Protein Nutrition and Malnutrition in CKD and ESRD

1
Department of Nephrology, Guizhou Provincial People’s Hospital, Guizhou 550002, China
2
Division of Nephrology and Hypertension, Department of Medicine, Mayo Clinic College of Medicine, 200 First Street SW, Rochester, MN 55905, USA
*
Author to whom correspondence should be addressed.
Nutrients 2017, 9(3), 208; https://doi.org/10.3390/nu9030208
Submission received: 14 December 2016
/
Accepted: 23 February 2017
/
Published: 27 February 2017
(This article belongs to the Special Issue Nutrition and Chronic Kidney Disease)
{kind=link}
{kind=link}
Abstract
:Elevated protein catabolism and protein malnutrition are common in patients with chronic kidney disease (CKD) and end-stage renal disease (ESRD). The underlying etiology includes, but is not limited to, metabolic acidosis intestinal dysbiosis; systemic inflammation with activation of complements, endothelin-1 and renin-angiotensin-aldosterone (RAAS) axis; anabolic hormone resistance; energy expenditure elevation; and uremic toxin accumulation. All of these derangements can further worsen kidney function, leading to poor patient outcomes. Many of these CKD-related derangements can be prevented and substantially reversed, representing an area of great potential to improve CKD and ESRD care. This review integrates known information and recent advances in the area of protein nutrition and malnutrition in CKD and ESRD. Management recommendations are summarized. Thorough understanding the pathogenesis and etiology of protein malnutrition in CKD and ESRD patients will undoubtedly facilitate the design and development of more effective strategies to optimize protein nutrition and improve outcomes.
1. Introduction
Chronic kidney disease (CKD) has become a worldwide epidemic with an occurrence rate in the population of approximately 5%–15% [1]. Prevalence of end-stage renal disease (ESRD) population relying on dialysis is also on the rise [2]. Suboptimal nutritional intake is common in the population of CKD and ESRD and poses a direct risk for protein malnutrition [3,4]. Suboptimal nutritional status has been related to multiple alterations including metabolic acidosis, bowel flora alteration and hormonal dysregulation, all of which could promote kidney disease progression and increase morbidity and mortality. This review presents updated information, intergrading previous knowledge with a specific focus on the unique aspect of protein balance and nutrition in CKD and ESRD. Current practice recommendations are presented.
2. Protein Nutrition in Healthy Adults and in CKD and ESRD
The USA Food and Nutrition Board of the National Academy of Sciences suggests that a minimum dietary protein requirement for a healthy adult in a stable non-pregnant, non-lactating and non-recovery condition is 0.6 g/kg/day. Considering a safety margin, the “Recommended Dietary Allowance” (RDA) of protein intake is 0.8 g/kg/day [5]. It is recommended that greater than half of the protein intake should be of a high biologic value (HBV, nitrogen incorporated into the body/total absorbed nitrogen >75%), such as proteins in eggs, fish, poultry, meat and dairy products. The key feature of HBV proteins is the presence of essential amino acids (the amino acids that are not produced by the body and are required from dietary intake). Studies have shown, however, that the daily protein consumption of an average American is approximately 1.3 g/kg/day, 1.25 and 1.36 g/kg/day for women and men, respectively. Even among individuals over age 75, daily protein consumption remains at approximately 1.1 g/kg/day, considerably higher than recommended intake [6].
Dietary proteins are digested to amino acids which can be further broken down to generate both acids and bases. Proteins from meat and dairy products (from a typical Western diet) generate predominantly acidic products including hydrogen chloride (HCl), sulfuric acid (H2SO4) and phosphoric acids (H3PO4). These acids are nonvolatile and rely on kidney for their excretion (primarily in the form of ammonium salts and phosphoric salts). A healthy individual generates net acids, approximately 1 mEq/kg/day (mmol/kg/day), referred to as NEAP (net endogenous acid production). These are rapidly buffered by sodium bicarbonate (NaHCO3) to form sodium salts. During this process, bicarbonate is consumed, which needs to be regenerated, a task accomplished by the kidneys. To achieve a steady acid–base balance, renal tubules must reabsorb ~4500 mEq of filtered HCO3− and generate (through H+ excretion) an additional ~70–80 mEq HCO3− daily, to neutralize the daily net acid generation [7]. In patients with reduced kidney function, nonvolatile acids can accumulate causing metabolic acidosis.
3. Metabolic and Regulatory Derangements in CKD and ESRD
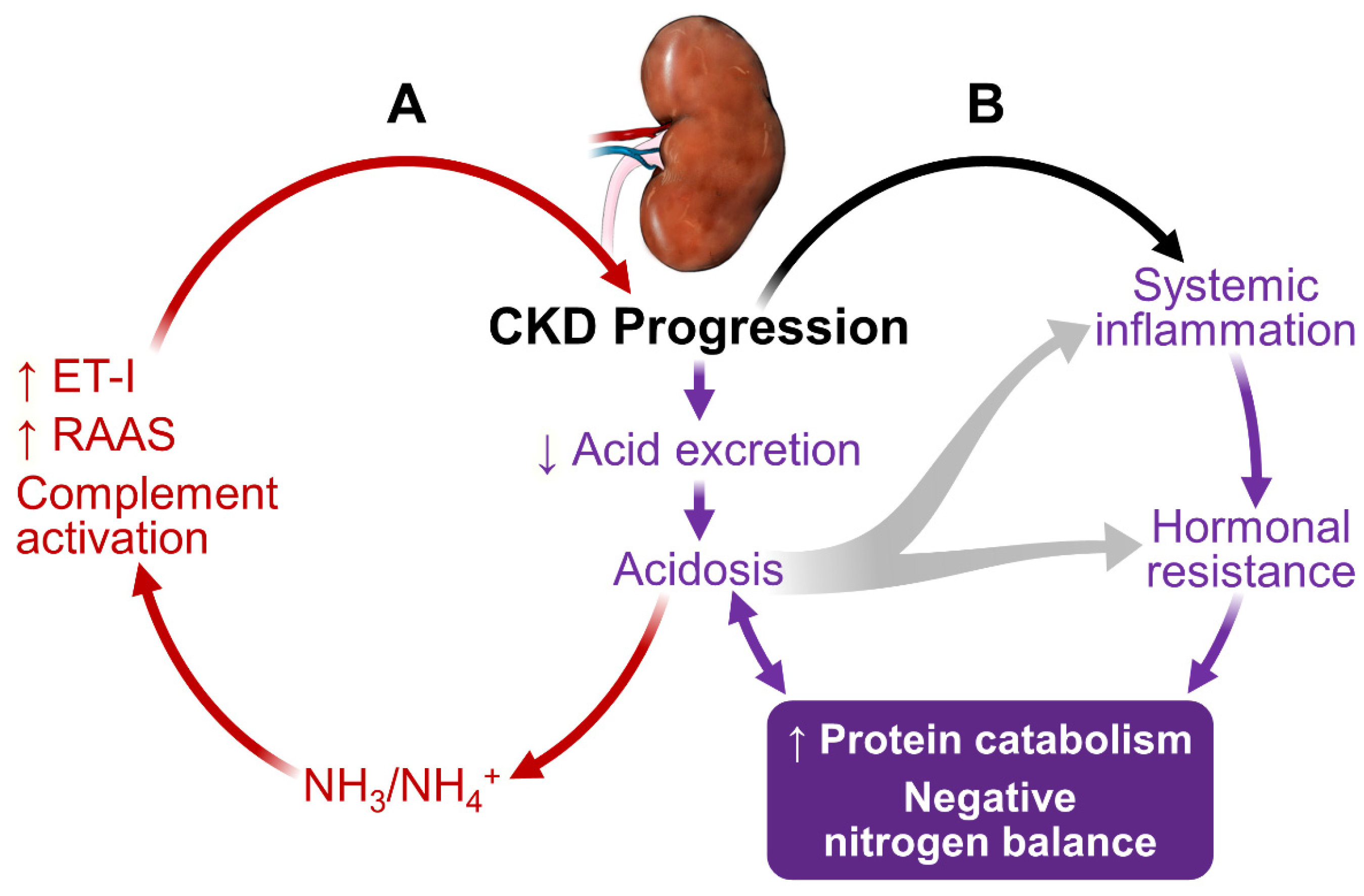
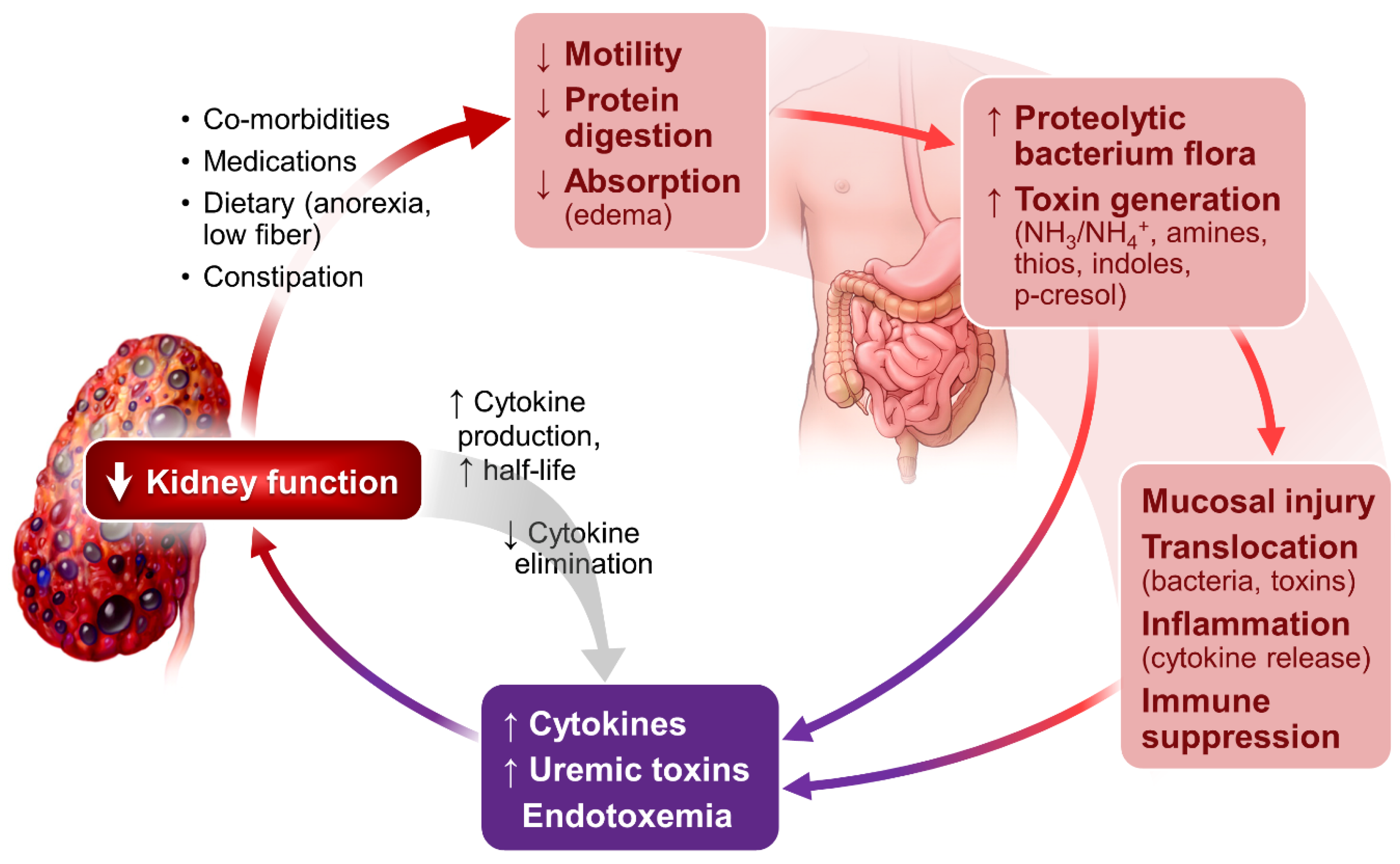
As shown in Figure 1, kidney dysfunction is associated with defects in acid excretion, systemic inflammation, end-organ hormone resistance and uremic toxin accumulation. These abnormalities can further worsen kidney function, creating a vicious circle, adversely affect patients’ outcome.
3.1. Metabolic Acidosis
In CKD, the reduced number of functioning nephrons compromises the kidney’s capacity to excrete acid [8]. Metabolic acidosis is the earliest and one of the most common manifestations of CKD. The acidosis worsens progressively as CKD progresses [9,10,11,12]. At the individual nephron level, each residual functioning nephron undergoes compensatory hypertrophy and generates an excess amount of NH3 in an attempt to excrete acid in the form of NH4+ [13,14]. NH3/NH4+ can lead to complement activation, resulting in tubule-interstitial inflammation, injury and fibrosis [15]. Acidosis also increases endothelin-1 and aldosterone production, furthering CKD progression [16,17].
Metabolic acidosis plays an important role in the accelerated protein catabolism, negative nitrogen balance and loss of lean body mass in CKD and ESRD [18,19]. Acidosis activates proteolysis through activating the ubiquitin-proteasome system (UPS) and caspase-3 [20]. Caspase-3 cleaves actomyosin and myofibrils, providing suitable substrates for UPS-mediated degradation. Caspase-3 also cleaves subunits (Rpt2 and 6) of 19S proteosome particle to activate the 26S proteosome-mediated protein degradation. Thus, acidosis in CKD can preferentially cause muscle protein breakdown to a much greater extent than mobilizing protein from other organs. Acidosis also contributes to insulin resistance, growth hormone resistance and glucocorticoid hypersecretion. These hormonal defects contribute to the protein catabolic state (detailed below).
It is not surprising that acidosis promotes CKD progression [21] and increases mortality [22]. Importantly, a normal range of serum HCO3− concentrations is associated with favorable clinical outcomes [23,24] and correction of acidosis corrects virtually all known adverse effects associated with acidosis in CKD and ESRD [15,25,26,27,28].
3.2. Sustained Inflammation
Sustained systemic and tissue inflammation is a prominent feature of CKD and ESRD [29,30]. It is related to a multitude of abnormalities in the setting of kidney failure. As illustrated in Figure 2, altered gut microbiome profile, evidenced in even the early stages of CKD [31] and in ESRD [32,33], plays an important role in the genesis of inflammation. Impaired protein digestion [34] increases intestinal protein fermentation by proteolytic bacteria and increases production of toxic metabolites including NH3/NH4OH, which are injurious to intestinal mucosa [35,36,37].
The intestinal epithelial injury results in the translocation of toxic metabolites and bacteria endotoxins from intestinal lumen to the circulation, stimulating production of inflammatory cytokines through binding the lipopolysaccharides and Toll-like receptors [38,39]. Impaired kidney elimination of uric acid also fosters the selection of gut bacteria that produce urease and uricase. Most of these bacteria are capable of generating toxins including indole and p-cresol, contributing to uremia in the setting of CKD and ESRD [40,41].
The generation and systemic accumulation of uremic toxins exemplify the importance of gut flora in the pathogenesis of uremic syndrome. Gut-derived indole and p-cresol are sulfonated in the liver, forming uremic toxins, indoxyl sulfate (IS) [42] and p-Cresyl sulfate (pCS). IS mediates renal tubulointerstitial fibrosis via upregulation and activation of TGF-β1 [43]. pCS increase the production of inflammatory cytokines and oxidative stress in CKD [44]. A recent in vitro study show that pCS induces macrophage activation but interfere with antigen processing, likely contributing to a compromised adaptive immune response [45]. Both IS and pCS activate RAAS, in additional to activating TGF/Smad pathways, and likely contribute to epithelial–mesangial transformation and CKD progression [46]. IS and pCS are associated with increased cardiovascular and all-cause mortality in patients with CKD and ESRD [47,48,49,50,51]. The readers are referred to several excellent comprehensive updates on uremic toxins [52,53] and the contribution of intestinal bacteria in the generation of uremic toxins and systemic inflammation [54].
In addition to gut source of inflammation, fat distribution is also associated with inflammatory state. Studies have shown that abdominal fat, not peripheral fat, is highly associated with inflammation, insulin resistance, dyslipidemia, and oxidative stress as well as cardiovascular events in CKD [55,56,57,58,59,60,61]. Dialysis and dialysis-related factors, both hemodialysis and peritoneal dialysis, are also a prominent source of inflammation [30,62,63].
Negative protein balance in the inflammatory state in CKD and ESRD can be ascribed to the activation of multiple cytokine (TNF, IL-1 and IL-6)-mediated mechanisms [64,65,66]. For instance, TWEAK (TNF-related weak inducer of apoptosis) is associated with pathways in the regulation of NF-kappa B (nuclear factor kappa light chain enhancer of activated B cells). It inhibits myogenesis and activates muscle protein degradative cascades [67]. TWEAK, as well as IL-6, is highly expressed in patients with kidney failure, and both are shown to be associated with reduced muscle strength in dialysis patients [68]. Myostatin, a TNF-beta superfamily protein, is upregulated in CKD and can be further activated by free radicals. Myostatin binds to muscle membrane ActRIIB (high-affinity type 2 Activin receptor), activates ALK-4 and ALK-5 (type I activin receptor serine kinases). These kinases trigger downstream phosphorylation of Smads 2 and 3, leading to signal activations that ultimately result in muscle degradation and atrophy. These effects are confirmed by multiple experimental loss-of-function myostatin mutations or deletions in animal models which consistently show that attenuation of myostatin leads to an increase in the size and number of skeletal muscle fibers and muscle mass [69,70], opposite to the observations in CKD and ESRD. Overexpression of an endogenous myostatin inhibitor (follistatin isoform) increases muscle mass and strength. Commensurate with gene manipulating studies, muscle levels of myostatin in the aging population and CKD patients are shown to be elevated [70], contributing to a negative muscle protein balance. Inhibiting cytokine pathways of myostatin in CKD can mitigate inflammation-associated muscle protein degradation, improve sensitivity to insulin/IGF-1 and reduce muscle protein breakdown, leading to increased muscle growth [71,72]. Moreover, exercise upregulates follistatin and the increase is associated with increased muscle strength and mass in patients with CKD [73,74].
Inflammation also induces multiple hormonal derangements including enhancing glucocorticoid-mediated effects and mitigating insulin/IGF-1 effects by inducing tissue resistance [75]. Muscle protein degrades, in part, through activation of intracellular NADPH oxidases [76,77,78]. IL-6 has also been shown to interact with serum amyloid A leading to impairment of insulin-IGF-1 signaling via activation of suppressor of cytokine signaling (SOCS3) and downstream loss of insulin receptor substrate 1 (IRS-1) in muscle [79]. Moreover, IL-6 mediated signaling impairs assimilation of endogenous amino acids for muscle protein synthesis and enhanced caspase-3 activity, further compromising protein nitrogen and muscle protein balance [80,81].
Collectively, inflammation, through a complex array of mechanisms, preferentially increases in muscle protein catabolism and suppresses muscle protein anabolism, leading to a net muscle protein loss in CKD and ESRD.
3.3. Hormonal Disorders
Hormonal disorders are prominent in CKD [82]. Acidosis, inflammation and uremic toxins have all been shown to contribute to the hormonal dysregulation in CKD and ESRD. Strong evidence demonstrates the existence of tissue resistance to insulin, growth hormone-insulin-like growth factor-insulin-like-growth factor binding protein (GH-IGF-IGFBP) axis, gonadal hormone (testosterone) and vitamin D. Catabolic activity of glucocorticoids, however, is elevated.
In CKD and ESRD, insulin resistance is associated with significantly elevated protein catabolism, due primarily to post-receptor defects and activation of UPS, leading to muscle protein degradation rather than reduced hepatic glucose intake. Importantly, the insulin resistance-related muscle protein breakdown in dialysis patients is seen not only in patients with diabetes but also in dialysis patients without overt diabetes. Notably, the negative nitrogen balance and hyperaminoacidemia resulting from elevated protein breakdown can be reversed through insulin administration [83]. Moreover, dialysis can correct, at least in part, diminished insulin response and improve tissue insulin sensitivity [84,85,86,87].
The GH-IGF-IGFBP axis plays an important role in kidney development and kidney diseases. Adequate activity of the axis enhances renal blood flow and GFR and can cause Na+ retention-mediated volume expansion [88]. CKD is associated with multiple derangements in the GH-IGF-IGFBP axis [89,90,91]. Although the plasma GH levels may be normal or elevated in CKD [90] due to limited GH clearance, at the tissue level, there is evidence of GH resistance, leading to insufficient downstream effects [92,93]. Serum IGF-1 levels may also be normal (or reduced in advanced CKD). Its circulating binding proteins, IGFBPs, tend to be elevated due to a heightened hepatic production and diminished renal clearance. IGFBPs bind to circulating IGF-1, which in most instances results in a decreased IGF bioavailability [94,95]. Tissue resistance to IGF-mediated effects has also been demonstrated in uremic rodents [96]. Urinary loss of IGF and IGFBP can also be significant in patients with nephrotic syndrome and contributes to the inadequate IGF-related functions [97].
Given the tissue resistance to GH and IGF-1, it should come as no surprise that children with kidney failure exhibit growth retardation. In adults, such hormone resistance manifests as an accelerated protein catabolism and protein malnutrition. Altered PI3-kinase/AKT activity, downstream signaling of growth factors (insulin and IGF-1), activates caspase-3 and enhances cleavage of actomyosin complexes and myofibrils [98] and is a critical step in muscle protein degradation. The signaling impairment also alters muscle satellite cell function [99], interfering with muscle injury repair and maintenance of muscle mass [79]. All of these degradative processes can be exacerbated in the context of insufficient energy provision. Thus, GH-IGF-IGFBP axis dysfunction plays an important role not only in growth but also in nitrogen and muscle protein balance.
Emerging studies have uncovered a novel regulatory signaling pathway of insulin/IGF mediated by a number of muscle specific micro-RNAs [100,101,102]. In CKD muscle, the microRNA expression pattern is altered [71]. For instance, mir-29 in CKD muscle is depressed, increasing YinYang1 protein and negatively regulating myogenesis [71]. Further studies in this area are necessary to improve our understanding of the complex mechanisms underlying muscle protein loss in CKD and ESRD. Manipulating muscle specific miroRNAs could constitute potentially useful novel targets for interventions to prevent and treat muscle protein loss and protein malnutrition.
Testosterone, a prototypical anabolic hormone, induces skeletal muscle hypertrophy and positive nitrogen balance under physiological conditions. Testosterone also inhibits expression of myostatin-mediated muscle protein degradation, induces muscle response to IGFs, promotes IGF-1 mRNA expression and recruits pluripotent stem cell differentiation into myocytes [103]. CKD patients have a blunted circulating testosterone level. This is due primarily to prolactin accumulation in CKD, leading to impaired gonadotropin releasing hormone secretion from the anterior pituitary, which, in turn, causes testosterone deficiency [104,105]. Dramatic reversal of this otherwise nearly universal occurrence of hypogonadism in dialysis patients after successful kidney transplantation [106,107] illustrates the highly toxic but reversible nature of the uremic milieu. Even at the early stages of CKD, a significant association of muscle mass loss with reduced endogenous testosterone has been demonstrated [108], a major component of negative protein balance patients with renal failure.
Another notable hormonal defect in CKD and ESRD is the heightened production and activity of glucocorticoids. Metabolic acidosis and inflammatory state in CKD pathologically enhances adrenal glucocorticoid production and activity. Glucocorticoids in CKD and ESRD, through activating the glucocorticoid receptor and binding to phosphatidylinositol 3-kinase, suppress Akt phosphorylation [75], a defect further magnified by the parallel presence of acidosis [109]. Reduction in phosphorylated Akt contributes to the muscle protein degradation through multiple mechanisms including upregulating proteolytic pathways and impairment of intracellular growth hormone (insulin/IGF-I)-mediated signaling pathways [98,110,111].
Suboptimal vitamin D status, common in patients with CKD and ESRD [112], has also been associated with muscle protein imbalance and catabolism. Tissue resistance to and reduced circulating 25-OH vitamin D, as well as increased fibroblast growth factor-23, worsens secondary hyperparathyroidism [113], which contributes to muscle degradation. Vitamin D has been shown to be involved in pathways of muscle regulation [114,115,116]. Moreover, replacement of active form of vitamin D (1,25-OH2 vitamin D) has been shown to improve muscle size and strength, markers of muscle metabolism as well as serum albumin concentration [115,117]. Vitamin D deficiency in CKD contributes to RAAS hyperactivation, causing multiple detrimental downstream effects including compromised mental status [118]. Altered mental status can lead to poor dietary intake, contributing to poor nutritional intake, intestinal dysmicrobia, accumulation of uremic toxins, and, ultimately, worsening protein malnutrition.
Taken together, in CKD and ESRD, there is enhanced activity of catabolic hormones and reduced activity and resistance to anabolic hormones. These changes together with other uremic conditions including acidemia, inflammation and decreased nutritional intake can work in concert to cause a persistent net negative nitrogen balance and loss of lean body mass.
4. Energy Prescription and Protein-Energy Wasting (PEW) in CKD and ESRD
Protein catabolism and nitrogen balance in CKD are tightly linked to energy intake [119]. A negative energy intake accelerates protein catabolism as protein being used from energy supply, leading to a negative nitrogen balance.
Multiple studies have shown that in patients with CKD and ESRD, their resting energy expenditure is increased compared to non-CKD individuals [120,121,122,123,124]. Inflammatory state and co-morbidities associated with CKD and ESRD such as cardiovascular disease, poorly controlled diabetes, and hyperparathyroidism can all contribute to the increased resting energy expenditure [121,123,124,125,126,127]. Resting energy expenditure is shown to increase from 12% to 20% during dialysis [128]. Thus, patients with renal failure require a higher amount of energy intake than healthy individuals. CKD and ESRD patients are, thus, susceptible to insufficient energy intake.
In non-dialysis CKD patients, a neutral or slightly positive nitrogen balance can be maintained with a low quantity (~0.6 g/kg/day) but high quality (HBV) protein diet and adequate energy intake (30–35 kcal/kg/day) [129]. With adequate energy intake (ketogenic diet) and supplemental amino acids, even with very low (0.3 g/kg/day) protein intake, CKD patients can maintain a neutral nitrogen balance and stable clinical status [130]. These dietary related favorable effects are allegedly derived from reduced generation of toxic waste products and enhanced insulin/IGF sensitivity. A recent meta-analysis by Jiang [131] adds weight to the existing impression that a low protein diet is effective in management of CKD without necessarily causing adverse safety and nutritional effects. An individualized meal plan should be devised under the supervision of a nephrology dietitian.
In patients on maintenance hemo- or peritoneal dialysis, their protein requirement is, on the contrary, much higher, 1.2–1.3 g/kg/day, based on KDOQI (Kidney Disease Outcomes Quality Initiative) Clinical Practice Guideline for Nutrition. The higher protein requirement is due to dialysis related protein loss, extra-energy expenditure and persistent inflammation [132]. Protein intake at <0.8 g/kg/day or >1.4 g/kg/day has been shown to be associated with increased mortality in dialysis patients [133].
Protein-energy wasting (PEW) denotes concurrent losses of protein and energy stores in patients with kidney dysfunction. It tends to develop and progress with CKD progression [134,135]. In the setting of suboptimal energy supply, CKD and ESRD patients catabolize muscle to provide needed energy, leading to protein malnutrition [136]. Limited physical activity in the setting of enhanced resting energy demand, although tempering the total body energy expenditure [137], would inevitably result in muscle atrophy and generalized deconditioning. The readers of this paper are referred to several detailed reviews on the topic [138,139,140,141].
5. Clinical Recommendations
An integrated multidisciplinary approach with targeted and individualized nutritional interventions based on the degree of kidney dysfunction, comorbidity, baseline nutritional status and physical functional capacity is necessary to improve outcomes for CKD and ESRD patients. Conditions that contribute to protein catabolism should be minimized or eliminated.
5.1. Optimizing Nutritional Therapy
Although randomized interventional patient-based trials are scarce, the general consensus is that active nutritional measures can mitigate a large number of the metabolic and hormonal derangements in CKD and ESRD. For patients with relatively stable health conditions, absence of active medical events and not on dialysis, nutritional assessment including intake and anthropometric measurements (body weight, BMI and valid indicators of nutritional status [142]) every 3–6 months is advisable. For patients with active medical issues such as surgical operation, acute infection, or cardiovascular events, more frequent nutritional analysis and modifications are required. Ongoing modification of nutritional parameters is necessary. Once a patient reaches ESRD, monthly review of the patient’s laboratory data and dietary intake by a renal dietitian or a nutritional professional is advised.
Dietary energy provision for both dialysis and non-dialysis CKD patients should be 30–35 kcal/kg (ideal body weight)/day. The recommended amount of protein intake for non-dialysis CKD patients is 0.6 to 0.8 g/kg/day with >50% HBV proteins. For patients on peritoneal dialysis and hemodialysis, dietary protein intake in the range of 1.0–1.2 g/kg/day is advised.
With a low protein and adequate energy diet, hyperphosphatemia can often be minimized in non-dialysis CKD patients. In dialysis patients requiring high protein intake, risk for hyperphosphatemia can be substantial and should be carefully monitored and when appropriate, phosphate binders should be promptly instituted [143]. A recent prospective study by Rhee et al. of a cohort (n = 110) of hypoalbuminemic hemodialysis patients showed beneficial effects of increasing serum albumin (≥0.2 mg/dL) and maintaining serum phosphorous levels within a target range (3.5 to <5.5 mg/dL) by providing high-protein meals during hemodialysis combined with lanthanum carbonate administration [144]. It should be noted, however, the occurrence of intradialytic hypotension associated with the feeding was not detailed. In patients with intercurrent acute illness that causes hypercatabolism, temporarily enhancing protein intake may be necessary to meet the demands. Protein, amino acids and ketoacid supplementation may be effective in improving protein energy wasting irrespective of the etiology [145]. It remains, however, to be tested as to whether such supplementation can translate to better clinical outcomes such as survival or reduced CKD progression.
5.2. Correcting Metabolic Acidosis
Metabolic acidosis should be corrected with sodium bicarbonate (NaHCO3). NaHCO3 corrects acidosis in children with renal tubular acidosis and stimulates growth in premature infants and children with kidney failure. NaHCO3 and potassium bicarbonate improve nitrogen balance in elderly with even mild metabolic acidosis [146,147]. Based on available evidence and while awaiting results from several larger sized randomized interventional trials [148,149,150], we suggest (1) increasing dietary alkali (fruits and vegetables) for patients in stages 3 and 4 CKD and with preserved NaHCO3 (22–24 mmol/L) [150], and (2) initiating oral NaHCO3 for patients with CKD and serum HCO3− < 22 mmol/L [151]. The HCO3− goal should be 24–26 mmol/L. Over correction of HCO3− to >26 mmol/L should be avoided [152,153,154].
In dialysis patients (both hemodialysis and peritoneal dialysis), correction of metabolic acidosis reduces protein degradation and negative nitrogen balance [25,155,156,157,158], and significantly improves virtually all hormonal alterations [159], signifying the importance of close monitoring and management of a patient acid–base status as it closely relates to the morbidity and mortality. Over and rapid correction could, however, be detrimental and should therefore be avoided [160]. Graded dialysate HCO3− concentrations during each dialysis might minimize the large acid–base fluctuation. Appropriately designed trials are needed to test this assumption.
5.3. Eliminating Correctible Inflammatory Factors
Dietary modifications including protein restrictions play a major role in minimizing inflammation in non-dialysis patients. Limiting protein waste products can lead to less uremic toxin elaboration and less toxin-induced inflammation. The positive effects of moderate dietary protein restriction on inflammatory state (IL-6) have been shown in proteinuric diabetic patients [161]. The intestinal source of inflammation can be minimized by preventing dysbiosis through increasing dietary fiber, appropriate treatment of constipation in addition to an adequate dietary provision of protein and energy. The use of probiotics and/or prebiotics is controversial and needs further study. Given the lack of substantial adverse effects, however, these supplementations could be considered when appropriate. Fecal transplantation is theoretically plausible as it might reestablish normal intestinal flora to minimize intestinal source of toxins and inflammation. It, however, needs study in CKD and ESRD population before it can be incorporated into clinical practice.
5.4. Minimizing Hormonal Alterations
Dietary modification, correcting acidosis and normalizing intestinal flora could all contribute to minimizing hormonal abnormalities seen in CKD and ESRD. In addition, insulin sensitizers (such as metformin, rosiglitazone and pioglitazone) used when appropriate and in patients with mild-to-moderate CKD (within CKD stage 3a, estimated glomerular filtration rate > 45 mL/min/1.73 m2), can be administered in conjunction with other diabetes treatment. Muscle protein may be preserved with heightened insulin sensitivity. For CKD patients with hypovitaminosis D, current KQIGO recommendation is to correct the circulating 25-hydroxyvitamin D to an adequate level (>30 ng/mL), which, in general, is the same as that for the general population [118]. It is, however, important to keep in mind that evidence supporting the recommendation is limited. Although correcting hypovitaminosis D has its intuitive appeal, controversy exists [164]. Ongoing clinical follow up and balance pros and cons of the vitamin D supplement are required. Further detailed, carefully designed longer-term interventional studies are needed.
Growth hormone and testosterone use in adult CKD and ESRD is controversial. GH has been used in children with renal failure to foster growth. In adult patients, several studies of GH (and IGF-1) administration in the last decade have been shown to reduce inflammation and muscle catabolism and improve nutritional status in ESRD patients [165,166,167]. Its use, however, has not been incorporated into routine practice. Similarly, several randomized interventional studies using androgen in ESRD patients have shown improvement in muscle mass and nutritional status [168,169,170]. Caution should be exercised, however, as testosterone treatment has been reported to induce a number of treatment-related complications [171]. Moreover, the precise formulation, strength and dosing intervals of anabolic hormones in patients with CKD and ESRD have not been established. Until further studies demonstrate consistent benefit and safety, anabolic hormone supplementation may not be used as a routine treatment modality for adult CKD and ESRD patients.
5.5. Increasing Physical Activity
After reaching appropriate amount of protein and energy intake, CKD and ESRD patients should be encouraged to be physically active. Exercise increases expression of the anti-inflammatory protein follistatin, improves sensitivity to IGFs and enhances muscle fiber generation [73,74]. In line with these observations, exercise in rodent CKD models and in limited studies of both pre-dialysis and dialysis patients has been shown to reduce and prevent muscle loss [172,173,174,175]. Although presumed beneficial effects of physical exercise are multiple, appropriate-sized interventional trials are lacking. In practice, physical activity is encouraged in general to preserve lean body mass and maintain protein nutrition balance.
6. Summary
In patients with CKD and ESRD, prominent metabolic and regulatory derangements occur including acidosis, systemic inflammation, and hormonal dysregulation that have been attributed to the development of hypercatabolism and risk for negative nitrogen balance. Worse yet, with often concurrent comorbidity and imposed dietary restriction and medications, CKD and ESRD patients commonly experience decreased appetite, anorexia, and a variety of gastrointestinal abnormalities including gastroparesis, slow intestinal transit, diarrhea/constipation and increased gut mucosal permeability. They are at a high risk for developing intestinal dysbiosis and increased intestinal bacteria derived cytokine and uremic toxins. If energy supply is less than optimal, an accelerated loss of lean body mass due to protein degradation will inevitably ensue, leading to increased morbidity and mortality. Existing evidence, although limited by sample size, often retrospective design and secondary analysis of clinical trials, supports the concept that CKD and dialysis patients can benefit from carefully designed nutritional therapy (sufficient energy and an appropriate amount of HBV protein). Further confirmation from prospective randomized controlled trials to examine clinical outcomes such as mortality from nutritional interventions in the CKD and ESRD population is required.
The elucidation of intestinal-kidney bidirectional and dynamic interactions has allowed us to better appreciate the role of diet and nutrition in the pathogenesis of protein energy alterations and maladaptation in CKD and ESRD. Nutritional intervention and manipulation of gut microbiome to obtain a desired array of microbial population in the intestine may represent a novel class of nontoxic and potentially effective strategies to prevent protein malnutrition in CKD and ESRD. Further research to thoroughly understand the complex intestinal-kidney interplay and patient-based trials in this area are needed. Current evidence supports the notion that dietary monitoring and modifications based on the patient clinical condition will likely enhance patient nutritional status and preserve a favorable bowel microbiome, lean body mass and kidney function. Thus, nutritional therapy should be undertaken as one of the critical and renal protective strategies in parallel with other measures.
Key Points:
- Protein malnutrition is common in patients with CKD and ESRD, a growing patient population worldwide.
- Protein malnutrition is associated with increased morbidity and mortality.
- Protein malnutrition can be prevented and substantially reversed with ongoing dietary monitoring and nutritional therapy.
- Protein intake of 0.6–0.8 g/kg/day for non-dialysis CKD patients and 1.0–1.2 g/kg/day for patients on peritoneal dialysis or hemodialysis, with >50% HBV proteins, are advised.
- Daily energy intake in CKD and ESRD patients should be 30–35 kcal/kg (ideal body weight).
- Metabolic acidosis is related to multiple metabolic derangements, adversely affects kidney and patient outcome, and should be corrected.
- In patients with stage 3 CKD and without evidence of metabolic acidosis, the dietary modification with increased basis such as vegetables and fruits may be initiated to prevent metabolic acidosis.
- Constipation and abnormal bowel habits can compromise gut epithelial cell integrity causing dysbiosis, promoting inflammation and uremic toxin accumulation.
- Exercise, based on patient capacity, should be incorporated in as a part of the CKD and ESRD management.
- Anabolic hormone replacement is controversial and has not been routinely used for adult CKD and ESRD patients.
- Current recommendation is to supplement 25(OH)-vitamin D for CKD and ESRD patients with suboptimal vitamin D status.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| UPS | Proteasome-ubiquitin system |
| CKD | Chronic kidney disease |
| ESRD | End-stage renal disease |
| HBV | High biological value |
| IS | Indoxyl sulfate |
| pCS | p-Cresyl sulfate |
| HCl | Hydrogen chloride |
| H2SO4 | Sulfuric acid |
| H3PO4 | Phosphoric acids |
| NaHCO3 | Sodium bicarbonate |
| TWEAK | TNF-related weak inducer of apoptosis |
| IL-1 | Interleukin-1 |
| IL-6 | Interleukin-6 |
| NF-kappa B | Nuclear factor kappa light chain enhancer of activated B cells |
| SOCS3 | Suppressor of cytokine signaling |
| IRS-1 | Insulin receptor substrate 1 |
| GH-IGF-IGFBP | Growth factor-insulin-like-growth factor binding protein |
| IGF-1 | Insulin like growth factor-1 |
| KDOQI | Kidney Disease Outcomes Quality Initiative |
| PEW | Protein-energy wasting |
References
- De Nicola, L.; Zoccali, C. Chronic kidney disease prevalence in the general population: Heterogeneity and concerns. Nephrol. Dial. Transplant. 2016, 31, 331–335. [Google Scholar] [CrossRef] [PubMed]
- 2015 USRDS Annual Data Report. Volume 2. Available online: https://www.usrds.org/2015/download/vol2_USRDS_ESRD_15.pdf (accessed on 14 January 2017).
- Ikizler, T.A.; Cano, N.J.; Franch, H.; Fouque, D.; Himmelfarb, J.; Kalantar-Zadeh, K.; Kuhlmann, M.K.; Stenvinkel, P.; TerWee, P.; Teta, D.; et al. Prevention and treatment of protein energy wasting in chronic kidney disease patients: A consensus statement by the International Society of Renal Nutrition and Metabolism. Kidney Int. 2013, 84, 1096–1107. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Iguacel, C.; González-Parra, E.; Pérez-Gómez, M.V.; Mahíllo, I.; Egido, J.; Ortiz, A.; Carrero, J.J. Prevalence of protein-energy wasting syndrome and its association with mortality in haemodialysis patients in a centre in Spain. Nefrologia 2013, 33, 495–505. [Google Scholar] [PubMed]
- Kovesdy, C.P.; Kopple, J.D.; Kalantar-Zadeh, K. Management of protein-energy wasting in non-dialysis-dependent chronic kidney disease: Reconciling low protein intake with nutritional therapy. Am. J. Clin. Nutr. 2013, 97, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.W.; Byham-Gray, L.D.; Scott Parrott, J.; Rigassio-Radler, D.; Mandayam, S.; Jones, S.L.; Mitch, W.E.; Gaber, A.O. The mean dietary protein intake at different stages of chronic kidney disease is higher than current guidelines. Kidney Int. 2013, 83, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, B.M. The kidney and acid-base regulation. Adv. Physiol. Educ. 2009, 33, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Wrong, O.; Davies, H.E. The excretion of acid in renal disease. Q. J. Med. 1959, 28, 259–313. [Google Scholar] [PubMed]
- Kraut, J.A.; Kurtz, I. Metabolic acidosis of CKD: Diagnosis, clinical characteristics, and treatment. Am. J. Kidney Dis. 2005, 45, 978–993. [Google Scholar] [CrossRef] [PubMed]
- Kopple, J.D.; Kalantar-Zadeh, K.; Mehrotra, R. Risks of chronic metabolic acidosis in patients with chronic kidney disease. Kidney Int. Suppl. 2005. [Google Scholar] [CrossRef] [PubMed]
- Raphael, K.L.; Zhang, Y.; Ying, J.; Greene, T. Prevalence of and risk factors for reduced serum bicarbonate in chronic kidney disease. Nephrology 2014, 19, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Driver, T.H.; Shlipak, M.G.; Katz, R.; Goldenstein, L.; Sarnak, M.J.; Hoofnagle, A.N.; Siscovick, D.S.; Kestenbaum, B.; de Boer, I.H.; Ix, J.H. Low serum bicarbonate and kidney function decline: The Multi-Ethnic Study of Atherosclerosis (MESA). Am. J. Kidney Dis. 2014, 64, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Baylis, C.; Verlander, J.W.; Han, K.H.; Reungjui, S.; Handlogten, M.E.; Weiner, I.D. Effect of reduced renal mass on renal ammonia transporter family, Rh C glycoprotein and Rh B glycoprotein, expression. Am. J. Physiol. Renal. Physiol. 2007, 293, F1238–F1247. [Google Scholar] [CrossRef] [PubMed]
- Karim, Z.; Attmane-Elakeb, A.; Bichara, M. Renal handling of NH4+ in relation to the control of acid-base balance by the kidney. J. Nephrol. 2002, 15 (Suppl. 5), S128–S134. [Google Scholar] [PubMed]
- Nath, K.A.; Hostetter, M.K.; Hostetter, T.H. Pathophysiology of chronic tubulo-interstitial disease in rats. Interactions of dietary acid load, ammonia, and complement component C3. J. Clin. Investig. 1985, 76, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Wesson, D.E.; Simoni, J. Acid retention during kidney failure induces endothelin and aldosterone production which lead to progressive GFR decline, a situation ameliorated by alkali diet. Kidney Int. 2010, 78, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Wesson, D.E.; Simoni, J.; Broglio, K.; Sheather, S. Acid retention accompanies reduced GFR in humans and increases plasma levels of endothelin and aldosterone. Am. J. Physiol. Renal. Physiol. 2011, 300, F830–F837. [Google Scholar] [CrossRef] [PubMed]
- Reaich, D.; Channon, S.M.; Scrimgeour, C.M.; Goodship, T.H. Ammonium chloride-induced acidosis increases protein breakdown and amino acid oxidation in humans. Am. J. Physiol. 1992, 263, E735–E739. [Google Scholar] [PubMed]
- Ballmer, P.E.; McNurlan, M.A.; Hulter, H.N.; Anderson, S.E.; Garlick, P.J.; Krapf, R. Chronic metabolic acidosis decreases albumin synthesis and induces negative nitrogen balance in humans. J. Clin. Investig. 1995, 95, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Pickering, W.P.; Price, S.R.; Bircher, G.; Marinovic, A.C.; Mitch, W.E.; Walls, J. Nutrition in CAPD: Serum bicarbonate and the ubiquitin-proteasome system in muscle. Kidney Int. 2002, 61, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Vallet, M.; Metzger, M.; Haymann, J.P.; Flamant, M.; Gauci, C.; Thervet, E.; Boffa, J.J.; Vrtovsnik, F.; Froissart, M.; Stengel, B.; et al. Urinary ammonia and long-term outcomes in chronic kidney disease. Kidney Int. 2015, 88, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.N.; Abramowitz, M.; Hostetter, T.H.; Melamed, M.L. Serum bicarbonate levels and the progression of kidney disease: A cohort study. Am. J. Kidney Dis. 2009, 54, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Raphael, K.L.; Wei, G.; Baird, B.C.; Greene, T.; Beddhu, S. Higher serum bicarbonate levels within the normal range are associated with better survival and renal outcomes in African Americans. Kidney Int. 2011, 79, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Kanda, E.; Ai, M.; Yoshida, M.; Kuriyama, R.; Shiigai, T. High serum bicarbonate level within the normal range prevents the progression of chronic kidney disease in elderly chronic kidney disease patients. BMC Nephrol. 2013, 14, 4. [Google Scholar] [CrossRef] [PubMed]
- De Brito-Ashurst, I.; Varagunam, M.; Raftery, M.J.; Yaqoob, M.M. Bicarbonate supplementation slows progression of CKD and improves nutritional status. J. Am. Soc. Nephrol. 2009, 20, 2075–2084. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, A.; Simoni, J.; Sheather, S.J.; Broglio, K.R.; Rajab, M.H.; Wesson, D.E. Daily oral sodium bicarbonate preserves glomerular filtration rate by slowing its decline in early hypertensive nephropathy. Kidney Int. 2010, 78, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Phisitkul, S.; Khanna, A.; Simoni, J.; Broglio, K.; Sheather, S.; Rajab, M.H.; Wesson, D.E. Amelioration of metabolic acidosis in patients with low GFR reduced kidney endothelin production and kidney injury, and better preserved GFR. Kidney Int. 2010, 77, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Abramowitz, M.K.; Melamed, M.L.; Bauer, C.; Raff, A.C.; Hostetter, T.H. Effects of oral sodium bicarbonate in patients with CKD. Clin. J. Am. Soc. Nephrol. 2013, 8, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Amdur, R.L.; Feldman, H.I.; Gupta, J.; Yang, W.; Kanetsky, P.; Shlipak, M.; Rahman, M.; Lash, J.P.; Townsend, R.R.; Ojo, A.; et al. Inflammation and Progression of CKD: The CRIC Study. Clin. J. Am. Soc. Nephrol. 2016, 11, 1546–1556. [Google Scholar] [CrossRef] [PubMed]
- Snaedal, S.; Qureshi, A.R.; Lund, S.H.; Germanis, G.; Hylander, B.; Heimbürger, O.; Carrero, J.J.; Stenvinkel, P.; Bárány, P. Dialysis modality and nutritional status are associated with variability of inflammatory markers. Nephrol. Dial. Transplant. 2016, 31, 1320–1327. [Google Scholar] [CrossRef] [PubMed]
- Ramezani, A.; Raj, D.S. The gut microbiome, kidney disease, and targeted interventions. J. Am. Soc. Nephrol. 2014, 25, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; DeSantis, T.Z.; Ni, Z.; Nguyen, T.H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Hida, M.; Aiba, Y.; Sawamura, S.; Suzuki, N.; Satoh, T.; Koga, Y. Inhibition of the accumulation of uremic toxins in the blood and their precursors in the feces after oral administration of Lebenin, a lactic acid bacteria preparation, to uremic patients undergoing hemodialysis. Nephron 1996, 74, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Bammens, B.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. Evidence for impaired assimilation of protein in chronic renal failure. Kidney Int. 2003, 64, 2196–2203. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Yuan, J.; Rahimi, A.; Ni, Z.; Said, H.; Subramanian, V.S. Disintegration of colonic epithelial tight junction in uremia: A likely cause of CKD-associated inflammation. Nephrol. Dial. Transplant. 2012, 27, 2686–2693. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Goshtasbi, N.; Yuan, J.; Jellbauer, S.; Moradi, H.; Raffatellu, M.; Kalantar-Zadeh, K. Uremic plasma impairs barrier function and depletes the tight junction protein constituents of intestinal epithelium. Am. J. Nephrol. 2012, 36, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Yuan, J.; Norris, K. Role of urea in intestinal barrier dysfunction and disruption of epithelial tight junction in chronic kidney disease. Am. J. Nephrol. 2013, 37, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Pugin, J.; Heumann, I.D.; Tomasz, A.; Kravchenko, V.V.; Akamatsu, Y.; Nishijima, M.; Glauser, M.P.; Tobias, P.S.; Ulevitch, R.J. CD14 is a pattern recognition receptor. Immunity 1994, 1, 509–516. [Google Scholar] [CrossRef]
- Freudenberg, M.A.; Tchaptchet, S.; Keck, S.; Fejer, G.; Huber, M.; Schütze, N.; Beutler, B.; Galanos, C. Lipopolysaccharide sensing an important factor in the innate immune response to Gram-negative bacterial infections: Benefits and hazards of LPS hypersensitivity. Immunobiology 2008, 213, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Hatch, M.; Vaziri, N.D. Enhanced enteric excretion of urate in rats with chronic renal failure. Clin. Sci. (Lond.) 1994, 86, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Piceno, Y.M.; Desantis, T.Z.; Pahl, M.; Andersen, G.L.; Vaziri, N.D. Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am. J. Nephrol. 2014, 39, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Ise, M.; Seo, H.; Niwa, T. Indoxyl sulfate increases the gene expressions of TGF-beta 1, TIMP-1 and pro-alpha 1(I) collagen in uremic rat kidneys. Kidney Int. Suppl. 1997, 62, S15–S22. [Google Scholar] [PubMed]
- Watanabe, H.; Miyamoto, Y.; Honda, D.; Tanaka, H.; Wu, Q.; Endo, M.; Noguchi, T.; Kadowaki, D.; Ishima, Y.; Kotani, S.; et al. p-Cresyl sulfate causes renal tubular cell damage by inducing oxidative stress by activation of NADPH oxidase. Kidney Int. 2013, 83, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, M.L.; Bonan, N.B.; Dias, G.; Brehm, F.; Steiner, T.M.; Souza, W.M.; Stinghen, A.E.; Barreto, F.C.; Elifio-Esposito, S.; Pecoits-Filho, R.; et al. p-Cresyl sulfate affects the oxidative burst, phagocytosis process, and antigen presentation of monocyte-derived macrophages. Toxicol. Lett. 2016, 263, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.Y.; Chang, S.C.; Wu, M.S. Uremic toxins induce kidney fibrosis by activating intrarenal renin-angiotensin-aldosterone system associated epithelial-to-mesenchymal transition. PLoS ONE 2012, 7, e34026. [Google Scholar] [CrossRef] [PubMed]
- Adijiang, A.; Higuchi, Y.; Nishijima, F.; Shimizu, H.; Niwa, T. Indoxyl sulfate, a uremic toxin, promotes cell senescence in aorta of hypertensive rats. Biochem. Biophys. Res. Commun. 2010, 399, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; European Uremic Toxin Work Group (EUTox). Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Bammens, B.; Evenepoel, P.; Keuleers, H.; Verbeke, K.; Vanrenterghem, Y. Free serum concentrations of the protein-bound retention solute p-cresol predict mortality in hemodialysis patients. Kidney Int. 2006, 69, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Liabeuf, S.; Barreto, D.V.; Barreto, F.C.; Meert, N.; Glorieux, G.; Schepers, E.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; et al. Free p-cresylsulphate is a predictor of mortality in patients at different stages of chronic kidney disease. Nephrol. Dial. Transplant. 2010, 25, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.K.; Claes, K.; Bammens, B.; de Loor, H.; Viaene, L.; Verbeke, K.; Kuypers, D.; Vanrenterghem, Y.; Evenepoel, P. p-Cresol and cardiovascular risk in mild-to-moderate kidney disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A.; European Uremic Toxin Work Group. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Mutsaers, H.A.; Engelke, U.F.; Wilmer, M.J.; Wetzels, J.F.; Wevers, R.A.; van den Heuvel, L.P.; Hoenderop, J.G.; Masereeuw, R. Optimized metabolomic approach to identify uremic solutes in plasma of stage 3–4 chronic kidney disease patients. PLoS ONE 2013, 8, e71199. [Google Scholar] [CrossRef] [PubMed]
- Ramezani, A.; Massy, Z.A.; Meijers, B.; Evenepoel, P.; Vanholder, R.; Raj, D.S. Role of the Gut Microbiome in Uremia: A Potential Therapeutic Target. Am. J. Kidney Dis. 2016, 67, 483–498. [Google Scholar] [CrossRef] [PubMed]
- Witasp, A.; Carrero, J.J.; Heimbürger, O.; Lindholm, B.; Hammarqvist, F.; Stenvinkel, P.; Nordfors, L. Increased expression of pro-inflammatory genes in abdominal subcutaneous fat in advanced chronic kidney disease patients. J. Intern. Med. 2011, 269, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Axelsson, J.; Rashid Qureshi, A.; Suliman, M.E.; Honda, H.; Pecoits-Filho, R.; Heimbürger, O.; Lindholm, B.; Cederholm, T.; Stenvinke, P. Truncal fat mass as a contributor to inflammation in end-stage renal disease. Am. J. Clin. Nutr. 2004, 80, 1222–1229. [Google Scholar] [PubMed]
- Gohda, T.; Gotoh, H.; Tanimoto, M.; Sato, M.; Io, H.; Kaneko, K.; Hamada, C.; Tomino, Y. Relationship between abdominal fat accumulation and insulin resistance in hemodialysis patients. Hypertens. Res. 2008, 31, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Odamaki, M.; Furuya, R.; Ohkawa, S.; Yoneyama, T.; Nishikino, M.; Hishida, A.; Kumagai, H. Altered abdominal fat distribution and its association with the serum lipid profile in non-diabetic haemodialysis patients. Nephrol. Dial. Transplant. 1999, 14, 2427–2432. [Google Scholar] [CrossRef] [PubMed]
- Odamaki, M.; Furuya, R.; Kinumura, Y.; Ikegaya, N.; Kumagai, H. Association between plasma adiponectin concentration and visceral fat accumulation in hemodialysis patients. Nephron. Clin. Pract. 2006, 102, c8–c13. [Google Scholar] [CrossRef] [PubMed]
- Witasp, A.; Carrero, J.J.; Hammarqvist, F.; Qureshi, A.R.; Heimbürger, O.; Schalling, M.; Lindholm, B.; Nordfors, L.; Stenvinkel, P. Expression of osteoprotegerin in human fat tissue; implications for chronic kidney disease. Eur. J. Clin. Investig. 2011, 41, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, M.A.; Carrero, J.J.; Canziani, M.E.; Watanabe, R.; Lemos, M.M.; Cuppari, L. Visceral obesity assessed by computed tomography predicts cardiovascular events in chronic kidney disease patients. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Luger, A.; Kovarik, J.; Stummvoll, H.K.; Urbanska, A.; Luger, T.A. Blood-membrane interaction in hemodialysis leads to increased cytokine production. Kidney Int. 1987, 32, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Borazan, A.; Ustün, H.; Ustundag, Y.; Aydemir, S.; Bayraktaroglu, T.; Sert, M.; Yilmaz, A. The effects of peritoneal dialysis and hemodialysis on serum tumor necrosis factor-alpha, interleukin-6, interleukin-10 and C-reactive-protein levels. Mediat. Inflamm. 2004, 13, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Carrero, J.J.; Stenvinkel, P. Inflammation in end-stage renal disease—What have we learned in 10 years? Semin. Dial. 2010, 23, 498–509. [Google Scholar] [CrossRef] [PubMed]
- Delano, M.J.; Moldawer, L.L. The origins of cachexia in acute and chronic inflammatory diseases. Nutr. Clin. Pract. 2006, 21, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Meuwese, C.L.; Stenvinkel, P.; Dekker, F.W.; Carrero, J.J. Monitoring of inflammation in patients on dialysis: Forewarned is forearmed. Nat. Rev. Nephrol. 2011, 7, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Dogra, C.; Changotra, H.; Wedhas, N.; Qin, X.; Wergedal, J.E.; Kumar, A. TNF-related weak inducer of apoptosis (TWEAK) is a potent skeletal muscle-wasting cytokine. FASEB J. 2007, 21, 1857–1869. [Google Scholar] [CrossRef] [PubMed]
- Carrero, J.J.; Ortiz, A.; Qureshi, A.R.; Martín-Ventura, J.L.; Bárány, P.; Heimbürger, O.; Marrón, B.; Metry, G.; Snaedal, S.; Lindholm, B.; et al. Additive effects of soluble TWEAK and inflammation on mortality in hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Dallmann, G.; Müller, G.; Patthy, L.; Soller, M.; Varga, L. A deletion in the myostatin gene causes the compact (Cmpt) hypermuscular mutation in mice. Mamm. Genome 1998, 9, 671–672. [Google Scholar] [CrossRef] [PubMed]
- Han, H.Q.; Mitch, W.E. Targeting the myostatin signaling pathway to treat muscle wasting diseases. Curr. Opin. Support Palliat. Care 2011, 5, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; Hu, Z.; Klein, J.D.; Zhang, L.; Fang, F.; Mitch, W.E. Decreased miR-29 suppresses myogenesis in CKD. J. Am. Soc. Nephrol. 2011, 22, 2068–2076. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Rajan, V.; Lin, E.; Hu, Z.; Han, H.Q.; Zhou, X.; Song, Y.; Min, H.; Wang, X.; Du, J.; Mitch, W.E. Pharmacological inhibition of myostatin suppresses systemic inflammation and muscle atrophy in mice with chronic kidney disease. FASEB J. 2011, 25, 1653–1663. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.; Brandt, C.; Nielsen, A.R.; Hojman, P.; Whitham, M.; Febbraio, M.A.; Pedersen, B.K.; Plomgaard, P. Exercise induces a marked increase in plasma follistatin: Evidence that follistatin is a contraction-induced hepatokine. Endocrinology 2011, 152, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Leehey, D.J.; Moinuddin, I.; Bast, J.P.; Qureshi, S.; Jelinek, C.S.; Cooper, C.; Edwards, L.C.; Smith, B.M.; Collins, E.G. Aerobic exercise in obese diabetic patients with chronic kidney disease: A randomized and controlled pilot study. Cardiovasc Diabetol. 2009, 8, 62. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Wang, H.; Lee, I.H.; Du, J.; Mitch, W.E. Endogenous glucocorticoids and impaired insulin signaling are both required to stimulate muscle wasting under pathophysiological conditions in mice. J. Clin. Investig. 2009, 119, 3059–3069. [Google Scholar] [CrossRef] [PubMed]
- Spindler, S.R. Caloric restriction: From soup to nuts. Ageing Res. Rev. 2010, 9, 324–353. [Google Scholar] [CrossRef] [PubMed]
- Finn, P.F.; Dice, J.F. Proteolytic and lipolytic responses to starvation. Nutrition 2006, 22, 830–844. [Google Scholar] [CrossRef] [PubMed]
- Keusch, G.T. The history of nutrition: Malnutrition, infection and immunity. J. Nutr. 2003, 133, 336S–340S. [Google Scholar] [PubMed]
- Zhang, L.; Du, J.; Hu, Z.; Han, G.; Delafontaine, P.; Garcia, G.; Mitch, W.E. IL-6 and serum amyloid A synergy mediates angiotensin II-induced muscle wasting. J. Am. Soc. Nephrol. 2009, 20, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Raj, D.S.; Moseley, P.; Dominic, E.A.; Onime, A.; Tzamaloukas, A.H.; Boyd, A.; Shah, V.O.; Glew, R.; Wolfe, R.; Ferrando, A. Interleukin-6 modulates hepatic and muscle protein synthesis during hemodialysis. Kidney Int. 2008, 73, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Boivin, M.A.; Battah, S.I.; Dominic, E.A.; Kalantar-Zadeh, K.; Ferrando, A.; Tzamaloukas, A.H.; Dwivedi, R.; Ma, T.A.; Moseley, P.; Raj, D.S. Activation of caspase-3 in the skeletal muscle during haemodialysis. Eur. J. Clin. Investig. 2010, 40, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Feneberg, R.; Schaefer, F.; Veldhuis, J.D. Neuroendocrine adaptations in renal disease. Pediatr. Nephrol. 2003, 18, 492–497. [Google Scholar] [CrossRef] [PubMed]
- LeRoith, D.; Taylor, S.I.; Olefsky, J.M. Physiological action of insulin. In Diabetes Mellitus: A Fundamental and Clinical Text; Leroith, D.T.S., Olefsky, J., Eds.; Williams & Wilkins: Philadelphia, PA, USA, 2000; pp. 148–161. [Google Scholar]
- DeFronzo, R.A.; Alvestrand, A.; Smith, D.; Hendler, R.; Hendler, E.; Wahren, J. Insulin resistance in uremia. J. Clin. Investig. 1981, 67, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Pupim, L.B.; Flakoll, P.J.; Majchrzak, K.M.; Aftab Guy, D.L.; Stenvinkel, P.; Ikizler, T.A. Increased muscle protein breakdown in chronic hemodialysis patients with type 2 diabetes mellitus. Kidney Int. 2005, 68, 1857–1865. [Google Scholar] [CrossRef] [PubMed]
- Siew, E.D.; Pupim, L.B.; Majchrzak, K.M.; Shintani, A.; Flakoll, P.J.; Ikizler, T.A. Insulin resistance is associated with skeletal muscle protein breakdown in non-diabetic chronic hemodialysis patients. Kidney Int. 2007, 71, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Koppe, L.; Pillon, N.J.; Vella, R.E.; Croze, M.L.; Pelletier, C.C.; Chambert, S.; Massy, Z.; Glorieux, G.; Vanholder, R.; Dugenet, Y. p-Cresyl sulfate promotes insulin resistance associated with CKD. J. Am. Soc. Nephrol. 2013, 24, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Moller, J. Effects of growth hormone on fluid homeostasis. Clinical and experimental aspects. Growth Horm. IGF Res. 2003, 13, 55–74. [Google Scholar] [CrossRef]
- Powell, D.R. Effects of renal failure on the growth hormone-insulin-like growth factor axis. J. Pediatr. 1997, 131, S13–S16. [Google Scholar] [CrossRef]
- Tonshoff, B.; Blum, W.F.; Mehls, O. Derangements of the somatotropic hormone axis in chronic renal failure. Kidney Int. Suppl. 1997, 58, S106–S113. [Google Scholar] [PubMed]
- Haffner, D.; Schaefer, F.; Girard, J.; Ritz, E.; Mehls, O. Metabolic clearance of recombinant human growth hormone in health and chronic renal failure. J. Clin. Investig. 1994, 93, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, F.; Chen, Y.; Tsao, T.; Nouri, P.; Rabkin, R. Impaired JAK-STAT signal transduction contributes to growth hormone resistance in chronic uremia. J. Clin. Investig. 2001, 108, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.F.; Zheng, Z.; Tummala, P.; Oh, J.; Schaefer, F.; Rabkin, R. Chronic uremia attenuates growth hormone-induced signal transduction in skeletal muscle. J. Am. Soc. Nephrol. 2004, 15, 2630–2636. [Google Scholar] [CrossRef] [PubMed]
- Ferry, R.J., Jr.; Cerri, R.W.; Cohen, P. Insulin-like growth factor binding proteins: New proteins, new functions. Horm. Res. 1999, 51, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Roelfsema, V.; Lane, M.H.; Clark, R.G. Insulin-like growth factor binding protein (IGFBP) displacers: Relevance to the treatment of renal disease. Pediatr. Nephrol. 2000, 14, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Mak, R.H.; Pak, Y.K. End-organ resistance to growth hormone and IGF-I in epiphyseal chondrocytes of rats with chronic renal failure. Kidney Int. 1996, 50, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Park, S.K.; Kim, J.S. Insulin-like growth factor-I (IGF-I) and IGF-binding proteins in children with nephrotic syndrome. J. Clin. Endocrinol. Metab. 1996, 81, 1856–1860. [Google Scholar] [PubMed]
- Du, J.; Wang, X.; Miereles, C.; Bailey, J.L.; Debigare, R.; Zheng, B.; Price, S.R.; Mitch, W.E. Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J. Clin. Investig. 2004, 113, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Price, F.; Rudnicki, M.A. Satellite cells and the muscle stem cell niche. Physiol. Rev. 2013, 93, 23–67. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Zhang, Y.; Ren, J. IGF-1 deficiency resists cardiac hypertrophy and myocardial contractile dysfunction: Role of microRNA-1 and microRNA-133a. J. Cell Mol. Med. 2012, 16, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Elia, L.; Contu, R.; Quintavalle, M.; Varrone, F.; Chimenti, C.; Russo, M.A.; Cimino, V.; De Marinis, L.; Frustaci, A.; Catalucci, D.; et al. Reciprocal regulation of microRNA-1 and insulin-like growth factor-1 signal transduction cascade in cardiac and skeletal muscle in physiological and pathological conditions. Circulation 2009, 120, 2377–2385. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.B.; Xu, H.; Xie, S.J.; Zhou, H.; Qu, L.H. Insulin-like growth factor-1 receptor is regulated by microRNA-133 during skeletal myogenesis. PLoS ONE 2011, 6, e29173. [Google Scholar] [CrossRef] [PubMed]
- Carrero, J.J.; Stenvinkel, P. The vulnerable man: Impact of testosterone deficiency on the uraemic phenotype. Nephrol. Dial. Transplant. 2012, 27, 4030–4041. [Google Scholar] [CrossRef] [PubMed]
- Handelsman, D.J.; Dong, Q. Hypothalamo-pituitary gonadal axis in chronic renal failure. Endocrinol. Metab. Clin. North Am. 1993, 22, 145–161. [Google Scholar] [PubMed]
- Holley, J.L. The hypothalamic-pituitary axis in men and women with chronic kidney disease. Adv. Chronic Kidney Dis. 2004, 11, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Phadke, A.G.; MacKinnon, K.J.; Dossetor, J.B. Male fertility in uremia: Restoration by renal allografts. Can. Med. Assoc. J. 1970, 102, 607–608. [Google Scholar] [PubMed]
- Lim, V.S.; Fang, V.S. Gonadal dysfunction in uremic men. A study of the hypothalamo-pituitary-testicular axis before and after renal transplantation. Am. J. Med. 1975, 58, 655–662. [Google Scholar] [CrossRef]
- Cigarran, S.; Pousa, M.; Castro, M.J.; González, B.; Martínez, A.; Barril, G.; Aguilera, A.; Coronel, F.; Stenvinkel, P.; Carrero, J.J. Endogenous testosterone, muscle strength, and fat-free mass in men with chronic kidney disease. J. Ren. Nutr. 2013, 23, e89–e95. [Google Scholar] [CrossRef] [PubMed]
- Mitch, W.E.; Bailey, J.L.; Wang, X.; Jurkovitz, C.; Newby, D.; Price, S.R. Evaluation of signals activating ubiquitin-proteasome proteolysis in a model of muscle wasting. Am. J. Physiol. 1999, 276, C1132–C1138. [Google Scholar] [PubMed]
- May, R.C.; Kelly, R.A.; Mitch, W.E. Metabolic acidosis stimulates protein degradation in rat muscle by a glucocorticoid-dependent mechanism. J. Clin. Investig. 1986, 77, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Ohkawa, S.; Li, H.; Roberts-Wilson, T.K.; Price, S.R. FOXO3a mediates signaling crosstalk that coordinates ubiquitin and atrogin-1/MAFbx expression during glucocorticoid-induced skeletal muscle atrophy. FASEB J. 2010, 24, 2660–2669. [Google Scholar] [CrossRef] [PubMed]
- Nigwekar, S.U.; Bhan, I.; Thadhani, R. Ergocalciferol and cholecalciferol in CKD. Am. J. Kidney Dis. 2012, 60, 139–156. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; David, V.; Quarles, L.D. Regulation and function of the FGF23/klotho endocrine pathways. Physiol. Rev. 2012, 92, 131–155. [Google Scholar] [CrossRef] [PubMed]
- Girgis, C.M.; Clifton-Bligh, R.J.; Mokbel, N.; Cheng, K.; Gunton, J.E. Vitamin D signaling regulates proliferation, differentiation, and myotube size in C2C12 skeletal muscle cells. Endocrinology 2014, 155, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Harter, H.R.; Birge, S.J.; Martin, K.J.; Klahr, S.; Karl, I.E. Effects of vitamin D metabolites on protein catabolism of muscle from uremic rats. Kidney Int. 1983, 23, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.M.; Tajar, A.; Pye, S.R.; Boonen, S.; Vanderschueren, D.; Bouillon, R.; O’Neill, T.W.; Bartfai, G.; Casanueva, F.F.; Finn, J.D.; et al. Association of hypogonadism with vitamin D status: The European Male Ageing Study. Eur. J. Endocrinol. 2012, 166, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Gordon, P.L.; Sakkas, G.K.; Doyle, J.W.; Shubert, T.; Johansen, K.L. Relationship between vitamin D and muscle size and strength in patients on hemodialysis. J. Ren. Nutr. 2007, 17, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Lin, J.; Qian, Q. Role of Vitamin D in Cognitive Function in Chronic Kidney Disease. Nutrients 2016. [Google Scholar] [CrossRef] [PubMed]
- Kopple, J.D.; Monteon, F.J.; Shaib, J.K. Effect of energy intake on nitrogen metabolism in nondialyzed patients with chronic renal failure. Kidney Int. 1986, 29, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Sciatti, E.; Lombardi, C.; Ravera, A.; Vizzardi, E.; Bonadei, I.; Carubelli, V.; Gorga, E.; Metra, M. Nutritional Deficiency in Patients with Heart Failure. Nutrients 2016, 8, 442. [Google Scholar] [CrossRef] [PubMed]
- Avesani, C.M.; Cuppari, L.; Silva, A.C.; Sigulem, D.M.; Cendoroglo, M.; Sesso, R.; Draibe, S.A. Resting energy expenditure in pre-dialysis diabetic patients. Nephrol. Dial. Transplant. 2001, 16, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Neyra, R.; Chen, K.Y.; Sun, M.; Shyr, Y.; Hakim, R.M.; Ikizler, T.A. Increased resting energy expenditure in patients with end-stage renal disease. JPEN J. Parenter. Enter. Nutr. 2003, 27, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.Y.; Sea, M.M.; Tang, N.; Sanderson, J.E.; Lui, S.F.; Li, P.K.; Woo, J. Resting energy expenditure and subsequent mortality risk in peritoneal dialysis patients. J. Am. Soc. Nephrol. 2004, 15, 3134–3143. [Google Scholar] [CrossRef] [PubMed]
- Cuppari, L.; de Carvalho, A.B.; Avesani, C.M.; Kamimura, M.A.; Dos Santos Lobão, R.R.; Draibe, S.A. Increased resting energy expenditure in hemodialysis patients with severe hyperparathyroidism. J. Am. Soc. Nephrol. 2004, 15, 2933–2939. [Google Scholar] [CrossRef] [PubMed]
- Utaka, S.; Avesani, C.M.; Draibe, S.A.; Kamimura, M.A.; Andreoni, S.; Cuppari, L. Inflammation is associated with increased energy expenditure in patients with chronic kidney disease. Am. J. Clin. Nutr. 2005, 82, 801–805. [Google Scholar] [PubMed]
- Kamimura, M.A.; Draibe, S.A.; Dalboni, M.A.; Cendoroglo, M.; Avesani, C.M.; Manfredi, S.R.; Canziani, M.E.; Cuppari, L. Serum and cellular interleukin-6 in haemodialysis patients: Relationship with energy expenditure. Nephrol. Dial. Transplant. 2007, 22, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Mafra, D.; Deleaval, P.; Teta, D.; Cleaud, C.; Arkouche, W.; Jolivot, A.; Fouque, D. Influence of inflammation on total energy expenditure in hemodialysis patients. J. Ren. Nutr. 2011, 21, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Kaysen, G.A.; Greene, T.; Daugirdas, J.T.; Kimmel, P.L.; Schulman, G.W.; Toto, R.D.; Levin, N.W.; Yan, G.; HEMO Study Group. Longitudinal and cross-sectional effects of C-reactive protein, equilibrated normalized protein catabolic rate, and serum bicarbonate on creatinine and albumin levels in dialysis patients. Am. J. Kidney Dis. 2003, 42, 1200–1211. [Google Scholar] [CrossRef] [PubMed]
- Tom, K.; Young, V.R.; Chapman, T.; Masud, T.; Akpele, L.; Maroni, B.J. Long-term adaptive responses to dietary protein restriction in chronic renal failure. Am. J. Physiol. 1995, 268, E668–E677. [Google Scholar] [PubMed]
- Masud, T.; Young, V.R.; Chapman, T.; Maroni, B.J. Adaptive responses to very low protein diets: The first comparison of ketoacids to essential amino acids. Kidney Int. 1994, 45, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Zhang, X.; Yang, L.; Li, Z.; Qin, W. Effect of restricted protein diet supplemented with keto analogues in chronic kidney disease: A systematic review and meta-analysis. Int. Urol. Nephrol. 2016, 48, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-Zadeh, K.; Block, G.; McAllister, C.J.; Humphreys, M.H.; Kopple, J.D. Appetite and inflammation, nutrition, anemia, and clinical outcome in hemodialysis patients. Am. J. Clin. Nutr. 2004, 80, 299–307. [Google Scholar] [PubMed]
- Shinaberger, C.S.; Kilpatrick, R.D.; Regidor, D.L.; McAllister, C.J.; Greenland, S.; Kopple, J.D.; Kalantar-Zadeh, K. Longitudinal associations between dietary protein intake and survival in hemodialysis patients. Am. J. Kidney Dis. 2006, 48, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Fouque, D.; Kalantar-Zadeh, K.; Kopple, J.; Cano, N.; Chauveau, P.; Cuppari, L.; Franch, H.; Guarnieri, G.; Ikizler, T.A.; Kaysen, G.; et al. A proposed nomenclature and diagnostic criteria for protein-energy wasting in acute and chronic kidney disease. Kidney Int. 2008, 73, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-Zadeh, K.; Ikizler, T.A.; Block, G.; Avram, M.M.; Kopple, J.D. Malnutrition-inflammation complex syndrome in dialysis patients: Causes and consequences. Am. J. Kidney Dis. 2003, 42, 864–881. [Google Scholar] [CrossRef] [PubMed]
- Ikizler, T.A.; Pupim, L.B.; Brouillette, J.R.; Levenhagen, D.K.; Farmer, K.; Hakim, R.M.; Flakoll, P.J. Hemodialysis stimulates muscle and whole body protein loss and alters substrate oxidation. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E107–E116. [Google Scholar] [PubMed]
- Avesani, C.M.; Trolonge, S.; Deléaval, P.; Baria, F.; Mafra, D.; Faxén-Irving, G.; Chauveau, P.; Teta, D.; Kamimura, M.A.; Cuppari, L.; et al. Physical activity and energy expenditure in haemodialysis patients: An international survey. Nephrol. Dial. Transplant. 2012, 27, 2430–2434. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, A.; Regolisti, G.; Karupaiah, T.; Sahathevan, S.; Sadu Singh, B.K.; Khor, B.H.; Salhab, N.; Karavetian, M.; Cupisti, A.; Fiaccadori, E. Protein-energy wasting and nutritional supplementation in patients with end-stage renal disease on hemodialysis. Clin. Nutr. 2016. [Google Scholar] [CrossRef] [PubMed]
- Pupim, L.B.; Caglar, K.; Hakim, R.M.; Shyr, Y.; Ikizler, T.A. Uremic malnutrition is a predictor of death independent of inflammatory status. Kidney Int. 2004, 66, 2054–2060. [Google Scholar] [CrossRef] [PubMed]
- Rambod, M.; Bross, R.; Zitterkoph, J.; Benner, D.; Pithia, J.; Colman, S.; Kovesdy, C.P.; Kopple, J.D.; Kalantar-Zadeh, K. Association of Malnutrition-Inflammation Score with quality of life and mortality in hemodialysis patients: A 5-year prospective cohort study. Am. J. Kidney Dis. 2009, 53, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Kopple, J.D. National kidney foundation K/DOQI clinical practice guidelines for nutrition in chronic renal failure. Am. J. Kidney Dis. 2001, 37 (Suppl. 2), S66–S70. [Google Scholar] [CrossRef] [PubMed]
- Dhondup, T.; Qian, Q. Electrolyte and Acid-base Disorders in Chronic Kidney Disease and End-stage Kidney Failure. Blood Purif. 2017. [Google Scholar] [CrossRef] [PubMed]
- Rhee, C.M.; You, A.S.; Koontz Parsons, T.; Tortorici, A.R.; Bross, R.; St-Jules, D.E.; Jing, J.; Lee, M.L.; Benner, D.; Kovesdy, C.P.; et al. Effect of high-protein meals during hemodialysis combined with lanthanum carbonate in hypoalbuminemic dialysis patients: Findings from the FrEDI randomized controlled trial. Nephrol. Dial. Transplant. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-Zadeh, K.; Cano, N.J.; Budde, K.; Chazot, C.; Kovesdy, C.P.; Mak, R.H.; Mehrotra, R.; Raj, D.S.; Sehgal, A.R.; Stenvinkel, P.; et al. Diets and enteral supplements for improving outcomes in chronic kidney disease. Nat. Rev. Nephrol. 2011, 7, 369–384. [Google Scholar] [CrossRef] [PubMed]
- McSherry, E.; Morris, R.C., Jr. Attainment and maintenance of normal stature with alkali therapy in infants and children with classic renal tubular acidosis. J. Clin. Investig. 1978, 61, 509–527. [Google Scholar] [CrossRef] [PubMed]
- Frassetto, L.; Morris, R.C., Jr.; Sebastian, A. Potassium bicarbonate reduces urinary nitrogen excretion in postmenopausal women. J. Clin. Endocrinol. Metab. 1997, 82, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Witham, M.D.; Band, M.M.; Littleford, R.C.; Avenell, A.; Soiza, R.L.; McMurdo, M.E.; Sumukadas, D.; Ogston, S.A.; Lamb, E.J.; Hampson, G.; et al. Does oral sodium bicarbonate therapy improve function and quality of life in older patients with chronic kidney disease and low-grade acidosis (the BiCARB trial)? Study protocol for a randomized controlled trial. Trials 2015, 16, 326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaggl, M.; Cejka, D.; Plischke, M.; Heinze, G.; Fraunschiel, M.; Schmidt, A.; Hörl, W.H.; Sunder-Plassmann, G. Effect of oral sodium bicarbonate supplementation on progression of chronic kidney disease in patients with chronic metabolic acidosis: Study protocol for a randomized controlled trial (SoBic-Study). Trials 2013, 14, 196. [Google Scholar] [CrossRef] [PubMed]
- Di Iorio, B.; Aucella, F.; Conte, G.; Cupisti, A.; Santoro, D. A prospective, multicenter, randomized, controlled study: The correction of metabolic acidosis with use of bicarbonate in Chronic Renal Insufficiency (UBI) Study. J. Nephrol. 2012, 25, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Goraya, N.; Simoni, J.; Jo, C.H.; Wesson, D.E. Treatment of metabolic acidosis in patients with stage 3 chronic kidney disease with fruits and vegetables or oral bicarbonate reduces urine angiotensinogen and preserves glomerular filtration rate. Kidney Int. 2014, 86, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Andrassy, K.M. Comments on ‘KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease’. Kidney Int. 2013, 84, 622–623. [Google Scholar] [CrossRef] [PubMed]
- Kovesdy, C.P.; Anderson, J.E.; Kalantar-Zadeh, K. Association of serum bicarbonate levels with mortality in patients with non-dialysis-dependent CKD. Nephrol. Dial. Transplant. 2009, 24, 1232–1237. [Google Scholar] [CrossRef] [PubMed]
- Dobre, M.; Yang, W.; Pan, Q.; Appel, L.; Bellovich, K.; Chen, J.; Feldman, H.; Fischer, M.J.; Ham, L.L.; Hostetter, T.; et al. Persistent high serum bicarbonate and the risk of heart failure in patients with chronic kidney disease (CKD): A report from the Chronic Renal Insufficiency Cohort (CRIC) study. J. Am. Heart Assoc. 2015, 4, e001599. [Google Scholar] [CrossRef] [PubMed]
- Reaich, D.; Channon, S.M.; Scrimgeour, C.M.; Daley, S.E.; Wilkinson, R.; Goodship, T.H. Correction of acidosis in humans with CRF decreases protein degradation and amino acid oxidation. Am. J. Physiol. 1993, 265, E230–E235. [Google Scholar] [PubMed]
- Graham, K.A.; Reaich, D.; Channon, S.M.; Downie, S.; Goodship, T.H. Correction of acidosis in hemodialysis decreases whole-body protein degradation. J. Am. Soc. Nephrol. 1997, 8, 632–637. [Google Scholar] [PubMed]
- Graham, K.A.; Reaich, D.; Channon, S.M.; Downie, S.; Gilmour, E.; Passlick-Deetjen, J.; Goodship, T.H. Correction of acidosis in CAPD decreases whole body protein degradation. Kidney Int. 1996, 49, 1396–1400. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.; Moorhouse, J.; Iles-Smith, H.; Baker, F.; Johnstone, J.; James, G.; Troughton, J.; Bircher, G.; Walls, J. Role of an improvement in acid-base status and nutrition in CAPD patients. Kidney Int. 1997, 52, 1089–1095. [Google Scholar] [CrossRef] [PubMed]
- Wiederkehr, M.R.; Kalogiros, J.; Krapf, R. Correction of metabolic acidosis improves thyroid and growth hormone axes in haemodialysis patients. Nephrol. Dial. Transplant. 2004, 19, 1190–1197. [Google Scholar] [CrossRef] [PubMed]
- Tentori, F.; Karaboyas, A.; Robinson, B.M.; Morgenstern, H.; Zhang, J.; Sen, A.; Ikizler, T.A.; Rayner, H.; Fissell, R.B.; Vanholder, R.; et al. Association of dialysate bicarbonate concentration with mortality in the Dialysis Outcomes and Practice Patterns Study (DOPPS). Am. J. Kidney Dis. 2013, 62, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Giordano, M.; Giordano, M.; Lucidi, P.; Ciarambino, T.; Gesuè, L.; Castellino, P.; Cioffi, M.; Gresele, P.; Paolisso, G.; De Feo, P. Effects of dietary protein restriction on albumin and fibrinogen synthesis in macroalbuminuric type 2 diabetic patients. Diabetologia 2008, 51, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Cheng, Z.; Qian, Q. Intravenous Fluids and Acute Kidney Injury. Blood Purif. 2017. [Google Scholar] [CrossRef] [PubMed]
- Niebauer, J.; Volk, H.D.; Kemp, M.; Dominguez, M.; Schumann, R.R.; Rauchhaus, M.; Poole-Wilson, P.A.; Coats, A.J.; Anker, S.D. Endotoxin and immune activation in chronic heart failure: A prospective cohort study. Lancet 1999, 353, 1838–1842. [Google Scholar] [CrossRef]
- Agarwal, R.; Georgianos, P.I. Con: Nutritional vitamin D replacement in chronic kidney disease and end-stage renal disease. Nephrol. Dial. Transplant. 2016, 31, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Kopple, J.D.; Cheung, A.K.; Christiansen, J.S.; Djurhuus, C.B.; El Nahas, M.; Feldt-Rasmussen, B.; Mitch, W.E.; Wanner, C.; Göthberg, M.; Ikizler, T.A. Opportunity & trade: A large-scale randomized clinical trial of growth hormone in hemodialysis patients. Nephrol. Dial. Transplant. 2011, 26, 4095–4103. [Google Scholar] [PubMed]
- Guebre-Egziabher, F.; Juillard, L.; Boirie, Y.; Laville, M.; Beaufrère, B.; Fouque, D. Short-term administration of a combination of recombinant growth hormone and insulin-like growth factor-I induces anabolism in maintenance hemodialysis. J. Clin. Endocrinol. Metab. 2009, 94, 2299–2305. [Google Scholar] [CrossRef] [PubMed]
- Feldt-Rasmussen, B.; Lange, M.; Sulowicz, W.; Gafter, U.; Lai, K.N.; Wiedemann, J.; Christiansen, J.S.; El Nahas, M.; APCD Study Group. Growth hormone treatment during hemodialysis in a randomized trial improves nutrition, quality of life, and cardiovascular risk. J. Am. Soc. Nephrol. 2007, 18, 2161–2171. [Google Scholar] [CrossRef] [PubMed]
- Gascon, A.; Belvis, J.J.; Berisa, F.; Iglesias, E.; Estopiñán, V.; Teruel, J.L. Nandrolone decanoate is a good alternative for the treatment of anemia in elderly male patients on hemodialysis. Geriatr. Nephrol. Urol. 1999, 9, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Barton Pai, A.; Chretien, C.; Lau, A.H. The effects of nandrolone decanoate on nutritional parameters in hemodialysis patients. Clin. Nephrol. 2002, 58, 38–46. [Google Scholar] [PubMed]
- Navarro, J.F.; Mora, C.; Macía, M.; García, J. Randomized prospective comparison between erythropoietin and androgens in CAPD patients. Kidney Int. 2002, 61, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Orr, R.; Singh, M.F. The anabolic androgenic steroid oxandrolone in the treatment of wasting and catabolic disorders: Review of efficacy and safety. Drugs 2004, 64, 725–750. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; Du, J.; Klein, J.D.; Bailey, J.L.; Mitch, W.E. Exercise ameliorates chronic kidney disease-induced defects in muscle protein metabolism and progenitor cell function. Kidney Int. 2009, 76, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Storer, T.W.; Casaburi, R.; Sawelson, S.; Kopple, J.D. Endurance exercise training during haemodialysis improves strength, power, fatigability and physical performance in maintenance haemodialysis patients. Nephrol. Dial. Transplant. 2005, 20, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Castaneda, C.; Gordon, P.L.; Parker, R.C.; Uhlin, K.L.; Roubenoff, R.; Levey, A.S. Resistance training to reduce the malnutrition-inflammation complex syndrome of chronic kidney disease. Am. J. Kidney Dis. 2004, 43, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Snijders, T.; Verdijk, L.B.; van Loon, L.J. The impact of sarcopenia and exercise training on skeletal muscle satellite cells. Ageing Res. Rev. 2009, 8, 328–338. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Interconnection of CKD progression with metabolic acidosis, inflammation, hormonal resistance and protein catabolism. (A) Kidney dysfunction limits proton (H+) excretion, resulting in a systemic metabolic acidosis. The acidosis causes activation of complement systems, renin angiotensin aldosterone systems and endothelin-1. These acidosis-mediated effects cause CKD progression, forming a viscous cycle; (B) Acidosis promotes inflammation and tissue resistance to multiple anabolic hormones and simultaneously enhances activity of catabolic corticosteroids. Protein catabolism generates acidic products, contributing to acidosis in the setting of CKD and ESRD. Collectively, these abnormalities give rise to a state of protein catabolism, causing sustained negative nitrogen balance, leading to muscle wasting.
Figure 1.
Interconnection of CKD progression with metabolic acidosis, inflammation, hormonal resistance and protein catabolism. (A) Kidney dysfunction limits proton (H+) excretion, resulting in a systemic metabolic acidosis. The acidosis causes activation of complement systems, renin angiotensin aldosterone systems and endothelin-1. These acidosis-mediated effects cause CKD progression, forming a viscous cycle; (B) Acidosis promotes inflammation and tissue resistance to multiple anabolic hormones and simultaneously enhances activity of catabolic corticosteroids. Protein catabolism generates acidic products, contributing to acidosis in the setting of CKD and ESRD. Collectively, these abnormalities give rise to a state of protein catabolism, causing sustained negative nitrogen balance, leading to muscle wasting.
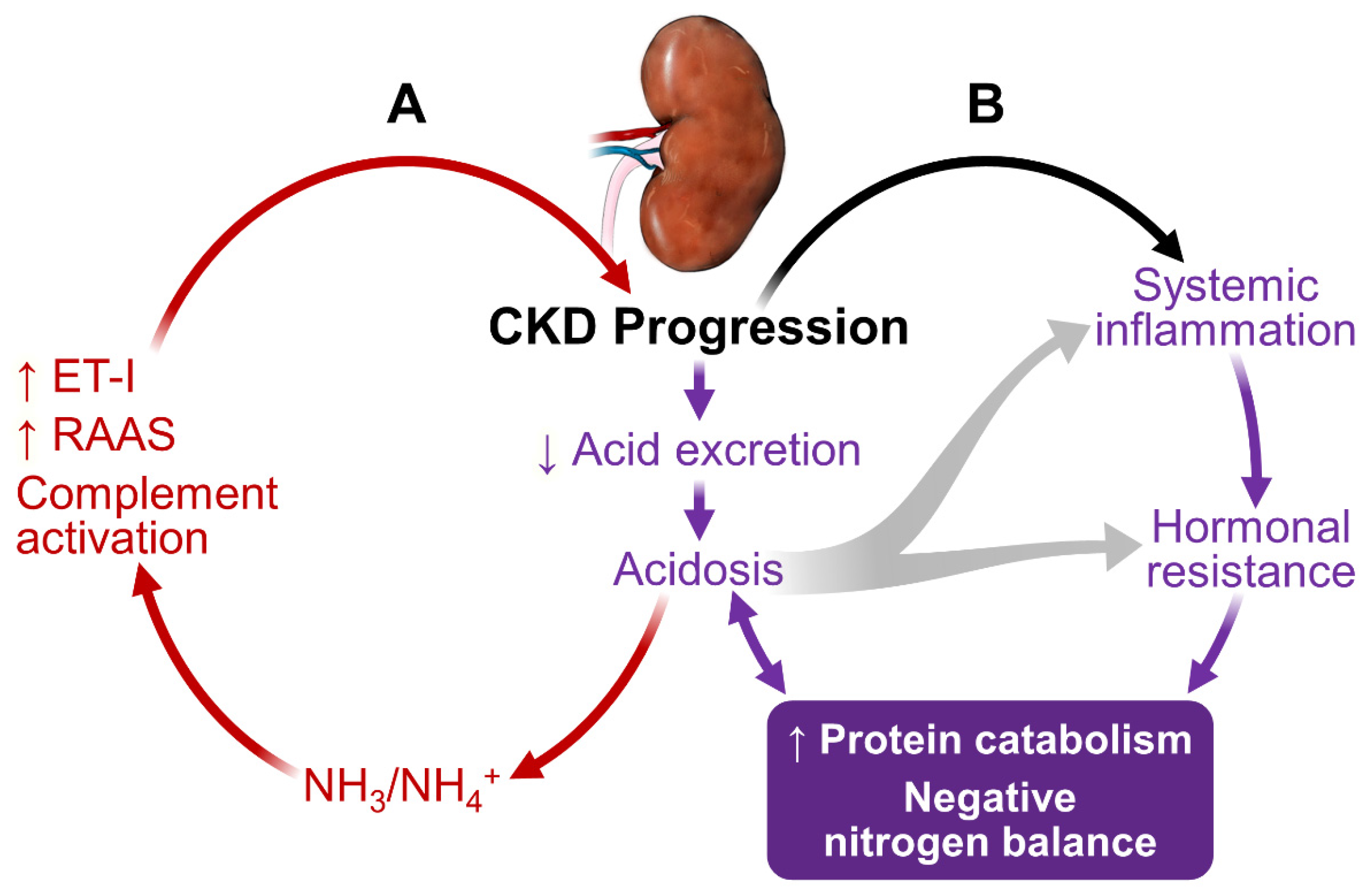
Figure 2.
Kidney-intestinal axis: Gastrointestinal-related inflammation and uremic toxin generation in CKD and ESRD. With progressive decline in kidney function, cytokine production and half-life increases while cytokine elimination decreases. CKD patients frequently have comorbidities (diabetes, hypertension, cardiovascular diseases, anemia and hyperphosphatemia) and are on multiple medications including diuretics (often with fluids restriction), iron preparations, phosphate binders and more that can slow bowel transit and cause constipation. With imposed dietary restrictions for CKD and ESRD, they tend to be on a diet low in fiber that can affect gut motility. Slow intestinal transit plus bowel mucosa edema (occurs often when patients with worsening kidney dysfunction and cardiovascular abnormalities) could alter protein digestion and bowel flora. Proteolytic bacteria floras are preferentially selected over the saccarolytic bacteria. The predominant proteolytic bacteria ferment luminal proteins and generate toxic metabolites and ammonia. The toxic metabolites would disrupt intestinal mucosal barriers causing increasing bacteria and toxin translocation. The systemic exposure and accumulation of these toxins and bacteria causes a low-grade but persistent endotoxemia. Adequate gut flora is known to enhance bowel integrity and promote immunoregulatory function. Disruption of normal flora could compromise gut immune function and, in the extreme, create a situation of “immunoparalysis”. The consequence of these effects is systemic cytokine activation, accumulation of uremic toxins and endotoxemia (purple rectangle).
Figure 2.
Kidney-intestinal axis: Gastrointestinal-related inflammation and uremic toxin generation in CKD and ESRD. With progressive decline in kidney function, cytokine production and half-life increases while cytokine elimination decreases. CKD patients frequently have comorbidities (diabetes, hypertension, cardiovascular diseases, anemia and hyperphosphatemia) and are on multiple medications including diuretics (often with fluids restriction), iron preparations, phosphate binders and more that can slow bowel transit and cause constipation. With imposed dietary restrictions for CKD and ESRD, they tend to be on a diet low in fiber that can affect gut motility. Slow intestinal transit plus bowel mucosa edema (occurs often when patients with worsening kidney dysfunction and cardiovascular abnormalities) could alter protein digestion and bowel flora. Proteolytic bacteria floras are preferentially selected over the saccarolytic bacteria. The predominant proteolytic bacteria ferment luminal proteins and generate toxic metabolites and ammonia. The toxic metabolites would disrupt intestinal mucosal barriers causing increasing bacteria and toxin translocation. The systemic exposure and accumulation of these toxins and bacteria causes a low-grade but persistent endotoxemia. Adequate gut flora is known to enhance bowel integrity and promote immunoregulatory function. Disruption of normal flora could compromise gut immune function and, in the extreme, create a situation of “immunoparalysis”. The consequence of these effects is systemic cytokine activation, accumulation of uremic toxins and endotoxemia (purple rectangle).
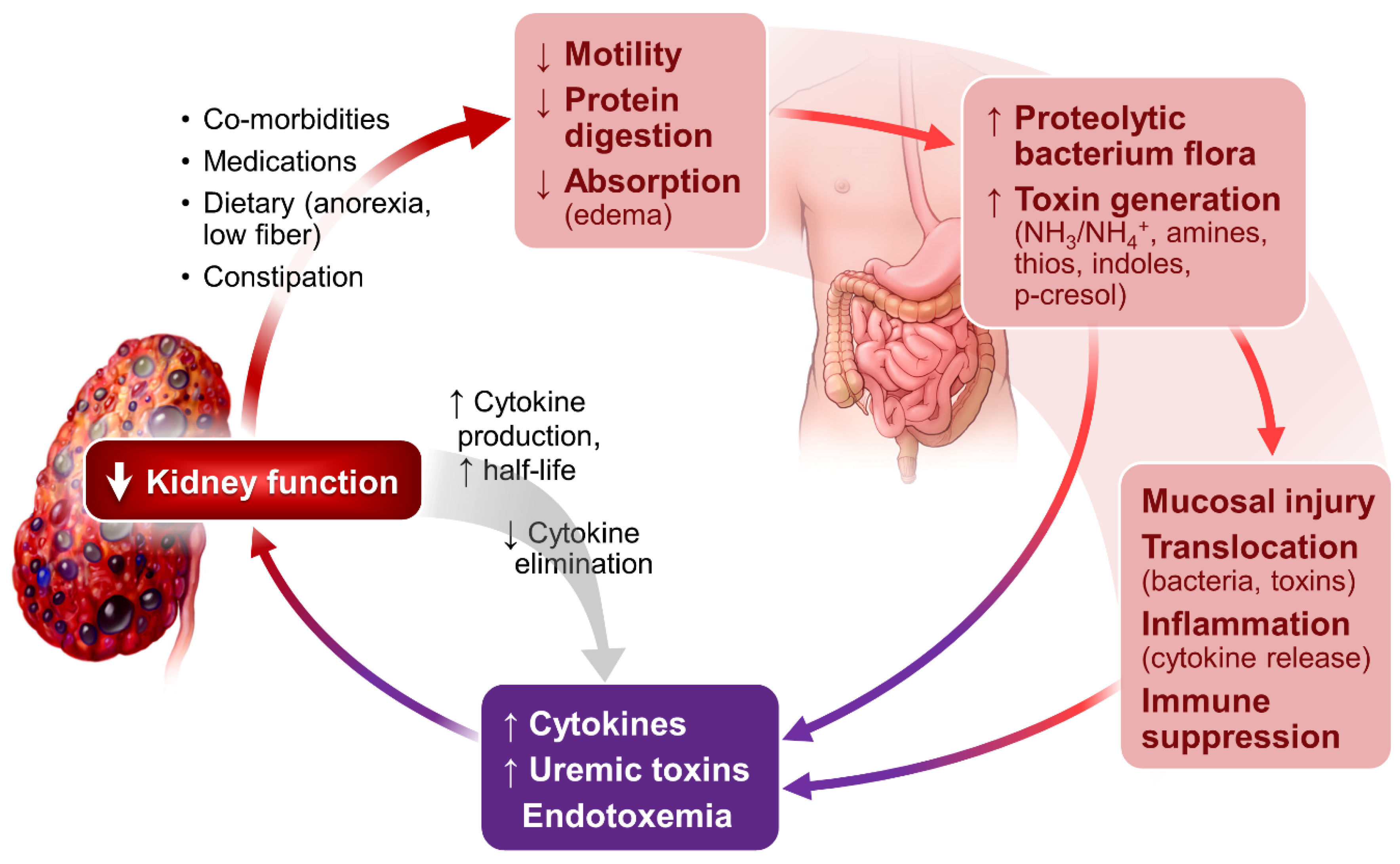
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zha, Y.; Qian, Q. Protein Nutrition and Malnutrition in CKD and ESRD. Nutrients 2017, 9, 208. https://doi.org/10.3390/nu9030208
AMA Style
Zha Y, Qian Q. Protein Nutrition and Malnutrition in CKD and ESRD. Nutrients. 2017; 9(3):208. https://doi.org/10.3390/nu9030208
Chicago/Turabian StyleZha, Yan, and Qi Qian. 2017. "Protein Nutrition and Malnutrition in CKD and ESRD" Nutrients 9, no. 3: 208. https://doi.org/10.3390/nu9030208
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.