The “Metabolic Memory” Theory and the Early Treatment of Hyperglycemia in Prevention of Diabetic Complications

 , , ,
, , ,  ,
, {kind=link}
Abstract
:1. Introduction
2. “Metabolic Memory”: Experimental Evidence Initially Pointed toward Oxidative Stress as Its Cause
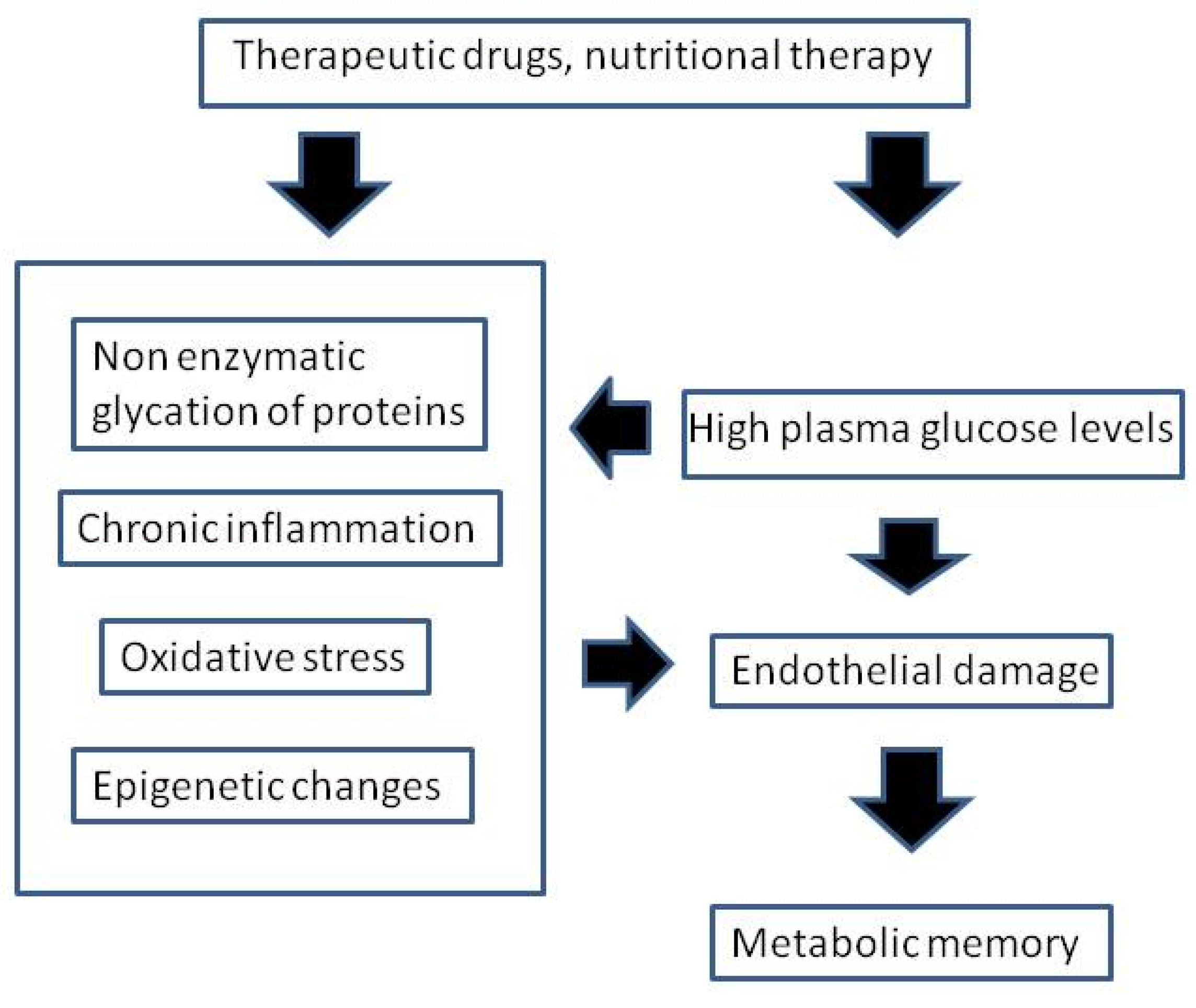
3. Theoretical Basis for “Metabolic Memory”
4. Therapeutic Implications and Prospects
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nathan, D.M.; Cleary, P.A.; Backlund, J.Y.; Genuth, S.M.; Lachin, J.M.; Orchard, T.J.; Raskin, P.; Zinman, B. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N. Engl. J. Med. 2005, 353, 2643–2653. [Google Scholar] [PubMed]
- The Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. Intensive diabetes therapy and carotid intima-media thickness in type 1 diabetes mellitus. N. Engl. J. Med. 2003, 348, 2294–2303. [Google Scholar]
- Holman, R.R.; Paul, S.K.; Bethel, M.A.; Matthews, D.R.; Neil, H.A. Long-term follow-up after tight control of blood pressure in type 2 diabetes. N. Engl. J. Med. 2008, 359, 1565–1576. [Google Scholar] [CrossRef] [PubMed]
- Engerman, R.L.; Kern, T.S. Progression of incipient diabetic retinopathy during good glycemic control. Diabetes 1987, 36, 808–812. [Google Scholar] [CrossRef] [PubMed]
- The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar]
- Writing Team for the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. Effect of intensive therapy on the microvascular complications of type 1 diabetes mellitus. JAMA 2002, 287, 2563–2569. [Google Scholar]
- Writing Team for the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. Sustained effect of intensive treatment of type 1 diabetes mellitus on development and progression of diabetic nephropathy: The Epidemiology of Diabetes Interventions and Complications (EDIC) study. JAMA 2003, 290, 2159–2167. [Google Scholar]
- The Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group; de Boer, I.H.; Sun, W.; Cleary, P.A.; Lachin, J.M.; Molitch, M.E.; Steffes, M.W.; Zinman, B. Intensive diabetes therapy and glomerular filtration rate in type 1 diabetes. N. Engl. J. Med. 2011, 365, 2366–2376. [Google Scholar] [PubMed]
- Holman, R.R.; Paul, S.K.; Bethel, M.A.; Matthews, D.R.; Neil, H.A. 10-Year follow-up of intensive glucose control in type 2 diabetes. N. Engl. J. Med. 2008, 359, 1577–1589. [Google Scholar] [CrossRef] [PubMed]
- Gaede, P.; Lund-Andersen, H.; Parving, H.H.; Pedersen, O. Effect of a multifactorial intervention on mortality in type 2 diabetes. N. Engl. J. Med. 2008, 358, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Duckworth, W.; Abraira, C.; Moritz, T.; Reda, D.; Emanuele, N.; Reaven, P.D.; Zieve, F.J.; Marks, J.; Davis, S.N.; Hayward, R.; et al. Glucose control and vascular complications in veterans with type 2 diabetes. N. Engl. J. Med. 2009, 360, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Reaven, P.D.; Moritz, T.E.; Schwenke, D.C.; Anderson, R.J.; Criqui, M.; Detrano, R.; Emanuele, N.; Kayshap, M.; Marks, J.; Mudaliar, S.; et al. Intensive glucose-lowering therapy reduces cardiovascular disease events in Veterans Affairs Diabetes Trial participants with lower calcified coronary atherosclerosis. Diabetes 2009, 58, 2642–2648. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A. The emerging challenge in diabetes: The “metabolic memory”. Vasc. Pharmacol. 2012, 57, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Russell, N.D.; Cooper, M.E. 50 years forward: Mechanisms of hyperglycaemia driven diabetic complications. Diabetologia 2015, 58, 1708–1714. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Sala, R.; Cagliero, E.; Lorenzi, M. Overexpression of fibronectin induced by diabetes or high glucose: Phenomenon with a memory. Proc. Natl. Acad. Sci. USA 1990, 87, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Engerman, R.; Bloodworth, J.M., Jr.; Nelson, S. Relationship of microvascular disease in diabetes to metabolic control. Diabetes 1977, 26, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.; Beebe, D.; Oates, P.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [PubMed]
- Kowluru, R.A.; Kanwar, M.; Kennedy, A. Metabolic memory phenomenon and accumulation of peroxynitrite in retinal capillaries. Exp. Diabetes Res. 2007, 2007, 21976. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Abbas, S.N.; Odenbach, S. Reversal of hyperglycemia and diabetic nephropathy: Effect of reinstitution of good metabolic control on oxidative stress in the kidney of diabetic rats. J. Diabetes Complicat. 2004, 18, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Esper, R.J.; Nordaby, R.A.; Vilariño, J.O.; Paragano, A.; Cacharrón, J.L.; Machado, R.A. Endothelial dysfunction: A comprehensive appraisal. Cardiovasc. Diabetol. 2006, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Esposito, K.; Ihnat, M.; Thorpe, J.; Giugliano, D. Long-term glycemic control influences the long-lasting effect of hyperglycemia on endothelial function in type 1 diabetes. J. Clin. Endocrinol. Metab. 2009, 94, 2751–2756. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Esposito, K.; Ihnat, M.; Thorpe, J.; Giugliano, D. Effect of acute hyperglycaemia, long-term glycaemic control and insulin on endothelial dysfunction and inflammation in type 1 diabetic patients with different characteristics. Diabet. Med. 2010, 27, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Ihnat, M.A. ‘Glycaemic variability’: A new therapeutic challenge in diabetes and the critical care setting. Diabet. Med. 2010, 27, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Schisano, B.; Tripathi, G.; McGee, K.; McTernan, P.G.; Ceriello, A. Glucose oscillations, more than constant high glucose, induce p53 activation and a metabolic memory in human endothelial cells. Diabetologia 2011, 54, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, G. Do advanced glycation end products contribute to the development of long-term diabetic complications? Nutr. Metab. Cardiovasc. Dis. 2008, 18, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Stirban, A.; Tschoepe, D.; Stratmann, B. Shifting the disease management paradigm from glucose. What are the pros? Diabetes Care 2009, 32, S349–S352. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Inhat, M.A.; Thorpe, J.E. The “metabolic memory”: Is more than just tight glucose control necessary to prevent diabetic complications? J. Clin. Endocrinol. Metab. 2009, 94, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Jax, T.W. Metabolic memory: A vascular perspective. Cardiovasc. Diabetol. 2010, 9, 51. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.A.; Zhang, E.; Natarajan, R. Epigenetic mechanisms in diabetic complications and metabolic memory. Diabetologia 2015, 58, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.A.; Natarajan, R. Epigenetic mechanisms in diabetic vascular complications. Cardiovasc. Res. 2011, 90, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Pirola, L.; Balcerczyk, A.; Okabe, J.; El-Osta, A. Epigenetic phenomena linked to diabetic complications. Nat. Rev. Endocrinol. 2010, 6, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Wegner, M.; Neddermann, D.; Piorunska-Stolzmann, M.; Jagodzinski, P.P. Role of epigenetic mechanisms in the development of chronic complications of diabetes. Diabetes Res. Clin. Pract. 2014, 105, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Brasacchio, D.; Okabe, J.; Tikellis, C.; Balcerczyk, A.; George, P.; Baker, E.K.; Calkin, A.C.; Brownlee, M.; Cooper, M.E.; El-Osta, A. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene activating epigenetic marks that coexist on the lysine tail. Diabetes 2009, 58, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Miao, F.; Gonzalo, I.G.; Lanting, L.; Natarajan, R. In vivo chromatin remodelling events leading to inflammatory gene transcription under diabetic conditions. J. Biol. Chem. 2004, 279, 18091–18097. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.A.; Natarajan, R. Role of epigenetic mechanisms in the vascular complications of diabetes. Subcell. Biochem. 2013, 61, 435–454. [Google Scholar] [PubMed]
- Al-Haddad, R.; Karnib, N.; Assaad, R.A.; Bilen, Y.; Emmanuel, N.; Ghanem, A.; Younes, J.; Zibara, V.; Stephan, J.S.; Sleiman, S.F. Epigenetic changes in diabetes. Neurosci. Lett. 2016, 625, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Baek, D.; Villén, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The impact of microRNAs on protein output. Nature 2008, 455, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Piovan, C.; Croce, C.M. Interplay between microRNAs and the epigenetic machinery: An intricate network. Biochim. Biophys. Acta 2010, 1799, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Breving, K.; Esquela-Kerscher, A. The complexities of microRNA regulation: Mirandering around the rules. Int. J. Biochem. Cell Biol. 2010, 42, 1316–1329. [Google Scholar] [CrossRef] [PubMed]
- Guay, C.; Regazzi, R. Circulating microRNAs as novel biomarkers for diabetes mellitus. Nat. Rev. Endocrinol. 2013, 9, 513–521. [Google Scholar] [CrossRef] [PubMed]
- McClelland, A.D.; Kantharidis, P. MicroRNA in the development of diabetic complications. Clin. Sci. 2014, 126, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Beltrami, C.; Angelini, T.G.; Emanueli, C. Noncoding RNAs in diabetes vascular complications. J. Mol. Cell. Cardiol. 2015, 89, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.A.; Webb, R.C. Potential role of Toll-like receptors in programming of vascular dysfunction. Clin. Sci. 2013, 125, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Guarner, V.; Rubio-Ruiz, M.E. Low-grade systemic inflammation connects aging, metabolic syndrome and cardiovascular disease. Interdiscip. Top. Gerontol. 2015, 40, 99–106. [Google Scholar] [PubMed]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced glycation end products: Sparking the development of diabetic vascular injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Hanefeld, M.; Leiter, L.; Monnier, L.; Moses, A.; Owens, D.; Tajima, N.; Tuomilehto, J. Postprandial glucose regulation and diabetic complications. Arch. Intern. Med. 2004, 164, 2090–2095. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A. Postprandial hyperglycemia and diabetes complications: Is it time to treat? Diabetes 2005, 54, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Quagliaro, L.; Piconi, L.; Assaloni, R.; Da Ros, R.; Maier, A.; Esposito, K.; Giugliano, D. Effect of postprandial hypertriglyceridemia and hyperglycemia on circulating adhesion molecules and oxidative stress generation and the possible role of simvastatin treatment. Diabetes 2004, 53, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Babaei-Jadidi, R.; Howell, S.K.; Thornalley, P.J.; Beisswenger, P.J. Glycated and oxidized protein degradation products are indicators of fasting and postprandial hyperglycemia in diabetes. Diabetes Care 2005, 28, 2465–2471. [Google Scholar] [CrossRef] [PubMed]
- Schiekofer, S.; Andrassy, M.; Chen, J.; Rudofsky, G.; Schneider, J.; Wendt, T.; Stefan, N.; Humpert, P.; Fritsche, A.; Stumvoll, M.; et al. Acute hyperglycemia causes intracellular formation of CML and activation of ras, p42/44 MAPK, and nuclear factor kappaB in PBMCs. Diabetes 2003, 52, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Rahbar, S.; Natarajan, R.; Yerneni, K.; Scott, S.; Gonzales, N.; Nadler, J.L. Evidence that pioglitazone, metformin and pentoxifylline are inhibitors of glycation. Clin. Chim. Acta 2000, 301, 65–77. [Google Scholar] [CrossRef]
- Miyata, T.; van Ypersele, D.S.; Ueda, Y.; Ichimori, K.; Inagi, R.; Onogi, H.; Ishikawa, N.; Nangaku, M.; Kurokawa, K. Angiotensin II receptor antagonists and angiotensin-converting enzyme inhibitors lower in vitro the formation of advanced glycation end products: Biochemical mechanisms. J. Am. Soc. Nephrol. 2002, 13, 2478–2487. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Yamagishi, S.; Nakamura, K.; Matsui, T.; Imaizumi, T.; Takeuchi, M.; Koga, H.; Ueno, T.; Sata, M. Telmisartan inhibits AGE-induced C-reactive protein production through downregulation of the receptor for AGE via peroxisome proliferator activated receptor-gamma activation. Diabetologia 2006, 49, 3094–3099. [Google Scholar] [CrossRef] [PubMed]
- Corgnali, M.; Piconi, L.; Ihnat, M.; Ceriello, A. Evaluation of gliclazide ability to attenuate the hyperglycaemic ‘memory’ induced by high glucose in isolated human endothelial cells. Diabetes Metab. Res. Rev. 2008, 24, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Swislocki, A.L.; Jialal, I. Glucagon-like peptide-1 receptor agonists and diabetic cardiovascular disease: Implications of the LEADER study. Metab. Syndr. Relat. Disord. 2016, 14, 375–377. [Google Scholar] [CrossRef] [PubMed]
- Geyer, P.E.; Wewer Albrechtsen, N.J.; Tyanova, S.; Grassl, N.; Iepsen, E.W.; Lundgren, J.; Madsbad, S.; Holst, J.J.; Torekov, S.S.; Mann, M. Proteomics reveals the effects of sustained weight loss on the human plasma proteome. Mol. Syst. Biol. 2016, 12, 901. [Google Scholar] [CrossRef] [PubMed]
- Kuwata, H.; Iwasaki, M.; Shimizu, S.; Minami, K.; Maeda, H.; Seino, S.; Nakada, K.; Nosaka, C.; Murotani, K.; Kurose, T.; et al. Meal sequence and glucose excursion, gastric emptying and incretin secretion in type 2 diabetes: A randomised, controlled crossover, exploratory trial. Diabetologia 2016, 59, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Little, T.J.; Bound, M.J.; Borg, M.; Zhang, X.; Deacon, C.F.; Horowitz, M.; Jones, K.L.; Rayner, C.K. A protein preload enhances the glucose-lowering efficacy of vildagliptin in type 2 diabetes. Diabetes Care 2016, 39, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.R.; Wang, Z.; Zhou, W.; Fan, S.R.; Ma, R.; Xue, L.; Yang, L.; Li, Y.S.; Tan, H.L.; Shao, Q.H.; et al. Epalrestat protects against diabetic peripheral neuropathy by alleviating oxidative stress and inhibiting polyol pathway. Neural Regen. Res. 2016, 11, 345–351. [Google Scholar] [PubMed]
- Janssen, H.L.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Dávalos, A.; Chroni, A. Antisense oligonucleotides, microRNAs, and antibodies. Handb. Exp. Pharmacol. 2015, 224, 649–689. [Google Scholar] [PubMed]
- Santulli, G.; Wronska, A.; Uryu, K.; Diacovo, T.G.; Gao, M.; Marx, S.O.; Kitajewski, J.; Chilton, J.M.; Akat, K.M.; Tuschl, T.; et al. A selective microRNA-based strategy inhibits restenosis while preserving endothelial function. J. Clin. Investig. 2014, 124, 4102–4114. [Google Scholar] [CrossRef] [PubMed]
- Hartig, S.M.; Hamilton, M.P.; Bader, D.A.; McGuire, S.E. The miRNA interactome in metabolic homeostasis. Trends Endocrinol. Metab. 2015, 26, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Krützfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Testa, R.; Bonfigli, A.R.; Prattichizzo, F.; La Sala, L.; De Nigris, V.; Ceriello, A. The “Metabolic Memory” Theory and the Early Treatment of Hyperglycemia in Prevention of Diabetic Complications. Nutrients 2017, 9, 437. https://doi.org/10.3390/nu9050437
Testa R, Bonfigli AR, Prattichizzo F, La Sala L, De Nigris V, Ceriello A. The “Metabolic Memory” Theory and the Early Treatment of Hyperglycemia in Prevention of Diabetic Complications. Nutrients. 2017; 9(5):437. https://doi.org/10.3390/nu9050437
Chicago/Turabian StyleTesta, Roberto, Anna Rita Bonfigli, Francesco Prattichizzo, Lucia La Sala, Valeria De Nigris, and Antonio Ceriello. 2017. "The “Metabolic Memory” Theory and the Early Treatment of Hyperglycemia in Prevention of Diabetic Complications" Nutrients 9, no. 5: 437. https://doi.org/10.3390/nu9050437