppl. 1), 10–12. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Arroyo, S.; Cuyàs, E.; Bosch-Barrera, J.; Alarcón, T.; Joven, J.; Menendez, J.A. Activation of the methylation cycle in cells reprogrammed into a stem cell-like state. Oncoscience 2016, 2, 958–967. [Google Scholar] [PubMed]
- Riera-Borrull, M.; Rodríguez-Gallego, E.; Hernández-Aguilera, A.; Luciano, F.; Ras, R.; Cuyàs, E.; Camps, J.; Segura-Carretero, A.; Menendez, J.A.; Joven, J.; et al. Exploring the Process of Energy Generation in Pathophysiology by Targeted Metabolomics: Performance of a Simple and Quantitative Method. J. Am. Soc. Mass. Spectrom. 2016, 27, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Romero, I.L.; Litchfield, L.M.; Lengyel, E.; Locasale, J.W. Metformin Targets Central Carbon Metabolism and Reveals Mitochondrial Requirements in Human Cancers. Cell Metab. 2016. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luciano-Mateo, F.; Hernández-Aguilera, A.; Cabre, N.; Camps, J.; Fernández-Arroyo, S.; Lopez-Miranda, J.; Menendez, J.A.; Joven, J. Nutrients in Energy and One-Carbon Metabolism: Learning from Metformin Users. Nutrients 2017, 9, 121. https://doi.org/10.3390/nu9020121
Luciano-Mateo F, Hernández-Aguilera A, Cabre N, Camps J, Fernández-Arroyo S, Lopez-Miranda J, Menendez JA, Joven J. Nutrients in Energy and One-Carbon Metabolism: Learning from Metformin Users. Nutrients. 2017; 9(2):121. https://doi.org/10.3390/nu9020121
Chicago/Turabian StyleLuciano-Mateo, Fedra, Anna Hernández-Aguilera, Noemi Cabre, Jordi Camps, Salvador Fernández-Arroyo, Jose Lopez-Miranda, Javier A. Menendez, and Jorge Joven. 2017. "Nutrients in Energy and One-Carbon Metabolism: Learning from Metformin Users" Nutrients 9, no. 2: 121. https://doi.org/10.3390/nu9020121