Cellular and Molecular Aspects of the β-N-Methylamino-l-alanine (BMAA) Mode of Action within the Neurodegenerative Pathway: Facts and Controversy

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
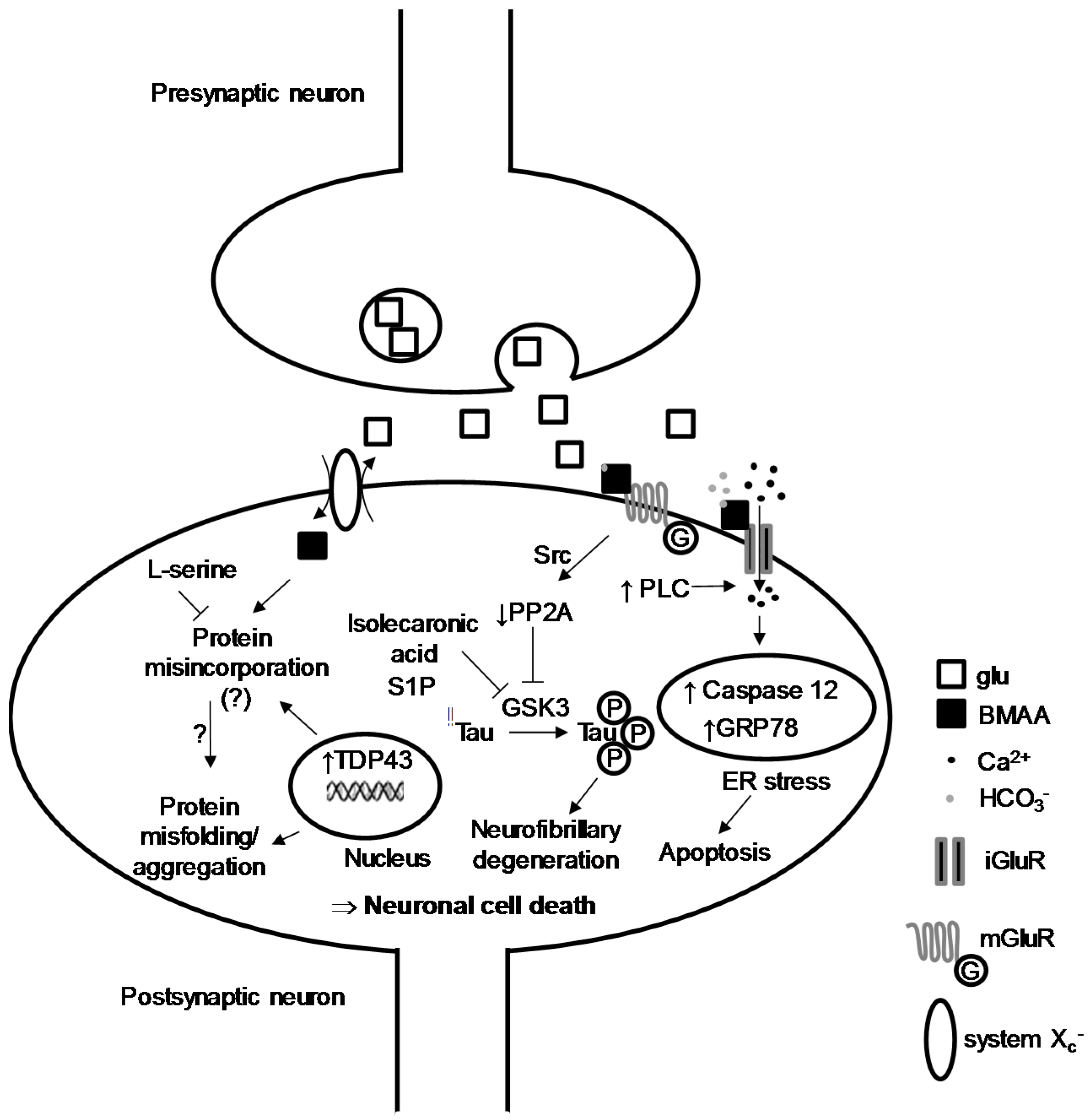
2. BMAA-Induced Excitotoxicity through Glutamate Receptors
3. Intracellular Actors Involved in BMAA-Induced Neurodegeneration
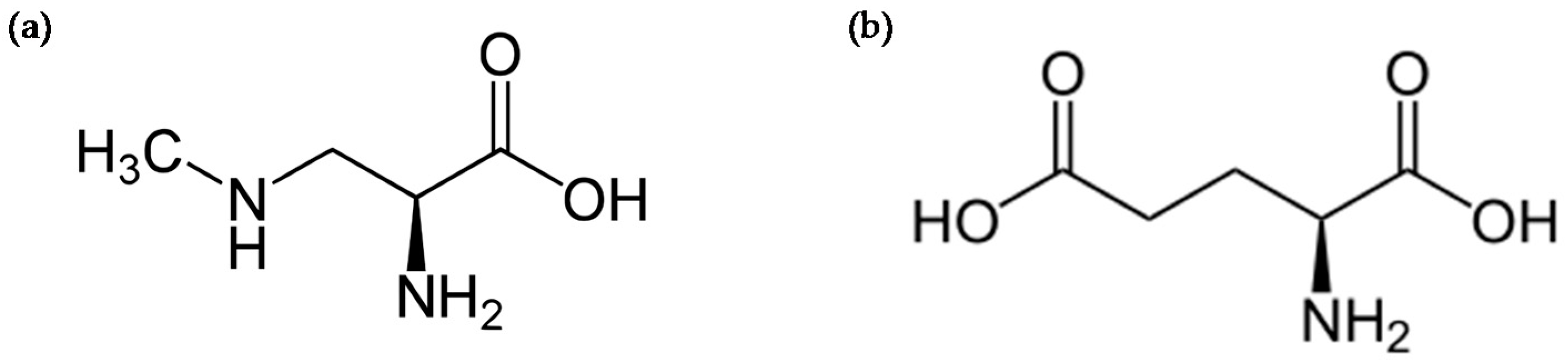
4. Protein Incorporation of BMAA and Cellular Stress
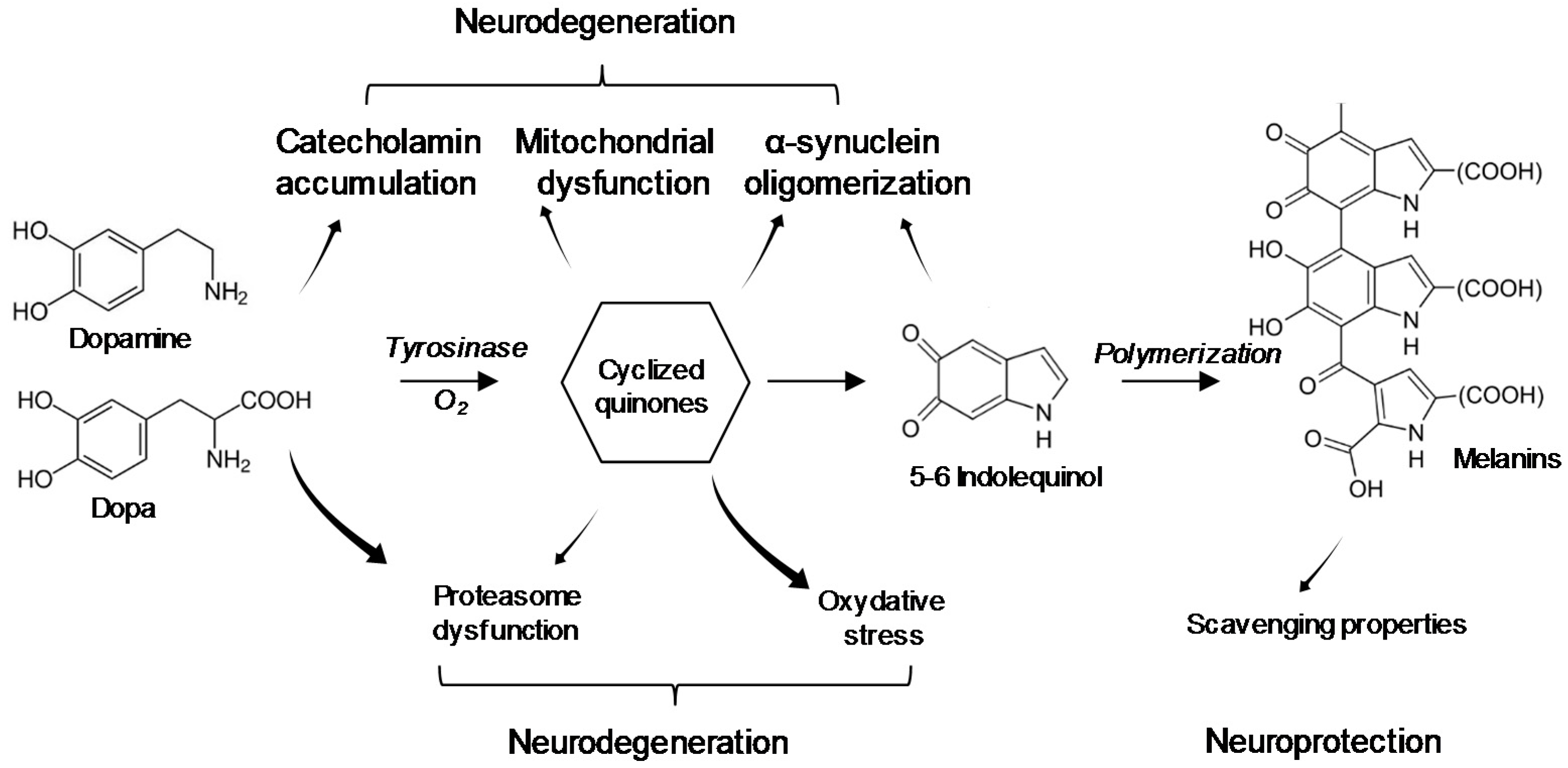
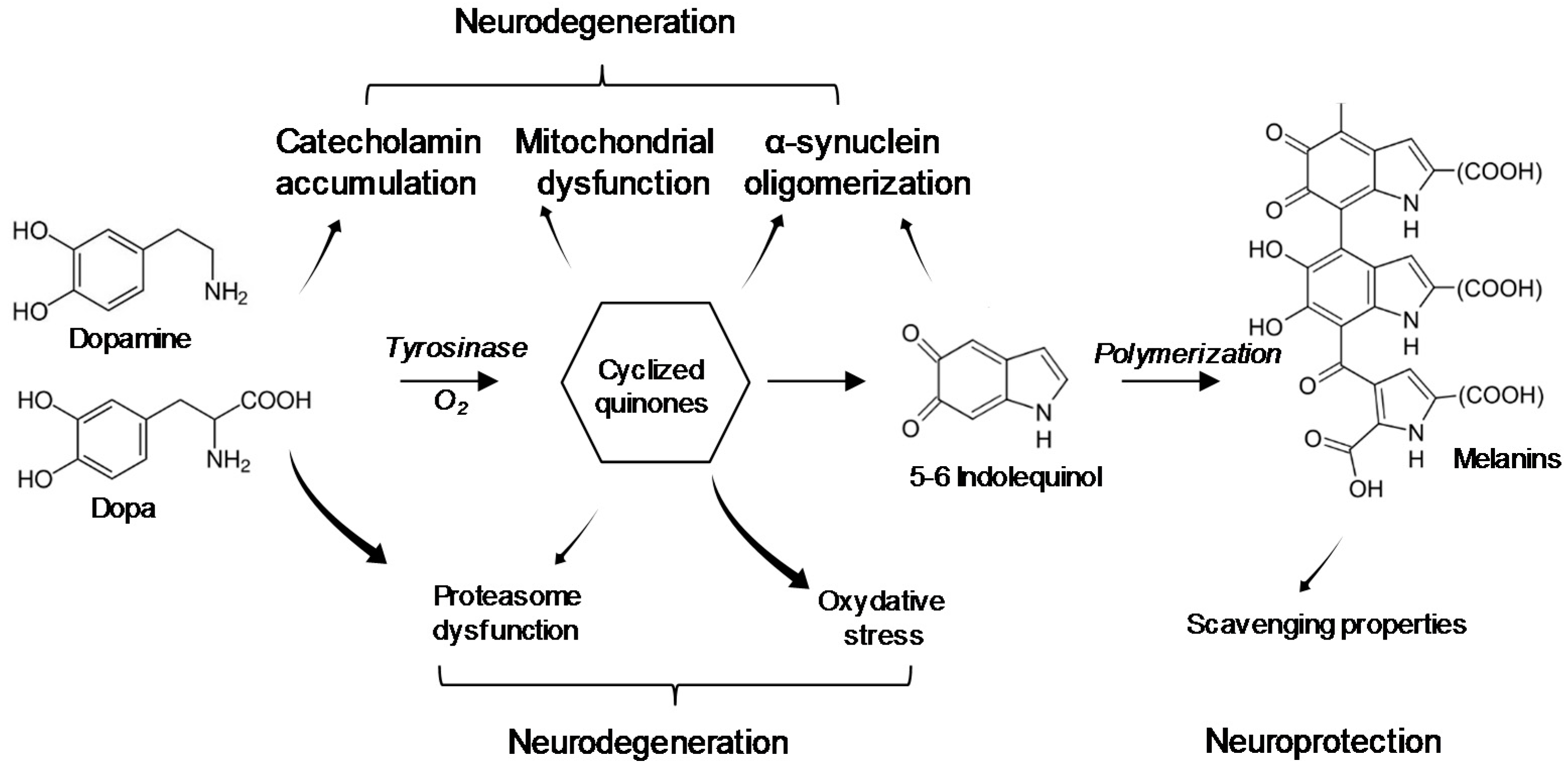
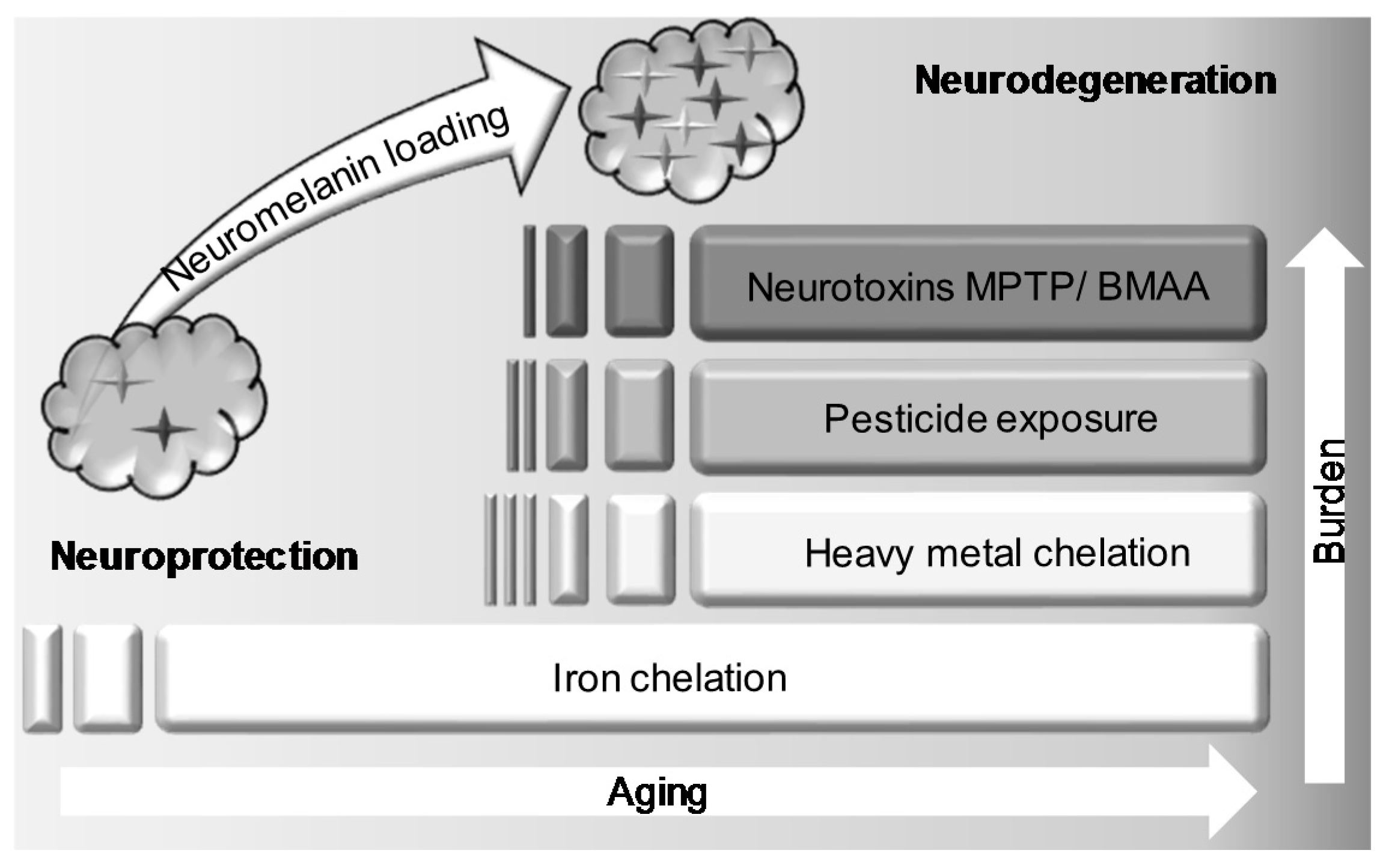
5. Interaction of BMAA with Neuromelanin
6. Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cox, P.A.; Banack, S.A.; Murch, S.J.; Rasmussen, U.; Tien, G.; Bidigare, R.R.; Metcalf, J.S.; Morrison, L.F.; Codd, G.A.; Bergman, B. Diverse taxa of cyanobacteria produce β-N-methylamino-l-alanine, a neurotoxic amino acid. Proc. Natl. Acad. Sci. USA 2005, 102, 5074–5078. [Google Scholar] [CrossRef] [PubMed]
- Downing, S.; Banack, S.A.; Metcalf, J.S.; Cox, P.A.; Downing, T.G. Nitrogen starvation of cyanobacteria results in the production of β-N-methylamino-l-alanine. Toxicon 2011, 58, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Eriksson, J.; Lage, S.; Jonasson, S.; Shams, S.; Mehine, M.; Ilag, L.L.; Rasmussen, U. Diatoms: A novel source for the neurotoxin BMAA in aquatic environments. PLoS ONE 2014, 9, e84578. [Google Scholar] [CrossRef] [PubMed]
- Réveillon, D.; Séchet, V.; Hess, P.; Amzil, Z. Production of BMAA and DAB by diatoms (Phaeodactylum tricornutum, Chaetoceros sp., Chaetoceros calcitrans and Thalassiosira pseudonana) and bacteria isolated from a diatom culture. Harmful Algae 2016, 58, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Lage, S.; Costa, P.R.; Moita, T.; Eriksson, J.; Rasmussen, U.; Rydberg, S.J. BMAA in shellfish from two Portuguese transitional water bodies suggests the marine dinoflagellate Gymnodinium catenatum as a potential BMAA source. Aquat. Toxicol. 2014, 152, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Banack, S.A.; Cox, P.A. Biomagnification of cycad neurotoxins in flying foxes: Implications for ALS-PDC in Guam. Neurology 2003, 61, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Cox, P.A.; Sacks, O.W. Cycad neurotoxins, consumption of flying foxes, and ALS-PDC disease in Guam. Neurology 2002, 58, 956–959. [Google Scholar] [CrossRef] [PubMed]
- Cox, P.A.; Banack, S.A.; Murch, S.J. Biomagnification of cyanobacterial neurotoxins and neurodegenerative disease among the Chamorro people of Guam. Proc. Natl. Acad. Sci. USA 2003, 100, 13380–13383. [Google Scholar] [CrossRef] [PubMed]
- Banack, S.A.; Metcalf, J.S.; Bradley, W.G.; Cox, P.A. Detection of cyanobacterial neurotoxin β-N-methylamino-l-alanine within shellfish in the diet of an ALS patient in Florida. Toxicon 2014, 90, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Réveillon, D.; Abadie, E.; Sechet, V.; Brient, L.; Savar, V.; Bardoui, M.; Hess, P.; Amzil, Z. Beta-N-methylamino-l-alanine: LC-MS/MS optimization, screening of cyanobacterial strains and occurrence in shellfish from Thau, a French Mediterranean lagoon. Mar. Drugs 2014, 12, 5441–5467. [Google Scholar] [CrossRef] [PubMed]
- Réveillon, D.; Abadie, E.; Séchet, V.; Masseret, E.; Hess, P.; Amzil, Z. β-N-methylamino-l-alanine (BMAA) and isomers: Distribution in different food web compartments of Thau lagoon, French Mediterranean Sea. Mar. Environ. Res. 2015, 110, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Réveillon, D.; Séchet, V.; Hess, P.; Amzil, Z. Systematic detection of BMAA (β-N-methylamino-l-alanine) and DAB (2,4-diaminobutyric acid) in mollusks collected in shellfish production areas along the French coasts. Toxicon 2016, 110, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Pablo, J.; Banack, S.A.; Cox, P.A.; Johnson, T.E.; Papapetropoulos, S.; Bradley, W.G.; Buck, A.; Mash, D.C. Cyanobacterial neurotoxin BMAA in ALS and Alzheimer’s disease. Acta Neurol. Scand. 2009, 120, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Meneely, J.P.; Chevallier, O.P.; Graham, S.; Greer, B.; Green, B.D.; Elliott, C.T. β-methylamino-l-alanine (BMAA) is not found in the brains of patients with confirmed Alzheimer’s disease. Sci. Rep. 2016, 6, 36363. [Google Scholar] [CrossRef] [PubMed]
- Berntzon, L.; Ronnevi, L.O.; Bergman, B.; Eriksson, J. Detection of BMAA in the human central nervous system. Neuroscience 2015, 292, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Van Den Bosch, L.; Van Damme, P.; Bogaert, E.; Robberecht, W. The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2006, 1762, 1068–1082. [Google Scholar] [CrossRef] [PubMed]
- Chiu, A.S.; Gehringer, M.M.; Braidy, N.; Guillemin, G.J.; Welch, J.H.; Neilan, B.A. Gliotoxicity of the cyanotoxin, β-methyl-amino-l-alanine (BMAA). Sci. Rep. 2013, 3, 1482. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.H.; Choi, D.W. Beta-N-methylamino-l-alanine neurotoxicity: Requirement for bicarbonate as a cofactor. Science 1988, 241, 973–975. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.H.; Christine, C.W.; Choi, D.W. Bicarbonate dependence of glutamate receptor activation by beta-N-methylamino-l-alanine: Channel recording and study with related compounds. Neuron 1989, 3, 321–326. [Google Scholar] [CrossRef]
- Richter, K.E.; Mena, E.E. l-beta-methylaminoalanine inhibits [3H]glutamate binding in the presence of bicarbonate ions. Brain Res. 1989, 492, 385–388. [Google Scholar] [CrossRef]
- Myers, T.G.; Nelson, S.D. Neuroactive carbamate adducts of beta-N-methylamino-l-alanine and ethylenediamine. J. Biol. Chem. 1990, 265, 10193–10195. [Google Scholar] [PubMed]
- Copani, A.; Canonico, P.L.; Catania, M.V.; Aronica, E.; Bruno, V.; Ratti, E.; van Amsterdam, F.T.; Gaviraghi, G.; Nicoletti, F. Interaction between beta-N-methylamino-l-alanine and excitatory amino acid receptors in brain slices and neuronal cultures. Brain Res. 1991, 558, 79–86. [Google Scholar] [CrossRef]
- Rakonczay, Z.; Matsuoka, Y.; Giacobini, E. Effects of l-beta-N-methylamino-l-alanine (L-BMAA) on the cortical cholinergic and glutamatergic systems of the rat. J. Neurosci. Res. 1991, 29, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Brownson, D.M.; Mabry, T.J.; Leslie, S.W. The cycad neurotoxic amino acid, β-N-methylamino-l-alanine (BMAA), elevates intracellular calcium levels in dissociated rat brain cells. J. Ethnopharmacol. 2002, 82, 159–167. [Google Scholar] [CrossRef]
- Buenz, E.J.; Howe, C.L. β-methylamino-alanine (BMAA) injures hippocampal neurons in vivo. Neurotoxicology 2007, 28, 702–704. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.Z.; Yu, S.; Hsu, C.I.; Liu, J.; Acab, A.; Wu, R.; Tao, A.; Chiang, B.J.; Weiss, J.H. Intrathecal infusion of BMAA induces selective motor neuron damage and astrogliosis in the ventral horn of the spinal cord. Exp. Neurol. 2014, 261, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.D.; Banack, S.A.; Cox, P.A.; Weiss, J.H. BMAA selectively injures motor neurons via AMPA/kainate receptor activation. Exp. Neurol. 2006, 201, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Lopicic, S.; Nedeljkov, V.; Cemerikic, D. Augmentation and ionic mechanism of effect of β-N-methylamino-l-alanine in presence of bicarbonate on membrane potential of Retzius nerve cells of the leech Haemopis sanguisuga. Comp. Biochem. Physiol. Part A 2009, 153, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, K.A.; Robertson, A. Domoic acid and human exposure risks: A review. Toxicon 2010, 56, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Gong, C.X.; Liu, F. Microtubule-associated protein tau as a therapeutic target in Alzheimer’s disease. Expert Opin. Ther. Targets 2014, 18, 307–318. [Google Scholar] [CrossRef] [PubMed]
- De Munck, E.; Muñoz-Sáez, E.; Miguel, B.G.; Solas, M.T.; Ojeda, I.; Martínez, A.; Gil, C.; Arahuetes, R.M. β-N-methylamino-l-alanine causes neurological and pathological phenotypes mimicking Amyotrophic Lateral Sclerosis (ALS): The first step towards an experimental model for sporadic ALS. Environ. Toxicol. Pharmacol. 2013, 36, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Sáez, E.; de Munck, E.; Arahuetes, R.M.; Solas, M.T.; Martínez, A.M.; Miguel, B.G. β-N-methylamino-l-alanine induces changes in both GSK3 and TDP-43 in human neuroblastoma. J. Toxicol. Sci. 2013, 38, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Arif, M.; Kazim, S.F.; Grundke-Iqbal, I.; Garruto, R.M.; Iqbal, K. Tau pathology involves protein phosphatase 2A in parkinsonism-dementia of Guam. Proc. Natl. Acad. Sci. USA 2014, 111, 1144–1149. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Rush, T.; Zapata, J.; Lobner, D. β-N-methylamino-l-alanine induces oxidative stress and glutamate release through action on system Xc-. Exp. Neurol. 2009, 217, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Sáez, E.; de Munck García, E.; Arahuetes Portero, R.M.; Vicente, F.; Ortiz-López, F.J.; Cantizani, J.; Gómez Miguel, B. Neuroprotective role of sphingosine-1-phosphate in L-BMAA treated neuroblastoma cells (SH-SY5Y). Neurosci. Lett. 2015, 593, 83–89. [Google Scholar] [CrossRef] [PubMed]
- De Munck, E.; Palomo, V.; Muñoz-Sáez, E.; Perez, D.I.; Gómez-Miguel, B.; Solas, M.T.; Gil, C.; Martínez, A.; Arahuetes, R.M. Small GSK-3 inhibitor shows efficacy in a motor neuron disease murine model modulating autophagy. PLoS ONE 2016, 11, e0162723. [Google Scholar] [CrossRef] [PubMed]
- De Pedro, N.; Cantizani, J.; Ortiz-López, F.J.; González-Menéndez, V.; Cautain, B.; Rodríguez, L.; Bills, G.F.; Reyes, F.; Genilloud, O.; Vicente, F. Protective effects of isolecanoric acid on neurodegenerative in vitro models. Neuropharmacology 2016, 101, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Baralle, F.E. The multiple roles of TDP-43 in pre-mRNA processing and gene expression regulation. RNA Biol. 2010, 7, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Vande Velde, C.; Bouchard, J.P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F.; et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Borroni, B.; Bonvicini, C.; Alberici, A.; Buratti, E.; Agosti, C.; Archetti, S.; Papetti, A.; Stuani, C.; Di Luca, M.; Gennarelli, M.; et al. Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum. Mutat. 2009, 30, E974–E983. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Sáez, E.; de Munck García, E.; Arahuetes Portero, R.M.; Martínez, A.; Solas Alados, M.T.; Miguel, B.G. Analysis of β-N-methylamino-l-alanine (L-BMAA) neurotoxicity in rat cerebellum. Neurotoxicology 2015, 48, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, O.; Berg, A.L.; Hanrieder, J.; Arnerup, G.; Lindström, A.K.; Brittebo, E.B. Intracellular fibril formation, calcification, and enrichment of chaperones, cytoskeletal, and intermediate filament proteins in the adult hippocampus CA1 following neonatal exposure to the nonprotein amino acid BMAA. Arch. Toxicol. 2015, 89, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Glover, W.B.; Mash, D.C.; Murch, S.J. The natural non-protein amino acid N-β-methylamino-l-alanine (BMAA) is incorporated into protein during synthesis. Amino Acids 2014, 46, 2553–2559. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, O.; Jiang, L.; Andersson, M.; Ilag, L.L.; Brittebo, E.B. Protein association of the neurotoxin and non protein amino acid BMAA (β-N-methylamino-l-alanine) in the liver and brain following neonatal administration in rats. Toxicol. Lett. 2014, 226, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Albano, R.; Lobner, D. Transport of BMAA into neurons and astrocytes by system Xc−. Neurotox. Res. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, R.A.; Cox, P.A.; Banack, S.A.; Rodgers, K.J. The non-protein amino acid BMAA is misincorporated into human proteins in place of l-serine causing protein misfolding and aggregation. PLoS ONE 2013, 8, e75376. [Google Scholar] [CrossRef] [PubMed]
- Perry, T.L.; Bergeron, C.; Biro, A.J.; Hansen, S. β-N-methylamino-l-alanine. Chronic oral administration is not neurotoxic to mice. J. Neurol. Sci. 1989, 94, 173–180. [Google Scholar] [CrossRef]
- Lobner, D.; Piana, P.M.; Salous, A.K.; Peoples, R.W. Beta-N-methylamino-l-alanine enhances neurotoxicity through multiple mechanisms. Neurobiol. Dis. 2007, 25, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Santucci, S.; Zsürger, N.; Chabry, J. β-N-methylamino-l-alanine induced in vivo retinal cell death. J. Neurochem. 2009, 109, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Q.; Rush, T.; Ciske, J.; Lobner, D. Selective death of cholinergic neurons induced by betamethylamino-l-alanine. Neuroreport 2010, 21, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, O.; Berg, A.L.; Lindström, A.K.; Hanrieder, J.; Arnerup, G.; Roman, E.; Bergquist, J.; Lindquist, N.G.; Brittebo, E.B.; Andersson, M. Neonatal exposure to the cyanobacterial toxin BMAA induces changes in protein expression and neurodegeneration in adult hippocampus. Toxicol. Sci. 2012, 130, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Okle, O.; Stemmer, K.; Deschl, U.; Dietrich, D.R. L-BMAA induced ER stress and enhanced caspase 12 cleavage in human neuroblastoma SH-SY5Y cells at low nonexcitotoxic concentrations. Toxicol. Sci. 2013, 131, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, O.; Kultima, K.; Wadensten, H.; Nilsson, A.; Roman, E.; Andrén, P.E.; Brittebo, E.B. Neurotoxin-induced neuropeptide perturbations in striatum of neonatal rats. J. Proteome Res. 2013, 12, 1678–1690. [Google Scholar] [CrossRef] [PubMed]
- Petrozziello, T.; Secondo, A.; Tedeschi, V.; Esposito, A.; Sisalli, M.; Scorziello, A.; Di Renzo, G.; Annunziato, L. ApoSOD1 lacking dismutase activity neuroprotects motor neurons exposed to beta-methylamino-l-alanine through the Ca2+/Akt/ERK1/2 prosurvival pathway. Cell Death Differ. 2017, 24, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Frøyset, A.K.; Khan, E.A.; Fladmark, K.E. Quantitative proteomics analysis of zebrafish exposed to sublethal dosages of β-methyl-amino-l-alanine (BMAA). Sci. Rep. 2016, 6, 29631. [Google Scholar] [CrossRef] [PubMed]
- Beri, J.; Nash, T.; Martin, R.M.; Bereman, M.S. Exposure to BMAA mirrors molecular processes linked to neurodegenerative disease. Proteomics 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Martin-Bastida, A.; Pietracupa, S.; Piccini, P. Neuromelanin in parkinsonian disorders: An update. Int. J. Neurosci. 2017, 127, 1116–1123. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.; Karlsson, O.; Banack, S.A.; Brandt, I. Transfer of developmental neurotoxin β-N-methylamino-l-alanine (BMAA) via milk to nursed offspring: Studies by mass spectrometry and image analysis. Toxicol. Lett. 2016, 258, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, O.; Jiang, L.; Ersson, L.; Malmström, T.; Ilag, L.L.; Brittebo, E.B. Environmental neurotoxin interaction with proteins: Dose-dependent increase of free and protein-associated BMAA (β-N-methylamino-l-alanine) in neonatal rat brain. Sci. Rep. 2015, 5, 15570. [Google Scholar] [CrossRef] [PubMed]
- Murch, S.J.; Cox, P.A.; Banack, S.A.; Steele, J.C.; and Sacks, O.W. Occurrence of beta-methylamino-l-alanine (BMAA) in ALS/PDC patients from Guam. Acta Neurol. Scand. 2004, 110, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Montine, T.J.; Li, K.; Perl, D.P.; Galasko, D. Lack of β-methylamino-l-alanine in brain from controls, AD, or Chamorros with PDC. Neurology 2005, 65, 768–769. [Google Scholar] [CrossRef] [PubMed]
- Isaias, I.U.; Trujillo, P.; Summers, P.; Marotta, G.; Mainardi, L.; Pezzoli, G.; Zecca, L.; Costa, A. Neuromelanin imaging and dopaminergic loss in Parkinson’s disease. Front. Aging Neurosci. 2016, 8, 196. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Zucca, F.A.; Wilms, H.; Sulzer, D. Neuromelanin of the substantia nigra: A neuronal black hole with protective and toxic characteristics. Trends Neurosci. 2003, 26, 578–580. [Google Scholar] [CrossRef] [PubMed]
- D’Amato, R.J.; Lipman, Z.P.; Snyder, S.H. Selectivity of the parkinsonian neurotoxin MPTP: Toxic metabolite MPP+ binds to neuromelanin. Science 1986, 231, 987–989. [Google Scholar] [CrossRef] [PubMed]
- D’Amato, R.J.; Alexander, G.M.; Schwartzman, R.J.; Kitt, C.A.; Price, D.L.; Snyder, S.H. Evidence for neuromelanin involvement in MPTP-induced neurotoxicity. Nature 1987, 327, 324–326. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, O.; Berg, C.; Brittebo, E.B.; Lindquist, N.G. Retention of the cyanobacterial neurotoxin β-N-methylamino-l-alanine in melanin and neuromelanin-containing cells—A possible link between Parkinson-dementia complex and pigmentary retinopathy. Pigment Cell Melanoma Res. 2009, 22, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Jimbow, K. Current update and trends in melanin pigmentation and melanin biology. Keio J. Med. 1995, 44, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Mårs, U.; Larsson, B.S. Pheomelanin as a binding site for drugs and chemicals. Pigment Cell Res. 1999, 12, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Tribl, F.; Arzberger, T.; Riederer, P.; Gerlach, M. Tyrosinase is not detected in human catecholaminergic neurons by immunohistochemistry and western blot analysis. J. Neural Transm. Suppl. 2007, 72, 51–55. [Google Scholar]
- Zucca, F.A.; Basso, E.; Cupaioli, F.A.; Ferrari, E.; Sulzer, D.; Casella, L.; Zecca, L. Neuromelanin of the human substantia nigra: An update. Neurotox. Res. 2014, 25, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Bellei, C.; Costi, P.; Albertini, A.; Monzani, E.; Casella, L.; Gallorini, M.; Bergamaschi, L.; Moscatelli, A.; Turro, N.J.; et al. New melanic pigments in the human brain that accumulate in aging and block environmental toxic metals. Proc. Natl. Acad. Sci. USA 2008, 105, 17567–17572. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, K.; Murase, T.; Zucca, F.A.; Zecca, L.; Ito, S. Biosynthetic pathway to neuromelanin and its aging process. Pigment Cell Melanoma Res. 2012, 25, 792–803. [Google Scholar] [CrossRef] [PubMed]
- Krone, B. The Why of Sporadic Motor Neuron Disease—Many Factors, Still a Mystery? J. Neurol. Neuromed. 2016, 1, 1–5. Available online: http://www.jneurology.com/articles/the-why-of-sporadic-motor-neuron-disease--many-factors-still-a-mystery.pdf (accessed on 21 December 2017). [CrossRef]
- McManus, M.J.; Murphy, M.P.; Franklin, J.L. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 15703–15715. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; Ulusoy, A.; Innamorato, N.G.; Sahin, G.; Rábano, A.; Kirik, D.; Cuadrado, A. α-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum. Mol. Genet. 2012, 21, 3173–3192. [Google Scholar] [CrossRef] [PubMed]
- Dariani, S.; Baluchnejadmojarad, T.; Roghani, M. Thymoquinone attenuates astrogliosis, neurodegeneration, mossy fiber sprouting, and oxidative stress in a model of temporal lobe epilepsy. J. Mol. Neurosci. 2013, 51, 679–686. [Google Scholar] [CrossRef] [PubMed]
- An, T.; Shi, P.; Duan, W.; Zhang, S.; Yuan, P.; Li, Z.; Wu, D.; Xu, Z.; Li, C.; Guo, Y. Oxidative stress and autophagic alteration in brainstem of SOD1-G93A mouse model of ALS. Mol. Neurobiol. 2014, 49, 1435–1448. [Google Scholar] [CrossRef] [PubMed]
- Eleuteri, S.; Di Giovanni, S.; Rockenstein, E.; Mante, M.; Adame, A.; Trejo, M.; Wrasidlo, W.; Wu, F.; Fraering, P.C.; Masliah, E.; et al. Novel therapeutic strategy for neurodegeneration by blocking Aβ seeding mediated aggregation in models of Alzheimer’s disease. Neurobiol. Dis. 2015, 74, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, N.G.; Larsson, B.S.; Lydén-Sokolowski, A. Autoradiography of [14C]paraquat or [14C]diquat in frogs and mice: Accumulation in neuromelanin. Neurosci. Lett. 1988, 93, 1–6. [Google Scholar] [CrossRef]
- Silva, B.A.; Breydo, L.; Fink, A.L.; Uversky, V.N. Agrochemicals, α-synuclein, and Parkinson’s disease. Mol. Neurobiol. 2013, 47, 598–612. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.; Sokoloski, T.D.; Patil, P.N. Binding of dopaminergic drugs by the neuromelanin of the substantia nigra, synthetic melanins and melanin granules. Fed. Proc. 1978, 37, 2403–2407. [Google Scholar] [PubMed]
- Enochs, W.S.; Sarna, T.; Zecca, L.; Swartz, H.M. The roles of neuromelanin binding of metal ions, and oxidative cytotoxicity in the pathogenesis of Parkinson’s disease: A hypothesis. J. Neural Transm. Park. Dis. Dement. Sect. 1994, 7, 83–100. [Google Scholar] [CrossRef] [PubMed]
- Zareba, M.; Bober, A.; Korytowski, W.; Zecca, L.; Sarna, T. The effect of a synthetic neuromelanin on yield of free hydroxyl radicals generated in model systems. Biochim. Biophys. Acta 1995, 1271, 343–348. [Google Scholar] [CrossRef]
- Ito, S.; Wakamatsu, K. Chemical degradation of melanins: Application to identification of dopamine-melanin. Pigment Cell Res. 1998, 11, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Forno, L.S.; Tetrud, J.; Reeves, A.G.; Kaplan, J.A.; Karluk, D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann. Neurol. 1999, 46, 598–605. [Google Scholar] [CrossRef]
- Wilms, H.; Zecca, L.; Rosenstiel, P.; Sievers, J.; Deuschl, G.; Lucius, R. Inflammation in Parkinson’s diseases and other neurodegenerative diseases: Cause and therapeutic implications. Curr. Pharm. Des. 2007, 13, 1925–1928. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Ballard, P. Parkinsonism induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): Implications for treatment and the pathogenesis of Parkinson’s disease. Can. J. Neurol. Sci. 1984, 11, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Duncan, M.W.; Villacreses, N.E.; Pearson, P.G.; Wyatt, L.; Rapoport, S.I.; Kopin, I.J.; Markey, S.P.; Smith, Q.R. 2-amino-3-(methylamino)-propanoic acid (BMAA) pharmacokinetics and blood-brain barrier permeability in the rat. J. Pharmacol. Exp. Ther. 1991, 258, 27–35. [Google Scholar] [PubMed]
- Lewerenz, J.; Maher, P. Chronic glutamate toxicity in neurodegenerative diseases-what is the evidence? Front. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef] [PubMed]
- Van Onselen, R.; Venables, L.; van de Venter, M.; Downing, T.G. β-N-Methylamino-l-Alanine Toxicity in PC12: Excitotoxicity vs. Misincorporation. Neurotox. Res. 2018, 33, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Beebe, K.; Nangle, L.A.; Jang, J.; Longo-Guess, C.M.; Cook, S.A.; Davisson, M.T.; Sundberg, J.P.; Schimmel, P.; Ackerman, S.L. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature 2006, 443, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Marsden, C.D. Pigmentation in the nucleus substantiae nigrae of mammals. J. Anat. 1961, 95, 256–261. [Google Scholar] [PubMed]
- Barden, H.; Levine, S. Histochemical observations on rodent brain melanin. Brain Res. Bull. 1983, 10, 847–851. [Google Scholar] [CrossRef]
- Zeevalk, G.D.; Nicklas, W.J. Acute excitotoxicity in chick retina caused by the unusual amino acids BOAA and BMAA: Effects of MK-801 and kynurenate. Neurosci. Lett. 1989, 102, 284–290. [Google Scholar] [CrossRef]
- Zeevalk, G.D.; Nicklas, W.J. Nitric oxide in retina: Relation to excitatory amino acids and excitotoxicity. Exp. Eye Res. 1994, 58, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.A.; McDarby, J.V.; Lavine, L.; Steele, J.C.; Calne, D.B. A retinopathy on Guam with high prevalence in Lytico-Bodig. Ophthalmology 1989, 96, 1731–1735. [Google Scholar] [CrossRef]
- Campbell, R.J.; Steele, J.C.; Cox, T.A.; Loerzel, A.J.; Belli, M.; Belli, D.D.; Kurland, L.T. Pathologic findings in the retinal pigment epitheliopathy associated with the amyotrophic lateral sclerosis/parkinsonism-dementia complex of Guam. Ophthalmology 1993, 100, 37–42. [Google Scholar] [CrossRef]
- Hanlon, S.D.; Steele, J.C. An unusual retinal pigment epitheliopathy endemic to the island of Guam. Optom. Vis. Sci. 1993, 70, 854–859. [Google Scholar] [CrossRef] [PubMed]
- Steele, J.C.; Wresch, R.; Hanlon, S.D.; Keystone, J.; Ben-Shlomo, Y. A unique retinal epitheliopathy is associated with amyotrophic lateral sclerosis/Parkinsonism-Dementia complex of Guam. Mov. Disord. 2015, 30, 1271–1275. [Google Scholar] [CrossRef] [PubMed]
- Dayhaw-Barker, P. Retinal pigment epithelium melanin and ocular toxicity. Int. J. Toxicol. 2002, 21, 451–454. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Steele, J.C. The ALS/PDC syndrome of Guam: Potential biomarkers for an enigmatic disorder. Prog. Neurobiol. 2011, 95, 663–669. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delcourt, N.; Claudepierre, T.; Maignien, T.; Arnich, N.; Mattei, C. Cellular and Molecular Aspects of the β-N-Methylamino-l-alanine (BMAA) Mode of Action within the Neurodegenerative Pathway: Facts and Controversy. Toxins 2018, 10, 6. https://doi.org/10.3390/toxins10010006
Delcourt N, Claudepierre T, Maignien T, Arnich N, Mattei C. Cellular and Molecular Aspects of the β-N-Methylamino-l-alanine (BMAA) Mode of Action within the Neurodegenerative Pathway: Facts and Controversy. Toxins. 2018; 10(1):6. https://doi.org/10.3390/toxins10010006
Chicago/Turabian StyleDelcourt, Nicolas, Thomas Claudepierre, Thomas Maignien, Nathalie Arnich, and César Mattei. 2018. "Cellular and Molecular Aspects of the β-N-Methylamino-l-alanine (BMAA) Mode of Action within the Neurodegenerative Pathway: Facts and Controversy" Toxins 10, no. 1: 6. https://doi.org/10.3390/toxins10010006