Survey of Candidate Genes for Maize Resistance to Infection by Aspergillus flavus and/or Aflatoxin Contamination

 ,
, {kind=link}
Abstract
:1. Introduction
2. Results
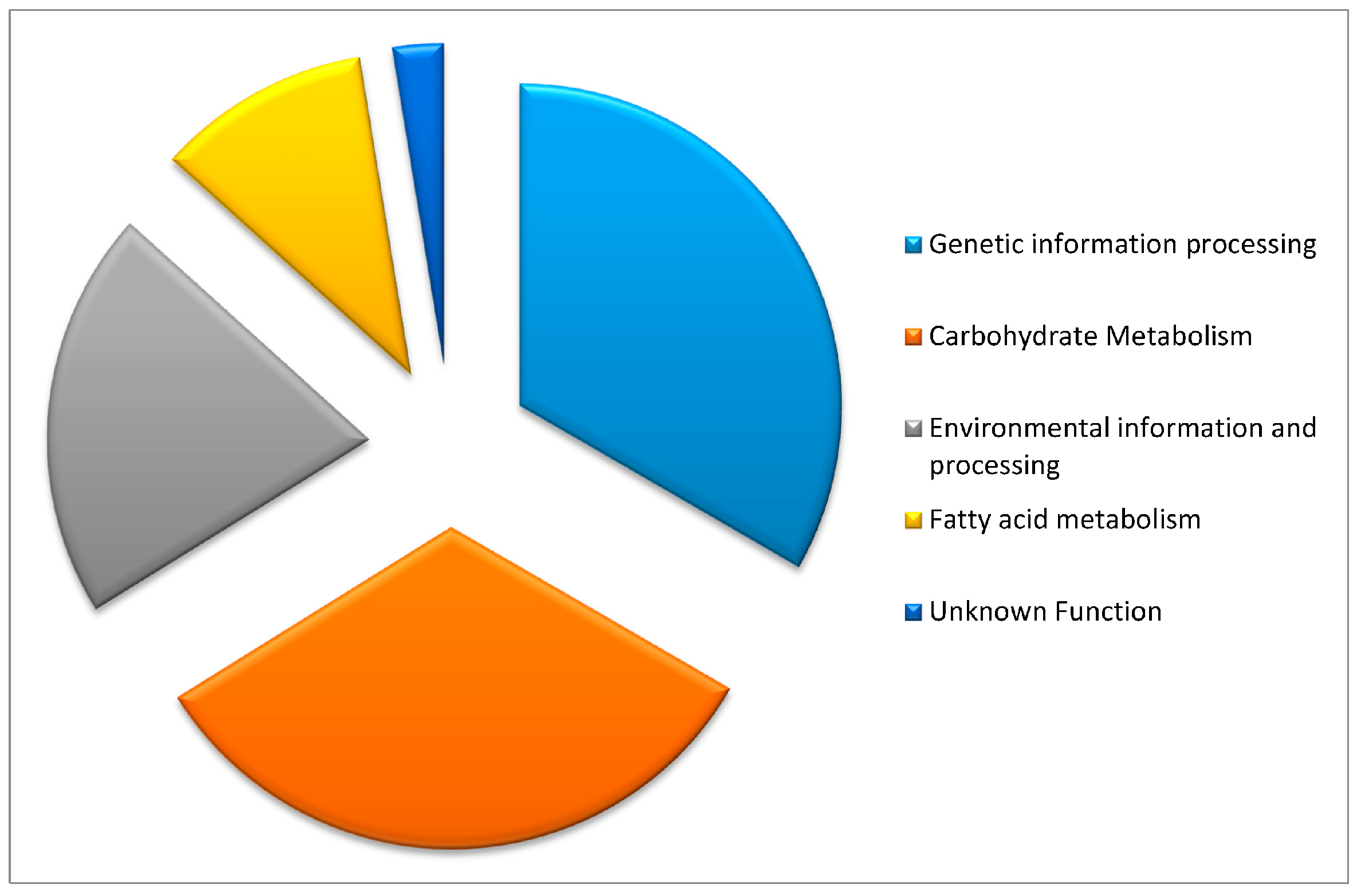
2.1. Candidate Gene Functions
2.2. Significant Associations or Linkages to Phenotypic Effects
3. Discussion and Conclusions
4. Materials and Methods
4.1. Classification of Candidate Genes
4.2. Materials and Methods
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Agricultural Supply and Demand Estimates. Available online: http://usda.mannlib.cornell.edu/usda/waob/wasde//2010s/2017/wasde-07-12-2017.pdf (accessed on 9 September 2017).
- Mitchell, N.; Bowers, E.J.; Hurburgh, C.; Wu, F. Potential economic losses to the US corn industry from aflatoxin contamination. Food Addit. Contam. Part A 2016, 33, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Warburton, M.L.; Williams, W.P.; Hawkins, L.; Bridges, S.; Gresham, C.; Harper, J.; Ozkan, S.; Mylroie, J.E.; Shan, X. A public platform for the verification of the phenotypic effect of candidate genes for resistance to aflatoxin accumulation and Aspergillus flavus infection in maize. Toxins 2011, 3, 754–765. [Google Scholar] [CrossRef] [PubMed]
- Warburton, M.; Williams, W. Aflatoxin resistance in maize: What have we learned lately? Adv. Bot. 2014. [Google Scholar] [CrossRef]
- Mideros, S.X.; Warburton, M.L.; Jamann, T.M.; Windham, G.L.; Williams, W.P.; Nelson, R.J. Quantitative trait loci influencing mycotoxin contamination of maize: Analysis of linkage mapping, characterization of near-isogenic lines, and meta-analysis. Crop Sci. 2014, 54, 127–142. [Google Scholar] [CrossRef]
- Kelley, R.Y.; Williams, W.P.; Mylroie, J.E.; Boykin, D.L.; Hawkins, L.K.; Windham, G.L.; Brooks, T.D.; Bridges, S.M.; Scheffler, B.E.; Wilkinson, J.K. Genomic profile of maize response to Aspergillus flavus infection. Toxin Rev. 2009, 28, 129–141. [Google Scholar] [CrossRef]
- Peethambaran, B.; Hawkins, L.K.; Windham, G.L.; Williams, W.P.; Luthe, D.S. Anti-fungal activity of maize silk proteins and role of chitinases in Aspergillus flavus resistance. Toxin Rev. 2009, 29, 27–39. [Google Scholar] [CrossRef]
- Pechanova, O.; Pechan, T. Maize-pathogen interactions: An ongoing combat from a proteomics perspective. Int. J. Mol. Sci. 2015, 16, 28429–48448. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.D.; Perkins, A.; Williams, W.P.; Warburton, M.L. Using genome-wide associations to identify metabolic pathways involved in maize aflatoxin accumulation resistance. BMC Genom. 2015, 16, 673. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.Y.; Gresham, C.; Harper, J.; Bridges, S.M.; Warburton, M.L.; Hawkins, L.K.; Pechanova, O.; Peethambaran, B.; Pechan, T.; Luthe, D.S.; et al. Integrated database for identifying candidate genes for Aspergillus flavus resistance in maize. BMC Bioinform. 2010, 11, S25. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, L.K.; Mylroie, J.E.; Oliveira, D.A.; Smith, J.S.; Ozkan, S.; Windham, G.L. Characterization of the maize chitinase genes and their effect on Aspergillus flavus and aflatoxin accumulation resistance. PLoS ONE 2015, 10, e0126185. [Google Scholar] [CrossRef] [PubMed]
- Ogunola, O.F.; Hawkins, L.K.; Mylroie, E.; Kolomiets, M.V.; Borrego, E.; Tang, J.D.; Williams, W.P.; Warburton, M.L. Characterization of the maize lipoxygenase gene family in relation to aflatoxin accumulation resistance. PLoS ONE 2017, 12, e0181265. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. Kegg: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- KEGG Pathway Database. Available online: http://www.genome.jp/kegg/pathway.html (accessed on 11 November 2017).
- Berger, S.; Sinha, A.; Roitsch, T. Plant physiology meets phytopathology: Plant primary metabolism and plant-pathogen interactionc. J. Exp. Bot. 2007, 58, 4019–4026. [Google Scholar] [CrossRef] [PubMed]
- Smeekens, S.; Ma, J.; Hanson, J.; Rolland, F. Sugar signals and molecular networks controlling plant growth. Curr. Opin. Plant Biol. 2010, 13, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Liu, H.; Deng, Y.; Xiao, J.; Li, X.; Wang, S. The WRKY45-2 WRKY13 WRKY42 transcriptional regulatory cascade is required for rice resistance to fungal pathogen. Plant Physiol. 2015, 167, 1087–1099. [Google Scholar] [CrossRef] [PubMed]
- Fountain, J.; Scully, B.; Ni, X.; Kemerait, R.; Lee, D.; Chen, Z.-Y.; Guo, B. Environmental influences on maize-Aspergillus flavus interactions and aflatoxin production. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Fountain, J.C.; Raruang, Y.; Luo, M.; Brown, R.L.; Guo, B.; Chen, Z.-Y. Potential role of WRKY transcription factors in regulating host defense responses during Aspergillus flavus infection of immature maize kernels. Physiol. Mol. Plant Pathol. 2014, 89, 31–40. [Google Scholar] [CrossRef]
- Fountain, J.C.; Khera, P.; Yang, L.; Nayak, S.N.; Scully, B.T.; Lee, R.D.; Chen, Z.-Y.; Kemerait, R.C.; Varshney, R.K.; Guo, B. Resistance to Aspergillus flavus in maize and peanut: Molecular biology, breeding, environmental stress, and future perspectives. Crop J. 2015, 3, 229–237. [Google Scholar] [CrossRef]
- Pandey, S.; Somssich, I. The role of WRKY transcription factors in plant immunity. Plant Physiol. 2009, 150, 1648–1655. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Feng, B.; Yang, S.; Huang, Y.; Tang, Y. The R2R3-MYB transcription factor gene family in maize. PLoS ONE 2012, 7, e37463. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.; Bevan, M.; Brutnell, T.; Buell, C.; Cone, K.; Hake, S.; Jackson, D.; Kellogg, E.; Lawrence, C.; McCouch, S.; et al. A recommendation for naming transcription factor proteins in grasses. Plant Physiol. 2009, 149, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, A., Jr.; Nishiyama, M.Y.; Fuentes, B.; Souza, G.; Janies, D.; Gray, J.; Grotewold, E. Grassius: A platform for comparative regulatory genomics across the grasses. Plant Physiol. 2009, 149, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Heine, G.; Malik, V.; Dias, A.; Grotewold, E. Expression and molecular characterization of zmmyb-if35 and related R2R2-MYB transcription factors. Mol. Biotechnol. 2007, 37, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, P.; Carrari, F. ASR1 transcription factor and its role in metabolism. Plant Signal. Behav. 2015, 10, e992751. [Google Scholar] [CrossRef] [PubMed]
- Virlouvet, L.; Jacquemot, M.; Gerentes, D.; Corti, H.; Bouton, S.; Gilard, F.; Valot, B.; Trouverie, J.; Tchezkez, G.; Falque, M.; et al. The ZMARS1 protein influences branched-chain amino acid biosynthesis and maintains kernel yield in maize under water-limited conditions. Plant Physiol. 2011, 157, 917–936. [Google Scholar] [CrossRef] [PubMed]
- Stemmer, C.; Schwander, A.; Bauw, G.; Fojan, P.; Grasser, K. Protein kinase CK2 differentially phosphorylates maize chromosomal high mobility group B (HMGB) proteins modulating their stability and DNA interactions. J. Biol. Chem. 2002, 277, 1092–1098. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, Z.; Liu, D.; Zhang, K.; Li, A.; Mao, L. Squamosa promoter-binding protein-like transcription factors: Star players for plant growth and development. J. Integr. Plant Biol. 2010, 52, 946–951. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Cheng, Y.; Cheng, X.; Jiang, H.; Zhu, S.; Cheng, B. Indentiification and characterization of dicer-like, argonaute, and RNA dependent RNA polymerase gene families in maize. Plant Cell Rep. 2011, 30, 1347. [Google Scholar] [CrossRef] [PubMed]
- Pegoraro, C.; Mertz, L.; da Maia, L.; Rombaldi, C.; de Oliveira, A. Importance of heat shock proteins in maize. J. Crop Sci. Biotech. 2011, 14, 85–95. [Google Scholar] [CrossRef]
- Garg, A.; Kim, J.-K.; Owens, T.; Ranwala, A.; Choi, Y.; Kochian, L.; Wu, R. Trehalose accumulation in rice plants conferes high tolerance to different abiotic stresses. Proc. Natl. Acad. Sci. USA 2002, 99, 15898–15903. [Google Scholar] [CrossRef] [PubMed]
- Bravo, J.M.; Campo, S.; Murillo, I.; Coca, M.; Segundo, B.S. Fungus- and wound-induced accumulation of mrna containing a class II chitinase of the pathogenesis-related protein 4 (PR-4) family of maize. Plant Mol. Biol. 2003, 52, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Cletus, J.; Balasubramanian, V.; Vashist, D.; Sakthivel, N. Transgenic expression of plant chitinases to enhance disease resistance. Biotechnol. Lett. 2013, 35, 1719–1732. [Google Scholar] [CrossRef] [PubMed]
- Cordero, M.; Raventos, D.; Segungo, B.S. Differential expression and induction of chitinases and β-1-3-glucanases in response to fungal infection during germination of maize seeds. MPMI 1994, 7, 23–31. [Google Scholar] [CrossRef]
- Dowd, P.F.; Naumann, T.A.; Price, N.P.J.; Johnson, E.T. Identification of a maize (Zea mays) chitinase allele sequence suitable for a role in ear rot fungal resistance. Agri Gene 2018, 7, 15–22. [Google Scholar] [CrossRef]
- Zhou, M.; Zhang, Q.; Sun, Z.; Chen, L.H.; Liu, B.X.; Zhang, K.X.; Zhu, X.M.; Shao, J.R.; Tang, Y.X.; Wu, Y.M. Trehalose metabolism-related genes in maize. J. Plant Growth Reg. 2014, 33, 256–271. [Google Scholar] [CrossRef]
- Rademacher, E.; Offringa, R. Evolutionay adaptation of plant AGC kinases: From light signaling to cell polarity regulation. Front. Plant Sci. 2012, 3, 250. [Google Scholar] [CrossRef] [PubMed]
- Luan, S.; Kudla, J.; Rodriguez-Concepcion, M.; Yalovsky, S.; Gruissem, W. Calmodulins and calcineurin b-like proteins. Plant Cell 2002, 14 (Suppl. 1), s389–s400. [Google Scholar] [CrossRef] [PubMed]
- Van Loon, L.; Van Strien, E. The families of pathogenesis-related proteins, their activities, and comparative analysis of PR-1 type proteins. Physiol. Mol. Plant Pathol. 1999, 55, 85–97. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Brown, R.L.; Damann, K.E.; Cleveland, T.E. Identification of maize kernel endosperm proteins associated with resistance to aflatoxin contamination by Aspergillus flavus. Phytopathology 2007, 97, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Zeier, J. New insights into the regulation of plant immunity by amino acid metabolic pathways. Plant Cell Environ. 2013, 36, 2085–2103. [Google Scholar] [CrossRef] [PubMed]
- Kadotani, N.; Akagi, A.; Takatsuji, H.; Miwa, T.; Igarashi, D. Exogenous proteinogenic amino acids induce systemic resistance in rice. BMC Plant Biol. 2016, 16, 60. [Google Scholar] [CrossRef] [PubMed]
- Black, C.A.; Karban, R.; Godfrey, L.D.; Granett, J.; Chaney, W.E. Jasmonic acid: A vaccine against leafminers (diptera: Agromyzidae) in celery. Environ. Entomol. 2003, 32, 1196–1202. [Google Scholar] [CrossRef]
- Tzin, V.; Fernandez-Pozo, N.; Richter, A.; Schmelz, E.; Shoettner, M.; Schafer, M.; Ahern, K.; Meihls, L.; Kaur, H.; Huffaker, A.; et al. Dynamic maize responses to aphid feeding are revealed by time series of transcriptomic and metabolomic assays. Plant Physiol. 2015, 169, 1727–1743. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Brefort, T.; Neidig, N.; Djamei, A.; Kahnt, J.; Vermerris, W.; Koenig, S.; Feussner, K.; Feussner, I.; Kahman, R. A secreted Ustilago maydis effector promotes virulence by targeting anthocyanin biosynthesis in maize. eLife 2014, 3, e01355. [Google Scholar] [CrossRef] [PubMed]
- Joshi, C.; Chiang, V. Conserved sequence motifs in plant s-adenosyl-l-methionine-dependent methyltransferases. Plant Mol. Biol. 1998, 37, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Olukolu, B.; Bian, Y.; De Vries, B.; Tracy, W.F.; Wisser, R.; Holland, J.; Balint-Kurti, P. The genetics of leaf flecking in maize and its relationship to plant defense and disease resistance. Plant Physiol. 2016, 172, 1787–1803. [Google Scholar] [CrossRef] [PubMed]
- Koo, A.J.K.; Howe, G.A. Role of peroxisomal B-oxidation in the production of plant signaling compounds. Plant Signal. Behav. 2007, 2, 20–22. [Google Scholar] [CrossRef] [PubMed]
- Warburton, M.; Tang, J.; Windham, G.; Hawkins, L.; Murray, S.; Xu, W.; Boykin, D.; Perkins, A.; Williams, W. Genome-wide association mapping of Aspergillus flavus and aflatoxin accumulation resistance in maize. Crop Sci. 2015, 55, 1857–1867. [Google Scholar] [CrossRef]
- Nolte, S.; Young, B.; Tolley, L.; Gibson, D.; Young, J.; Lightfoot, D. Translocation and metabolic fingerprint effects in GDHA-transformed tobacco. Crop Sci. 2017, 57, 350–364. [Google Scholar] [CrossRef]
- Joshi, R.; Wani, S.H.; Singh, B.; Bohra, A.; Dar, Z.A.; Lone, A.A.; Pareek, A.; Singla-Pareek, S.L. Transcription factors and plants response to drought stress: Current understandings and future directions. Front. PLant Sci. 2016, 7, 1029. [Google Scholar] [CrossRef] [PubMed]
- Rojas, C.M.; Senthil-Kumar, M.; Tzin, V.; Mysore, K.S. Regulation of primary plant metabolism during plant-pathogen interactions and its contribution to plant defense. Front. Plant Sci. 2017, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.; Chen, Z.; Warburton, M.; Luo, M.; Menkir, A.; Fakhoury, A.; Bhatnagar, D. Discovery and characterization of proteins associated with aflatoxin-resistance: Evaluating their potential as breeding markers. Toxins 2010, 2, 919–933. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, P.; Ware, D.; Ni, J.; Chang, K.; Zhao, W.; Schmidt, S.; Pan, S.; Clark, K.; Teytelman, L.; Cartinhour, S.; et al. Gramene: Development and integration of trait and gene ontologies for rice. Comp. Funct. Genom. 2002, 3, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Bastian, F.; Chibucos, M.; Gaudet, P.; Giglio, M.; Holliday, G.; Huang, H.; Lewis, S.; Niknejad, A.; Orchard, S.; Poux, S.; et al. The confidence information ontology: A step toward a standard for asserting confidence in annotations. Database 2015. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.U. Uniprot: A hub for protein information. Nucleic Acids Res. 2014, 43, D204–D212. [Google Scholar] [CrossRef] [PubMed]
- Goodstein, D.; Shu, S.; Howson, R.; Neupane, R.; Haye, R.; Faso, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.; Coggill, P.; Eberhardt, R.; Eddy, S.; Mistry, J.; Mitchel, A.; Potter, S.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Jiang, C.; Li, X.; Sheng, L.; Dong, Q.; Peng, X.; Li, Q.; Zhao, Y.; Jiang, H.; Cheng, B. Pigd: A database for intronless genes in the poaceae. BMC Genom. 2014, 15, 832. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Kegg as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Willcox, M.C.; Davis, G.L.; Windham, G.L.; Abbas, H.K.; Betrán, J.; Holland, J.B.; Williams, W.P.; Warburton, M.L. Confirming quantitiatve trait loci for aflatoxin resistance from mp313e in different genetic backgrounds. Mol. Breed. 2013, 32, 15–26. [Google Scholar] [CrossRef]
- Brooks, T.; Williams, W.P.; Windham, G.L.; Willcox, M.; Abbas, H.K. Quantitative trait loci contributing resistance to aflatoxin accumulation in the maize inbred mp313e. Crop Sci. 2005, 45, 171–174. [Google Scholar]
- Warburton, M.; Brooks, T.; Windham, G.; Williams, W. Identification of QTL contributing resistance to aflatoxin accumulation in maize. Mol. Breed. 2011, 27, 491–499. [Google Scholar] [CrossRef]
- Warburton, M.L.; Brooks, T.D.; Krakowsky, M.D.; Shan, X.; Windham, G.L.; Williams, W.P. Identification and mapping of new sources of resistance to aflatoxin accumulation in maize. Crop Sci. 2009, 49, 1403–1408. [Google Scholar] [CrossRef]
- Zummo, N.; Scott, G.E. Cob and kernel infection by Aspergillus flavus and Fusarium moniliforme in inoculated, field-grown maize ears. Plant Dis. 1990, 74, 627–630. [Google Scholar] [CrossRef]
- Windham, G.L.; Williams, W.P. Aspergillus flavus infection and aflatoxin accumulation in resistant and susceptible maize hybrids. Plant Dis. 1998, 82, 281–284. [Google Scholar] [CrossRef]
- Ooijen, J.V. Joinmap 4: Software for the Calculation of Genetic Linkage Maps in Experimental Populations; Kyazma, B.B., Ed.; ResearchGate: Wageningen, The Netherlands, 2006. [Google Scholar]
- Wang, S.; Basten, C.; Zeng, Z.-B. Windows QTL Cartographer 2.5; North Carolina State University: Raleigh, NC, USA, 2012. [Google Scholar]
- Doerge, R.; Churchill, G. Permutation tests for multiple loci affecting a quantitative character. Genetics 1996, 142, 285–294. [Google Scholar] [PubMed]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Pritcharsd, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Liu, K.; Muse, V. PowerMarker: Integrated analysis environment for genetic marker data. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Sekhon, R.; Lin, H.; Childs, K.; Hansey, C.; Buell, C.; deLeon, N.; Kaeppler, S. Genome-wide atlas of transcription during maize development. Plant J. 2011, 66, 553–563. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hawkins, L.K.; Warburton, M.L.; Tang, J.D.; Tomashek, J.; Alves Oliveira, D.; Ogunola, O.F.; Smith, J.S.; Williams, W.P. Survey of Candidate Genes for Maize Resistance to Infection by Aspergillus flavus and/or Aflatoxin Contamination. Toxins 2018, 10, 61. https://doi.org/10.3390/toxins10020061
Hawkins LK, Warburton ML, Tang JD, Tomashek J, Alves Oliveira D, Ogunola OF, Smith JS, Williams WP. Survey of Candidate Genes for Maize Resistance to Infection by Aspergillus flavus and/or Aflatoxin Contamination. Toxins. 2018; 10(2):61. https://doi.org/10.3390/toxins10020061
Chicago/Turabian StyleHawkins, Leigh K., Marilyn L. Warburton, Juliet D. Tang, John Tomashek, Dafne Alves Oliveira, Oluwaseun F. Ogunola, J. Spencer Smith, and W. Paul Williams. 2018. "Survey of Candidate Genes for Maize Resistance to Infection by Aspergillus flavus and/or Aflatoxin Contamination" Toxins 10, no. 2: 61. https://doi.org/10.3390/toxins10020061