Peripheral 5-HT3 Receptors Are Involved in the Antinociceptive Effect of Bunodosine 391


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
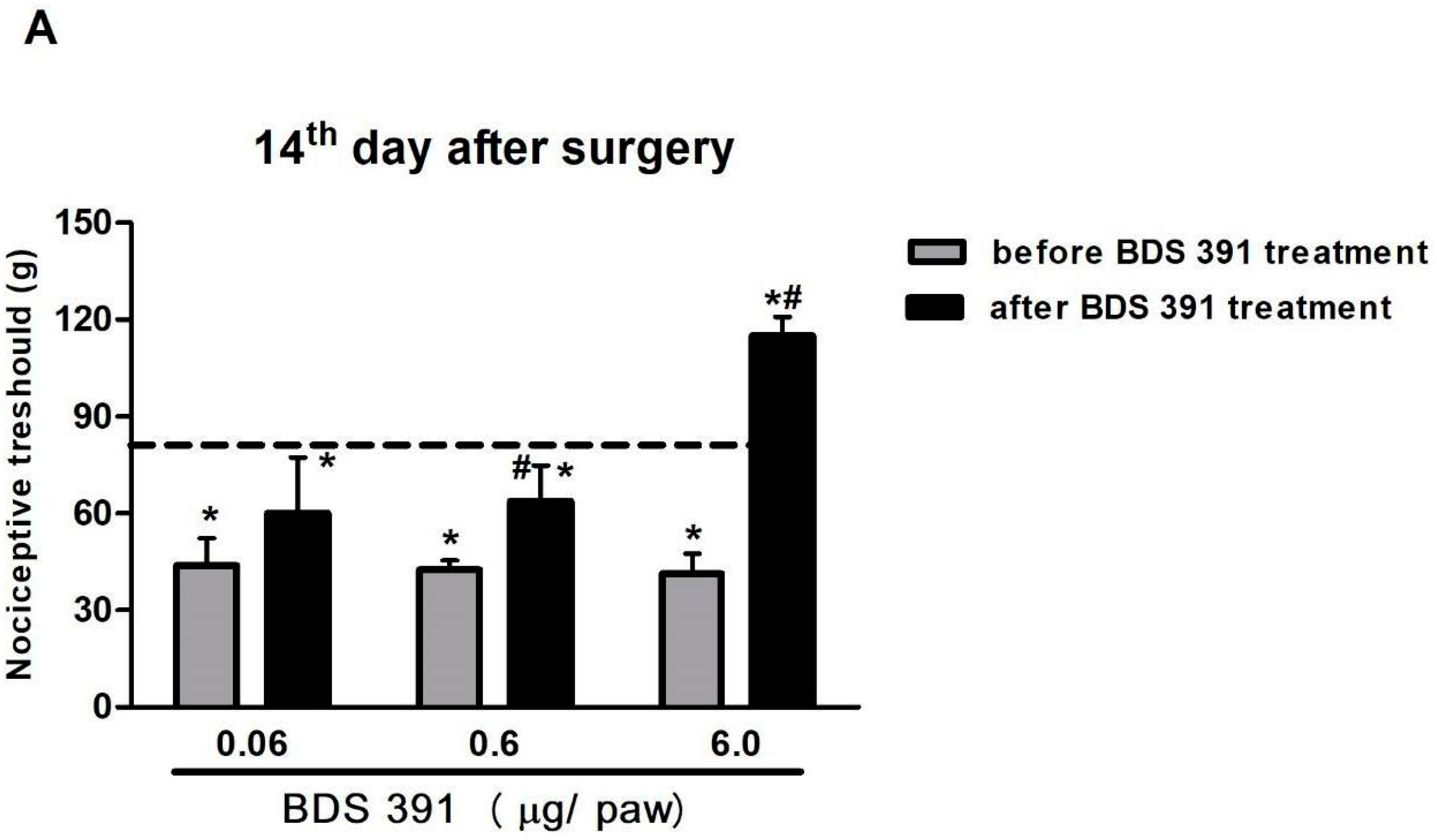
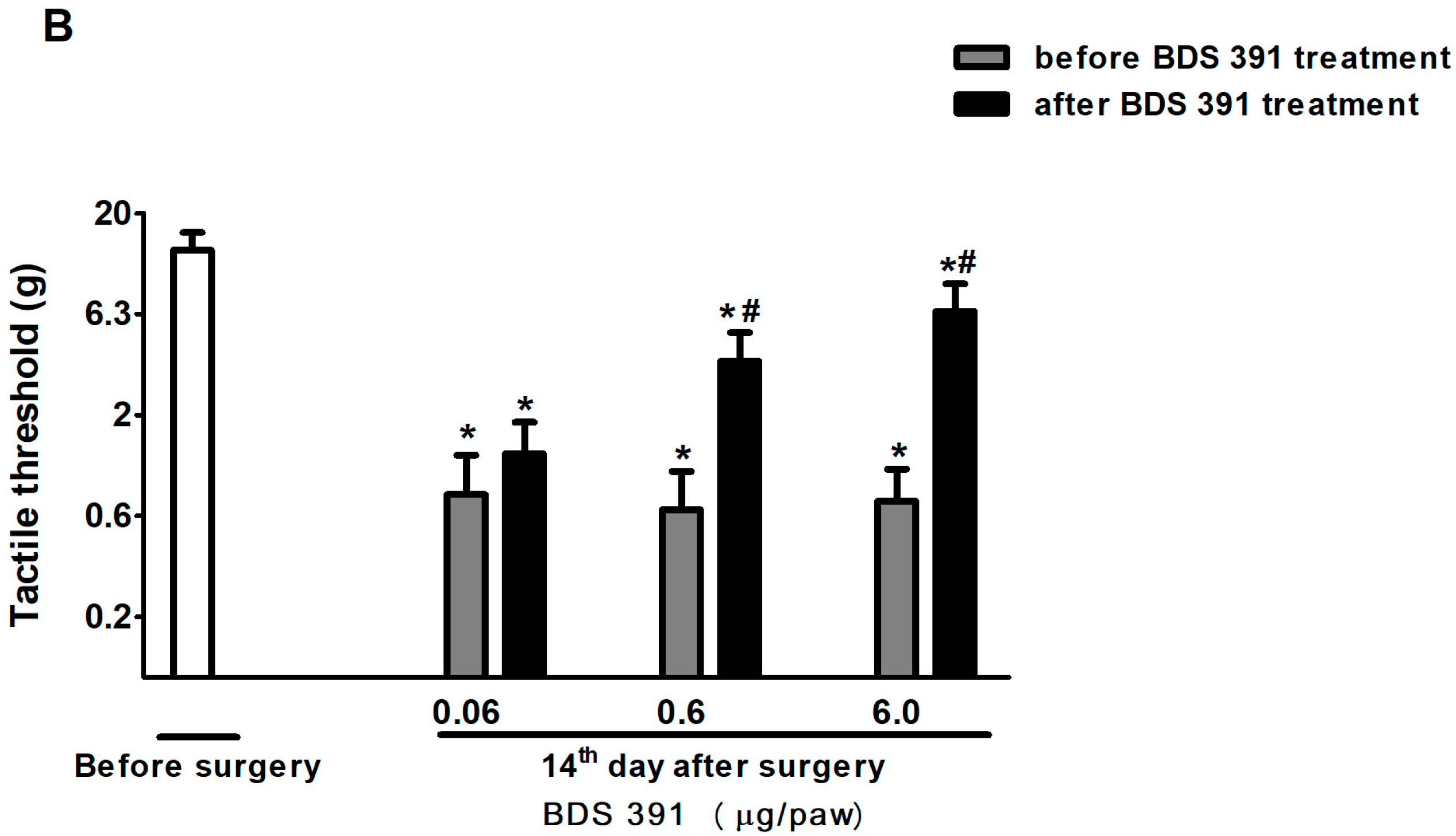
2.1. Characterization of the Antinociceptive Effect of BDS 391
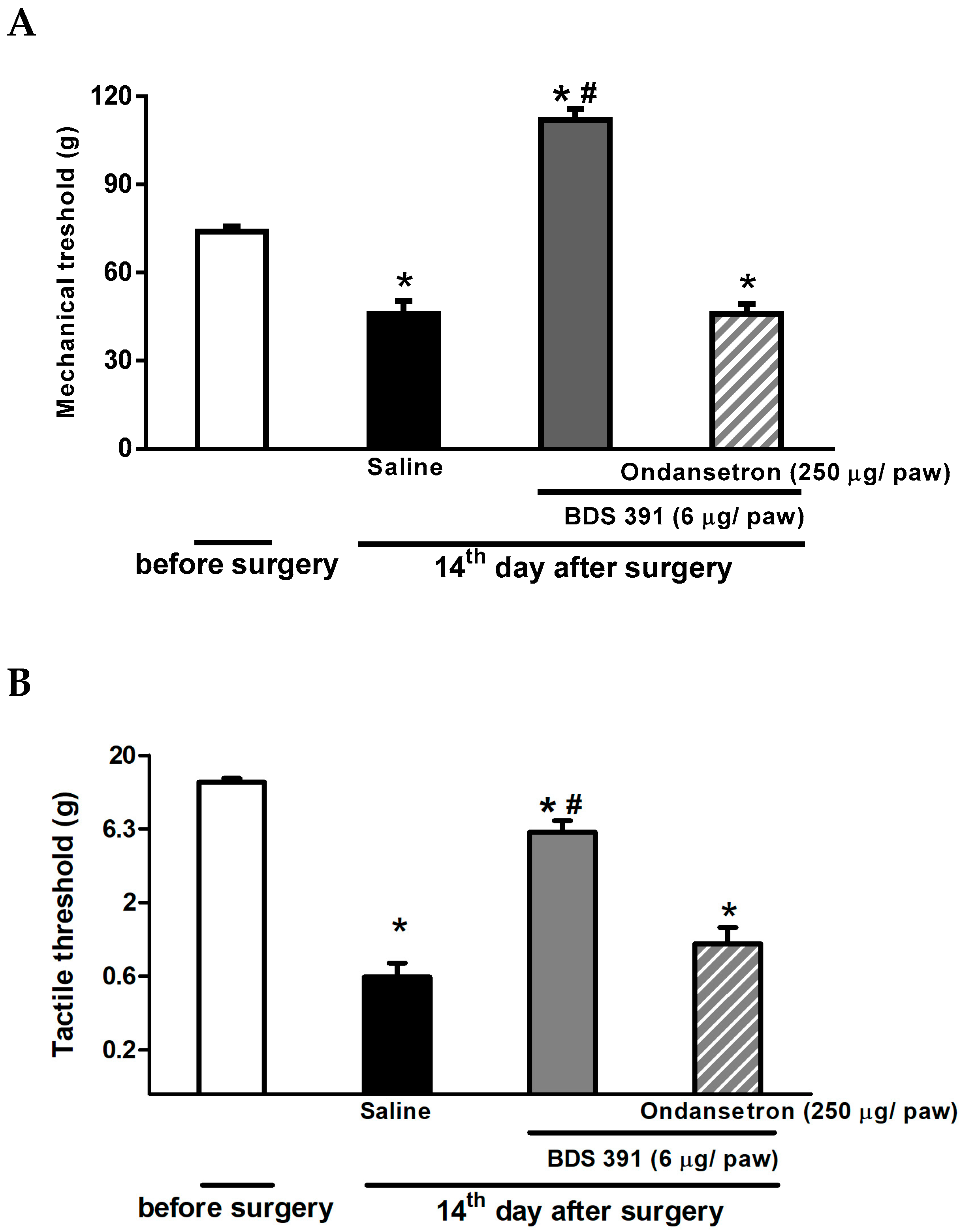
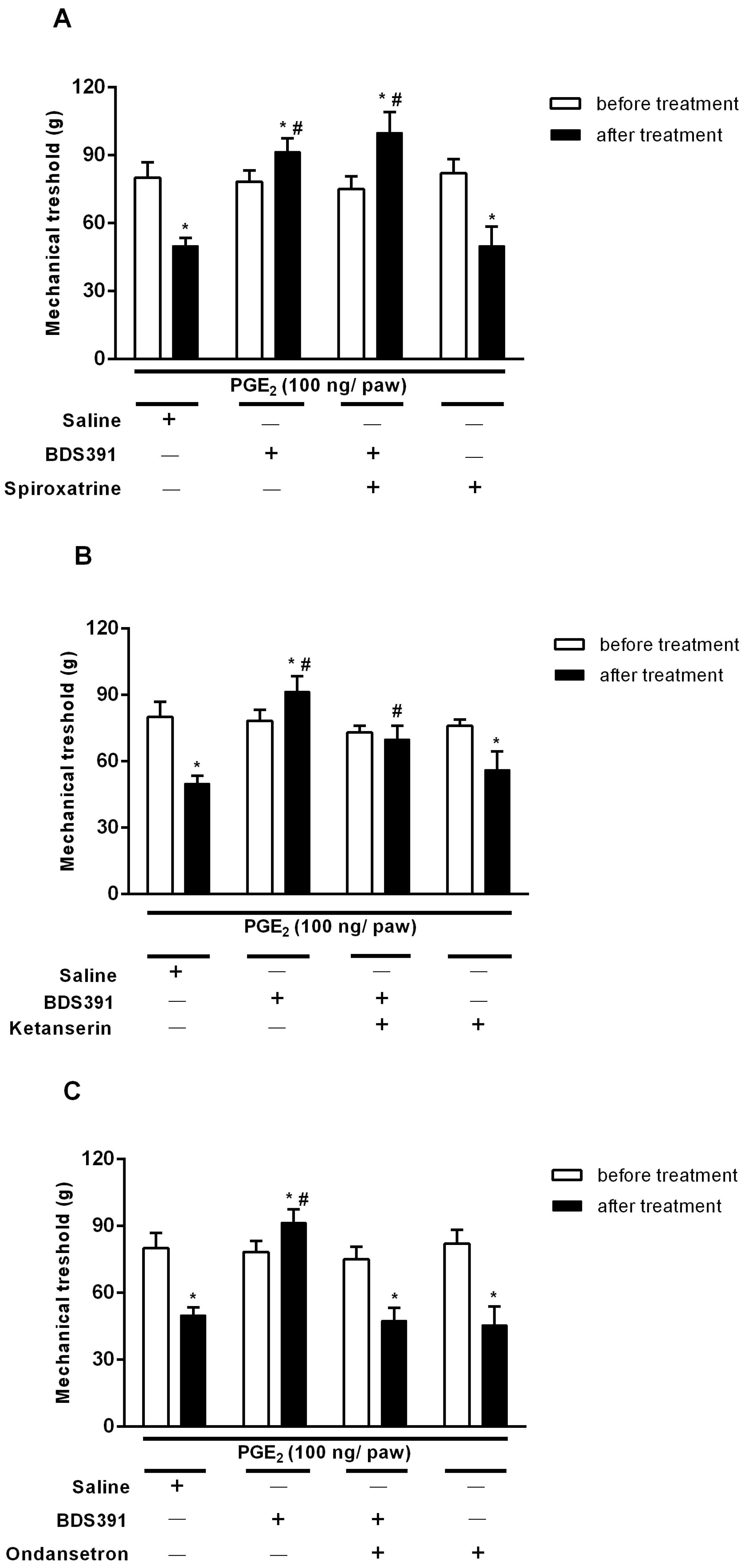
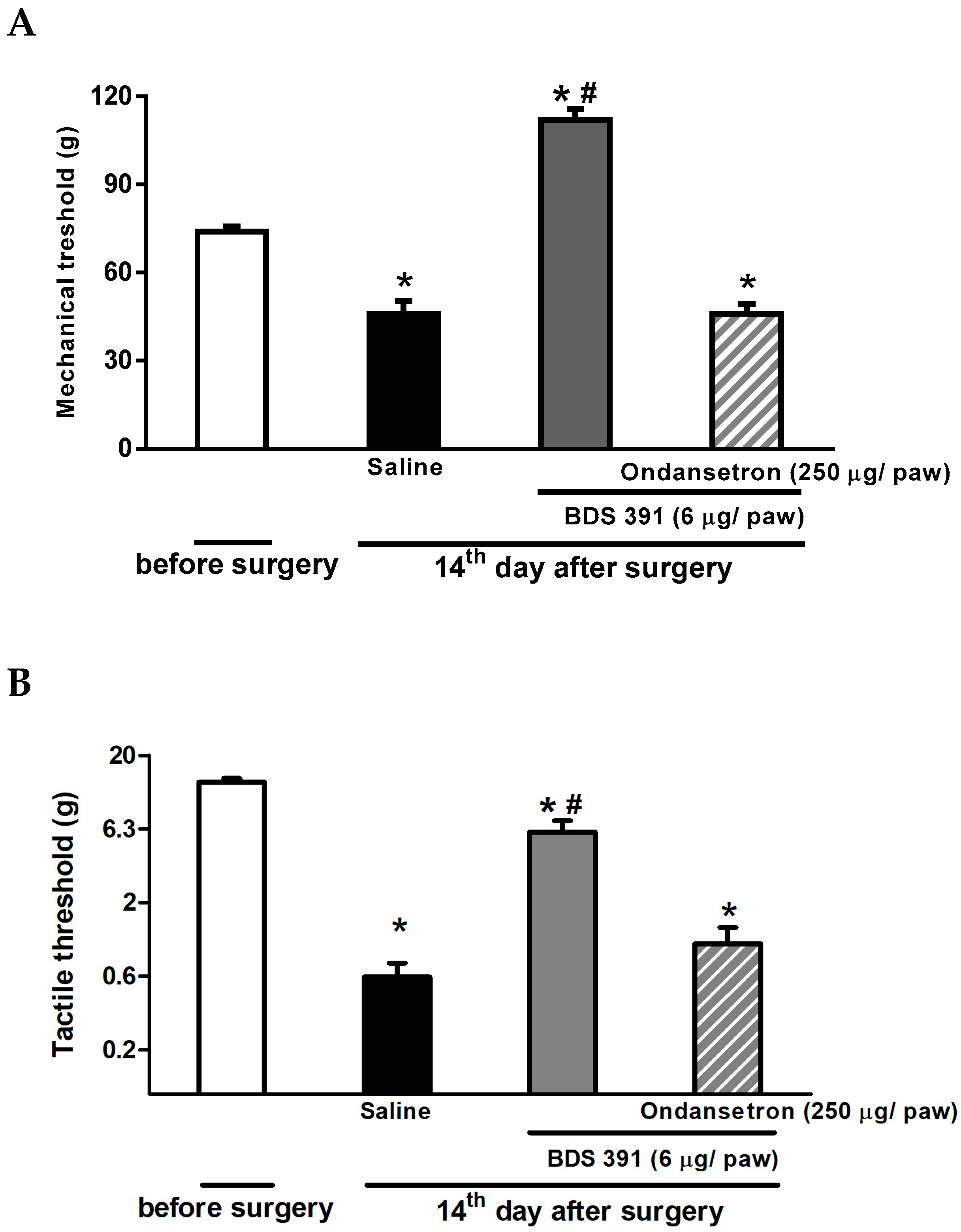
2.2. Serotonin Receptors Are Involved in the Antinociceptive Effect of BDS 391
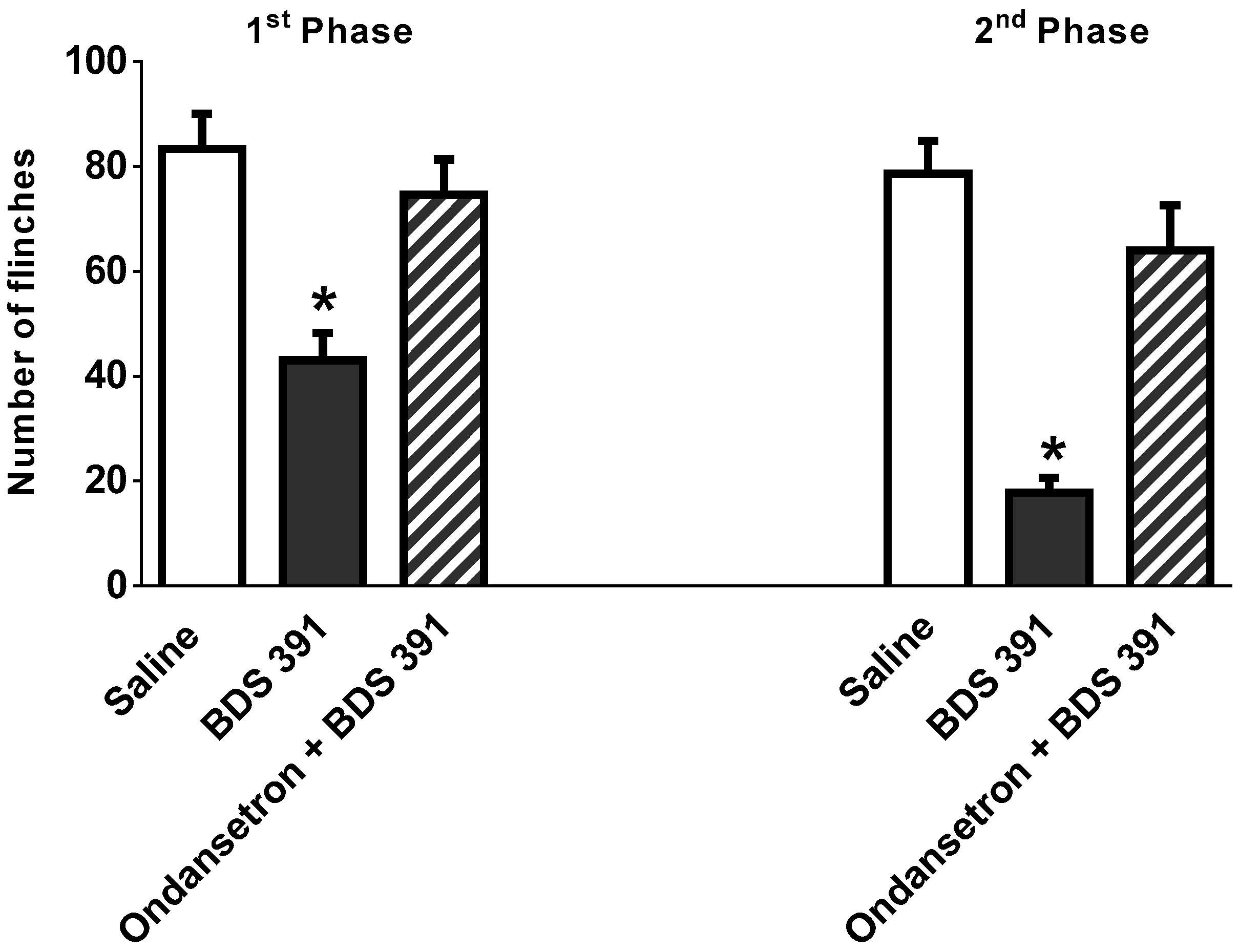
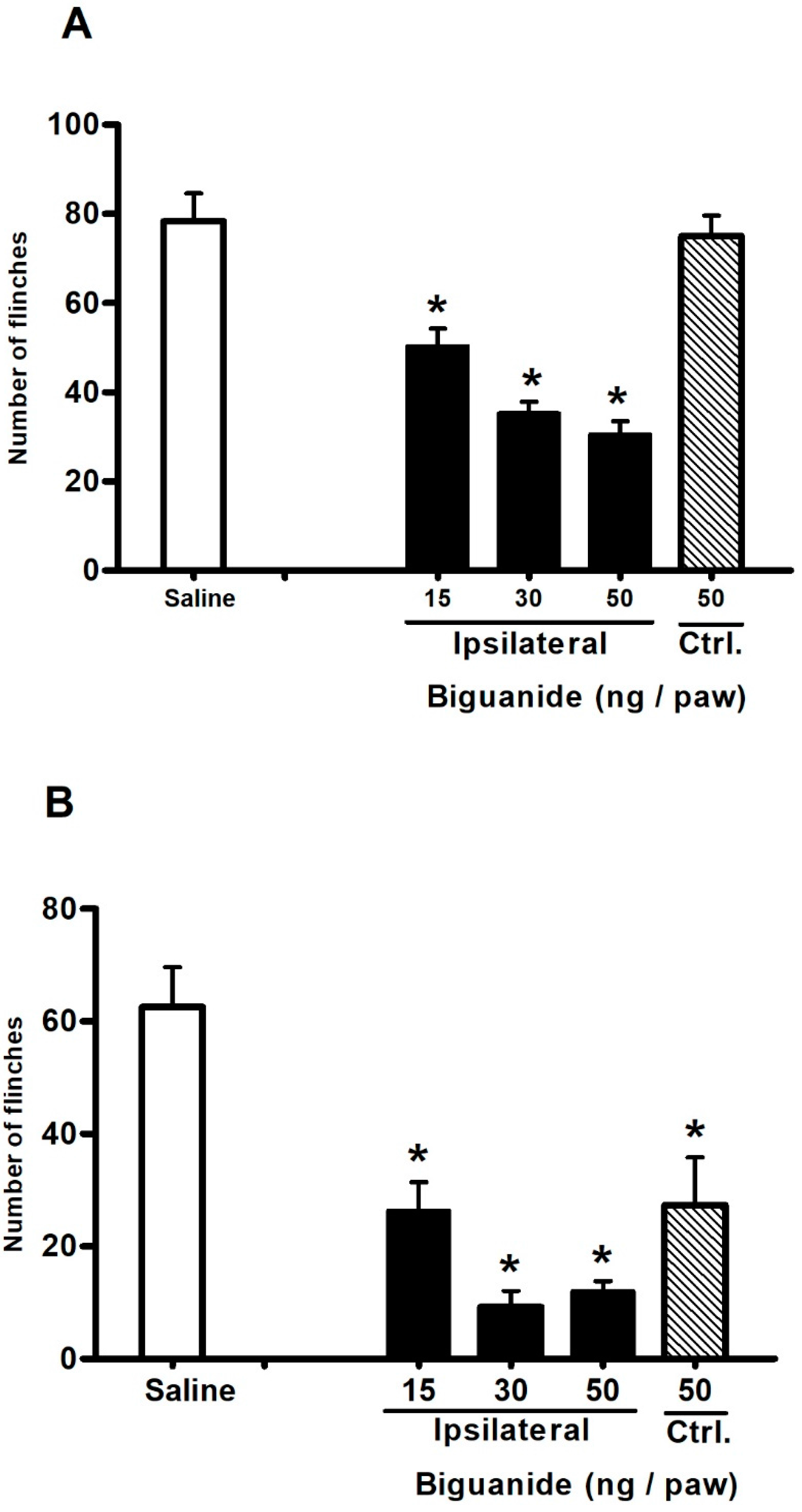
2.3. Antinociceptive Effect of Biguanide, a 5-HT3 Receptor Agonist
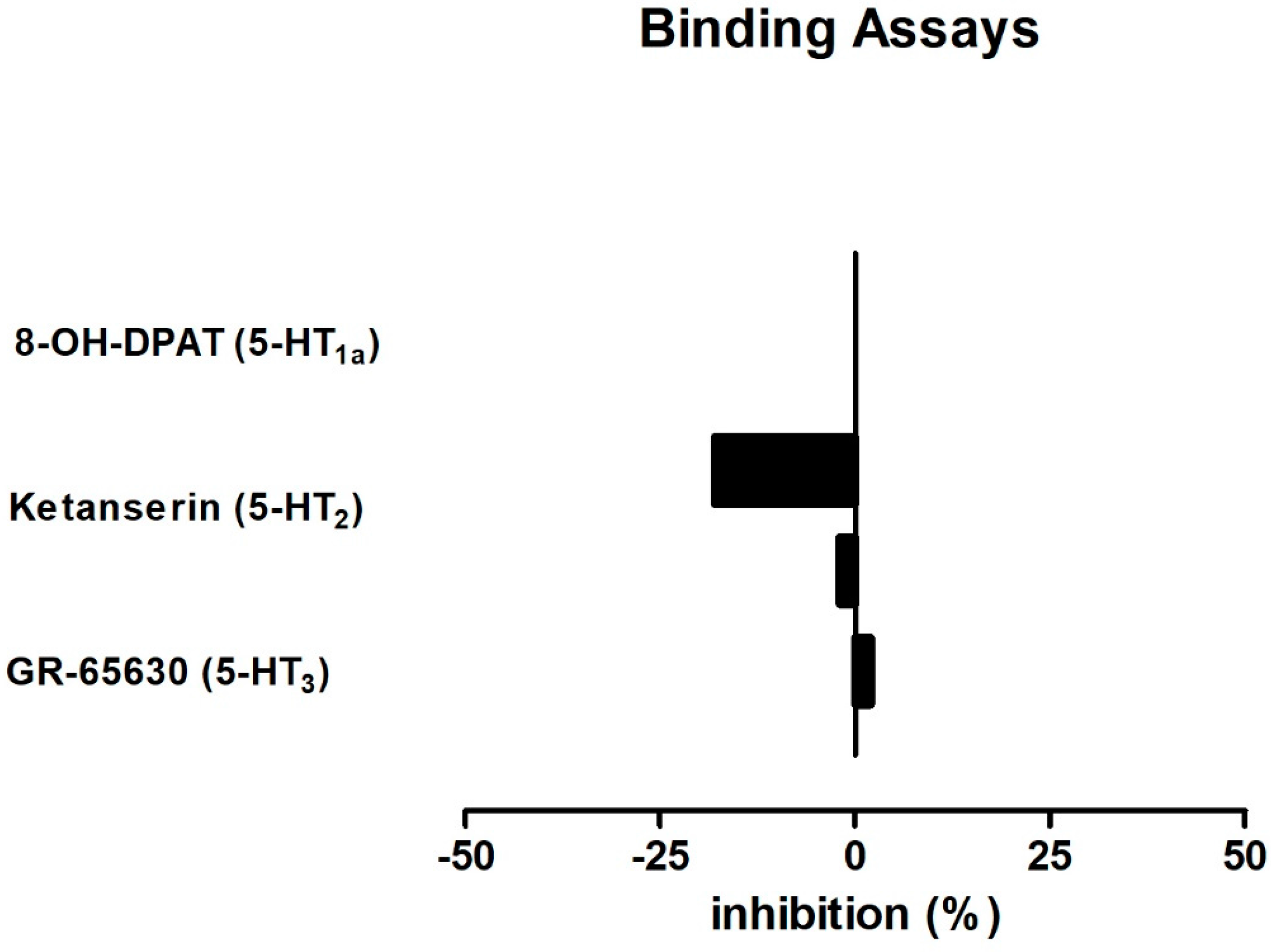
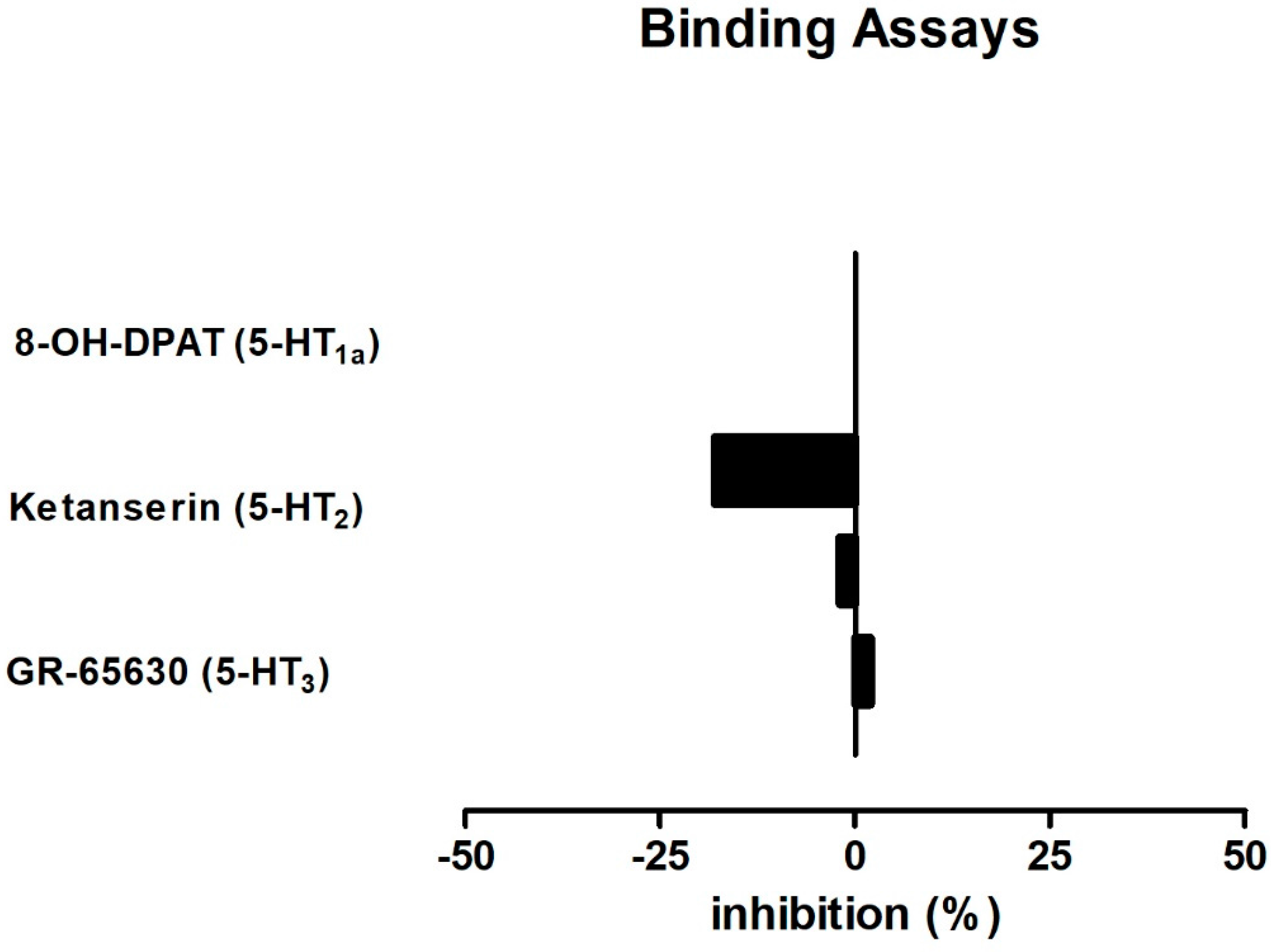
2.4. BDS 391 Did Not Directly Bind to 5-HT3 Receptors
3. Discussion
4. Material and Methods
4.1. Animals
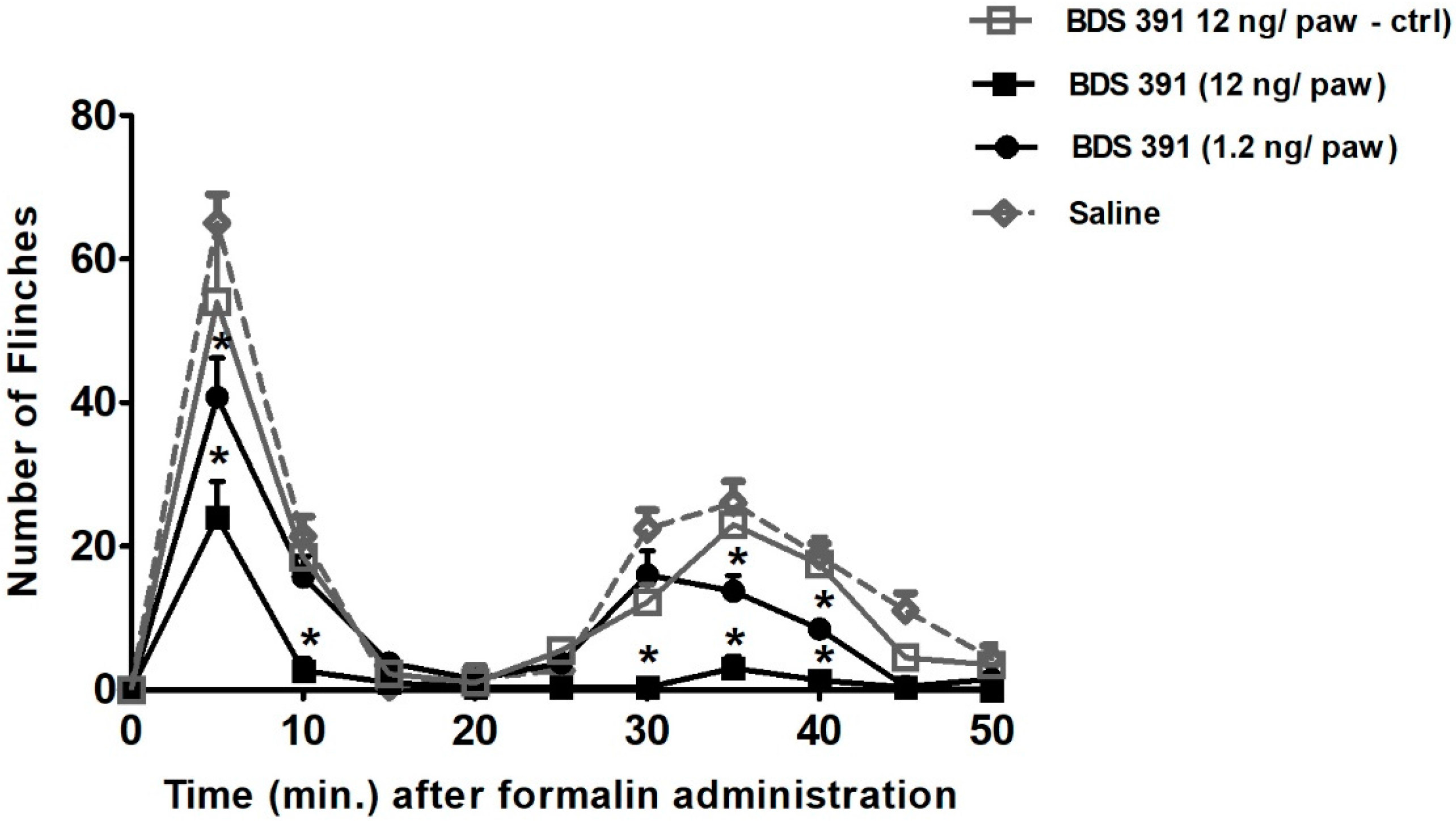
4.2. Formalin Test
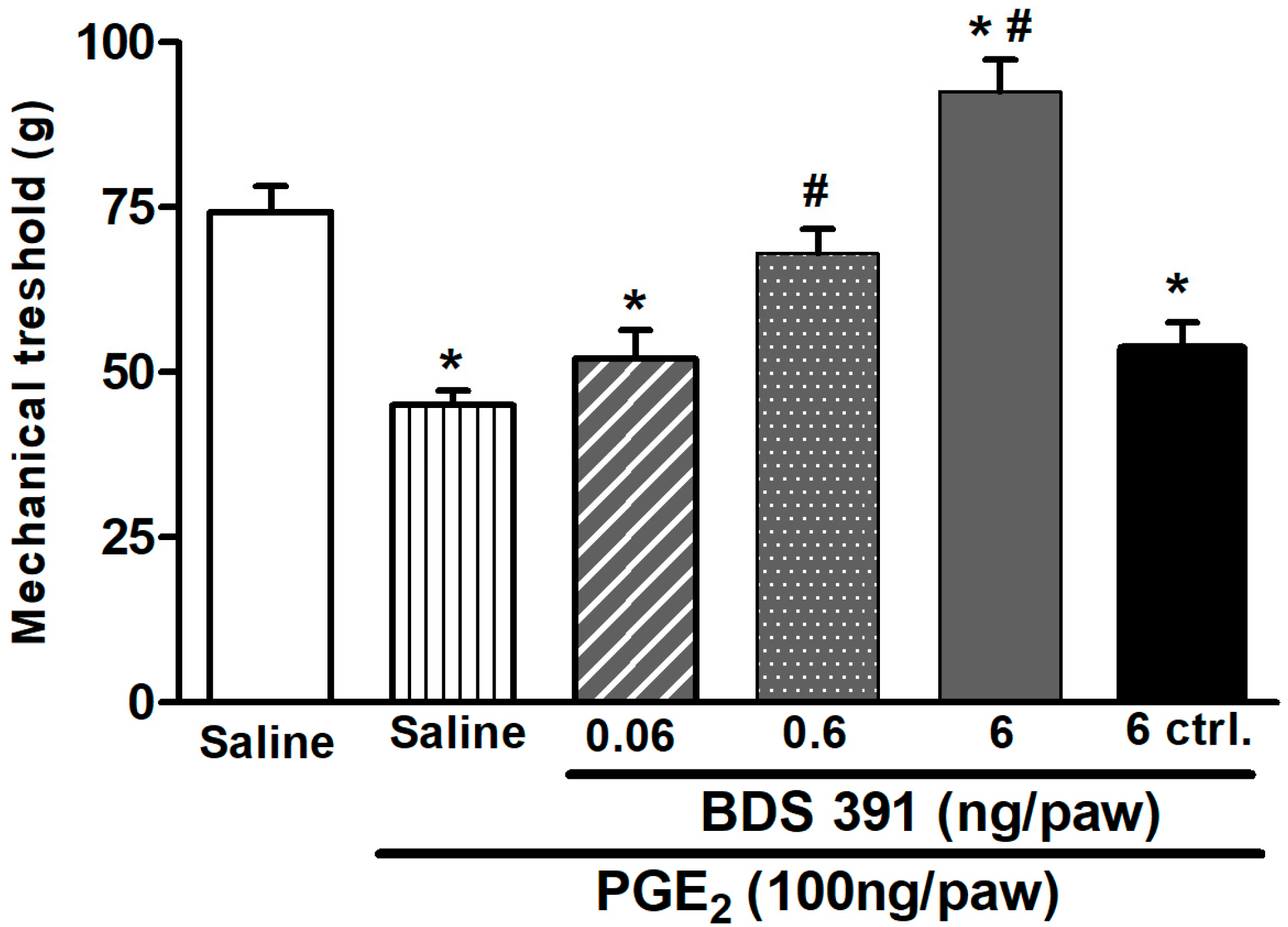
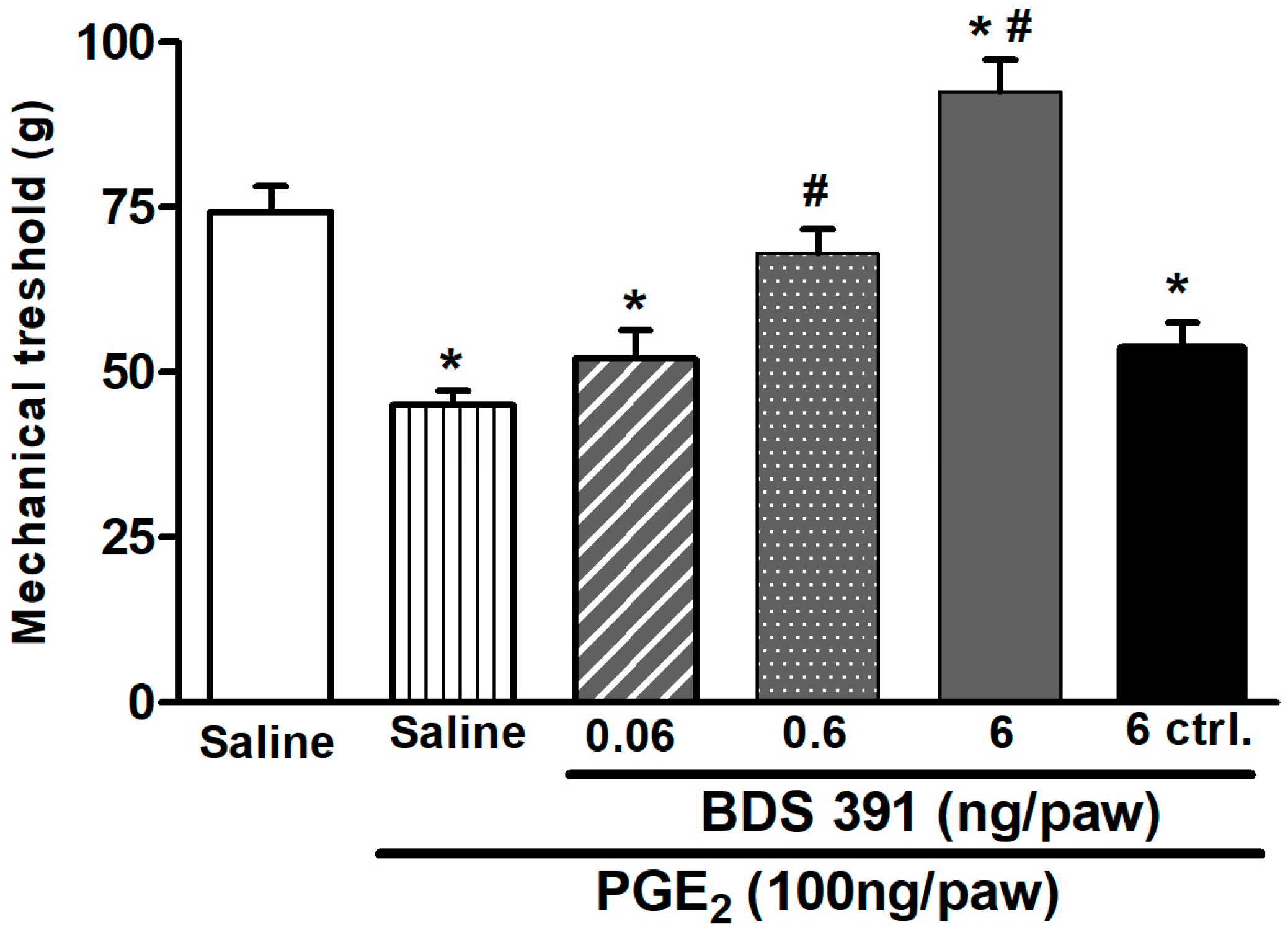
4.3. Prostaglandin E2-Induced Hyperalgesia
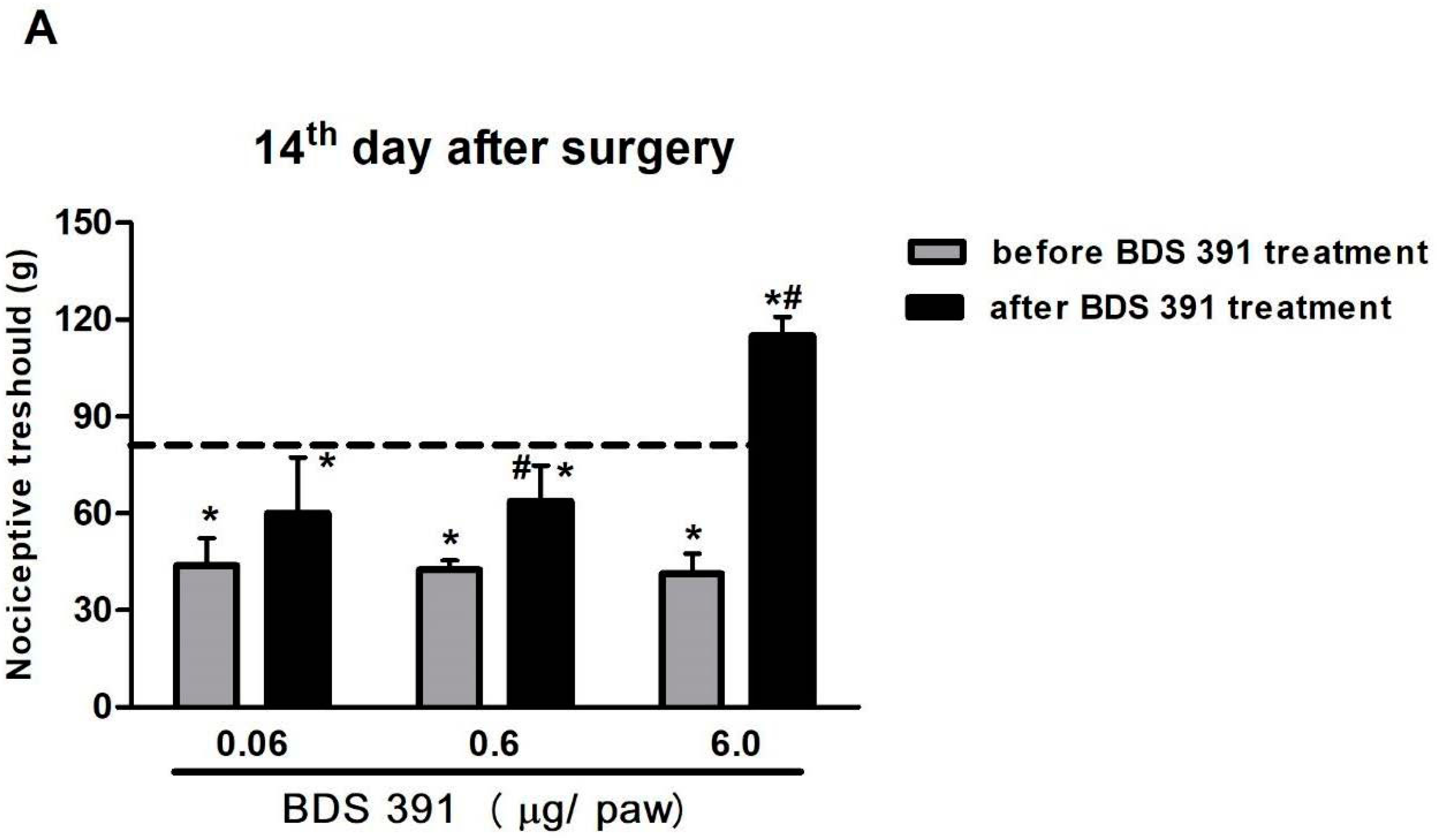
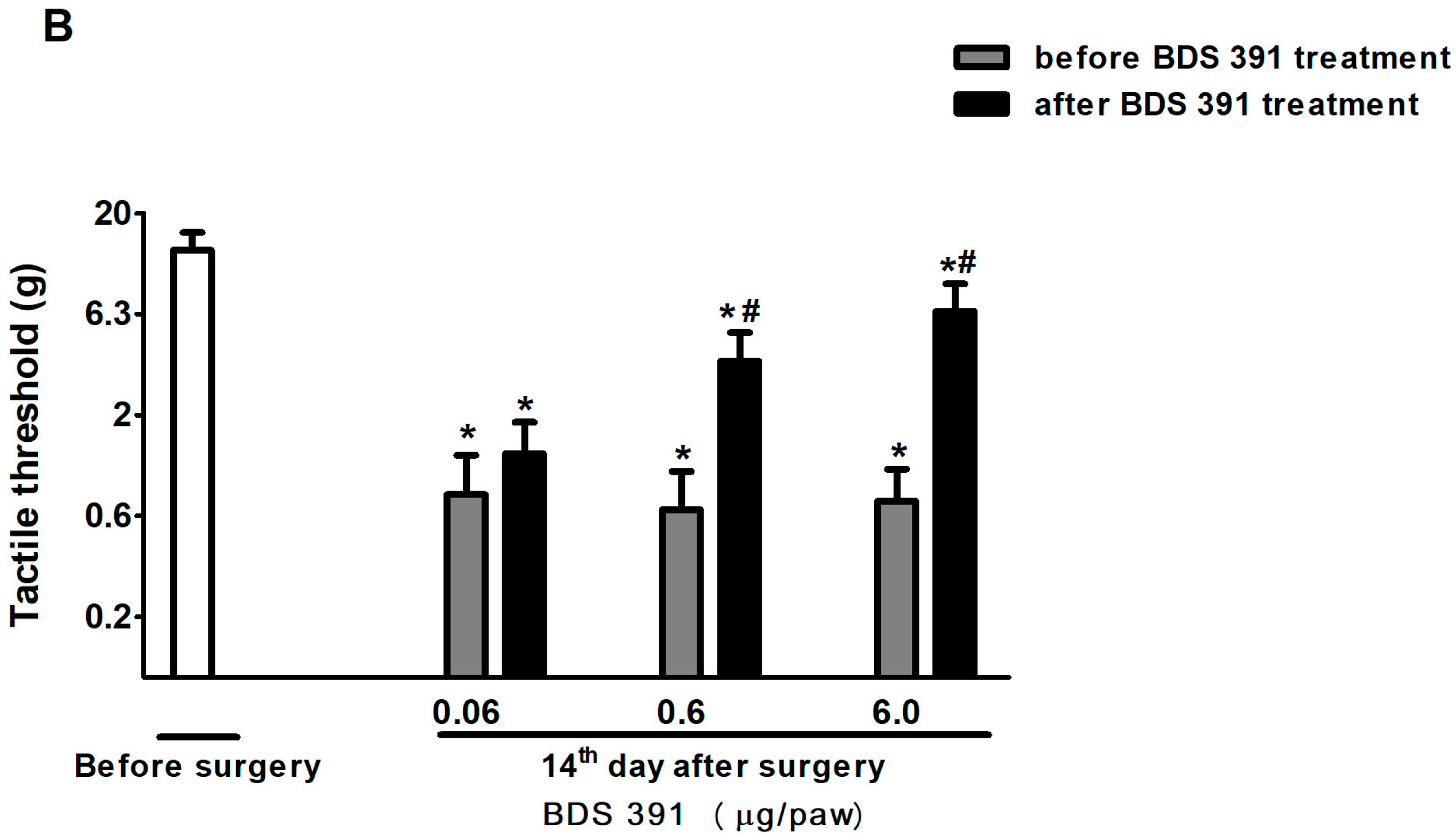
4.4. Chronic Constriction Injury
4.5. Evaluation of Mechanical Hyperalgesia
4.6. Evaluation of Low Threshold Mechanical Allodynia
4.7. Pharmacological Treatments
4.8. Statistical Analysis
4.9. Radioligand Binding Assay
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Malpezzi, E.L.; Freitas, J.C. Hemolytic activity of the nematocyst venom from the sea anemone Bunodosoma caissarum. Braz. J. Med. Biol. Res. 1991, 24, 1245–1249. [Google Scholar] [PubMed]
- Lagos, P.; Duran, R.; Cervenansky, C.; Freitas, J.C.; Silveira, R. Identification of hemolytic and neuroactive fractions in the venom of the sea anemone Bunodosoma cangicum. Braz. J. Med. Biol. Res. 2001, 34, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Anderluh, G.; Macek, P. Cytolytic peptide and protein toxins from sea anemones (Anthozoa: Actiniaria). Toxicon 2002, 40, 111–124. [Google Scholar] [CrossRef]
- Gladkikh, I.; Monastyrnaya, M.; Zelepuga, E.; Sintsova, O.; Tabakmakher, V.; Gnedenko, O.; Ivanov, A.; Hua, K.F.; Kozlovskaya, E. New Kunitz-Type HCRG Polypeptides from the Sea Anemone Heteractis crispa. Mar. Drugs 2015, 13, 6038–6063. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Fernandez, R.; Ziegelmüller, P.; González, L.; Mansur, M.; Machado, Y.; Redecke, L.; Hahn, U.; Betzel, C.; de los Ángeles Chávez, M. Two variants of the major serine protease inhibitor from the sea anemone Stichodactyla helianthus, expressed in Pichia pastoris. Protein Expr. Purif. 2016, 123, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Fernandez, R.; Peigneur, S.; Pons, T.; Alvarez, C.; González, L.; Chávez, M.A.; Tytgat, J. The Kunitz-Type Protein ShPI-1 Inhibits Serine Proteases and Voltage-Gated Potassium Channels. Toxins 2016, 8, 110. [Google Scholar] [CrossRef] [PubMed]
- Beress, L.; Beress, R.; Wunderer, G. Isolation and characterisation of three polypeptides with neurotoxic activity from Anemonia sulcata. FEBS Lett. 1975, 50, 311–314. [Google Scholar] [CrossRef]
- Kalman, K.; Pennington, M.W.; Lanigan, M.D.; Nguyen, A.; Rauer, H.; Mahnir, V.; Paschetto, K.; Kem, W.R.; Grissmer, S.; Gutman, G.A.; et al. ShK-Dap22, a potent Kv1.3-specific immunosuppressive polypeptide. J. Biol. Chem. 1998, 273, 32697–32707. [Google Scholar] [PubMed]
- Yan, L.; Herrington, J.; Goldberg, E.; Dulski, P.M.; Bugianesi, R.M.; Slaughter, R.S.; Banerjee, P.; Brochu, R.M.; Priest, B.T.; Kaczorowski, G.J.; et al. Stichodactyla helianthus peptide, a pharmacological tool for studying Kv3.2 channels. Mol. Pharmacol. 2005, 67, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Diochot, S.; Loret, E.; Bruhn, T.; Béress, L.; Lazdunski, M. APETx1, a new toxin from the sea anemone Anthopleura elegantissima, blocks voltage-gated human ether-a-go-go-related gene potassium channels. Mol. Pharmacol. 2003, 64, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Diochot, S.; Baron, A.; Rash, L.D.; Deval, E.; Escoubas, P.; Scarzello, S.; Salinas, M.; Lazdunski, M. A new sea anemone peptide, APETx2, inhibits ASIC3, a major acid-sensitive channel in sensory neurons. EMBO J. 2004, 23, 1516–1525. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.S.; Redaelli, E.; Zaharenko, A.J.; Cassulini, R.R.; Konno, K.; Pimenta, D.C.; Freitas, J.C.; Clare, J.J.; Wanke, E. Binding specificity of sea anemone toxins to Nav 1.1-1.6 sodium channels: Unexpected contributions from differences in the IV/S3-S4 outer loop. J. Biol. Chem. 2004, 279, 33323–33335. [Google Scholar] [PubMed]
- Garateix, A.; Flores, A.; García-Andrade, J.M.; Palmero, A.; Aneiros, A.; Vega, R.; Soto, E. Antagonism of glutamate receptors by a chromatographic fraction from the exudate of the sea anemone Phyllactis flosculifera. Toxicon 1996, 34, 443–450. [Google Scholar] [CrossRef]
- Zaharenko, A.J.; Picolo, G.; Ferreira, W.A., Jr.; Murakami, T.; Kazuma, K.; Hashimoto, M.; Cury, Y.; de Freitas, J.C.; Satake, M.; Konno, K. Bunodosine 391: An analgesic acylamino acid from the venom of the sea anemone Bunodosoma cangicum. J. Nat. Prod. 2011, 74, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Sommer, C. Serotonin in pain and analgesia: Actions in the periphery. Mol. Neurobiol. 2004, 30, 117–125. [Google Scholar] [CrossRef]
- Loyd, D.R.; Chen, P.B.; Hargreaves, K.M. Anti-hyperalgesic effects of anti-serotonergic compounds on serotonin- and capsaicin-evoked thermal hyperalgesia in the rat. Neuroscience 2012, 203, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Eide, P.K.; Hole, K. The role of 5-hydroxytryptamine (5-HT) receptor subtypes and plasticity in the 5-HT systems in the regulation of nociceptive sensitivity. Cephalalgia 1993, 13, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Diniz, D.A.; Petrocchi, J.A.; Navarro, L.C.; Souza, T.C.; Castor, M.G.; Perez, A.C.; Duarte, I.D.; Romero, T.R. Serotonin induces peripheral mechanical antihyperalgesic effects in mice. Eur. J. Pharmacol. 2015, 767, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, D.; Hannon, J.P.; Martin, G.R. Molecular, pharmacological and functional diversity of 5-HT receptors. Pharmacol. Biochem. Behav. 2002, 71, 533–554. [Google Scholar] [CrossRef]
- Parada, C.A.; Tambeli, C.H.; Cunha, F.Q.; Ferreira, S.H. The major role of peripheral release of histamine and 5-hydroxytryptamine in formalin-induced nociception. Neuroscience 2001, 102, 937–944. [Google Scholar] [CrossRef]
- Zeitz, K.P.; Guy, N.; Malmberg, A.B.; Dirajlal, S.; Martin, W.J.; Sun, L.; Bonhaus, D.W.; Stucky, C.L.; Julius, D.; Basbaum, A.I. The 5-HT3 subtype of serotonin receptor contributes to nociceptive processing via a novel subset of myelinated and unmyelinated nociceptors. J. Neurosci. 2002, 22, 1010–1019. [Google Scholar] [PubMed]
- Bravo-Hernandez, M.; Cervantes-Durán, C.; Pineda-Farias, J.B.; Barragán-Iglesias, P.; López-Sánchez, P.; Granados-Soto, V. Role of peripheral and spinal 5-HT receptors in development and maintenance of formalin-induced long-term secondary allodynia and hyperalgesia. Pharmacol. Biochem. Behav. 2012, 101, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, E.B., Jr.; Seniuk, J.G.T.; Godin, A.M.; Ferreira, W.C.; Dutra, M.B.; Oliveira, A.C.P.; Bastos, L.F.; Fiebich, B.L.; Coelho, M.M. Peripheral 5-HT1B and 5-HT2A receptors mediate the nociceptive response induced by 5-hydroxytryptamine in mice. Pharmacol. Biochem. Behav. 2011, 99, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Duran, C.; Vidal-Cantú, G.C.; Barragán-Iglesias, P.; Pineda-Farias, J.B.; Bravo-Hernández, M.; Murbartián, J.; Granados-Soto, V. Role of peripheral and spinal 5-HT2B receptors in formalin-induced nociception. Pharmacol. Biochem. Behav. 2012, 102, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Godinez-Chaparro, B.; López-Santillán, F.J.; Orduna, P.; Granados-Soto, V. Secondary mechanical allodynia and hyperalgesia depend on descending facilitation mediated by spinal 5-HT(4), 5-HT(6) and 5-HT(7) receptors. Neuroscience 2012, 222, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Godinez-Chaparro, B.; López-Santillán, F.J.; Argüelles, C.F.; Villalón, C.M.; Granados-Soto, V. Role of 5-HT(1)B/(1)D receptors in the reduction of formalin-induced nociception and secondary allodynia/hyperalgesia produced by antimigraine drugs in rats. Life Sci. 2013, 92, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Godinez-Chaparro, B.; Barragán-Iglesias, P.; Castañeda-Corral, G.; Rocha-González, H.I.; Granados-Soto, V. Role of peripheral 5-HT(4), 5-HT(6), and 5-HT(7) receptors in development and maintenance of secondary mechanical allodynia and hyperalgesia. Pain 2011, 152, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.R.; Choo, H.; Nam, G. Neuropathic pain-alleviating effects of pyrazole-conjugated arylsulfonamides as 5-HT6 receptor antagonists. Bioorg. Med. Chem. Lett. 2017, 27, 4146–4149. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, M.; Vogel, C.; Blenk, K.H.; Jänig, W. Algesics excite axotomised afferent nerve fibres within the first hours following nerve transection in rats. Pain 1997, 72, 347–354. [Google Scholar] [CrossRef]
- Kress, M.; Reeh, P.W.; Vyklicky, L. An interaction of inflammatory mediators and protons in small diameter dorsal root ganglion neurons of the rat. Neurosci. Lett. 1997, 224, 37–40. [Google Scholar] [CrossRef]
- Tzeng, J.I.; Chiu, C.C.; Wang, J.J.; Chen, Y.W.; Hung, C.H. Isobolographic analysis of the cutaneous antinociceptive interaction between bupivacaine co-injected with serotonin in rats. Pharmacol. Rep. 2017, 69, 846–850. [Google Scholar] [CrossRef] [PubMed]
- Bingham, S.; Davey, P.T.; Sammons, M.; Raval, P.; Overend, P.; Parsons, A.A. Inhibition of inflammation-induced thermal hypersensitivity by sumatriptan through activation of 5-HT(1B/1D) receptors. Exp. Neurol. 2001, 167, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Millan, M.J. Descending control of pain. Prog. Neurobiol. 2002, 66, 355–474. [Google Scholar] [CrossRef]
- Peng, Y.B.; Lin, Q.; Willis, W.D. The role of 5-HT3 receptors in periaqueductal gray-induced inhibition of nociceptive dorsal horn neurons in rats. J. Pharmacol. Exp. Ther. 1996, 276, 116–124. [Google Scholar] [PubMed]
- Kiefel, J.M.; Cooper, M.L.; Bodnar, R.J. Serotonin receptor subtype antagonists in the medial ventral medulla inhibit mesencephalic opiate analgesia. Brain Res. 1992, 597, 331–338. [Google Scholar] [CrossRef]
- Nadeson, R.; Goodchild, C.S. Antinociceptive role of 5-HT1A receptors in rat spinal cord. Br. J. Anaesth. 2002, 88, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Santello, M.; Bisco, A.; Nevian, N.E.; Lacivita, E.; Leopoldo, M.; Nevian, T. The brain-penetrant 5-HT7 receptor agonist LP-211 reduces the sensory and affective components of neuropathic pain. Neurobiol. Dis. 2017, 106, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Okubo, M.; Castro, A.; Guo, W.; Zou, S.; Ren, K.; Wei, F.; Keller, A.; Dubner, R. Transition to persistent orofacial pain after nerve injury involves supraspinal serotonin mechanisms. J. Neurosci. 2013, 33, 5152–5161. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.D.; DeWitte, C.; Ness, T.J.; Robbins, M.T. Serotonin enhances urinary bladder nociceptive processing via a 5-HT3 receptor mechanism. Neurosci. Lett. 2015, 604, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, M.; Gebhart, G.F. Spinal serotonin receptors mediate descending facilitation of a nociceptive reflex from the nuclei reticularis gigantocellularis and gigantocellularis pars alpha in the rat. Brain Res. 1991, 550, 35–48. [Google Scholar] [CrossRef]
- Dray, A. Inflammatory mediators of pain. Br. J. Anaesth. 1995, 75, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, A. Prostaglandin E2 and pain—An update. Biol. Pharm. Bull. 2011, 34, 1170–1173. [Google Scholar] [CrossRef] [PubMed]
- Cunha, F.Q.; Ferreira, S.H. Peripheral hyperalgesic cytokines. Adv. Exp. Med. Biol. 2003, 521, 22–39. [Google Scholar] [PubMed]
- Pierce, P.A.; Xie, G.X.; Levine, J.D.; Peroutka, S.J. 5-Hydroxytryptamine receptor subtype messenger RNAs in rat peripheral sensory and sympathetic ganglia: A polymerase chain reaction study. Neuroscience 1996, 70, 553–559. [Google Scholar] [CrossRef]
- Carlton, S.M.; Coggeshall, R.E. Immunohistochemical localization of 5-HT2A receptors in peripheral sensory axons in rat glabrous skin. Brain Res. 1997, 763, 271–275. [Google Scholar] [CrossRef]
- Meller, S.T.; Lewis, S.J.; Brody, M.J.; Gebhart, G.F. The peripheral nociceptive actions of intravenously administered 5-HT in the rat requires dual activation of both 5-HT2 and 5-HT3 receptor subtypes. Brain Res. 1991, 561, 61–68. [Google Scholar] [CrossRef]
- Hu, W.P.; Guan, B.C.; Ru, L.Q.; Chen, J.G.; Li, Z.W. Potentiation of 5-HT3 receptor function by the activation of coexistent 5-HT2 receptors in trigeminal ganglion neurons of rats. Neuropharmacology 2004, 47, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Dworkin, R.H.; Backonja, M.; Rowbotham, M.C.; Allen, R.R.; Argoff, C.R.; Bennett, G.J.; Bushnell, M.C.; Farrar, J.T.; Galer, B.S.; Haythornthwaite, J.A.; et al. Advances in neuropathic pain: Diagnosis, mechanisms, and treatment recommendations. Arch. Neurol. 2003, 60, 1524–2534. [Google Scholar] [CrossRef] [PubMed]
- Panczyk, K.; Golda, S.; Waszkielewicz, A.; Zelaszczyk, D.; Gunia-Krzyzak, A.; Marona, H. Serotonergic system and its role in epilepsy and neuropathic pain treatment: A review based on receptor ligands. Curr. Pharm. Des. 2015, 21, 1723–1740. [Google Scholar] [CrossRef] [PubMed]
- Tjolsen, A.; Berge, O.G.; Hunskaar, S.; Rosland, J.H.; Hole, K. The formalin test: An evaluation of the method. Pain 1992, 51, 5–17. [Google Scholar] [CrossRef]
- Puig, S.; Sorkin, L.S. Formalin-evoked activity in identified primary afferent fibers: Systemic lidocaine suppresses phase-2 activity. Pain 1996, 64, 345–355. [Google Scholar] [CrossRef]
- Hunskaar, S.; Hole, K. The formalin test in mice: Dissociation between inflammatory and non-inflammatory pain. Pain 1987, 30, 103–114. [Google Scholar] [CrossRef]
- Shibata, M.; Ohkubo, T.; Takahashi, H.; Inoki, R. Modified formalin test: Characteristic biphasic pain response. Pain 1989, 38, 347–352. [Google Scholar] [CrossRef]
- Wheeler-Aceto, H.; Porreca, F.; Cowan, A. The rat paw formalin test: Comparison of noxious agents. Pain 1990, 40, 229–238. [Google Scholar] [CrossRef]
- Doak, G.J.; Sawynok, J. Formalin-induced nociceptive behavior and edema: Involvement of multiple peripheral 5-hydroxytryptamine receptor subtypes. Neuroscience 1997, 80, 939–949. [Google Scholar] [CrossRef]
- Giordano, J.; Rogers, L.V. Peripherally administered serotonin 5-HT3 receptor antagonists reduce inflammatory pain in rats. Eur. J. Pharmacol. 1989, 170, 83–86. [Google Scholar] [CrossRef]
- Nakajima, K.; Obata, H.; Ito, N.; Goto, F.; Saito, S. The nociceptive mechanism of 5-hydroxytryptamine released into the peripheral tissue in acute inflammatory pain in rats. Eur. J. Pain 2009, 13, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Lochner, M.; Lummis, S.C. Agonists and antagonists bind to an A-A interface in the heteromeric 5-HT3AB receptor. Biophys. J. 2010, 98, 1494–1502. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.J.; Lummis, S.C. 5-HT3 receptors. Curr. Pharm. Des. 2006, 12, 3615–3630. [Google Scholar] [CrossRef] [PubMed]
- Faerber, L.; Drechsler, S.; Ladenburger, S.; Gschaidmeier, H.; Fischer, W. The neuronal 5-HT3 receptor network after 20 years of research—Evolving concepts in management of pain and inflammation. Eur. J. Pharmacol. 2007, 560, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Fozard, J.R. MDL 72222: A potent and highly selective antagonist at neuronal 5-hydroxytryptamine receptors. Naunyn-Schmiedebergs Arch. Pharmacol. 1984, 326, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.; Hawthorn, J.; Pople, A.; Gazet, J.C.; Ford, H.T.; Challoner, T.; Coombes, R.C. Prevention of emesis in patients receiving cytotoxic drugs by GR38032F, a selective 5-HT3 receptor antagonist. Lancet 1987, 1, 1461–1463. [Google Scholar] [CrossRef]
- Juarez, E.H.; Ochoa-Cortés, F.; Miranda-Morales, M.; Espinosa-Luna, R.; Montano, L.M.; Barajas-López, C. Selectivity of antagonists for the Cys-loop native receptors for ACh, 5-HT and GABA in guinea-pig myenteric neurons. Auton. Autacoid Pharmacol. 2014, 34, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Maricq, A.V.; Peterson, A.S.; Brake, A.J.; Myers, R.M.; Julius, D. Primary structure and functional expression of the 5HT3 receptor, a serotonin-gated ion channel. Science 1991, 254, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Macor, J.E.; Gurley, D.; Lanthorn, T.; Loch, J.; Mack, R.A.; Mullen, G.; Tran, O.; Wright, N.; Gordon, J.C. The 5-HT3 antagonist tropisetron (ICS 205-930) is a potent and selective alpha7 nicotinic receptor partial agonist. Bioorg. Med. Chem. Lett. 2001, 11, 319–321. [Google Scholar] [CrossRef]
- Rothlin, C.V.; Lioudyno, M.I.; Silbering, A.F.; Plazas, P.V.; Casati, M.E.G.; Katz, E.; Guth, P.S.; Elgoyhen, A.B. Direct interaction of serotonin type 3 receptor ligands with recombinant and native alpha 9 alpha 10-containing nicotinic cholinergic receptors. Mol. Pharmacol. 2003, 63, 1067–1074. [Google Scholar] [CrossRef] [PubMed]
- Papke, R.L.; Schiff, H.C.; Jack, B.A.; Horenstein, N.A. Molecular dissection of tropisetron, an alpha7 nicotinic acetylcholine receptor-selective partial agonist. Neurosci. Lett. 2005, 378, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Kilpatrick, G.J.; Butler, A.; Burridge, J.; Oxford, A.W. 1-(m-chlorophenyl)-biguanide, a potent high affinity 5-HT3 receptor agonist. Eur. J. Pharmacol. 1990, 182, 193–197. [Google Scholar] [CrossRef]
- Miles, T.F.; Lester, H.A.; Dougherty, D.A. Allosteric activation of the 5-HT3AB receptor by mCPBG. Neuropharmacology 2015, 91, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Andreev, Y.A.; Kozlov, S.A.; Koshelev, S.G.; Ivanova, E.A.; Monastyrnaya, M.M.; Kozlovskaya, E.P.; Grishin, E.V. Analgesic compound from sea anemone Heteractis crispa is the first polypeptide inhibitor of vanilloid receptor 1 (TRPV1). J. Biol. Chem. 2008, 283, 23914–23921. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain 1983, 16, 109–110. [Google Scholar] [CrossRef]
- Dubuisson, D.; Dennis, S.G. The formalin test: A quantitative study of the analgesic effects of morphine, meperidine, and brain stem stimulation in rats and cats. Pain 1977, 4, 161–174. [Google Scholar] [CrossRef]
- Picolo, G.; Cury, Y. Peripheral neuronal nitric oxide synthase activity mediates the antinociceptive effect of Crotalus durissus terrificus snake venom, a delta- and kappa-opioid receptor agonist. Life Sci. 2004, 75, 559–573. [Google Scholar] [CrossRef] [PubMed]
- Bennett, G.J.; Xie, Y.K. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 1988, 33, 87–107. [Google Scholar] [CrossRef]
- Bennett, G. An animal model of neuropathic pain: A review. Muscle Nerve 1993, 16, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Randall, L.O.; Selitto, J.J. A method for measurement of analgesia activity on inflamed tissue. Arch. Inst. Pharmacodyn. 1957, 111, 209–219. [Google Scholar]
- Gutierrez, V.P.; Konno, K.; Chacur, M.; Sampaio, S.C.; Picolo, G.; Brigatte, P.; Zambelli, V.O.; Cury, Y. Crotalphine induces potent antinociception in neuropathic pain by acting at peripheral opioid receptors. Eur. J. Pharmacol. 2008, 594, 84–92. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira Junior, W.A.; Zaharenko, A.J.; Kazuma, K.; Picolo, G.; Gutierrez, V.P.; De Freitas, J.C.; Konno, K.; Cury, Y. Peripheral 5-HT3 Receptors Are Involved in the Antinociceptive Effect of Bunodosine 391. Toxins 2018, 10, 12. https://doi.org/10.3390/toxins10010012
Ferreira Junior WA, Zaharenko AJ, Kazuma K, Picolo G, Gutierrez VP, De Freitas JC, Konno K, Cury Y. Peripheral 5-HT3 Receptors Are Involved in the Antinociceptive Effect of Bunodosine 391. Toxins. 2018; 10(1):12. https://doi.org/10.3390/toxins10010012
Chicago/Turabian StyleFerreira Junior, Wilson Alves, Andre Junqueira Zaharenko, Kohei Kazuma, Gisele Picolo, Vanessa Pacciari Gutierrez, Jose Carlos De Freitas, Katsuhiro Konno, and Yara Cury. 2018. "Peripheral 5-HT3 Receptors Are Involved in the Antinociceptive Effect of Bunodosine 391" Toxins 10, no. 1: 12. https://doi.org/10.3390/toxins10010012