Choline Supplementation Normalizes Fetal Adiposity and Reduces Lipogenic Gene Expression in a Mouse Model of Maternal Obesity


Abstract
:1. Introduction
2. Materials and Methods
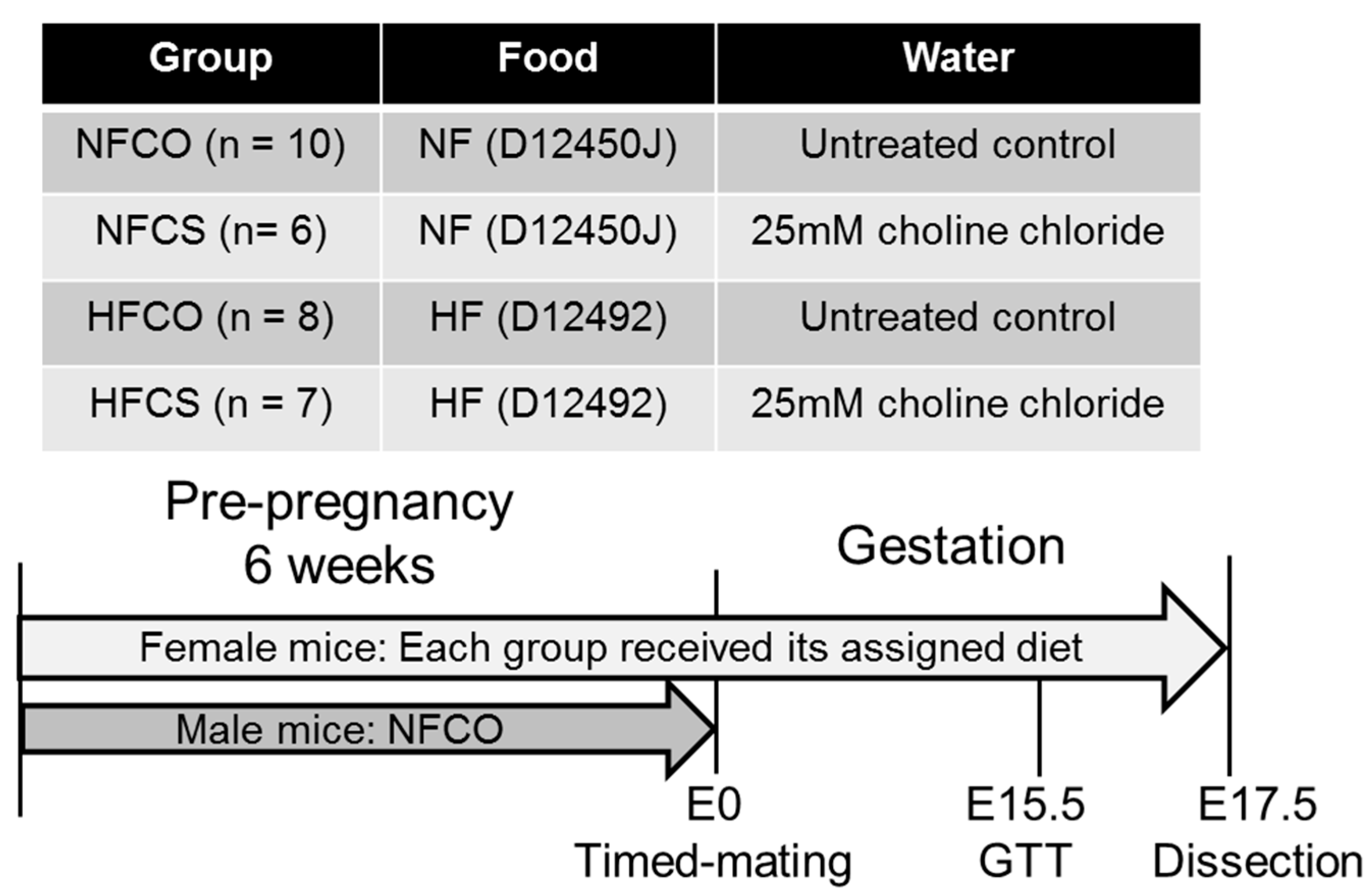
2.1. Animals and Diets
2.2. Intraperitoneal Glucose Tolerance (IGT) Tests
2.3. Tissue Collection
2.4. Analytical Measurements
2.4.1. Embryo Sexing
2.4.2. Fetal Adiposity Assessment
2.4.3. Serum Insulin, Glucose, Triglyceride, and Free Fatty Acid (FFA) Measurements
2.4.4. Homeostasis Model Assessment of Insulin Resistance (HOMA-IR) Index
2.4.5. Choline Measurements
2.4.6. RNA Extraction and Quantitative Real-Time PCR
2.4.7. Maternal and Fetal Liver Triglyceride Measurements
2.4.8. Statistical Analyses
3. Results
3.1. Food and Choline Intake
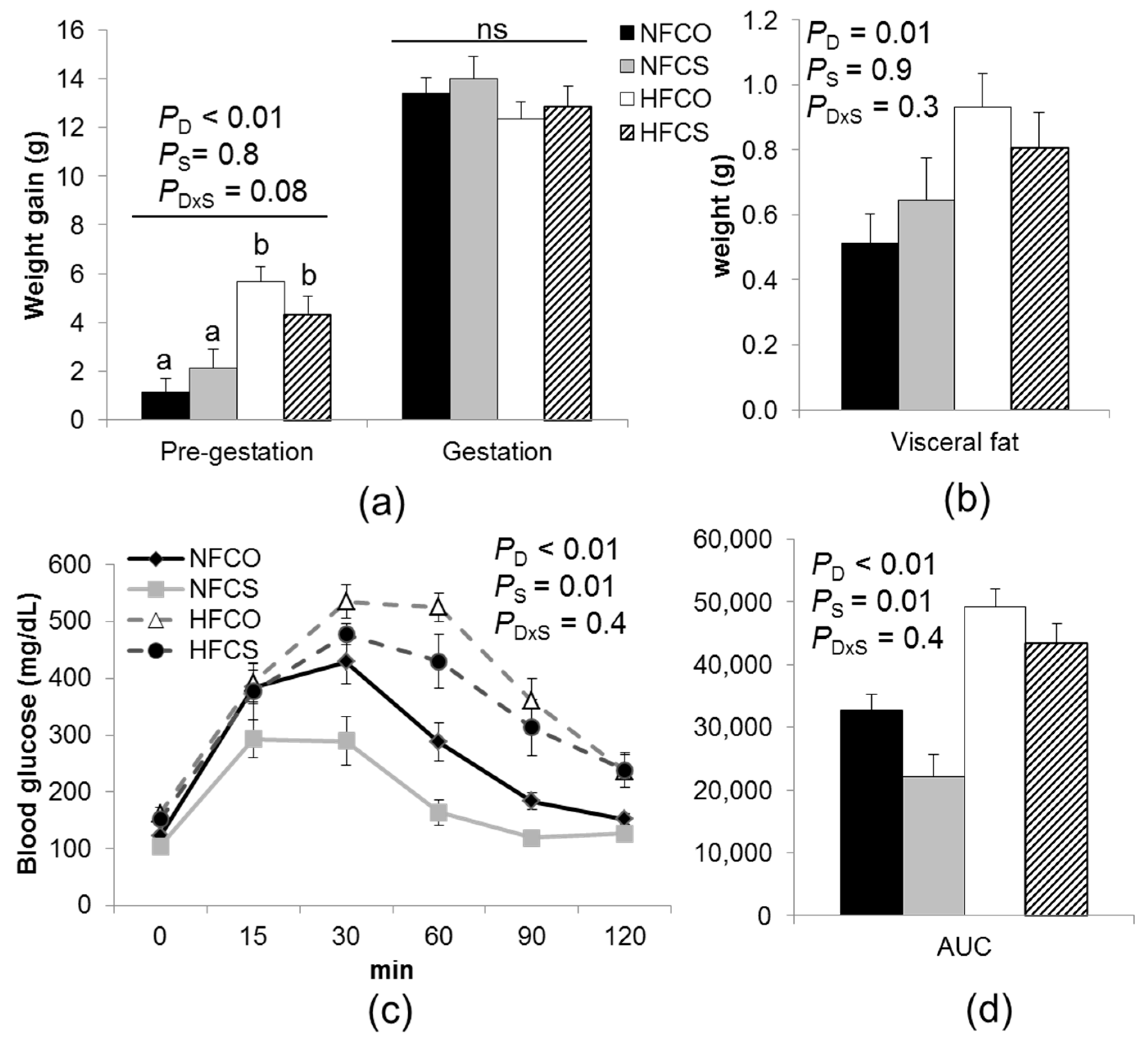
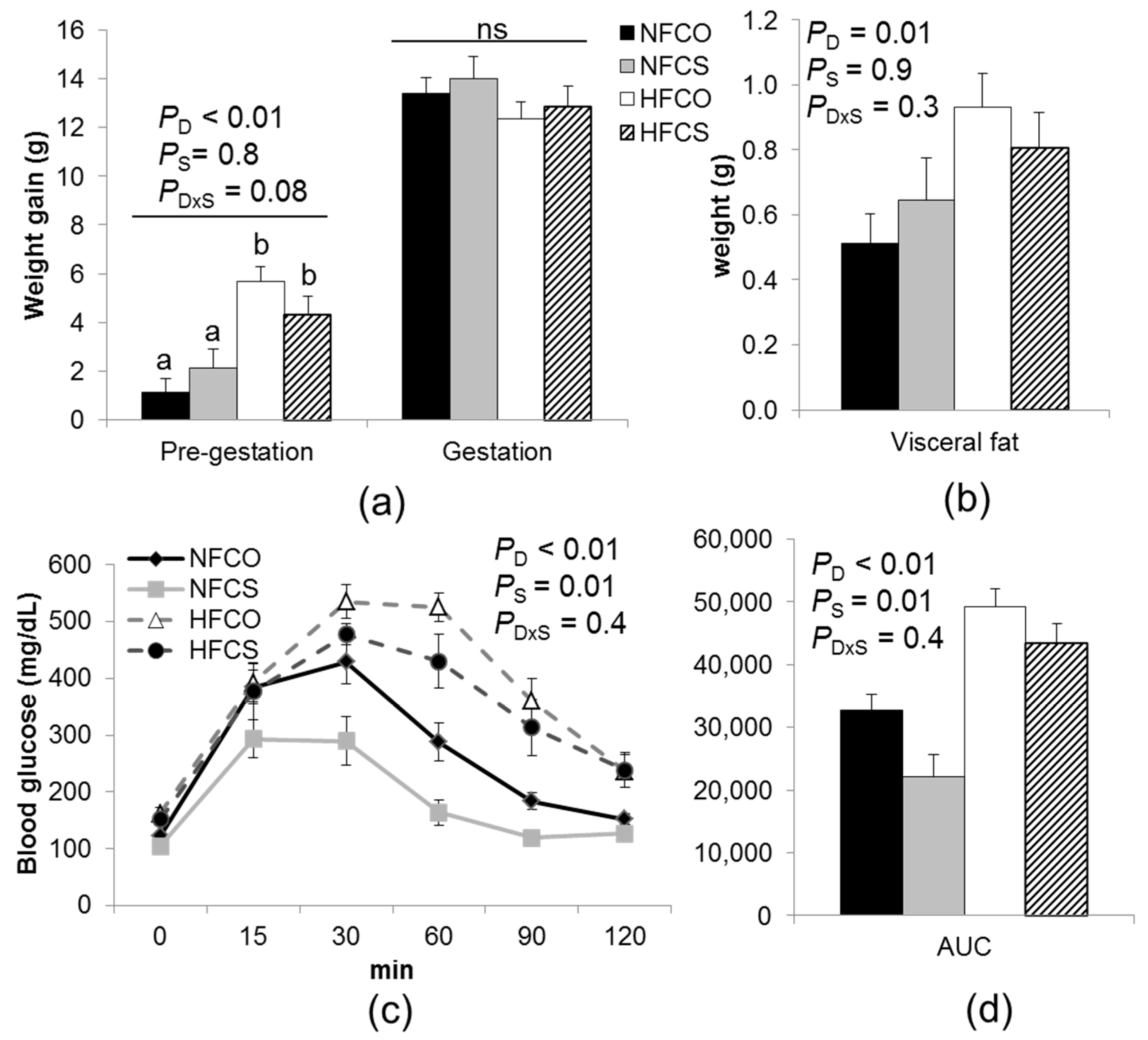
3.2. Maternal Weight, Abdominal Adiposity, Glucose Tolerance, and Serum Biomarkers
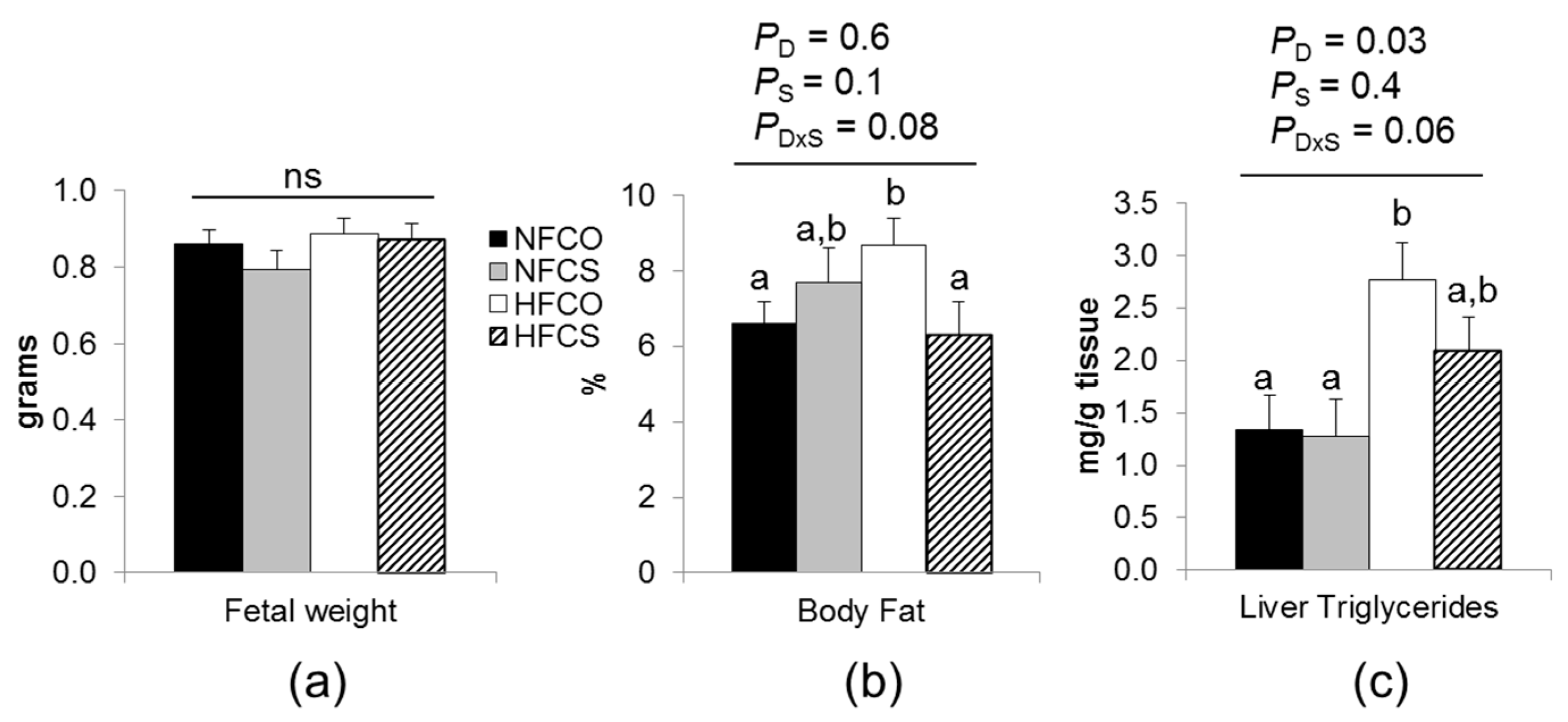
3.3. Placental and Embryonic Outcomes
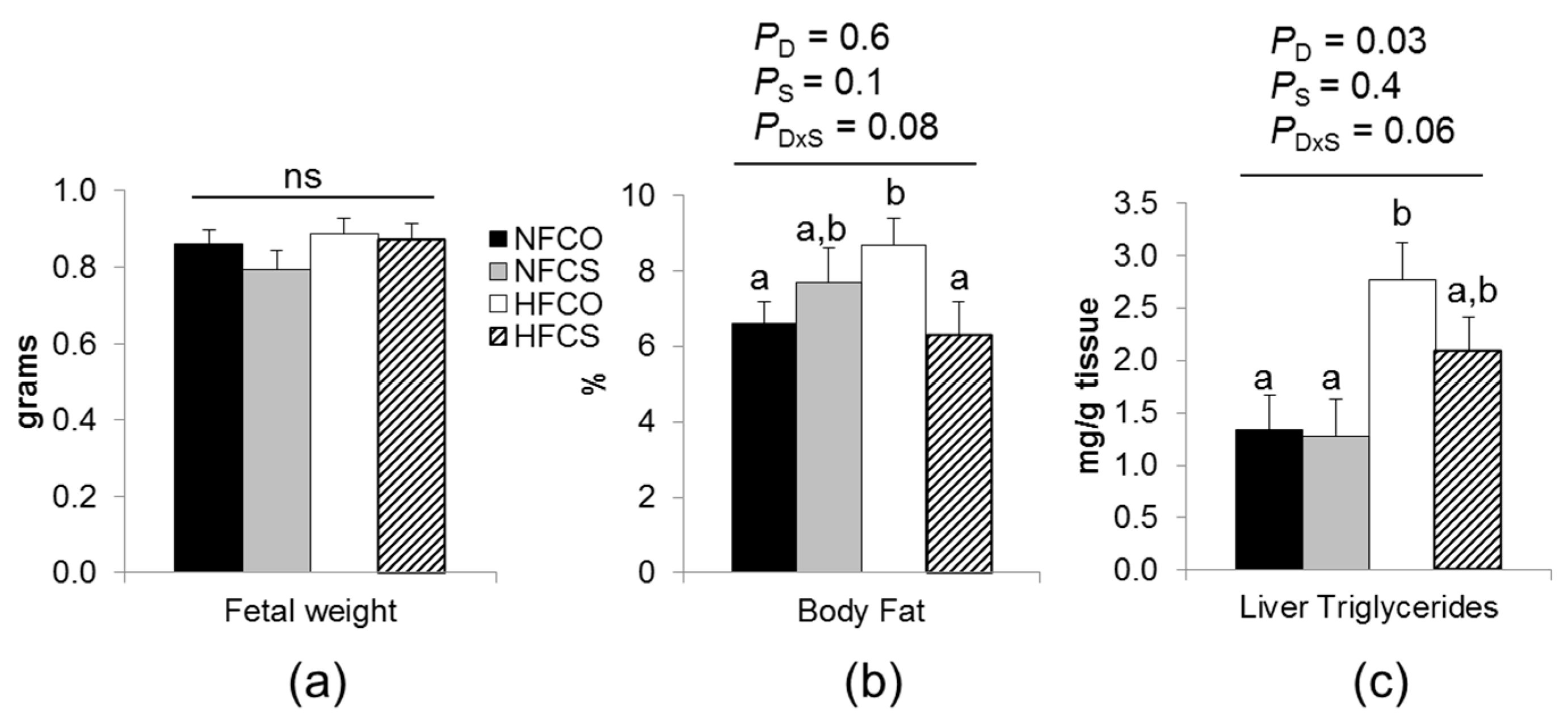
3.3.1. Maternal Choline Supplementation during HF Feeding Did Not Affect Fetal Weight but Reduced Fetal Adiposity
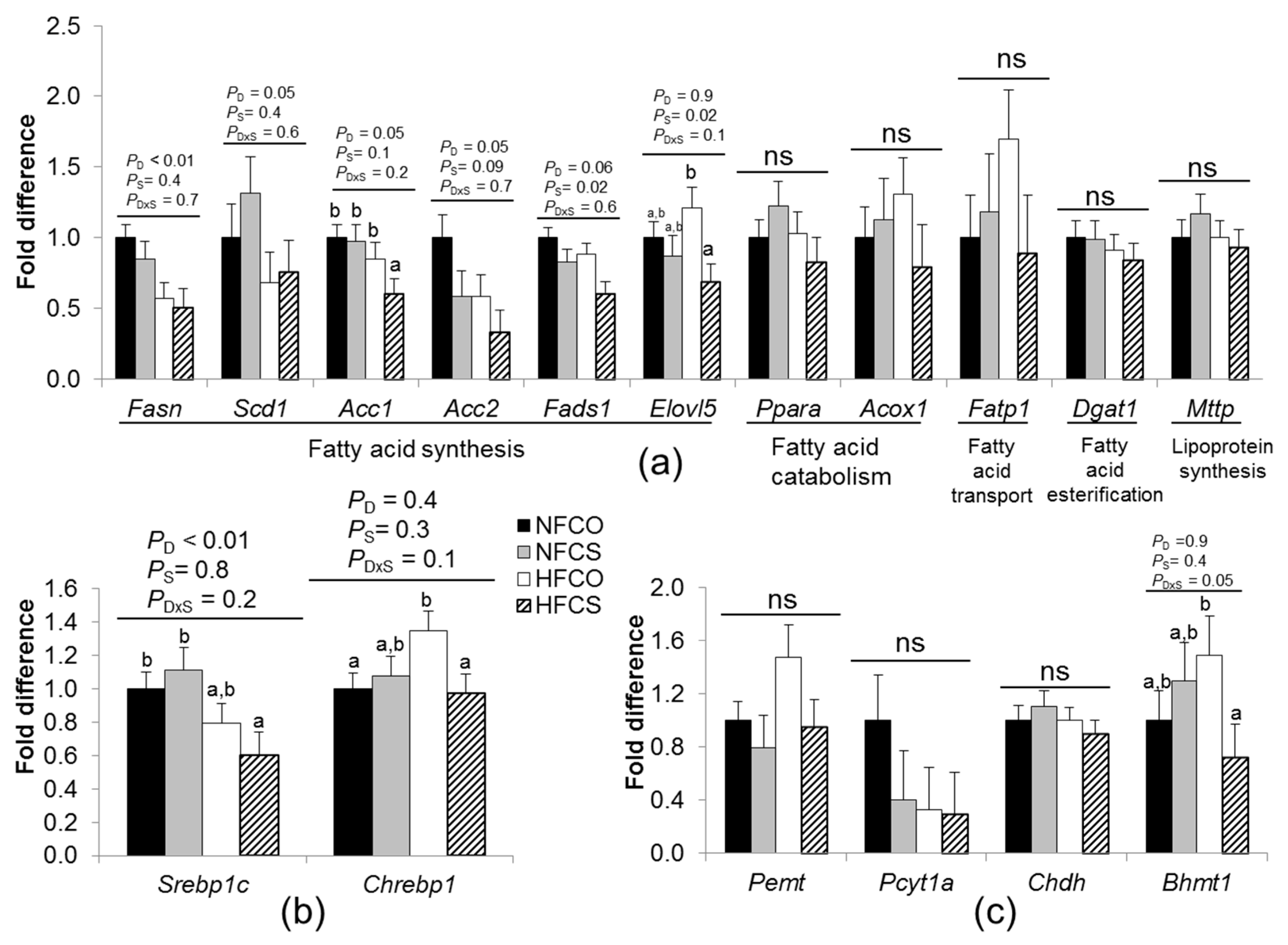
3.3.2. Maternal Choline Supplementation during HF Feeding Downregulated Lipogenic Gene Expression in Fetal Livers
3.3.3. HF Feeding and Maternal Choline Supplementation Altered Fetal Hepatic Choline Metabolism
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Catalano, P.M.; Shankar, K. Obesity and pregnancy: Mechanisms of short term and long term adverse consequences for mother and child. BMJ 2017, 356. [Google Scholar] [CrossRef] [PubMed]
- Van Hoorn, J.; Dekker, G.; Jeffries, B. Gestational diabetes versus obesity as risk factors for pregnancy-induced hypertensive disorders and fetal macrosomia. Aust. N. Z. J. Obstet. Gynaecol. 2002, 42, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Kamana, K.C.; Shakya, S.; Zhang, H. Gestational diabetes mellitus and macrosomia: A literature review. Ann. Nutr. Metab. 2015, 66, 14–20. [Google Scholar]
- Wang, Y.; Gao, E.; Wu, J.; Zhou, J.; Yang, Q.; Walker, M.C.; Mbikay, M.; Sigal, R.J.; Nair, R.C.; Wen, S.W. Fetal macrosomia and adolescence obesity: Results from a longitudinal cohort study. Int. J. Obes. 2009, 33, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Hermann, G.M.; Dallas, L.M.; Haskell, S.E.; Roghair, R.D. Neonatal macrosomia is an independent risk factor for adult metabolic syndrome. Neonatology 2010, 98, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Szostak-Węgierek, D.; Szamotulska, K. Fetal development and risk of cardiovascular diseases and diabetes type 2 in adult life. Med. Wieku Rozwoj. 2011, 15, 203–215. [Google Scholar] [PubMed]
- Sewell, M.F.; Huston-Presley, L.; Super, D.M.; Catalano, P. Increased neonatal fat mass, not lean body mass, is associated with maternal obesity. Am. J. Obstet. Gynecol. 2006, 195, 1100–1103. [Google Scholar] [CrossRef] [PubMed]
- Hull, H.R.; Thornton, J.C.; Ji, Y.; Paley, C.; Rosenn, B.; Mathews, P.; Navder, K.; Yu, A.; Dorsey, K.; Gallagher, D. Higher infant body fat with excessive gestational weight gain in overweight women. Am. J. Obstet. Gynecol. 2011, 205, 211.e1–211.e7. [Google Scholar] [CrossRef] [PubMed]
- Cetin, I.; Alvino, G.; Cardellicchio, M. Long chain fatty acids and dietary fats in fetal nutrition. J. Physiol. 2009, 587, 3441–3451. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Guo, Z.; Bosco, C.; Guidotti, S.; Wang, Y.; Wang, M.; Parast, M.; Schaack, J.; Hay, W.W.; Moore, T.R.; et al. Maternal high-fat feeding increases placental lipoprotein lipase activity by reducing SIRT1 expression in mice. Diabetes 2015, 64, 3111–3120. [Google Scholar] [CrossRef] [PubMed]
- Caudill, M.A. Pre- and postnatal health: Evidence of increased choline needs. J. Am. Diet. Assoc. 2010, 110, 1198–1206. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S.H.; Da Costa, K.A.; Franklin, P.D.; Alexander, E.A.; Lamont, J.T.; Sheard, N.F.; Beiser, A. Choline, an essential nutrient for humans. FASEB J. 1991, 5, 2093–2098. [Google Scholar] [CrossRef]
- Hebbard, L.; George, J. Animal models of nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.M.; Vance, D.E. The active synthesis of phosphatidylcholine is required for very low density lipoprotein secretion from rat hepatocytes. J. Biol. Chem. 1988, 263, 2998–3004. [Google Scholar] [PubMed]
- Chakravarthy, M.V.; Lodhi, I.J.; Yin, L.; Malapaka, R.R.; Xu, H.E.; Turk, J.; Semenkovich, C.F. Identification of a physiologically relevant endogenous ligand for PPARα in liver. Cell 2009, 138, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Lee, Y.K.; Mamrosh, J.L.; Busby, S.A.; Griffin, P.R.; Pathak, M.C.; Ortlund, E.A.; Moore, D.D. A nuclear-receptor-dependent phosphatidylcholine pathway with antidiabetic effects. Nature 2011, 474, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.K.; Jacobs, R.L.; Watts, J.L.; Rottiers, V.; Jiang, K.; Finnegan, D.M.; Shioda, T.; Hansen, M.; Yang, F.; Niebergall, L.J.; et al. A conserved SREBP-1/phosphatidylcholine feedback circuit regulates lipogenesis in metazoans. Cell 2011, 147, 840–852. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.F.; Iwasaki, Y.; Zhe, W.; Nishiyama, M.; Taguchi, T.; Tsugita, M.; Kambayashi, M.; Hashimoto, K.; Terada, Y. Hormonal regulation of acetyl-CoA carboxylase isoenzyme gene transcription. Endocr. J. 2010, 57, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.B.; Laffitte, B.A.; Patel, P.H.; Watson, M.A.; Matsukuma, K.E.; Walczak, R.; Collins, J.L.; Osborne, T.F.; Tontonoz, P. Direct and indirect mechanisms for regulation of fatty acid synthase gene expression by liver X receptors. J. Biol. Chem. 2002, 277, 11019–11025. [Google Scholar] [CrossRef] [PubMed]
- Varin, A.; Thomas, C.; Ishibashi, M.; Ménégaut, L.; Gautier, T.; Trousson, A.; Bergas, V.; de Barros, J.P.; Narce, M.; Lobaccaro, J.M.; et al. Liver X receptor activation promotes polyunsaturated fatty acid synthesis in macrophages: Relevance in the context of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Dalen, K.T.; Gustafsson, J.A.; Nebb, H.I. Regulation of hepatic fatty acid elongase 5 by LXRalpha-SREBP-1c. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2009, 1791, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Cordero, P.; Gomez-Uriz, A.M.; Campion, J.; Milagro, F.I.; Martinez, J.A. Dietary supplementation with methyl donors reduces fatty liver and modifies the fatty acid synthase DNA methylation profile in rats fed an obesogenic diet. Genes Nutr. 2013, 8, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yao, T.; Pini, M.; Zhou, Z.; Fantuzzi, G.; Song, Z. Betaine improved adipose tissue function in mice fed a high-fat diet: A mechanism for hepatoprotective effect of betaine in nonalcoholic fatty liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G634–G642. [Google Scholar] [CrossRef] [PubMed]
- Corbin, K.D.; Zeisel, S.H. Choline metabolism provides novel insights into nonalcoholic fatty liver disease and its progression. Curr. Opin. Gastroenterol. 2012, 28, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Fujiki, K.; Kano, F.; Shiota, K.; Murata, M. Expression of the peroxisome proliferator activated receptor gamma gene is repressed by DNA methylation in visceral adipose tissue of mouse models of diabetes. BMC Biol. 2009, 7. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Jia, Y.; Lu, J.; Yuan, M.; Sui, S.; Song, H.; Zhao, R. Maternal dietary betaine supplementation modifies hepatic expression of cholesterol metabolic genes via epigenetic mechanisms in newborn piglets. Br. J. Nutr. 2014, 112, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Wankhade, U.D.; Zhong, Y.; Kang, P.; Alfaro, M.; Chintapalli, S.V.; Thakali, K.M.; Shankar, K. Enhanced offspring predisposition to steatohepatitis with maternal high-fat diet is associated with epigenetic and microbiome alterations. PLoS ONE 2017, 12, 12. [Google Scholar] [CrossRef] [PubMed]
- Chmurzynska, A.; Stachowiak, M.; Gawecki, J.; Pruszynska-Oszmalek, E.; Tubacka, M. Protein and folic acid content in the maternal diet determine lipid metabolism and response to high-fat feeding in rat progeny in an age-dependent manner. Genes Nutr. 2012, 7, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Dhar, M.S.; Sommardahl, C.S.; Kirkland, T.; Nelson, S.; Donnell, R.; Johnson, D.K.; Castellani, L.W. Mice heterozygous for Atp10c, a putative amphipath, represent a novel model of obesity and type 2 diabetes. J. Nutr. 2004, 134, 799–805. [Google Scholar] [PubMed]
- Pruznak, A.M.; Kazi, A.A.; Frost, R.A.; Vary, T.C.; Lang, C.H. Activation of AMP-activated protein kinase by 5-aminoimidazole-4-carboxamide-1-beta-D-ribonucleoside prevents leucine-stimulated protein synthesis in rat skeletal muscle. J. Nutr. 2008, 138, 1887–1894. [Google Scholar] [PubMed]
- Nam, J.; Greenwald, E.; Jack-Roberts, C.; Ajeeb, T.T.; Malysheva, O.V.; Caudill, M.A.; Axen, K.; Saxena, A.; Semernina, E.; et al. Choline prevents fetal overgrowth and normalizes placental fatty acid and glucose metabolism in a mouse model of maternal obesity. J. Nutr. Biochem. 2017, in press. [Google Scholar] [CrossRef]
- Tai, M.M. A mathematical model for the determination of total area under glucose tolerance and other metabolic curves. Diabetes Care 1994, 17, 152–154. [Google Scholar] [CrossRef] [PubMed]
- McClive, P.J.; Sinclair, A.H. Rapid DNA extraction and PCR-sexing of mouse embryos. Mol. Reprod. Dev. 2001, 60, 225–226. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane-Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Yan, J.; Jiang, X.; West, A.A.; Perry, C.A.; Malysheva, O.V.; Devapatla, S.; Pressman, E.; Vermeylen, F.; Stabler, S.P.; Allen, R.H.; et al. Maternal choline intake modulates maternal and fetal biomarkers of choline metabolism in humans. Am. J. Clin. Nutr. 2012, 95, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Chew, T.W.; Jiang, X.; Yan, J.; Wang, W.; Lusa, A.L.; Carrier, B.J.; West, A.A.; Malysheva, O.V.; Brenna, J.T.; Gregory, J.F.; et al. Folate intake, MTHFR genotype, and sex modulate choline metabolism in mice. J. Nutr. 2011, 141, 1475–1481. [Google Scholar] [CrossRef] [PubMed]
- Aldrich, M.C.; Kumar, R.; Colangelo, L.A.; Williams, L.K.; Sen, S.; Kritchevsky, S.B.; Meibohm, B.; Galanter, J.; Hu, D.; Gignoux, C.R.; et al. Genetic ancestry-smoking interactions and lung function in African Americans: A cohort study. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thanopoulou, A.; Karamanos, B.; Archimandritis, A. Glycated hemoglobin, diabetes, and cardiovascular risk in nondiabetic adults. N. Engl. J. Med. 2010, 362, 2030–2031. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Yang, H.; Ramani, K.; Ara, A.I.; Chen, H.; Mato, J.M.; Lu, S.C. Inhibition of human betaine-homocysteine methyltransferase expression by S-adenosylmethionine and methylthioadenosine. Biochem. J. 2007, 401, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S.H. Metabolic crosstalk between choline/1-carbon metabolism and energy homeostasis. Clin. Chem. Lab. Med. 2013, 51, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Herrera, E.; Amusquivar, E.; López-Soldado, I.; Ortega, H. Maternal lipid metabolism and placental lipid transfer. Horm. Res. 2006, 65, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.W.; Ellis, J.M.; Coleman, R.A.; Zeisel, S.H. Mouse betaine-homocysteine S-methyltransferase deficiency reduces body fat via increasing energy expenditure and impairing lipid synthesis and enhancing glucose oxidation in white adipose tissue. J. Biol. Chem. 2012, 287, 16187–16198. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.C.; Qing, K.; Chen, Y. Diet-induced changes in stearoyl-CoA desaturase 1 expression in obesity-prone and -resistant mice. Obes. Res. 2004, 12, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Oosterveer, M.H.; van Dijk, T.H.; Tietge, U.J.; Boer, T.; Havinga, R.; Stellaard, F.; Groen, A.K.; Kuipers, F.; Reijngoud, D.J. High fat feeding induces hepatic fatty acid elongation in mice. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Zhao, F.; Mancuso, A.; Gruber, J.J.; Thompson, C.B. The glucose-responsive transcription factor ChREBP contributes to glucose-dependent anabolic synthesis and cell proliferation. Proc. Natl. Acad. Sci. USA 2009, 106, 21660–21665. [Google Scholar] [CrossRef] [PubMed]
- Raubenheimer, P.J.; Nyirenda, M.J.; Walker, B.R. A choline-deficient diet exacerbates fatty liver but attenuates insulin resistance and glucose intolerance in mice fed a high-fat diet. Diabetes 2006, 55, 2015–2020. [Google Scholar] [CrossRef] [PubMed]
- Caputo, M.; De Rosa, M.C.; Rescigno, T.; Zirpoli, H.; Vassallo, A.; De Tommasi, N.; Torino, G.; Tecce, M.F. Binding of polyunsaturated fatty acids to LXRα and modulation of SREBP-1 interaction with a specific SCD1 promoter element. Cell Biochem. Funct. 2014, 32, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Howard, T.D.; Mathias, R.A.; Seeds, M.C.; Herrington, D.M.; Hixson, J.E.; Shimmin, L.C.; Hawkins, G.A.; Sellers, M.; Ainsworth, H.C.; Sergeant, S.; et al. DNA methylation in an enhancer region of the FADS cluster is associated with FADS activity in human liver. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Bar, H.Y.; Yan, J.; Jones, S.; Brannon, P.M.; West, A.A.; Perry, C.A.; Ganti, A.; Pressman, E.; Devapatla, S.; et al. A higher maternal choline intake among third-trimester pregnant women lowers placental and circulating concentrations of the antiangiogenic factor fms-like tyrosine kinase-1 (sFLT1). FASEB J. 2013, 27, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Jones, S.; Andrew, B.Y.; Ganti, A.; Malysheva, O.V.; Giallourou, N.; Brannon, P.M.; Roberson, M.S.; Caudill, M.A. Choline inadequacy impairs trophoblast function and vascularization in cultured human placental trophoblasts. J. Cell. Physiol. 2014, 229, 1016–1027. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Food and Choline Intake | NFCO | NFCS | HFCO | HFCS | p Value | ||
|---|---|---|---|---|---|---|---|
| (n = 10) | (n = 6) | (n = 8) | (n = 7) | D | S | D × S | |
| Food intake (g/week) | 17.4 ± 0.2 | 17.4 ± 0.3 | 15.5 + 0.3 | 14.9 ± 0.3 | <0.01 | 0.3 | 0.4 |
| Caloric intake (kcal/week) | 67 ± 1 | 66 ± 2 | 81 ± 1 | 78 ± 1 | <0.01 | 0.3 | 0.3 |
| Water intake (mL/week) | 20.2 ± 0.4 a | 21.7 ± 0.8 a | 21.2 ± 0.4 a | 20.2 ± 0.5 a | 0.6 | 0.7 | 0.02 |
| Choline intake from food (µmol/week) | 132 ± 2 | 133 ± 4 | 160 ± 3 | 155 ± 3 | <0.01 | 0.5 | 0.4 |
| Choline intake from water (µmol/week) | N/A | 542 ± 16 | N/A | 501 ± 13 | 0.08 | N/A | N/A |
| Total choline intake (µmol/week) | 132 ± 2 a | 663 ± 12 c | 160 ± 3 b | 664 ± 12 c | 0.4 | 0.01 | 0.1 |
| Maternal Biomarkers | NFCO | NFCS | HFCO | HFCS | p Value | ||
|---|---|---|---|---|---|---|---|
| (n = 8) | (n = 6) | (n = 8) | (n = 7) | D | S | D × S | |
| Serum insulin (ng/mL) | 0.63 ± 0.05 a | 0.81± 0.06 b | 0.57 ± 0.05 a | 0.57 ± 0.05 a | 0.01 | 0.08 | 0.1 |
| Serum glucose (mg/dL) | 103.5 ± 5.9 | 88.9± 6.9 | 103.7 ± 5.9 | 94.1 ± 6.4 | 0.7 | 0.07 | 0.7 |
| HOMA-IR index | 28.0 ± 2.9 | 30.3 ± 3.4 | 24.9 ± 2.9 | 23.2 ± 3.1 | 0.1 | 0.9 | 0.5 |
| Serum triglycerides (mg/dL) | 37.9 ± 3.5 | 46.6 ± 5.2 | 31.2 ± 3.7 | 50.6 ± 4.1 | 0.7 | <0.01 | 0.5 |
| Serum FFAs (mmol/L) | 0.52± 0.12 | 0.74 ± 0.2 | 0.64 ± 0.14 | 0.80 ± 0.16 | 0.9 | 0.06 | 0.4 |
| Liver triglycerides (mg/g tissue) | 17.8 ± 4.9 a | 9.3 ± 6.4 a | 37.3 ± 5.5 b | 13.9 ± 5.5 a | 0.04 | <0.01 | 0.2 |
| Metabolites (nmol/g Tissue) | NFCO | NFCS | HFCO | HFCS | p Value | ||
|---|---|---|---|---|---|---|---|
| (n = 9) | (n = 6) | (n = 7) | (n = 6) | D | S | D × S | |
| Choline | 172 ± 12 | 196 ± 13 | 144 ± 13 | 169 ± 13 | 0.05 | 0.07 | 0.9 |
| Betaine | 719 ± 89 a | 1290 ± 102 b | 957 ± 103 a | 1740 ± 103 c | <0.01 | <0.01 | 0.2 |
| Methionine | 139 ± 12 | 158 ± 14 | 134 ± 14 | 134 ± 14 | 0.3 | 0.5 | 0.5 |
| Lysophosphatidylcholine | 253 ± 28 | 235 ± 33 | 273 ± 32 | 177 ± 33 | 0.6 | 0.08 | 0.3 |
| Glycerophosphocholine | 633 ± 60 | 735 ± 70 | 610 ± 70 | 587 ± 69 | 0.2 | 0.6 | 0.4 |
| Phosphocholine | 537 ± 54 | 687 ± 62 | 486 ± 62 | 629 ± 62 | 0.02 | 0.4 | 0.9 |
| Phosphatidylcholine | 22,006 ± 914 | 21,511 ± 1055 | 21,515 ± 1055 | 19,317 ± 1055 | 0.2 | 0.2 | 0.4 |
| Sphingomyelin | 3707 ± 228 | 3423 ± 263 | 3794± 263 | 3474 ± 263 | 0.8 | 0.2 | 0.9 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jack-Roberts, C.; Joselit, Y.; Nanobashvili, K.; Bretter, R.; Malysheva, O.V.; Caudill, M.A.; Saxena, A.; Axen, K.; Gomaa, A.; Jiang, X. Choline Supplementation Normalizes Fetal Adiposity and Reduces Lipogenic Gene Expression in a Mouse Model of Maternal Obesity. Nutrients 2017, 9, 899. https://doi.org/10.3390/nu9080899
Jack-Roberts C, Joselit Y, Nanobashvili K, Bretter R, Malysheva OV, Caudill MA, Saxena A, Axen K, Gomaa A, Jiang X. Choline Supplementation Normalizes Fetal Adiposity and Reduces Lipogenic Gene Expression in a Mouse Model of Maternal Obesity. Nutrients. 2017; 9(8):899. https://doi.org/10.3390/nu9080899
Chicago/Turabian StyleJack-Roberts, Chauntelle, Yaelle Joselit, Khatia Nanobashvili, Rachel Bretter, Olga V. Malysheva, Marie A. Caudill, Anjana Saxena, Kathleen Axen, Ahmed Gomaa, and Xinyin Jiang. 2017. "Choline Supplementation Normalizes Fetal Adiposity and Reduces Lipogenic Gene Expression in a Mouse Model of Maternal Obesity" Nutrients 9, no. 8: 899. https://doi.org/10.3390/nu9080899