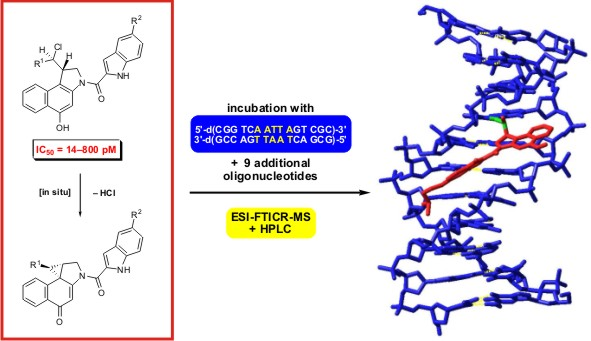
The
seco-drugs
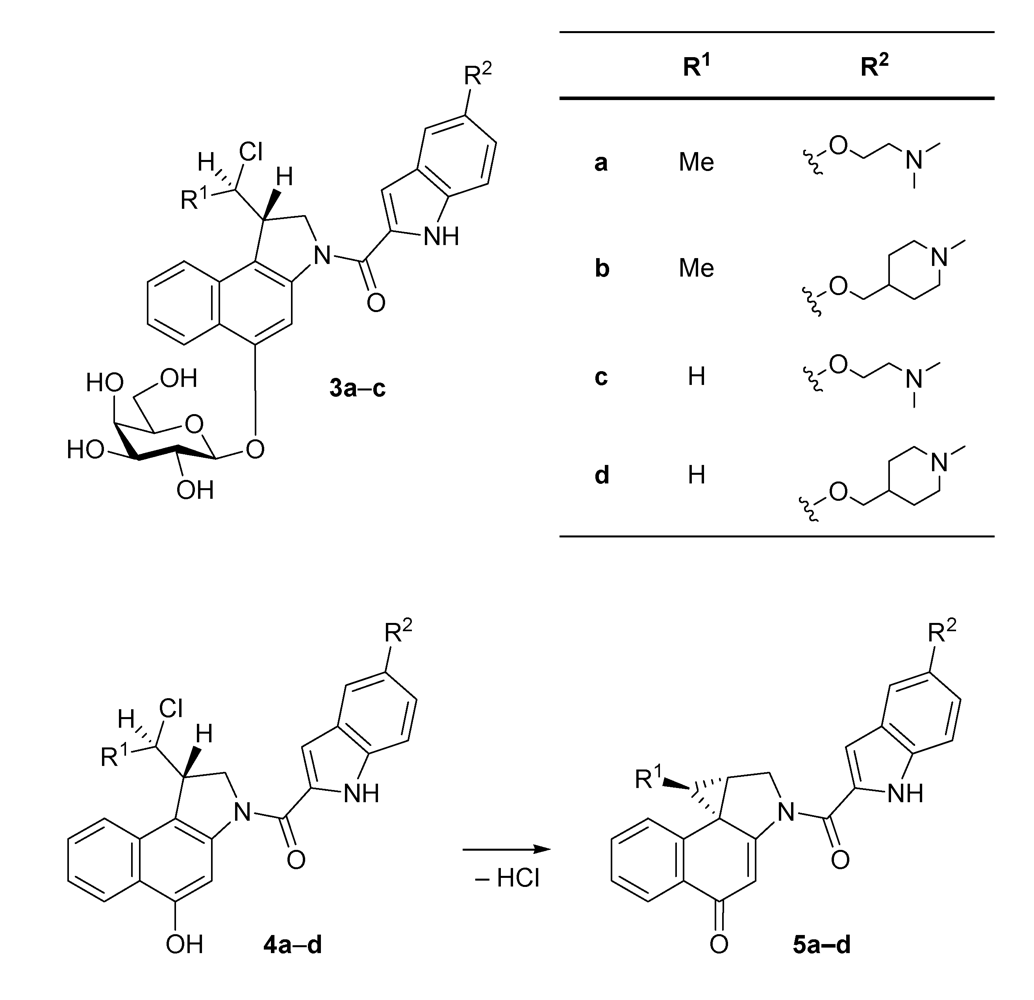
4c and
4d containing a hydrogen atom at the pharmacophoric unit (R
1 = H) were found to show a much higher cytotoxicity than their analogues
4a and
4b containing a methyl group (R
1 = Me) at the same position. This is presumably due to the fact that an attack of the cellular target, namely
N3 of the nucleobase adenine, at the electrophilic cyclopropane moiety of the corresponding drugs
5a-
d resulting in a subsequent formation of the corresponding DNA adducts, is less hindered in the absence of the sterically more demanding methyl group. Additionally, the lower hydrolytic stability of the drugs
5a and
5b bearing a methyl group decreases their concentration in cell culture media [
16,
17], which would lower the cytotoxicity of these drugs determined in cell culture assays. The different side chains (R
2) in
4a and
4b on the one hand and
4c and
4d on the other hand seem to have only a small influence on the drugs’ cytotoxicity since
4a and
4b as well as
4c and
4d, respectively, show cytotoxicities within the same order of magnitude. This is understandable because both side chains contain a tertiary amine and have approximately the same size. Denny
et al. as well as Boger
et al. have previously reported that substituents at C-5 of the DNA binding subunit, which are here represented by R
2, have a pronounced effect on the rate and efficiency of the DNA alkylation and the resulting biological potency of CC‑1065 analogues [
25,
26,
27,
28], but the exact type of the substituent does not seem to be as important as its length.
2.1. Investigations on the Reactivity and Sequence Selectivity of 4a-d against DNA Oligomers
The reactivity and sequence selectivity of
4a-
d against ten different synthetic double-stranded DNA oligomers (ds-
1-ds‑
10) and against the synthetic single-stranded DNA oligonucleotide
ON-
1 of ds‑
1 (
Figure 3) was investigated using ESI-FTICR MS in order to rationalise differences in the compounds’ cytotoxicities.
An alkylation of the DNA oligonucleotides
ON-
1 and
ON-
2 by the drugs
5a-
d results in the formation of covalent adducts denoted by
ON-
1*a-
d and
ON-
2*a-
d, respectively (
Figure 4). In the absence of suitable nucleophiles like DNA, the drugs are partially hydrolysed to give the inactive alcohols
6a-
d.
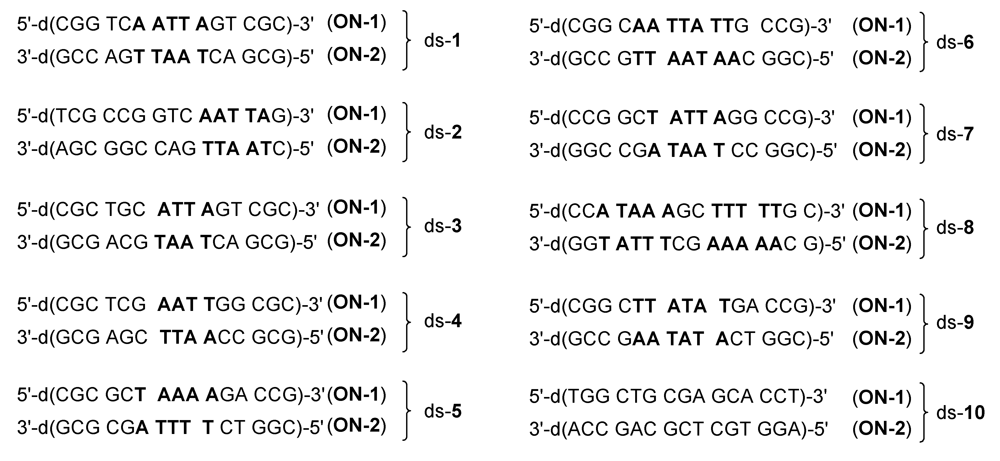
Figure 3.
Base sequences of the ten double-stranded DNA oligonucleotides used in the present study. Double-stranded DNA oligonucleotides are denoted with the praefix “ds-“. Oligonucleotides with lower molecular mass are denoted with the abbreviation “ON-1” and oligonucleotides with higher molecular mass with the abbreviation “ON-2”. A: Adenine, G: Guanine, C: Cytosine, T: Thymine.
Figure 3.
Base sequences of the ten double-stranded DNA oligonucleotides used in the present study. Double-stranded DNA oligonucleotides are denoted with the praefix “ds-“. Oligonucleotides with lower molecular mass are denoted with the abbreviation “ON-1” and oligonucleotides with higher molecular mass with the abbreviation “ON-2”. A: Adenine, G: Guanine, C: Cytosine, T: Thymine.
Figure 4.
Reaction of the drugs 5a-d with the DNA oligonucleotides ON-1 or ON-2 under formation of the alkylated oligonucleotides ON-1*a-d and ON-2*a-d, respectively, or hydrolysis of the drugs to give the alcohols 6a-d.
Figure 4.
Reaction of the drugs 5a-d with the DNA oligonucleotides ON-1 or ON-2 under formation of the alkylated oligonucleotides ON-1*a-d and ON-2*a-d, respectively, or hydrolysis of the drugs to give the alcohols 6a-d.
For the mass spectrometric investigations, the hydrochlorides of
seco-drugs
4a-
d were incubated with the DNA oligonucleotides in water for 24 h at 25 °C. The samples were then diluted with equivalent amounts of methanol and investigated directly by means of ESI-FTICR mass spectrometry without preliminary purification or enrichment of the products as described previously [
16,
18,
19]. Covalent adducts were identified by comparison of calculated and found masses and alkylation positions were determined by identification of the characteristic fragment ions formed from the alkylated oligonucleotides applying capillary skimmer dissociation (CSD) [
16,
18,
19,
29,
30,
31,
32].
If only one of the two oligonucleotides of the double-stranded DNA was alkylated, the percentage of alkylation of ON-1 or ON-2 was calculated based on the relative peak intensities of the corresponding isotope peaks of the unreacted oligonucleotides of ON-1 and ON-2 after 24 h of incubation as compared to the same ratio after 0 h of incubation. In case both oligonucleotides were alkylated, the preferred alkylation of one of the two oligonucleotides was determined based on the relative peak intensities of the covalent adducts. Analogously, if an oligonucleotide was alkylated in more than one position, the preferred binding site was determined based on the relative peak intensities of the respective characteristic fragment ions. The percentage of alkylation of the single-stranded oligonucleotide ON-1 of ds-1 was determined by calculating the ratio of the peak intensity of the covalent adduct as compared to the unmodified oligonucleotide.
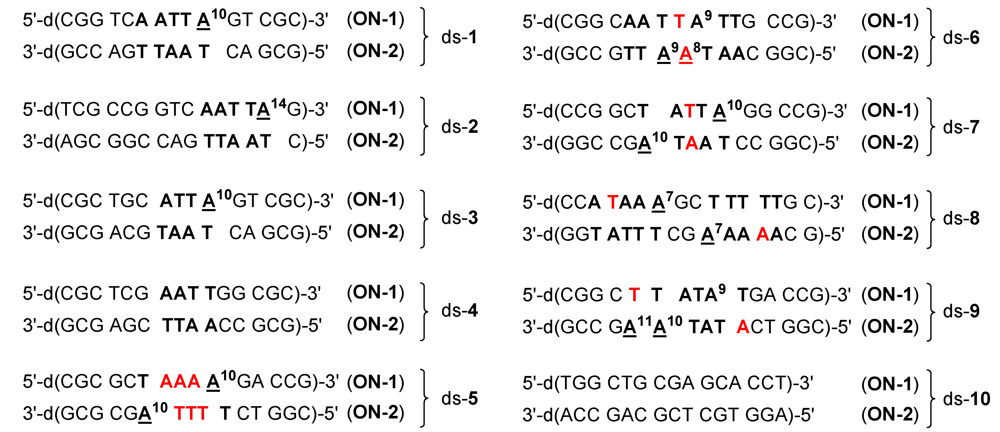
Figure 5.
Base sequences of the ten double-stranded DNA oligonucleotides used in the present study with alkylation positions marked by underlines. Potential alkylation positions in the oligonucleotides are denoted with a superscript and differing nucleobases in the 5'‑direction of potential competing binding sites are marked in red.
Figure 5.
Base sequences of the ten double-stranded DNA oligonucleotides used in the present study with alkylation positions marked by underlines. Potential alkylation positions in the oligonucleotides are denoted with a superscript and differing nucleobases in the 5'‑direction of potential competing binding sites are marked in red.
Figure 5 and
Table 2 show the results of the mass spectrometric investigations (as described above) in terms of the main alkylation site and the percentage of alkylation. In
Figure 5, alkylation positions are denoted by underlines and important differences in the base sequence in the 5'‑direction of potential competing binding sites of
ON-
1 as compared to
ON-
2 are marked in red. Notably, all drugs alkylated mainly the nucleobase adenine (A) in AT-rich DNA regions of at least four consecutive AT base pairs with the alkylated adenine situated at the 3'-end of this sequence. This sequence selectivity could also be observed for the natural products CC‑1065 and duocarmycin SA [
5] and the alkylation of the oligonucleotide
ON-
1 of ds-
4 by the drug
5d is the only exception hereof. Furthermore, all drugs prefer the nucleobase adenine (A) over thymine (T) in the first, second and third position in the 5'-direction of the binding site as is obvious from the preferred binding positions in the oligonucleotides ds-
6, ds-
7 and ds-
8, respectively (
Figure 5 and
Table 3). Hence, the drugs preferably alkylate the oligonucleotides
ON-
2 of these double-stranded oligonucleotides instead of the oligonucleotides
ON-
1.
Table 2.
Alkylation efficiency and alkylation position after incubation of the DNA oligonucleotides ds‑1-ds‑10 and ON-1 of ds-1 with the hydrochlorides of 4a-d in a 1:1 ratio for 24 h at 25 °C.
Table 2.
Alkylation efficiency and alkylation position after incubation of the DNA oligonucleotides ds‑1-ds‑10 and ON-1 of ds-1 with the hydrochlorides of 4a-d in a 1:1 ratio for 24 h at 25 °C.
| | Alkylation [%] | Alkylation position |
|---|
| DNA | 4a | 4b | 4c | 4d | 4a | 4b | 4c | 4d |
| ds-1 | 75 | 53 | 46 | 28 | A10 (ON-1) |
| ds-2 | 60 | 66 | 9 | 31 | A14 (ON-1) |
| ds-3 | 55 | 67 | 29 | 32 | A10 (ON-1) |
| ds-4 | - a | - a | - a | 32 | - b | ON-1 a |
| ds-5 | ON-1 and ON-2 c | 6 | A10 (ON-1) ≥ A10 (ON-2) | A10 (ON-1) |
| ds-6 | 30 | 20 | 17 | 13 | A8 (ON-2) ≥ A9 (ON-2) |
| ds-7 | ON-1 and ON-2 c | A10 (ON-2) ≥ A10 (ON-1) |
| ds-8 | ON-1 and ON-2 c | A7 (ON-2) ≥ A7 (ON-1) |
| ds-9 | 77 | 82 | 21 | 30 | A10 (ON-2) ≥ A11 (ON-2) |
| ds-10 | - a | - a | - a | - a | - b |
| ON-1 of ds-1 | 20 | 25 | - a | - a | - b |
In addition, all drugs show a preference for purine bases (A or G) over pyrimidine bases (C or T) in the first position in the 3'-direction of the binding site. This results for example in the alkylation of A
8 of
ON-
2 in ds‑
6 additionally to A
9 of the same DNA oligonucleotide (
Figure 5,
Table 3).
Table 3.
Prefered nucleobases in the vicinity of the alkylation position (0) as observed after incubation of the hydrochlorides of seco-drugs 4a-d with the DNA oligonucleotides ds‑1-ds‑10.
Table 3.
Prefered nucleobases in the vicinity of the alkylation position (0) as observed after incubation of the hydrochlorides of seco-drugs 4a-d with the DNA oligonucleotides ds‑1-ds‑10.
| | 5' | 4 | 3 | 2 | 1 | 0 | -1 | 3' |
|---|
| 4a, 4c, 4b, 4d | | A/T > G/C, G/C > A/T | A > T | A > T | A > T | A | A/G > C/T | |
Interestingly, the
seco-drugs
4a and
4c containing a right hand dimethylamino-ethoxyindole subunit prefer AT base pairs in the fourth position in the 5'-direction of the binding site with the AT-rich sequence being located in the middle of the double strand. This is obvious due to the lower alkylation efficiencies of the double-strands ds-
2 and ds-
3 as compared to ds‑
1. In contrast, the
seco-drugs
4b and
4d containing a right hand morpholinoindole subunit prefer CG base pairs in the fourth position in the 5'-direction of the binding site and AT-rich sequences located at the end of the double strand. This can be seen by the higher alkylation efficiencies of the double-strands ds-
6 and ds-
7 in comparison to ds-
1. Furthermore, all drugs showed a much higher reactivity against the double-stranded DNA oligonucleotide ds‑
1 than against the respective single-stranded oligonucleotide
ON-
1 of ds-
1 (
Table 2).
In summary, all drugs show a high selectivity for adenines in AT-rich DNA regions and differ only slightly in the preferred base sequence. Thus, differences in cytotoxicity are presumably not due to differences in sequence selectivity. Unexpectedly, also the alkylation efficiency regarding the same DNA oligonucleotide does not seem to correlate with the cytotoxicity of the drugs because, with the exception of ds-
4,
4a and
4b alkylated the DNA with a significantly higher efficiency than the
seco-drugs
4c and
4d even though they show a lower cytotoxicity as observed in the cell culture assays than the latter (
Table 2).
2.2. Investigations on the Reaction Kinetics of the Seco-Drugs
Consequently other factors seem to influence the biological activity of these alkylating agents and it could be argued that differences in the rate of the cyclisation reaction of the
seco-drugs
4a-
d to give the drugs
5a-
d or maybe differences in the rate of formation of the DNA adducts might play a crucial role. Therefore, the hydrochlorides of
seco-drugs
4a-
d were incubated with the double-stranded DNA oligonucleotides in buffer or water for up to 24 h at 25 °C and the kinetics of the reactions were investigated using HPLC. Since covalent and non-covalent adducts of the DNA and the drugs elute with the same retention time under the conditions of these measurements [
16], mixtures of both covalent and non-covalent adducts are denoted ds‑
x(*)a-
d in the following whereas purely covalently bound adducts as detected by means of mass spectrometry are denoted ds‑
x*a-
d. For evaluation, the area under the curve (AuC) in the HPLC chromatograms of the respective
seco-drugs and their derivatives was determined based on their absorption of light at
λ = 350 nm because at this wavelength the absorption of unmodified DNA can be neglected. It should be noted that the results obtained are only semi-quantitative, since we did not correct the calculated concentrations according to the molar extinction coefficients of the
seco-drugs and their derivatives; however, it can be assumed that the extinction coefficients are approximately the same. Indeed, the results allow a good and straightforward comparison of the reactivity of the
seco-drugs
4a-
d and their drugs
5a-
d, respectively.
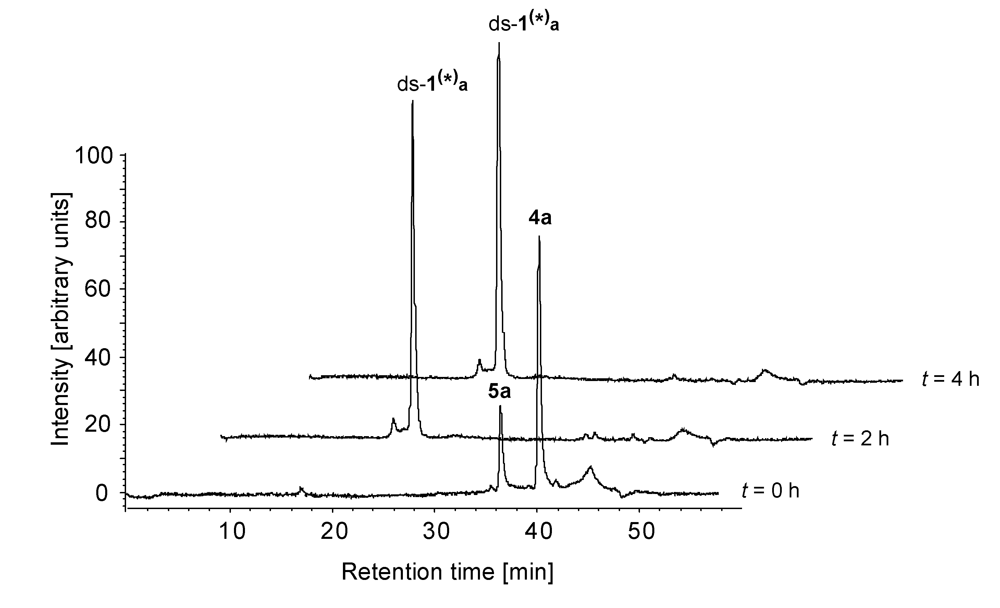
Figure 6 shows a series of HPLC chromatograms obtained at 0-4 h after starting the incubation of the hydrochloride of
seco-drug
4a with ds-
1 in phosphate buffer (pH 7). As can clearly be seen, the
seco-drug cyclises to give the corresponding drug
5a very rapidly and the formation of the respective DNA-adduct ds-
1(*)a has finished already after 2 h.
Table 4 shows the resulting AuCs determined after up to 6 hours of incubation of a 1:1 mixture of the hydrochlorides of the
seco-drugs
4a-
d with the DNA in phosphate buffer (pH 7).
The seco-drugs 4a and 4b cyclise rapidly to give the corresponding drugs 5a and 5b which subsequently form the respective DNA adducts ds-1(*)a and ds-1(*)b within two hours. Hence, no free seco-drugs or drugs can be observed after two hours of incubation. In contrast, the more cytotoxic seco-drugs 4c and 4d cyclise to the corresponding drugs 5c and 5d with a much lower reaction rate and, moreover, the DNA adduct formation proceeds quite slowly. Thus, after six hours of incubation, DNA adduct formation is not completed and therefore, seco-drugs 4c and 4d can still be detected in considerable amounts.
Figure 6.
HPLC chromatograms (λ = 350 nm)obtained 0-4 h after starting the incubation of the hydrochloride of seco-drug 4a with ds-1 in phosphate buffer (pH 7).
Figure 6.
HPLC chromatograms (λ = 350 nm)obtained 0-4 h after starting the incubation of the hydrochloride of seco-drug 4a with ds-1 in phosphate buffer (pH 7).
Table 4.
AuC after indicated times of incubation of the DNA oligonucleotide ds-1 with the the hydrochlorides of seco-drugs 4a-d in phosphate buffer (pH 7).
Table 4.
AuC after indicated times of incubation of the DNA oligonucleotide ds-1 with the the hydrochlorides of seco-drugs 4a-d in phosphate buffer (pH 7).
| | AuC [%] after the indicated time of incubation |
|---|
| Reaction mixture | Species | 0 h | 2 h | 4 h | 6 h |
| ds-1/4a | 4a | 78 | - | - | - |
| 5a | 22 | - | - | - |
| ds-1(*)a | - | 100 | 100 | 100 |
| ds-1/4b | 4b | 89 | - | - | - |
| 5b | 11 | - | - | - |
| ds-1(*)b | - | 100 | 100 | 100 |
| ds-1/4c | 4c | 74 | 45 | 19 | 12 |
| 5c | 26 | 20 | 14 | - |
| ds-1(*)c | - | 35 | 67 | 88 |
| ds-1/4d | 4d | 76 | 51 | 34 | 19 |
| 5d | 24 | 20 | 17 | 19 |
| ds-1(*)d | - | 29 | 49 | 62 |
Since
seco-drugs such as
4a-
d cyclise to the corresponding drugs nearly quantitatively in less than 90 minutes in buffer without DNA [
16,
17],
4c and
4d obviously are stabilised by an interaction with the DNA. This interaction is relatively weak because it can be disrupted under conditions of chromatography to give back the free
seco-drugs
4c and
4d as well as unchanged DNA. In addition, the formation of stable non-covalent and covalent complexes of the respective drugs
5c and
5d with the DNA oligonucleotides seems to be disfavoured because the amounts of ds-
1(*)c ds-
1(*)d increase only slowly and after prolonged incubation times, free drugs
5c (up to 4 h) and
5d (up to 6 h) can still be detected. Interestingly, the products of hydrolysis
6a-
d are not observed, indicating that the rate of nucleophilic attack by water is reduced in the presence of DNA.
Further, we investigated whether there might be differences in
seco-drug stabilisation or DNA adduct formation correlating with the base sequence. Using again HPLC, the interaction of the four
seco-drugs
4a-
d with the DNA oligonucleotides ds-
1-ds-
10 was analysed.
Table 5 and
Table 6 show the respective results obtained after 24 hours of incubation of a 1:1 mixture of DNA with the hydrochlorides of
4a-
d in water at 25 °C.
Table 5.
AuC after 24 h of incubation of the DNA oligonucleotides ds-1-ds-10 with the hydrochlorides of seco-drugs 4a and 4b in water (pH 7).
Table 5.
AuC after 24 h of incubation of the DNA oligonucleotides ds-1-ds-10 with the hydrochlorides of seco-drugs 4a and 4b in water (pH 7).
| | ds-x/4a | ds-x/4b |
|---|
| ds-x | 4a [%] | 5a [%] | ds-x(*)a [%] | 4b [%] | 5b [%] | ds-x(*)b[%] |
| ds-1 | - | - | 100 | 2 | 1 | 97 |
| ds-2 | - | 2 | 98 | 9 | 2 | 89 |
| ds-3 | - | 1 | 99 | - | 4 | 96 |
| ds-4 | - | 64 | 36 | - | 62 | 38 |
| ds-5 | - | 1 | 99 | 3 | 1 | 96 |
| ds-6 | - | 27 | 73 | - | 40 | 60 |
| ds-7 | - | 3 | 97 | - | 3 | 97 |
| ds-8 | - | - | 100 | 2 | 1 | 97 |
| ds-9 | - | 1 | 99 | - | 1 | 99 |
| ds-10 | - | 63 | 37 | - | 40 | 60 |
After 24 hours of incubation, only traces (≤9%) of the
seco-drugs
4a and
4b were detected. Furthermore, with the exception of ds-
4,ds-
6 and ds-
10, nearly all detectable amounts of the drugs
5a and
5b which had been formed during the time of incubation, were converted to the corresponding DNA adducts ds‑
x(*)a and ds‑
x(*)b, respectively. Additionally, even in those cases where only small amounts of DNA adduct were formed (ds-
4,ds-
6 undds-
10), no hydrolysis of the drugs
5a and
5b to the hydroxylated derivatives
6a and
6b (
Figure 4) occurred in contrast to a notable hydrolysis of these drugs in the absence of DNA [
16]. This indicates a weak interaction of the drugs with the DNA oligonucleotides that stabilises the drugs against hydrolysis on the one hand, but disfavours the alkylation reaction on the other hand. Consistent with this observation, the formation of covalent adducts as determined by the mass spectrometric investigations is low regarding ds-
4,ds-
6 and ds-
10.
Table 6.
AuC after 24 h of incubation of the DNA oligonucleotides ds-1-ds-10 with the seco-drugs 4c and 4d in water (pH 7).
Table 6.
AuC after 24 h of incubation of the DNA oligonucleotides ds-1-ds-10 with the seco-drugs 4c and 4d in water (pH 7).
| | ds-x/4c | ds-x/4d |
|---|
| ds-x | 4c [%] | 5c [%] | ds-x(*)c [%] | DNA fragments [%] | 4d [%] | 5d [%] | ds-x(*)d [%] | DNA fragments [%] |
|---|
| ds-1 | 1 | - | 99 | - | 45 | 3 | 52 | - |
| ds-2 | 13 | - | 87 | - | 17 | 2 | 81 | - |
| ds-3 | 8 | - | 92 | - | 19 | 3 | 78 | - |
| ds-4 | 15 | 68 | 17 | - | 19 | 68 | 13 | - |
| ds-5 | 59 | - | 41 | - | 63 | 3 | 34 | - |
| ds-6 | 4 | 13 | 53 | 30 | 27 | 8 | 48 | 17 |
| ds-7 | 3 | 11 | 73 | 13 | 7 | 2 | 84 | 7 |
| ds-8 | 5 | - | 95 | - | 30 | 3 | 67 | - |
| ds-9 | 2 | - | 83 | 15 | 21 | 3 | 70 | 6 |
| ds-10 | 29 | 53 | 18 | - | 33 | 41 | 26 | - |
Notably, the effect of stabilisation of the
seco-drugs and drugs by the DNA is much more pronounced in the case of
4c and
4d as well as
5c and
5d, in comparison to that of
4a and
4b as well as
5a and
5b. Though in the absence of DNA all
seco-drugs
4a-
d cyclise quantitatively within 90 min in buffer or cell culture media [
16,
17], in the presence of DNA, the
seco-drugs
4c and
4d cyclise to give the corresponding drugs
5c and
5d to a much lower extent than their methylated analogues
4a and
4b. Since the formation of the drugs is the prerequisite for the alkylation reaction, this might explain why the DNA alkylation is lower in case of
4c and
4d as compared to
4a and
4b. Interestingly, the extent of drug formation depends on the kind of double-stranded DNA oligonucleotide used, indicating a specific interaction of the
seco-drugs with the DNA. The most pronounced stabilisation of the
seco-drugs can be observed using ds-
5, in the presence of which only about 40% of
4c and
4d, respectively, are converted to the corresponding drugs in 24 hours. Nevertheless, with the exception of ds-
4,ds-
6, ds-
10 and partially also ds‑
7, most of
5c and
5d reacted with the DNA oligonucleotides under formation of the non-covalent and covalent DNA adducts
ds-
x(*)c and
ds-
x(*)d as was already found for
5a and
5b. However, additional DNA fragments indicative of strand cleavage could be observed in case of ds-
6,ds-
7 and ds-
9 after incubation with
4c and
4d in contrast to the respective incubations with
4a and
4b.
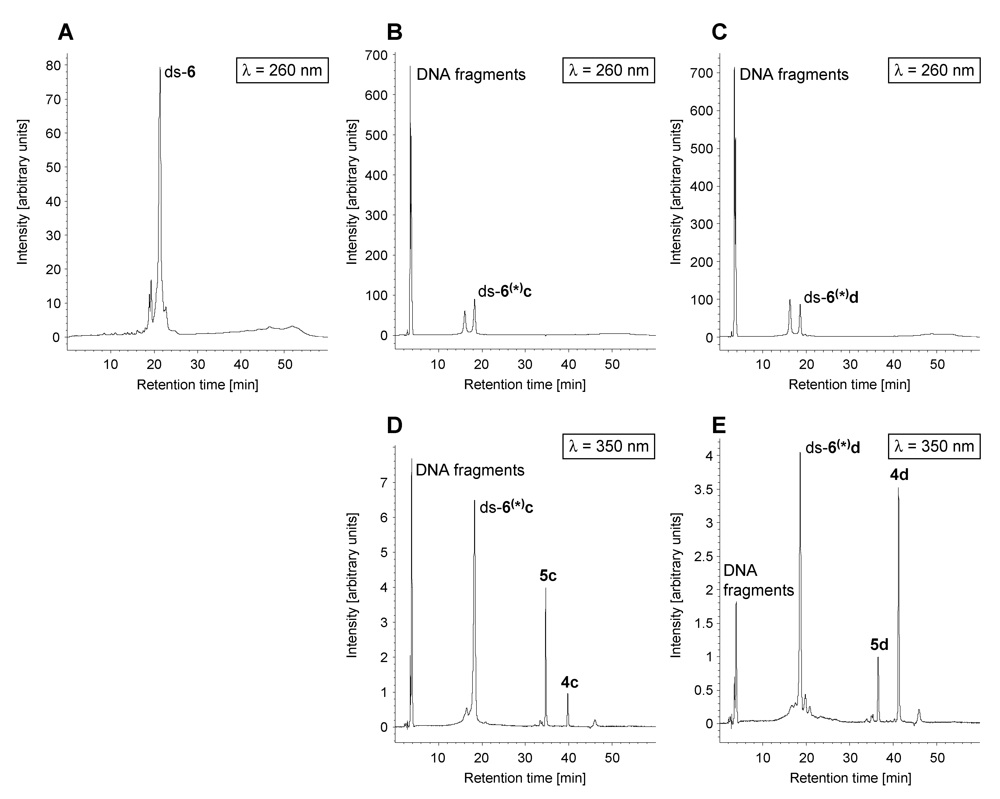
Figure 7 displays HPLC chromatograms showing the DNA fragmentation exemplarily for the reaction of
seco-drugs
4c and
4d with the oligonucleotide ds-
6. DNA was detected using a wavelength of
λ = 260 nm and the
seco-drugs
4c and
4d as well as all their derivatives including DNA adducts were detected using a wavelength of
λ = 350 nm. As can clearly be seen, the drugs
5c and
5d induce cleavage of the intact DNA to form DNA fragments a part of which is still covalently bound to the drug.
Figure 7.
HPLC chromatograms (λ = 260 nm and λ = 350 nm) of ds-6 in water (Α), a solution of DNA oligonucleotide ds-6 incubated for 24 h with 4c·HCl in water (pH 7; B and D) and a solution of DNA oligonucleotide ds-6 incubated for 24 h with 4d·HCl in water (pH 7; C and E).
Figure 7.
HPLC chromatograms (λ = 260 nm and λ = 350 nm) of ds-6 in water (Α), a solution of DNA oligonucleotide ds-6 incubated for 24 h with 4c·HCl in water (pH 7; B and D) and a solution of DNA oligonucleotide ds-6 incubated for 24 h with 4d·HCl in water (pH 7; C and E).
The high cytotoxicity of
4c and
4d might thus also be modulated by DNA strand cleavage. DNA lesions such as single- and double-strand breaks have been reported previously to be caused by analogues of CC-1065 and the duocarmycins such as adozelesin and bizelesin [
33]. These damages can lead to cell death in case they cannot be repaired by the cellular repair machinery [
34].




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




