The Impact of Uremic Toxins on Cerebrovascular and Cognitive Disorders
by
 and
and
Maryam Assem
1, 
Mathilde Lando
1,
Maria Grissi
2,
Saïd Kamel
2,3,
Ziad A. Massy
4,5,
Jean-Marc Chillon
2,6,* and
Lucie Hénaut
2 1
Nephrology Department, Amiens University Hospital, 80054 Amiens, France
2
MP3CV, EA7517, CURS, Jules Verne University of Picardie, 80054 Amiens, France
3
Biochemistry Laboratory, Amiens University Hospital, 80054 Amiens, France
4
Division of Nephrology, Ambroise Paré University Hospital, APHP, Boulogne-Billancourt, 92100 Paris, France
5
Inserm U1018, Team 5, CESP, UVSQ, Paris-Saclay University, 94800 Villejuif, France
6
DRCI, Amiens University Hospital, 80054 Amiens, France
*
Author to whom correspondence should be addressed.
Toxins 2018, 10(7), 303; https://doi.org/10.3390/toxins10070303
Submission received: 20 June 2018
/
Revised: 17 July 2018
/
Accepted: 19 July 2018
/
Published: 22 July 2018
(This article belongs to the Special Issue Uremia and Cardiovascular Disease)
{kind=link}
{kind=link}
Abstract
:Individuals at all stages of chronic kidney disease (CKD) have a higher risk of developing cognitive disorders and dementia. Stroke is also highly prevalent in this population and is associated with a higher risk of neurological deterioration, in-hospital mortality, and poor functional outcomes. Evidence from in vitro studies and in vivo animal experiments suggests that accumulation of uremic toxins may contribute to the pathogenesis of stroke and amplify vascular damage, leading to cognitive disorders and dementia. This review summarizes current evidence on the mechanisms by which uremic toxins may favour the occurrence of cerebrovascular diseases and neurological complications in CKD.
Key Contribution: This review summarizes our current knowledge about the involvement of uremic toxins in cerebrovascular and cognitive disorders in patients with chronic kidney disease. We specifically focused on the effects of uremic toxins, such as indoxyl sulfate, and oxidative stress on cerebrovascular and cognitive disorders.
1. Introduction
Chronic kidney disease (CKD) is associated with alterations of vascular functions, blood chemistry, and red blood cell production [1]. These dysfunctions impair blood perfusion, and this can subsequently affect brain functions. Indeed, cerebrovascular diseases are common in CKD patients, who display an increased rate of cognitive disorders and dementia [2,3] and a greater burden of abnormal brain white matter disease [4]. Cognitive impairments in CKD can be partly related to vascular dementia, as dialysis patients with cognitive disorders also present numerous cortical ischemic lesions [5,6,7]. Indeed, the risk of transient ischemic attack and stroke increases with progressive kidney function decline [8,9,10,11,12] and stroke in these patients is associated with more severe neurological deterioration, poorer functional recovery [13,14], and increased mortality [15,16].
In addition to traditional cardiovascular risk factors, such as hypertension, diabetes, inflammation, and dyslipidemia, non-traditional risk factors related to kidney injury may predispose CKD patients to cerebrovascular diseases. These non-traditional risk factors include the CKD-associated disorders of bone and mineral metabolism (CKD-MBD), inflammation, and oxidative stress. Oxidative stress is a key inducer of peripheral vascular dysfunction, characterized by impaired endothelial function, increased atherosclerosis, and vascular calcification. These vascular dysfunctions, which alter the general hemodynamics, increase the risk of cerebrovascular events, such as stroke. In addition, reactive oxygen species (ROS) exert damaging effects on the cerebral vasculature during stroke by increasing vasodilation, altering vascular reactivity, and destroying the blood-brain barrier (BBB) [17,18]. Oxidative entities also contribute to post-stroke DNA damage and cell death during ischemic stroke [19]. In patients with neurodegenerative diseases, ROS production exacerbates the expression of inflammatory mediators [20]. This neuroinflammation, together with the increased oxidative stress, contributes to the pathogenesis of neuronal degeneration [21] and can cause cell membrane damage as a result of lipid peroxidation, changes in protein structure and function due to protein oxidation, and structural DNA damage, hallmarks of several neurodegenerative diseases [22,23].
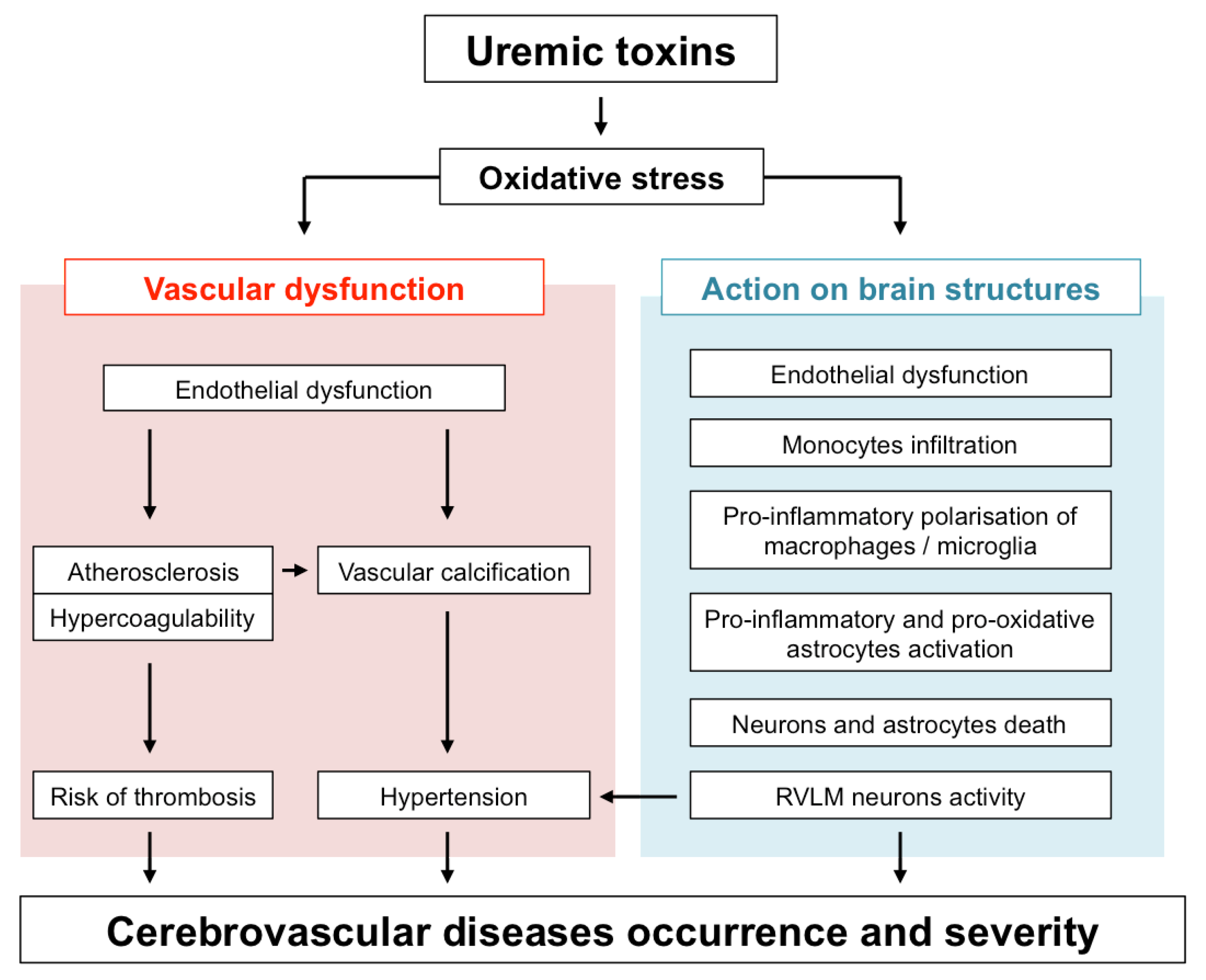
Protein-bound uremic toxins (UTs) such as indoxyl sulfate (IS) or paracresyl sulfate (PCS) are strong inducers of oxidative stress (for review see [24]). IS, PCS and guanidino compounds are highly expressed in uremic brains [25,26]. In addition, cerebrospinal fluid and brain levels of certain guanidine compounds, such as creatinine and methylguanidine, are substantially elevated in uremic patients [26]. Interestingly, these high toxin concentrations (up to 10-fold higher in CKD patients than in controls) were found in brain regions that play a determinant role in cognition, such as the thalamus, mammillary bodies, and cerebral cortex [26]. In this context, it is tempting to speculate that UTs-induced oxidative stress in these brain regions may directly impact the local microcirculation as well as brain-resident cells, therefore contributing to cognitive disorders and poor stroke recovery in CKD patients. This review summarizes our current knowledge on the mechanisms by which UTs-induced oxidative stress may promote both large vessels and microvascular dysfunction and how their subsequent effect on infiltrated macrophages, microglia, astrocytes, and neurons promotes brain damage. An overview of these mechanisms is provided in Figure 1.
2. Impact of Uremic Toxins on Large Vessels Functionality
2.1. Regulation of Blood Pressure
2.1.1. IS, Uric Acid, and Methylguanidine
Hypertension is the main risk factor for stroke [27]. Patients with CKD often present hypertension, but the cause of this hypertension, apart from sodium and fluid retention, has not been fully elucidated. The rostral ventrolateral medulla (RVLM), which contains presympathetic neurons, is known to be a pivotal region that regulates blood pressure [28]. According to a recent study, superfusion of bulbospinal RVLM neurons with uric acid, IS, and methylguanidine increased their activity, as evidenced by depolarization and an increased number of action potentials [29]. In this study, UT-induced activities of the RVLM neurons were suppressed by the addition of an antioxidant drug, suggesting that UT-induced oxidative stress plays a key role in bulbospinal RVLM neuron activation. The authors concluded that ROS production by UTs may cause hypertension by activating RVLM neurons [29]. Since antihypertensive drugs may decrease age-related cognitive decline and dementia [30,31], and given the positive correlation observed between hemodynamic impairment and cognitive impairment in the early stages of dementia [32,33], targeting UT-induced activation of RVLM neurons could be a promising approach to reduce hypertension and subsequent neurological complications.
2.1.2. Lanthionine
Uremic patients undergoing hemodialysis display high concentrations of the sulfur amino acid derivative lanthionine, which has been recently proposed as a UT [34]. Lanthionine, a natural nonproteogenic amino acid, is an analog of cysteine [35], which is considered as a stable product of hydrogen sulphide (H2S) metabolism and an indirect biomarker of its production [36,37]. Cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE), two key enzymes in H2S production, utilize as substrates cysteine and homocysteine, whose levels are both increased in uremic patients [36]. Interestingly, lanthionine is involved in CBS inhibition [38], which impairs H2S production in cell cultures [34].
In recent years, H2S, which is considered as the third gaseous mediator with nitric oxide (NO) and carbon monoxide (CO), has become recognized as an important endogenous vasodilator [39,40,41]. In the first report on this subject, H2S relaxed rat aortic tissues in vitro [42]. In this study, exposure to H2S enhanced NO-induced smooth muscle relaxation in the thoracic aorta, suggesting that the endogenous H2S may regulate smooth muscle tone in synergy with NO. In a subsequent study, an intravenous bolus injection of H2S was shown to transiently decrease rats’ blood pressure [43]. In this study, a small portion of the H2S-induced vasorelaxation was attenuated by either removal of the endothelium or the application of L-NAME (an inhibitor of NO synthase) in the presence of the endothelium, suggesting that a part of the vasodilator actions of H2S results from NO production. Confirming these data, mice displaying decreased H2S production due to genetic deletion of CSE showed significant hypertension and diminished endothelial vasorelaxation [44]. In this context, it is conceivable that the hypertension observed in CKD may be linked, at least in part, to lanthionine-induced CBS inhibition and subsequently impaired H2S production. Further studies will be needed to clarify this concern.
2.2. Vascular Dysfunction
Evidence from in vitro and clinical studies suggests that the actions of UTs on the vascular system may predispose to neurological disorders. For instance, UT-induced oxidative stress accelerates the progression of atherosclerosis [45,46] and endothelial dysfunction [47,48,49,50,51], both of which are associated with an increased risk of dementia and stroke [52,53]. In addition, inorganic phosphate (Pi), IS, TNF-α, IL-6, and advanced glycation end-products have been reported to promote the development of vascular calcification [54,55], at least in part through increased oxidative stress. This phenomenon rigidifies arteries and contributes to the onset of hypertension, the main risk factor for stroke. Of interest, the presence of intracranial artery calcification has been reported to be associated with mortality and vascular events after hospital discharge in patients with ischemic stroke [56]. Increased intracranial artery calcification in response to UTs in CKD could, therefore, be responsible for the poor stroke outcomes observed in CKD patients.
2.3. Hemostasis
Thrombosis and hemostasis impact both the occurrence and outcomes of cerebrovascular disease [57]. Of interest, patients with end-stage renal disease (ESRD) exhibit features of a hypercoagulable state [58]. Normal endothelium exerts anticoagulant and antithrombotic effects and, therefore, plays a pivotal role in hemostasis [59]. Indeed, platelet adhesion and aggregation are inhibited by glycosaminoglycan, NO, prostacyclin, endothelin-1, and ectonucleotidases present in endothelial cells. Interestingly, UTs such as IS have been reported to be associated with enhanced hemostatic disorders and may, therefore, represent interesting therapeutic targets in the prevention of cerebrovascular disease.
2.3.1. Indoxyl Sulfate
In 2011, Kaminski and colleagues reported the existence of a correlation between hemostatic factors such as tissue factor, von Willebrand factor, thrombomodulin, soluble urokinase-type plasminogen activator receptor, soluble intercellular adhesion molecule-1, and soluble vascular cell adhesion protein and the fraction of IS in CKD patients not undergoing hemodialysis [60]. In this study, levels of IS were independently associated with markers of impaired endothelial function (thrombomodulin, adhesion molecules), oxidative stress (superoxide-dismutase), and monocyte activation (neopterin). Interestingly, parameters that correlated with the levels of IS (von Willebrand factor, soluble urokinase-type plasminogen activator receptor, soluble intercellular adhesion molecule-1) were positively associated with the prevalence of cardiovascular disease in CKD patients. The authors concluded that IS may be one of the key links existing between impaired renal function and the prevalence of cardiovascular events, especially hemostatic disorders, through altered monocyte activation, intensified inflammatory processes, and augmented oxidative stress.
2.3.2. Homocysteine
Similarly, hyperhomocysteinemia, which is common in CKD patients [61], has a direct prothrombotic effect on the vascular system and may therefore lead to both large- and small-vessel disease [62]. Homocysteine is a thiol-containing amino acid derived from the metabolism of dietary methionine. Moderately elevated plasma homocysteine levels are an important independent risk factor for arterial and venous thrombosis [63,64]. Numerous mechanisms have been postulated by which hyperhomocysteinemia may induce thrombosis. Indeed, early studies performed in rabbits showed that hyperhomocysteinemia is associated with abnormalities in the key coagulation protein fibrinogen [65,66]. This acquired dysfibrinogenemia is characterized by formation of clots composed of abnormally thin, tightly packed fibers with an increased resistance to fibrinolysis. These data suggested that hyperhomocysteinemia might directly promote thrombosis by interfering with the normal process by which intravascular clots are removed [67]. Most hypotheses also involve injury to the vascular endothelium [68,69] or some alteration in endothelial function, such as decreased expression of the anticoagulant regulatory protein thrombomodulin [70], anticoagulant heparans [71], or binding sites for tissue plasminogen activator [72,73]. In addition, data from in vitro studies reported that the induction of tissue factor expression by cultured umbilical vein endothelial cells [74] and circulating monocytes might be a plausible mechanism by which homocysteine may induce thrombosis [75]. Deficiency of folic acid is a treatable cause of hyperhomocysteinemia. Interestingly, the decrease in homocysteine observed after folic acid treatment in human is accompanied by a decrease in the procoagulatory potential, characterized by decreased fibrinogen (with a procoagulation potential) and increased plasminogen (with an anti-coagulatory potential) [76]. Confirming the potential role of homocysteine-induced thrombosis on the onset of cerebrovascular disease, the China Stroke Primary Prevention Trial recently showed the benefits of folic acid, as a mean to reduce homocysteinemia and to prevent stroke in Chinese adults with hypertension [77].
2.3.3. Other UTs
Guanidino compounds, that are known to be associated with cognitive disorders as discussed below, have been shown to induce elevation of serum homocysteine [78]. In the same manner, CBS inhibition by lanthionine might be responsible for the degree of hyperhomocysteinemia observed in uremia. This suggests that these UTs may also impair brain function indirectly through dysregulated hemostasis. Therefore, targeting lanthionine and guanidino compounds may represent a promising strategy to reduce the hyperhomocysteinema and subsequent dysregulated hemostasis observed in CKD. The hypercoagulability observed in ESRD patients could also be linked to the kynurenine (KYN) pathway since KYN metabolites were reported to be significantly associated with elevated prothrombin factors 1 + 2 in dialysis patients [58].
2.4. Atrial Fibrillation
CKD is associated with a higher incidence of atrial fibrillation [79], which increases the risk of thromboembolic stroke, heart failure, and mortality [80]. However, the arrhythmogenic mechanisms linked to CKD are not fully elucidated.
Indoxyl Sulfate
In ex vivo experiments, exposure to IS increased rabbits pulmonary vein and left atrium arrhythmogenesis and reduced sinoatrial nodes pacemaker activity through oxidative stress [81]. In addition, data obtained from in vitro experiments reported that exposure to IS significantly increased neonatal rat cardiac fibroblast collagen synthesis and myocyte hypertrophy [82]. In this study, IS also stimulated TNF-α, IL-6, and IL-1β mRNA expression in THP-1 cells, which demonstrated, for the first time, that IS has pro-fibrotic, pro-hypertrophic, and pro-inflammatory effects and might, therefore, play an important role in adverse cardiac remodeling. These effects of IS on cardiac remodeling and cardiac electrophysiology, together with its previously discussed thrombogenic properties, may contribute to the higher prevalence rate of atrial fibrillations and subsequent stroke occurrence in CKD patients. Further studies will be needed to clarify this concern.
3. Impact of Uremic Toxins on Brain Microcirculation
3.1. Endothelial Cells
Over recent years, the major impact of small vessel disease (SVD) on cognitive impairment has been clearly recognized. SVD is a systemic disease, probably related to diffuse endothelial dysfunction, which affects the perforating arterioles, capillaries, and venules in the brain. Cerebral SVD causes focal lacunar infarction and more diffuse ischemia, referred to as leukoaraiosis [83]. Although often asymptomatic, it is responsible for almost half of all cases of dementia and for a significant proportion of stroke cases [84].
3.1.1. Uremic Toxins and Vasoreactivity
Phosphate and Indoxyl Sulfate
As discussed below, UTs are strong inducers of large vessel endothelial dysfunction. In addition, patients with renal disease are also prone to peripheral microvascular dysfunction [85]. Therefore, in 2011, our group evaluated the impact of uremia on the function of the cerebral microcirculation in a mouse model [86]. In this study, we demonstrated that the endothelium-dependent relaxation of cerebral microvessels was impaired during CKD while the endothelium-independent relaxation was not. In addition, we observed that plasma concentration of asymmetric dimethyl arginine (ADMA), an UT and endogenous inhibitor of eNOS, was elevated in CKD mice as compared to SHAM-operated mice, which suggested that these alterations were related, at least in part, to a decrease in NO production. In line with this hypothesis, we subsequently reported that the in vitro exposure to IS or high-Pi induced cerebral endothelial cells dysfunction by decreasing NO levels due to enhanced oxidative stress [87]. Interestingly, infusion of ADMA in 10 healthy volunteers was reported to decrease cerebral perfusion and arterial compliance [88]. Altogether, these data suggest that UTs may induce endothelial dysfunction not only in large vessels, but also in cerebral microvessels via increased ROS production, a phenomenon that may directly promote SVD and, subsequently, stroke and cognition. In line with these data, spatial working memory was reported to be impaired in mice subjected to eight weeks of CKD, which is known to exhibit increased circulating levels of UTs and increased oxidative DNA damage [89]. However, there is no direct evidence of a potential link between UT-induced ROS production, SVD, and subsequent cognitive impairment or stroke at the present time.
Homocysteine
Elevated homocysteine levels are observed in 85% of dialysis patients, but only in 10% of the general population [61]. In a prospective cohort study, plasma homocysteine was reported to be an independent risk factor for dementia [90]. In addition, elevated plasma homocysteine concentrations are associated with an increased risk for Alzheimer’s disease [91]. Furthermore, recent cross-sectional data provide evidence that a higher level of total homocysteine is associated with white matter hyper-intensity volume, suggesting that total homocysteine is a risk factor for white matter damage [92]. Interestingly, data obtained from in vitro [69,93] and in vivo studies [94,95] suggest that the effect of homocysteine on the number and progression of white matter lesions might be mediated through direct endothelial damage [6]. Indeed, in vitro exposure of endothelial cells to homocysteine had been reported to reduce cell adherence and to decrease the bioavailability of endothelium-derived NO [69,93]. In line with these data endothelium-dependent vasodilation, measured with high-resolution ultrasonography, was reported to be significantly impaired in hyperhomocysteinemic subjects compared with control subjects both in middle-aged [95] and elderly people [94], suggesting that the bioavailability of NO is decreased in hyperhomocysteinemic humans. Therefore, in 2004, Hassan and colleagues intended to determine whether elevated homocysteine levels is a risk factor for SVD and whether this association was mediated by endothelial dysfunction as assessed by circulating endothelial markers [83]. In their study, performed on 172 Caucasian patients with SVD and 172 community controls of similar age and sex, mean homocysteine levels were higher in SVD patients than in control subjects and homocysteine was a stronger risk factor in those with ischemic leukoaraiosis in comparison with isolated lacunar infarction. In addition, homocysteine levels were associated with the markers of endothelial dysfunction intercellular adhesion molecule 1 (ICAM-1) and thrombomodulin. Inclusion of these markers as covariates reduced the association with homocysteine, but improved the overall logistic regression model for prediction of SVD. These findings are consistent with the hypothesis that endothelial dysfunction is an important mechanism through which homocysteine mediates its effects in SVD, particularly in ischemic leukoaraiosis. Together, these data suggest that homocysteine-lowering therapy may be promising to reduce SVD in patients with CKD.
3.1.2. Uremic Toxins and Endothelial Cell Integrity
Phosphate, Indoxyl Sulfate, Oxalic Acid, and Homocysteine
UTs have been reported to directly alter the integrity of both large- and small-vessels endothelial cells. For instance, Pi increases the expression of adhesion molecules, such as vascular cell adhesion molecule 1 (VCAM-1) and ICAM-1 [96], two inflammation markers involved in leukocyte adhesion and rolling on endothelial cells. Phosphate may, therefore, predispose to local cerebral inflammation via increased VCAM-1 and ICAM-1 expression in cerebral endothelial cells, thereby contributing to neuroinflammatory diseases in CKD patients. Similarly, administration of IS to normal rats [97] or nephrectomized mice [98] induces leukocyte adhesion to the vascular wall. IS has also been reported to promote senescence of large-vessel endothelial cells via activation of p53 and ROS production [99]. Interestingly, IS also generates disruption of contact between pulmonary artery endothelial cells via phosphorylation of myosin light chain kinase (MLCK) and myosin light chains (MLC) and activation of ERK1/ERK2 [100]. Exposure to uremic levels of oxalic acid for a few days inhibits replication and migration of large-vessel endothelial cells in a concentration- and time-dependent manner [101]. Interestingly, in vitro studies have shown that homocysteine damages endothelial cells by increasing H2O2 production [102], affecting antioxidant defence systems [103] and triggering apoptosis via mitochondrial oxidant production [104]. Low concentrations of homocysteine were also reported to decrease endothelial cells proliferation [105]. According to several investigators, the toxic effects of homocysteine may be caused, at least in part, by the oxidant species H2O2, which is generated when homocysteine auto-oxidizes to the disulphide homocysteine or when it forms mixed disulphides with other thiols [69]. Altogether, these data suggest that, by exerting a similar action on both large vessels and microvessels, UTs such as IS, Pi, oxalic acid and homocysteine may predispose to cerebral inflammation by increasing BBB disruption, leukocyte adhesion, rolling and extravasation in brain tissue.
Lanthionine
In mice that underwent transient medial cerebral artery occlusion, administration of H2S donors decreased the infarction volume and improved neurological deficits [106]. In this model, the beneficial effects of H2S donors were related to a reduction of BBB permeability, brain edema and preserved expression of tight junction proteins in the ischemic brain. The authors demonstrated that H2S donors protected BBB integrity following experimental stroke possibly by acting through NF-κB inhibition to suppress neuroinflammation induction of MMP9 and NOX4-derived free radicals [106]. Therefore, the possibility that lanthionine-induced impaired H2S production may be involved in the poor post-stroke functional outcomes observed in CKD patients through disruption of BBB integrity cannot be ruled out.
3.1.3. Uremic Toxins and Angiogenesis
Angiogenesis is considered as a natural defence mechanism helping to restore oxygen and nutrient supply to the ischemic brain tissue [107]. Therefore, greater microvessels density in the ischemic border correlates with longer survival in stroke patients [108]. As discussed previously, the pathological mechanism of vascular cognitive impairment involves ischemic lesions in the hippocampus. Vascular endothelial growth factor (VEGF) is known to promote angiogenesis and enhances blood flow to ischemic regions. Indeed, induction of VEGF expression in rats with vascular cognitive impairment was reported to increase the number of blood vessels in the hippocampal region and to improve cognitive function and neuronal cell loss [109]. Vascular endothelial growth factor was also reported to promote angiogenesis and functional recovery in rats with stroke [110].
Lanthionine
Hydrogen sulphide was reported to stimulate the proliferation and migration of endothelial cells cultured in vitro [111,112]. In these cells, H2S also significantly increased tube-like structure formation in in vitro matrigel assays. Interestingly, H2S has been shown to induce angiogenesis indirectly through the release of VEGF from hypoxic smooth muscle cells [113]. In line with this observation, exposure of endothelial cells to VEGF increased the production of H2S and endogenously produced H2S was shown to participate in the angiogenic signalling of VEGF [111]. As a consequence, intraperitoneal administration of the H2S donor NaHS in mice increased neovascularization [112]. Therefore, the possibility that lanthionine-induced impaired H2S production may be involved in the poor functional post-stroke recovery and cognitive disorders observed in CKD through impaired angiogenesis cannot be ruled out. Further studies will be needed to clarify this concern.
3.2. Monocytes/Macrophages
Inflammation is enhanced in CKD patients [114,115,116] and inflammation markers are associated with increased morbidity and mortality in ESRD [117,118]. In dialysis patients, C-reactive protein (CRP) is predictive of stroke and death [119]. Several UTs, such as IS and PCS, exert pro-inflammatory effects and their serum concentrations are correlated with inflammatory markers in CKD patients [116]. Some of these inflammatory markers, such as TNF-α, IL-6, and IL-1β, are currently considered to be UTs [120]. This enhanced state of inflammation appears to play a major role in the neurological complications associated with CKD [24]. A recent review described the various mediators of post-ischemic inflammation [121]. Some of these mediators, such as IL-1α, IL-1β, and TNF-α, are involved in initiation of neuroinflammation, whereas others, including IL-1, 6, 10, 17, 20, and once again TNF-α, contribute to the amplification of neuroinflammation. In contrast, factors such as TGF-β, IL-10, 17, and 23 contribute to the resolution of neuroinflammation [121]. Data from in vitro studies and animal models suggest that the inflammation induced by UTs, such as IS, symmetric dimethylarginine (SDMA), guanidino compounds, or quinolinic acid (QUIN) may contribute to the increased risk of stroke and cognitive impairment observed in CKD patients.
3.2.1. Indoxyl Sulfate
As discussed above, the increased expression of ICAM-1, VCAM-1 and concomitant alteration of endothelial cell interactions observed in response to UTs raise the hypothesis that UTs may promote infiltration of inflammatory monocytes in brain tissue. For example, IS administration in mice has been reported to enhance TNF-α-induced recruitment of leukocytes to the vascular wall [122]. Confirming this observation, data from in vitro studies indicated that IS dose-dependently increased THP-1 monocyte adhesion to IL-1β-activated human endothelial cells under physiological flow conditions [123]. Leukocyte adhesion to the inflamed endothelium involves the β2-integrin family of receptors (which share a common β2 subunit), such as LFA-1 (CD11a/CD18), Mac-1 (CD11b/CD18), p150,95/CR4 (CD11c/CD18), and CD11d/CD18 [124,125]. Mac-1 is a receptor for ICAM-1 and extracellular matrices, abundantly present in injured tissue. Mac-1 expression and ROS production have been reported to be significantly higher in peripheral blood monocytes of subtotal nephrectomised CKD mice than in sham-operated mice [123]. In this model, treatment with AST-120, an oral adsorbent used in the clinic to reduce plasma IS levels, significantly decreased both Mac-1 expression and ROS production, raising the possibility that IS-induced Mac-1 expression may promote leukocyte recruitment to the vascular wall, thereby promoting inflammation.
Depending on the tissue microenvironment, macrophages can be driven to a classically activated pro-inflammatory phenotype (M1) by stimuli such as interferon-γ (IFN-γ), or an alternatively activated anti-inflammatory phenotype (M2) by factors including IL-4 and IL-13. A recent study reported that uremia increased M1 and impaired M2 polarization of macrophages by inhibition of the adenosine monophosphate (AMP)-activated protein kinase (AMPK) [126]. These data suggest that the UTs that accumulate in brain tissue during CKD might impact the polarization of infiltrated monocytes/macrophages. In line with this observation, IS has been reported to directly induce monocyte-mediated inflammation and ROS production in THP-1 monocytes via the NADPH oxidase and MAPK pathways [123] and display direct pro-inflammatory effects in vitro via activation of the NF-kB and MAPK pathways in macrophages differentiated from THP-1 cells [127]. In addition, in a recent study performed on in vitro THP-1 cell cultures, IS was reported to promote CD163 expression and transition to macrophages with the hallmarks of classical M1 (IL-6, CCL2, COX2) and alternative M2 (IL-10, PPARγ, TGF-β, TIMP-1) phenotypes via AhR/Nrf2 activation. The authors concluded that IS may skew monocyte differentiation toward low-inflammatory, profibrotic macrophages and may therefore contribute to persistence of chronic inflammation [128]. Post-stroke intracerebral monocyte recruitment following BBB disruption largely contributes to increased cerebral inflammation and subsequent aggravation of ischemic lesions. It is, therefore, conceivable that IS may amplify post-stroke brain inflammation by promoting monocyte recruitment and macrophage-induced inflammation after BBB disruption and this hypothesis should be tested in future studies.
3.2.2. Dimethylarginines
High serum levels of dimethylarginines (DMA) (both symmetric and asymmetric) are associated with greater stroke severity and deleterious stroke outcomes [129,130,131]. Of interest, SDMA has been reported to play a role in the inflammatory state observed in CKD by activating NF-κB, thereby increasing the expression of pro-inflammatory cytokines, such as IL-6 and TNF-α. Consequently, SDMA levels are associated with inflammatory markers, such as CRP in CKD patients [132]. Induction of inflammation and leukocyte activation by SDMA contributes to cardiovascular complications in uremic patients. Interestingly, SDMA levels have recently been reported to be associated with mediators of inflammation after acute stroke [133], suggesting a possible association between these compounds, inflammation, and stroke. Further work is needed to elucidate the impact of SDMA-induced inflammation on ischemic stroke lesions and subsequent recovery.
3.2.3. Guanidino Compounds
Guanidino compounds appear to exert dual effects on inflammation, as methylguanidine and guanidinoacetic acid increase TNF-α production in monocytes, while guanidinosuccinic acid exerts an inhibitory effect on TNF-α production in monocytes [134]. Whether these dual effects play a role in modulating neuroinflammatory diseases in CKD needs to be clarified.
3.2.4. Quinolinic Acid
During inflammation, the KYN pathway can be activated by cytokines, particularly IFN-γ, leading to the production of the protein-bound uremic excitotoxin QUIN by monocyte lineage cells [135,136]. Cerebral intravascular infusion of QUIN has been reported to enhance permeability of rat brain microvessels to plasma albumin [137]. The extracellular tissue concentration of albumin was consequently increased in the hippocampus proper and striatum, an effect associated with more severe neuronal loss. A vicious circle may therefore exist, in which cerebral inflammation amplifies cerebral inflammation via increased monocyte-induced QUIN secretion and subsequent microvessel permeability. In line with these data, QUIN neurotoxicity has been shown to be involved in the pathogenesis of several age-related neurodegenerative processes associated with neuroinflammation, including Alzheimer’s disease [138,139]. Indeed, decreased 3-hydroxykynurenine and QUIN concentrations have been observed in models of Huntington’s disease [140] and depression [141]. Schizophrenia and autism have been associated with increased kynurenic acid [142], and increased 3-hydroxykynurenine and decreased KYN and kynurenic acid concentrations have been observed in Parkinson’s disease [143]. In this context, QUIN might represent an important target to cure neuroinflammatory diseases in CKD. Further studies are needed to clarify this issue.
3.2.5. Homocysteine
Exposure of cultured primary human monocytes to homocysteine has been reported to increase protein secretion and mRNA expression, as well as activity of monocyte chemoattractant protein-1 (MCP-1) and IL-8, an effect mediated by ROS through NAD(P)H oxidase [144]. MCP-1 is considered as the strongest monocyte chemoattractant and IL-8 is known to be an important chemotactic factor for neutrophils. Therefore, the hyperhomocysteinemia observed in CKD patients may have a key role in leucocyte trafficking at the BBB.
3.2.6. Lanthionine
Hydrogen sulphide is an important inhibitor of acute inflammation, acting at the leukocyte-endothelium interface. Indeed, the administration of the H2S donor NaHS in rats suppressed nonsteroidal anti-inflammatory drug-induced granulocyte infiltration, expression of endothelial and leukocyte adhesion molecules, and expression of TNF-α [145]. Administration of H2S donors to rats also inhibited aspirin-induced leukocyte adherence and infiltration as well as carrageenan-induced paw edema [146]. In addition, H2S has been reported to induce neutrophil apoptosis, thereby contributing to resolution of inflammatory reactions [147]. In this context, targeting lanthionine, which impairs the production of H2S, appears as a promising strategy to reduce both the endothelial dysfunction and the inflammatory processes related to cerebrovascular diseases.
3.2.7. Other UTs
Other UTs may also be involved in stroke severity in CKD patients by increasing pro-inflammatory pathways. This is the case for uric acid that activates pro-inflammatory pathways, especially MAPK signalling in vascular smooth muscle cells [148], phenylacetic acid that promotes inflammation, increasing the risk of cardiovascular disease in the presence of uremia [149], and KYN that are associated with inflammation in CKD patients and which may play a role in cardiovascular disease, including stroke [150]. High circulating levels of β-2-microglobulin (B2M) have also recently been reported to be associated with an increased risk of ischemic stroke in women [151]. Since B2M levels correlate with CRP, TNF-α, IL-6, and cardiovascular risk factors in hemodialysis patients [152], this UT could possibly increase the risk of stroke by acting on inflammation.
4. Impact of Uremic Toxins on Brain Resident Cells
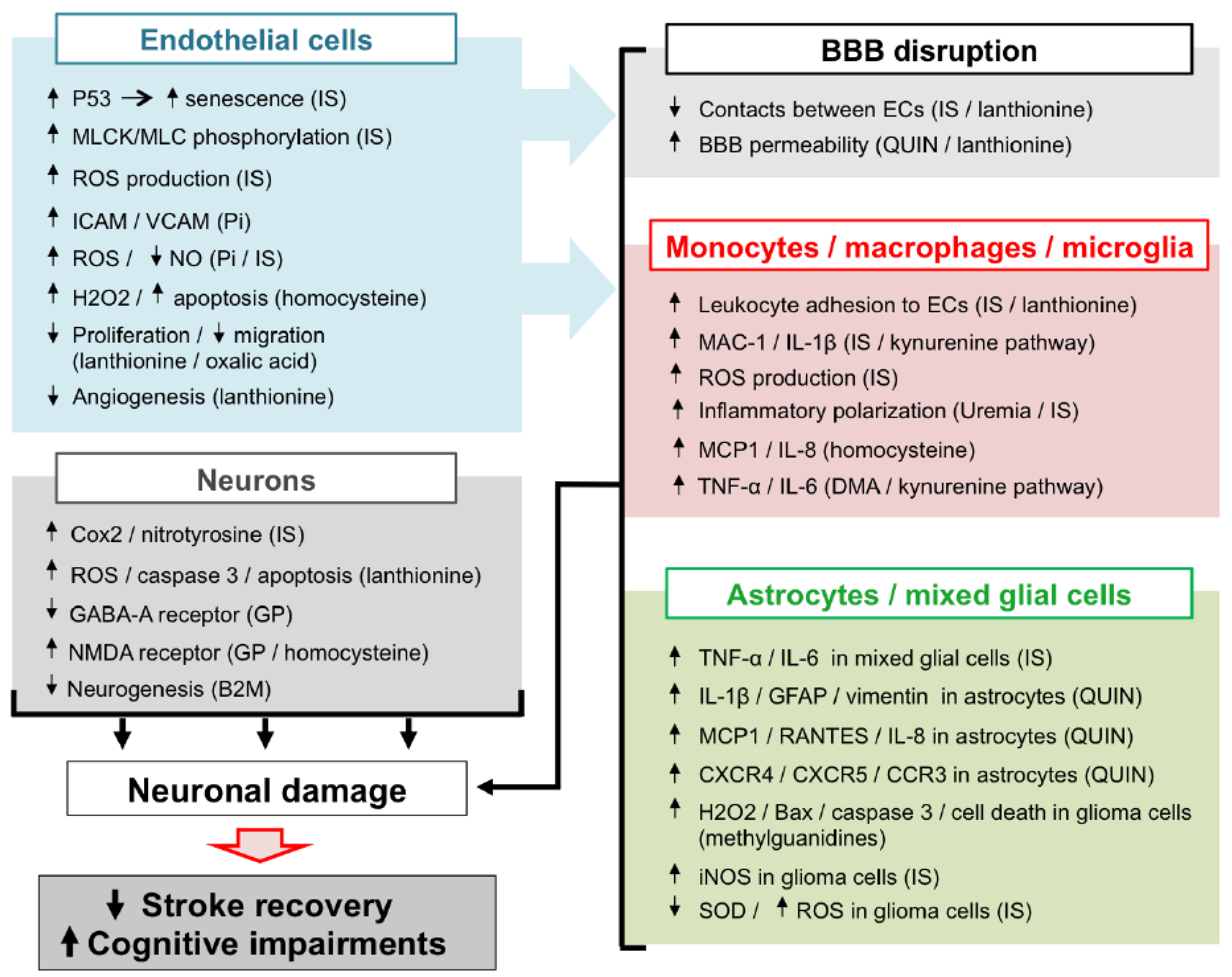
Renal impairment is associated with the accumulation of UTs within the cerebrospinal fluid and the brain tissue [25,26]. The accumulation of UTs within brain structures is thought to be linked to the expression of their transporter at the BBB and the blood-cerebrospinal fluid barrier (BCSFB) [153,154,155,156,157,158]. Indeed, recent studies show that IS undergoes efflux transport at the BBB via OAT3 [153,159]. In the same manner, guanidino compounds were reported to undergo efflux transport at the BCSFB via OCT3 [153,157,160]. Evidence obtained from in vivo and in vitro studies suggest that, once infiltrated within brain structures, these UTs may have deleterious impact on brain resident cells such as microglia, astrocytes, and neurons.
4.1. Microglia
Circulating macrophages that infiltrate into the inflamed central nervous system exert their effects in combination with resident microglia, the major immune cells present in the brain. Like macrophages, microglia, are highly plastic and can assume different functional phenotypes within brain lesions that critically influence the damage and the tissue repair that occurs following injury [161,162]. It has been clearly established that local microglia and newly-recruited macrophages assume a M2 phenotype at early stages of ischemic stroke, but are gradually transformed into the M1 phenotype in peri-infarct regions [163]. M1-polarized microglia/macrophages exacerbate neuronal death and are deleterious to tissue recovery following injury, while M2-polarized microglia/macrophages protect neurons against ischemia and enhance post-injury tissue repair [161,163,164,165]. Since UTs, such as IS, promote macrophage polarization toward the M1 phenotype, they may also concomitantly promote the polarization of resident microglia toward a pro-inflammatory phenotype. Further studies are needed to clarify this possibility.
Kynurenine Pathway
Kynurenine pathway metabolic balance has recently been reported to influence microglia activity [166]. The KYN pathway consists of two functionally distinct branches that generate both neuroactive and oxidatively reactive metabolites. In the brain, the rate-limiting enzyme for one of these branches, kynurenine 3-monooxygenase (KMO), is predominantly expressed in microglia and has emerged as a pivotal point of metabolic regulation [167]. In vitro exposure of murine microglia to lipopolysaccharide (LPS) promoted a dose-dependent increase in mRNA expression of IL-1β, IL-6, TNF-α, and inducible NO synthase (iNOS), together with the rate-limiting enzymes of the oxidative KYN pathway, indoleamine-2,3-dioxygenase (IDO)-1, and KMO [166]. Kynurenine and QUIN levels were increased in the medium 24 hours post-LPS. Inhibition of KMO by Ro 61-8048 following LPS challenge attenuated extracellular nitrite accumulation and LPS-induced expression of KMO and TNF-α. Similarly, primary microglia isolated from KMO-/- mice exhibited a significantly reduced pro-inflammatory response to LPS compared to WT controls. Altogether, these data suggested that targeting KMO may dampen neuroinflammation, which may impact the subsequent neurological disorders observed in CKD.
4.2. Astrocytes
In healthy neural tissue, astrocytes play critical roles in energy provision, regulation of blood flow, homeostasis of extracellular fluid, ions and transmitters, regulation of synapse function, and synaptic remodeling. Of interest, astrocytes respond to all forms of central nervous system insults, such as trauma, ischemia, or neurodegenerative diseases, by a process commonly referred to as reactive astrogliosis. Reactive astrogliosis involves changes in astrocyte molecular expression and morphology and, in severe cases, scar formation [168]. Reactive astrogliosis can exert both beneficial and detrimental effects in a context-dependent manner determined by specific molecular signalling cascades.
Astrocyte degeneration has been described in several pathological conditions, such as depressive disorders or dementia [169]. Reactive astrogliosis and dystrophy accompany these diseases and may directly contribute to the early alterations in synaptic transmission and cognitive processes that occur prior to neurodegeneration. In addition, astroglial cell loss described at later stages of several neurodegenerative diseases is also likely to indirectly alter neuronal function and survival by compromising glial physiological support and modulating neuronal activity, and may consequently accelerate the course of the disease [169,170].
4.2.1. Methylguanidines
Dementia is a neurological disease observed more commonly in uremic patients than in the general population and several types of dementia are associated with astroglial apoptosis. In a rat glioma cell line (C6) cultured in vitro, pre-incubation with methylguanidine significantly increased H2O2-induced cell death [171]. In this study, the fluorescent dye FURA 2-AM test showed significant elevation of [Ca2+]i in C6 cells co-treated with methylguanidine and H2O2. This effect was associated with a significant increase in H2O2-induced Bax expression and activation of caspase-3 and PARP in C6 cells. The authors concluded that methylguanidine could contribute to neurodegeneration associated with uremia by enhancing the pro-apoptotic effect of H2O2 and via alteration of mitochondrial calcium homeostasis in glial cells. Further studies are needed to evaluate whether the effect of methylguanidine on oxidative stress-induced astrocyte apoptosis is involved in CKD-related dementia. Recent evidence suggests that oxidative stress also compromises neurological recovery after stroke by causing glial cell death [19]. In future studies, it would be interesting to evaluate whether the oxidative stress-induced astrocyte apoptosis that occurs in response to methylguanidine alters post-stroke recovery in CKD.
4.2.2. Indoxyl Sulfate
The inflammatory reaction that occurs after stroke is known to progressively induce astrogliosis [172,173]. These reactive astrocytes then release growth-inhibitory molecules that chemically prevent axonal extensions and secrete ROS, pro-inflammatory cytokines and matrix metalloproteinases that accentuate ischemic stroke damage. In a recent study, IS was reported to promote iNOS and cyclooxygenase-2 (COX-2) expression, together with TNF-α and IL-6 release and nitrotyrosine formation in primary mouse astrocytes and mixed glial cells [174]. The same group reported also that IS decreased the expression of superoxide dismutase (SOD), an enzyme known for its antioxidant properties, in the C6 glioma cell line cultured in vitro, an effect associated with increased ROS production [174]. Altogether, these data suggest that IS-induced oxidative stress and subsequent astrogliosis generate a neurotoxic environment. Further studies will be needed to investigate the impact of IS-induced glial cell activation and subsequent inflammation on the stroke damage and neurodegeneration observed in CKD [174].
4.2.3. Quinolinic Acid
Quinolinic acid has been reported to increase IL-1β production in human astrocytes and concomitantly induce a marked increase in glial fibrillary acid protein levels and a reduction of vimentin levels, features consistent with astrogliosis [135]. Quinolinic acid also induces astrocytes to produce large quantities of MCP-1 (CCL2), RANTES (CCL5), and IL-8 (CXCL8) and upregulate the expression of chemokine receptors CXCR4, CCR5, and CCR3 [175]. Most of these effects are comparable to those induced by the classical mediators of inflammation, such as TNF-α, IL-1β, and IFN-γ [175]. These results suggest that this UT might be critical in the amplification of brain inflammation in CKD.
4.3. Neurons
Cognitive dysfunction is associated with a neuronal apoptosis response. Similarly, ischemic stroke triggers complex cellular events that progressively lead to both apoptotic and necrotic neuronal cell death. Oxidative stress is involved in nearly all neurological disorders [176] and several studies have suggested that accumulation of ROS results in neuronal damage and triggers apoptosis [177]. Uremic toxin-induced oxidative stress has been reported to promote cell death and cell senescence in various cell types [55,178,179,180,181]. UTs may, therefore, also promote cognitive dysfunction or impair post-stroke recovery via a direct neurotoxic action induced by increased oxidative stress. In line with this hypothesis, increased neuronal damage has been observed in mice following IS injection, a phenomenon associated with increased COX-2 expression and nitrotyrosine formation in brain tissue [174].
4.3.1. Guanidino Compounds
4.3.2. Homocysteine
Apart from its previously discussed role on coagulation and endothelial dysfunction, hyperhomocysteinemia could also impair neuronal pathways and display direct neurotoxic effects. Indeed, in cultures of cortical neurons, homocysteine caused direct neurotoxicity by activating the NMDA subtype of glutamate receptor [184]. Excessive stimulation of these receptors is known to mediate brain damage in focal ischemia [185,186]. Thus, homocysteine may not only be associated with the vascular injury leading to stroke, but may also participate in the ensuing neurotoxic response in the brain. As discussed previously, a prospective cohort study reported that plasma homocysteine was an independent risk factor for dementia [90]. In this study, higher plasma homocysteine levels were associated with smaller brain volume and the presence of silent brain infarcts, even in healthy, middle-aged adults. Clinical studies have also shown that elevated plasma homocysteine concentrations are associated with an increased risk of Alzheimer's disease [91]. Whether the cognitive disorders observed in response to hyperhomocysteinema appear as a consequence of NDMA receptors activation needs to be elucidated. Further studies are needed to evaluate the impact of hyperhomocysteinemia in the neurological damage associated with CKD.
4.3.3. β-2-Microglobulin
β-2-microglobulin accumulation in blood with aging has been reported to promote age-related cognitive dysfunction and impaired neurogenesis [187], a key phenomenon allowing brain parenchyma regeneration [188]. Neurogenesis is primordial to promote stroke recovery [189,190] and neurogenesis deficiency is considered to be a major cause of cognitive impairment [191]. Therefore, the possibility that B2M-induced inhibition of neurogenesis may account for CKD-induced poor stroke recovery cannot be ruled out and should be investigated.
4.3.4. Lanthionine
Cystathionine γ-lyase appears to be the predominant enzymatic source of H2S in the vasculature and heart [39], whereas in the central nervous system CBS predominates [39,41,192]. Since CBS is expressed and produces H2S in the brain and because the endogenous concentration of H2S in the brain is relatively high, it has been suggested that H2S may play a role in synaptic transmission [41]. Interestingly, physiological concentrations of H2S selectively enhance NMDA receptor-mediated responses and facilitate the induction of hippocampal long-term potentiation, suggesting that H2S functions as a neuromodulator involved in associative learning [193]. In addition, a growing body of evidence has shown that H2S mediates signals between neuronal cells and astrocytes by increasing Ca2+-influx in order to maintain calcium homeostasis and regulate synaptic activity [193,194]. Previous studies have indicated that H2S is involved in the pathophysiological process of ischemia-reperfusion injury and shock, and exerts protective effects on neurons. Indeed, H2S was reported to prevent oxygen-glucose deprivation/reoxygenation-induced apoptosis via improving mitochondrial dysfunction and suppressing an ROS-mediated caspase-3 pathway in mouse cortical neurons [195]. In other studies, H2S was reported to offer neuroprotection against traumatic brain injury in rats [196] and mice [197,198] through decreased oxidative stress and reduced apoptosis. Exogenous H2S administration in rats also protected against global cerebral ischemia/reperfusion injury via its anti-oxidative, anti-inflammatory and anti-apoptotic effects in rats [199]. These data suggest that lanthionine-induced impaired H2S production may play a central role in the onset and worsening of CKD-related cerebrovascular diseases. Further studies will be needed to clarify this concern.
5. Conclusions
In CKD patients, the accumulation of UTs is responsible for peripheral vascular dysfunction (endothelial dysfunction, atherosclerosis, vascular calcification, hypertension due to overactivation of RVLM neurons), which alters the general hemodynamic and favours the occurrence of cerebrovascular diseases. In addition, evidence from in vitro studies and in vivo animal experiments suggests that UTs may display direct deleterious effect in the brain microenvironment (Figure 2). Among the main mechanisms involved in these local effects, the endothelial dysfunction induced by molecules, such as Pi and IS may lead to BBB disruption and leukocyte adhesion, resulting in increased leukocyte infiltration into the damaged brain. Subsequent exposure of both macrophages and astrocytes to UTs amplifies the release of inflammatory cytokines and oxidative stress in the brain microenvironment. These deleterious mechanisms, together with the direct neurotoxic properties of certain UTs, promote neuronal death. Altogether, these mechanisms may account for the accelerated cognitive decline and poor stroke outcomes observed in CKD patients. However, to the best of our knowledge, evidence on the existence of a causal link between increased neurological disorders and UT-induced brain damage is still lacking. It should be noted that no animal models are currently available to study stroke and cognitive impairment in CKD. Such models need to be developed in order to advance research in this field. This review highlights the fact that IS is currently the most extensively studied UT in the context of cerebral disorders. In particular, IS is the only UT that has been reported to promote endothelial cell dysfunction, oxidative stress and inflammation and to induce glial cell activation and neuronal death. IS has also been reported to induce senescence of both vascular [99] and tubular cells [200] in the CKD setting. Cerebral cell senescence could, therefore, occur as a result of IS accumulation in the brain. In addition, uremic toxins such as IS are known to participate in the pathogenesis of stroke by amplifying atherosclerosis [46,201] and hypertension [29]. In vitro evidence suggests that reduction of IS by AST-120 attenuates monocyte inflammation [123], oxidative stress and endothelial dysfunction [202,203] related to CKD. Targeting IS, therefore, appears to be a promising strategy to protect CKD patients from brain damage. Interestingly, in a retrospective analysis, 3 to 5 years of treatment with AST-120 decreased the prevalence of stroke events in pre-dialysis CKD patients [204]. However, interventional studies designed to evaluate the impact of AST-120 on stroke outcomes in animals or patients with CKD are lacking and, therefore, need to be conducted. In the future, it would also be interesting to evaluate the impact of AST-120 administration in CKD animals presenting various neurological disorders, such as stroke or cognitive impairment. As discussed throughout this review, oxidative stress plays a central role in IS-induced endothelial cell dysfunction, inflammation, astrocyte activation, and neuronal death and is also a key mediator in the effects of other UTs. Studies on the impact of inhibition of pro-oxidative enzymes, such as NADPH oxidase [205], or myeloperoxidase [18], or increased antioxidant enzyme expression [206] in uremic animals or CKD patients should, therefore, also be envisaged.
Funding
This research received no external funding.
Conflicts of Interest
Z.A. Massy reports grants for CKD REIN and other research projects from Amgen, Baxter, Fresenius Medical Care, GlaxoSmithKline, Merck Sharp and Dohme-Chibret, Sanofi-Genzyme, Lilly, Otsuka, and the French government, as well as fees and grants to charities from Amgen, Bayer, and Sanofi-Genzyme. These sources of funding are not necessarily related to the content of the present manuscript. M. Assem, M. Lando, M. Grissi, S. Kamel, J.M. Chillon, and L. Hénaut declare that they have no conflicts of interest.
References
- Moodalbail, D.G.; Reiser, K.A.; Detre, J.A.; Schultz, R.T.; Herrington, J.D.; Davatzikos, C.; Doshi, J.J.; Erus, G.; Liu, H.S.; Radcliffe, J.; et al. Systematic review of structural and functional neuroimaging findings in children and adults with CKD. Clin. J. Am. Soc. Nephrol. 2013, 8, 1429–1448. [Google Scholar] [CrossRef] [PubMed]
- Seliger, S.L.; Siscovick, D.S.; Stehman-Breen, C.O.; Gillen, D.L.; Fitzpatrick, A.; Bleyer, A.; Kuller, L.H. Moderate renal impairment and risk of dementia among older adults: The cardiovascular health cognition study. J. Am. Soc. Nephrol. 2004, 15, 1904–1911. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.M. Cognitive impairment in the aging dialysis and chronic kidney disease populations: An occult burden. Adv. Chronic Kidney Dis. 2008, 15, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Weiner, D.E.; Gaussoin, S.A.; Nord, J.; Auchus, A.P.; Chelune, G.J.; Chonchol, M.; Coker, L.; Haley, W.E.; Killeen, A.A.; Kimmel, P.L.; et al. Cognitive function and kidney disease: Baseline data from the systolic blood pressure intervention trial (sprint). Am. J. Kidney Dis. 2017, 70, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Lass, P.; Buscombe, J.R.; Harber, M.; Davenport, A.; Hilson, A.J. Cognitive impairment in patients with renal failure is associated with multiple-infarct dementia. Clin. Nucl. Med. 1999, 24, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Bugnicourt, J.M.; Godefroy, O.; Chillon, J.M.; Choukroun, G.; Massy, Z.A. Cognitive disorders and dementia in ckd: The neglected kidney-brain axis. J. Am. Soc. Nephrol. 2013, 24, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Chillon, J.M.; Massy, Z.A.; Stengel, B. Neurological complications in chronic kidney disease patients. Nephrol. Dial. Transplant. 2015, 31, 1606–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, C.C.; Lee, C.T.; Ho, S.C.; Kuo, H.W.; Wu, T.N.; Yang, C.Y. Haemodialysis and the risk of stroke: A population-based cohort study in taiwan, a country of high incidence of end-stage renal disease. Nephrology 2012, 17, 243–248. [Google Scholar] [CrossRef] [PubMed]
- El Husseini, N.; Kaskar, O.; Goldstein, L.B. Chronic kidney disease and stroke. Adv. Chronic Kidney Dis. 2014, 21, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, K.; Ninomiya, T. Stroke and cerebrovascular diseases in patients with chronic kidney disease. Lancet Neurol. 2014, 13, 823–833. [Google Scholar] [CrossRef]
- Hojs Fabjan, T.; Hojs, R. Stroke and renal dysfunction. Eur. J. Intern. Med. 2014, 25, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Koren-Morag, N.; Goldbourt, U.; Tanne, D. Renal dysfunction and risk of ischemic stroke or tia in patients with cardiovascular disease. Neurology 2006, 67, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Kumai, Y.; Kamouchi, M.; Hata, J.; Ago, T.; Kitayama, J.; Nakane, H.; Sugimori, H.; Kitazono, T. Proteinuria and clinical outcomes after ischemic stroke. Neurology 2012, 78, 1909–1915. [Google Scholar] [CrossRef] [PubMed]
- Sozio, S.M.; Armstrong, P.A.; Coresh, J.; Jaar, B.G.; Fink, N.E.; Plantinga, L.C.; Powe, N.R.; Parekh, R.S. Cerebrovascular disease incidence, characteristics, and outcomes in patients initiating dialysis: The choices for healthy outcomes in caring for esrd (choice) study. Am. J. Kidney Dis. 2009, 54, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Hojs Fabjan, T.; Hojs, R.; Tetickovic, E.; Pecovnik Balon, B. Ischaemic stroke--impact of renal dysfunction on in-hospital mortality. Eur. J. Neurol. 2007, 14, 1351–1356. [Google Scholar] [CrossRef] [PubMed]
- Tsagalis, G.; Akrivos, T.; Alevizaki, M.; Manios, E.; Stamatellopoulos, K.; Laggouranis, A.; Vemmos, K.N. Renal dysfunction in acute stroke: An independent predictor of long-term all combined vascular events and overall mortality. Nephrol. Dial. Transplant. 2009, 24, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.L.; Bayraktutan, U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int. J. Stroke 2009, 4, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Liang, Y.; Zheng, S.; Zhang, H. Inhibition of myeloperoxidase by n-acetyl lysyltyrosylcysteine amide reduces oxidative stress-mediated inflammation, neuronal damage, and neural stem cell injury in a murine model of stroke. J. Pharmacol. Exp. Ther. 2018, 364, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Stetler, R.A.; Leak, R.K.; Shi, Y.; Li, Y.; Yu, W.; Bennett, M.V.L.; Chen, J. Oxidative stress and DNA damage after cerebral ischemia: Potential therapeutic targets to repair the genome and improve stroke recovery. Neuropharmacology 2018, 134, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.L.; Yang, C.M. Role of redox signaling in neuroinflammation and neurodegenerative diseases. BioMed Res. Int. 2013, 2013, 18. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.T.; Yu, J.; Grass, D.; de Beer, F.C.; Kindy, M.S. Inflammation-dependent cerebral deposition of serum amyloid a protein in a mouse model of amyloidosis. J. Neurosci. 2002, 22, 5900–5909. [Google Scholar] [CrossRef] [PubMed]
- Frank-Cannon, T.C.; Alto, L.T.; McAlpine, F.E.; Tansey, M.G. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol. Neurodegener. 2009, 4, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.A.; Richey, P.L.; Taneda, S.; Kutty, R.K.; Sayre, L.M.; Monnier, V.M.; Perry, G. Advanced maillard reaction end products, free radicals, and protein oxidation in alzheimer’s disease. Ann. N.Y. Acad. Sci. 1994, 738, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Miyamoto, Y.; Otagiri, M.; Maruyama, T. Update on the pharmacokinetics and redox properties of protein-bound uremic toxins. J. Pharm. Sci. 2011, 100, 3682–3695. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Saigusa, D.; Mishima, E.; Uchida, T.; Miura, D.; Morikawa-Ichinose, T.; Kisu, K.; Sekimoto, A.; Saito, R.; Oe, Y.; et al. Impact of the oral adsorbent ast-120 on organ-specific accumulation of uremic toxins: LC-MS/MS and MS imaging techniques. Toxins 2017, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- De Deyn, P.P.; Vanholder, R.; Eloot, S.; Glorieux, G. Guanidino compounds as uremic (neuro)toxins. Semin. Dial. 2009, 22, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Johansson, B.B. Hypertension mechanisms causing stroke. Clin. Exp. Pharmacol. Physiol. 1999, 26, 563–565. [Google Scholar] [CrossRef] [PubMed]
- Madden, C.J.; Sved, A.F. Rostral ventrolateral medulla C1 neurons and cardiovascular regulation. Cell Mol. Neurobiol. 2003, 23, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Oshima, N.; Onimaru, H.; Matsubara, H.; Uchida, T.; Watanabe, A.; Takechi, H.; Nishida, Y.; Kumagai, H. Uric acid, indoxyl sulfate, and methylguanidine activate bulbospinal neurons in the rvlm via their specific transporters and by producing oxidative stress. Neuroscience 2015, 304, 133–145. [Google Scholar] [CrossRef] [PubMed]
- In't Veld, B.A.; Ruitenberg, A.; Hofman, A.; Stricker, B.H.; Breteler, M.M. Antihypertensive drugs and incidence of dementia: The rotterdam study. Neurobiol. Aging 2001, 22, 407–412. [Google Scholar] [CrossRef]
- Duron, E.; Hanon, O. Antihypertensive treatments, cognitive decline, and dementia. J. Alzheimers Dis. 2010, 20, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Stefani, A.; Sancesario, G.; Pierantozzi, M.; Leone, G.; Galati, S.; Hainsworth, A.H.; Diomedi, M. Csf biomarkers, impairment of cerebral hemodynamics and degree of cognitive decline in alzheimer’s and mixed dementia. J. Neurol. Sci. 2009, 283, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Vicenzini, E.; Ricciardi, M.C.; Altieri, M.; Puccinelli, F.; Bonaffini, N.; Di Piero, V.; Lenzi, G.L. Cerebrovascular reactivity in degenerative and vascular dementia: A transcranial doppler study. Eur. Neurol. 2007, 58, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Perna, A.F.; Di Nunzio, A.; Amoresano, A.; Pane, F.; Fontanarosa, C.; Pucci, P.; Vigorito, C.; Cirillo, G.; Zacchia, M.; Trepiccione, F.; et al. Divergent behavior of hydrogen sulfide pools and of the sulfur metabolite lanthionine, a novel uremic toxin, in dialysis patients. Biochimie 2016, 126, 97–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perna, A.F.; Zacchia, M.; Trepiccione, F.; Ingrosso, D. The sulfur metabolite lanthionine: Evidence for a role as a novel uremic toxin. Toxins 2017, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Chiku, T.; Padovani, D.; Zhu, W.; Singh, S.; Vitvitsky, V.; Banerjee, R. H2s biogenesis by human cystathionine gamma-lyase leads to the novel sulfur metabolites lanthionine and homolanthionine and is responsive to the grade of hyperhomocysteinemia. J. Biol. Chem. 2009, 284, 11601–11612. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Padovani, D.; Leslie, R.A.; Chiku, T.; Banerjee, R. Relative contributions of cystathionine beta-synthase and gamma-cystathionase to h2s biogenesis via alternative trans-sulfuration reactions. J. Biol. Chem. 2009, 284, 22457–22466. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.X.; Wang, Y.B.; Peng, L.; Ge, X.Z.; Zhang, J.; Liu, S.S.; Zhang, X.N.; Xu, Z.H.; Chen, Z.; Luo, J.H. Lanthionine synthetase c-like protein 1 interacts with and inhibits cystathionine beta-synthase: A target for neuronal antioxidant defense. J. Biol. Chem. 2012, 287, 34189–34201. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Two’s company, three's a crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J. 2002, 16, 1792–1798. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, M. Hydrogen sulfide as a vasodilator. IUBMB Life 2005, 57, 603–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, K.; Kimura, H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Hosoki, R.; Matsuki, N.; Kimura, H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem. Biophys. Res. Commun. 1997, 237, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zhang, J.; Lu, Y.; Wang, R. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J. 2001, 20, 6008–6016. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine gamma-lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S. Molecular mechanisms underlying uremic toxin-related systemic disorders in chronic kidney disease: Focused on β2-microglobulin-related amyloidosis and indoxyl sulfate-induced atherosclerosis-Oshima Award Address 2016. Clin. Exp. Nephrol. 2018, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Zuo, Y.; Ma, J.; Yancey, P.G.; Hunley, T.E.; Motojima, M.; Fogo, A.B.; Linton, M.F.; Fazio, S.; Ichikawa, I.; et al. Oral activated charcoal adsorbent (AST-120) ameliorates extent and instability of atherosclerosis accelerated by kidney disease in apolipoprotein e-deficient mice. Nephrol. Dial. Transplant. 2011, 26, 2491–2497. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.C.; Li, L.C.; Chen, J.B.; Chang, H.W. Indoxyl sulfate-induced oxidative stress, mitochondrial dysfunction, and impaired biogenesis are partly protected by vitamin C and N-acetylcysteine. Sci. World J. 2015, 2015, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Carmona, A.; Guerrero, F.; Buendia, P.; Obrero, T.; Aljama, P.; Carracedo, J. Microvesicles derived from indoxyl sulfate treated endothelial cells induce endothelial progenitor cells dysfunction. Front. Physiol. 2017, 8, 666. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Wu, C.J.; Wu, P.C.; Pan, C.F.; Wang, T.J.; Sun, F.J.; Liu, H.L.; Chen, H.H.; Yeh, H.I. Indoxyl sulfate impairs endothelial progenitor cells and might contribute to vascular dysfunction in patients with chronic kidney disease. Kidney Blood Press. Res. 2016, 41, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.K.; Van Kerckhoven, S.; Verbeke, K.; Dehaen, W.; Vanrenterghem, Y.; Hoylaerts, M.F.; Evenepoel, P. The uremic retention solute p-cresyl sulfate and markers of endothelial damage. Am. J. Kidney Dis. 2009, 54, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, K.; Domaniewski, T.; Mysliwiec, M.; Pawlak, D. Kynurenines and oxidative status are independently associated with thrombomodulin and von willebrand factor levels in patients with end-stage renal disease. Thromb. Res. 2009, 124, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Romero, J.R.; Beiser, A.; Seshadri, S.; Benjamin, E.J.; Polak, J.F.; Vasan, R.S.; Au, R.; DeCarli, C.; Wolf, P.A. Carotid artery atherosclerosis, MRI indices of brain ischemia, aging, and cognitive impairment: The framingham study. Stroke 2009, 40, 1590–1596. [Google Scholar] [CrossRef] [PubMed]
- Vidal, J.S.; Sigurdsson, S.; Jonsdottir, M.K.; Eiriksdottir, G.; Thorgeirsson, G.; Kjartansson, O.; Garcia, M.E.; van Buchem, M.A.; Harris, T.B.; Gudnason, V.; et al. Coronary artery calcium, brain function and structure: The ages-reykjavik study. Stroke 2010, 41, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Henaut, L.; Chillon, J.M.; Kamel, S.; Massy, Z.A. Updates on the mechanisms and the care of cardiovascular calcification in chronic kidney disease. Semin. Nephrol. 2018, 38, 233–250. [Google Scholar] [CrossRef] [PubMed]
- Henaut, L.; Mary, A.; Chillon, J.M.; Kamel, S.; Massy, Z.A. The impact of uremic toxins on vascular smooth muscle cell function. Toxins 2018, 10, 218. [Google Scholar] [CrossRef] [PubMed]
- Bugnicourt, J.M.; Leclercq, C.; Chillon, J.M.; Diouf, M.; Deramond, H.; Canaple, S.; Lamy, C.; Massy, Z.A.; Godefroy, O. Presence of intracranial artery calcification is associated with mortality and vascular events in patients with ischemic stroke after hospital discharge: A cohort study. Stroke 2011, 42, 3447–3453. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.J. Brain regulation of thrombosis and hemostasis: From theory to practice. Stroke 2013, 44, 3275–3285. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, K.; Mysliwiec, M.; Pawlak, D. Hypercoagulability is independently associated with kynurenine pathway activation in dialysed uraemic patients. Thromb. Haemost. 2009, 102, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Vallet, B.; Wiel, E. Endothelial cell dysfunction and coagulation. Crit. Care Med. 2001, 29, S36–S41. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, T.W.; Pawlak, K.; Karbowska, M.; Mysliwiec, M.; Pawlak, D. Indoxyl sulfate—The uremic toxin linking hemostatic system disturbances with the prevalence of cardiovascular disease in patients with chronic kidney disease. BMC Nephrol. 2017, 18, 35. [Google Scholar] [CrossRef] [PubMed]
- Massy, Z.A. Importance of homocysteine, lipoprotein (a) and non-classical cardiovascular risk factors (fibrinogen and advanced glycation end-products) for atherogenesis in uraemic patients. Nephrol. Dial. Transplant. 2000, 15 (Suppl. 5), 81–91. [Google Scholar] [CrossRef] [PubMed]
- Fassbender, K.; Mielke, O.; Bertsch, T.; Nafe, B.; Froschen, S.; Hennerici, M. Homocysteine in cerebral macroangiography and microangiopathy. Lancet 1999, 353, 1586–1587. [Google Scholar] [CrossRef]
- Den Heijer, M.; Koster, T.; Blom, H.J.; Bos, G.M.; Briet, E.; Reitsma, P.H.; Vandenbroucke, J.P.; Rosendaal, F.R. Hyperhomocysteinemia as a risk factor for deep-vein thrombosis. N. Engl. J. Med. 1996, 334, 759–762. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Hennekens, C.H.; Selhub, J.; Miletich, J.P.; Malinow, M.R.; Stampfer, M.J. Interrelation of hyperhomocyst(e)inemia, factor v leiden, and risk of future venous thromboembolism. Circulation 1997, 95, 1777–1782. [Google Scholar] [CrossRef] [PubMed]
- Sauls, D.L.; Boyd, L.C.; Allen, J.C.; Hoffman, M. Differences in the metabolic response to exogenous homocysteine in juvenile and adult rabbits. J. Nutr. Biochem. 2004, 15, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Sauls, D.L.; Wolberg, A.S.; Hoffman, M. Elevated plasma homocysteine leads to alterations in fibrin clot structure and stability: Implications for the mechanism of thrombosis in hyperhomocysteinemia. J. Thromb. Haemost. 2003, 1, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Sauls, D.L.; Arnold, E.K.; Bell, C.W.; Allen, J.C.; Hoffman, M. Pro-thrombotic and pro-oxidant effects of diet-induced hyperhomocysteinemia. Thromb. Res. 2007, 120, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Harker, L.A.; Slichter, S.J.; Scott, C.R.; Ross, R. Homocystinemia. Vascular injury and arterial thrombosis. N. Engl. J. Med. 1974, 291, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Wall, R.T.; Harlan, J.M.; Harker, L.A.; Striker, G.E. Homocysteine-induced endothelial cell injury in vitro: A model for the study of vascular injury. Thromb. Res. 1980, 18, 113–121. [Google Scholar] [CrossRef]
- Lentz, S.R.; Sadler, J.E. Inhibition of thrombomodulin surface expression and protein c activation by the thrombogenic agent homocysteine. J. Clin. Investig. 1991, 88, 1906–1914. [Google Scholar] [CrossRef] [PubMed]
- Nishinaga, M.; Ozawa, T.; Shimada, K. Homocysteine, a thrombogenic agent, suppresses anticoagulant heparan sulfate expression in cultured porcine aortic endothelial cells. J. Clin. Investig. 1993, 92, 1381–1386. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, K.A. Homocysteine-induced modulation of tissue plasminogen activator binding to its endothelial cell membrane receptor. J. Clin. Investig. 1993, 91, 2873–2879. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, K.A.; Mauri, L.; Jacovina, A.T.; Zhong, F.; Mirza, U.A.; Padovan, J.C.; Chait, B.T. Tissue plasminogen activator binding to the annexin ii tail domain. Direct modulation by homocysteine. J. Biol. Chem. 1998, 273, 9987–9993. [Google Scholar] [CrossRef] [PubMed]
- Fryer, R.H.; Wilson, B.D.; Gubler, D.B.; Fitzgerald, L.A.; Rodgers, G.M. Homocysteine, a risk factor for premature vascular disease and thrombosis, induces tissue factor activity in endothelial cells. Arterioscler.Thromb. Vasc. Biol. 1993, 13, 1327–1333. [Google Scholar] [CrossRef]
- Khajuria, A.; Houston, D.S. Induction of monocyte tissue factor expression by homocysteine: A possible mechanism for thrombosis. Blood 2000, 96, 966–972. [Google Scholar] [PubMed]
- Mayer, O.; Filipovsky, J.; Hromadka, M.; Svobodova, V.; Racek, J.; Mayer, O., Jr.; Stehlik, P.; Trefil, L.; Zarybnicka, M. Treatment of hyperhomocysteinemia with folic acid: Effects on homocysteine levels, coagulation status, and oxidative stress markers. J. Cardiovasc. Pharm. 2002, 39, 851–857. [Google Scholar] [CrossRef]
- Huo, Y.; Li, J.; Qin, X.; Huang, Y.; Wang, X.; Gottesman, R.F.; Tang, G.; Wang, B.; Chen, D.; He, M.; et al. Efficacy of folic acid therapy in primary prevention of stroke among adults with hypertension in China: The CSPPT randomized clinical trial. JAMA 2015, 313, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Perna, A.F.; Ingrosso, D.; Satta, E.; Lombardi, C.; Galletti, P.; D’Aniello, A.; De Santo, N.G. Plasma protein aspartyl damage is increased in hemodialysis patients: Studies on causes and consequences. J. Am. Soc. Nephrol. 2004, 15, 2747–2754. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Watanabe, T.; Sasaki, S.; Nagai, K.; Roden, D.M.; Aizawa, Y. Close bidirectional relationship between chronic kidney disease and atrial fibrillation: The niigata preventive medicine study. Am. Heart J. 2009, 158, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Beyerbach, D.M.; Zipes, D.P. Mortality as an endpoint in atrial fibrillation. Heart Rhythm 2004, 1, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.T.; Chen, Y.C.; Hsieh, M.H.; Huang, S.Y.; Kao, Y.H.; Chen, Y.A.; Lin, Y.K.; Chen, S.A.; Chen, Y.J. The uremic toxin indoxyl sulfate increases pulmonary vein and atrial arrhythmogenesis. J. Cardiovasc. Electrophysiol. 2015, 26, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Lekawanvijit, S.; Adrahtas, A.; Kelly, D.J.; Kompa, A.R.; Wang, B.H.; Krum, H. Does indoxyl sulfate, a uraemic toxin, have direct effects on cardiac fibroblasts and myocytes? Eur. Heart J. 2010, 31, 1771–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, A.; Hunt, B.J.; O’Sullivan, M.; Bell, R.; D’Souza, R.; Jeffery, S.; Bamford, J.M.; Markus, H.S. Homocysteine is a risk factor for cerebral small vessel disease, acting via endothelial dysfunction. Brain 2004, 127, 212–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibuya, M.; Leite, C.D.C.; Lucato, L.T. Neuroimaging in cerebral small vessel disease: Update and new concepts. Dement. Neuropsychol. 2017, 11, 336–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, J.; Harakalova, M.; den Ruijter, H.; Pasterkamp, G.; Duncker, D.J.; Verhaar, M.C.; Asselbergs, F.W.; Cheng, C. Cardiorenal disease connection during post-menopause: The protective role of estrogen in uremic toxins induced microvascular dysfunction. Int. J. Cardiol. 2017, 238, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Bugnicourt, J.M.; Da Silveira, C.; Bengrine, A.; Godefroy, O.; Baumbach, G.; Sevestre, H.; Bode-Boeger, S.M.; Kielstein, J.T.; Massy, Z.A.; Chillon, J.M. Chronic renal failure alters endothelial function in cerebral circulation in mice. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1143–H1152. [Google Scholar] [CrossRef] [PubMed]
- Stinghen, A.E.; Chillon, J.M.; Massy, Z.A.; Boullier, A. Differential effects of indoxyl sulfate and inorganic phosphate in a murine cerebral endothelial cell line (bend.3). Toxins 2014, 6, 1742–1760. [Google Scholar] [CrossRef] [PubMed]
- Kielstein, J.T.; Donnerstag, F.; Gasper, S.; Menne, J.; Kielstein, A.; Martens-Lobenhoffer, J.; Scalera, F.; Cooke, J.P.; Fliser, D.; Bode-Boger, S.M. Adma increases arterial stiffness and decreases cerebral blood flow in humans. Stroke 2006, 37, 2024–2029. [Google Scholar] [CrossRef] [PubMed]
- Fujisaki, K.; Tsuruya, K.; Yamato, M.; Toyonaga, J.; Noguchi, H.; Nakano, T.; Taniguchi, M.; Tokumoto, M.; Hirakata, H.; Kitazono, T. Cerebral oxidative stress induces spatial working memory dysfunction in uremic mice: Neuroprotective effect of tempol. Nephrol. Dial. Transplant. 2014, 29, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, S.; Wolf, P.A.; Beiser, A.S.; Selhub, J.; Au, R.; Jacques, P.F.; Yoshita, M.; Rosenberg, I.H.; D’Agostino, R.B.; DeCarli, C. Association of plasma total homocysteine levels with subclinical brain injury: Cerebral volumes, white matter hyperintensity, and silent brain infarcts at volumetric magnetic resonance imaging in the framingham offspring study. Arch. Neurol. 2008, 65, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, S. Elevated plasma homocysteine levels: Risk factor or risk marker for the development of dementia and alzheimer’s disease? J. Alzheimers Dis. 2006, 9, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.B.; Paik, M.C.; Brown, T.R.; Stabler, S.P.; Allen, R.H.; Sacco, R.L.; DeCarli, C. Total homocysteine is associated with white matter hyperintensity volume: The northern manhattan study. Stroke 2005, 36, 1207–1211. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.S.; Osborne, J.A.; Jaraki, O.; Rabbani, L.E.; Mullins, M.; Singel, D.; Loscalzo, J. Adverse vascular effects of homocysteine are modulated by endothelium-derived relaxing factor and related oxides of nitrogen. J. Clin. Investig. 1993, 91, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Tawakol, A.; Omland, T.; Gerhard, M.; Wu, J.T.; Creager, M.A. Hyperhomocyst(e)inemia is associated with impaired endothelium-dependent vasodilation in humans. Circulation 1997, 95, 1119–1121. [Google Scholar] [CrossRef] [PubMed]
- Woo, K.S.; Chook, P.; Lolin, Y.I.; Cheung, A.S.; Chan, L.T.; Sun, Y.Y.; Sanderson, J.E.; Metreweli, C.; Celermajer, D.S. Hyperhomocyst(e)inemia is a risk factor for arterial endothelial dysfunction in humans. Circulation 1997, 96, 2542–2544. [Google Scholar] [CrossRef] [PubMed]
- Six, I.; Maizel, J.; Barreto, F.C.; Rangrez, A.Y.; Dupont, S.; Slama, M.; Tribouilloy, C.; Choukroun, G.; Maziere, J.C.; Bode-Boeger, S.; et al. Effects of phosphate on vascular function under normal conditions and influence of the uraemic state. Cardiovasc. Res. 2012, 96, 130–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pletinck, A.; Glorieux, G.; Schepers, E.; Cohen, G.; Gondouin, B.; Van Landschoot, M.; Eloot, S.; Rops, A.; Van de Voorde, J.; De Vriese, A.; et al. Protein-bound uremic toxins stimulate crosstalk between leukocytes and vessel wall. J. Am. Soc. Nephrol. 2013, 24, 1981–1994. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Osaka, M.; Higuchi, Y.; Nishijima, F.; Ishii, H.; Yoshida, M. Indoxyl sulfate induces leukocyte-endothelial interactions through up-regulation of e-selectin. J. Biol. Chem. 2010, 285, 38869–38875. [Google Scholar] [CrossRef] [PubMed]
- Adelibieke, Y.; Shimizu, H.; Muteliefu, G.; Bolati, D.; Niwa, T. Indoxyl sulfate induces endothelial cell senescence by increasing reactive oxygen species production and p53 activity. J. Ren. Nutr. 2012, 22, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.S.; Lin, Y.T.; Chen, Y.; Hung, K.Y.; Wang, S.M. Effects of indoxyl sulfate on adherens junctions of endothelial cells and the underlying signaling mechanism. J. Cell. Biochem. 2012, 113, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Levin, R.I.; Kantoff, P.W.; Jaffe, E.A. Uremic levels of oxalic acid suppress replication and migration of human endothelial cells. Arteriosclerosis 1990, 10, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Starkebaum, G.; Harlan, J.M. Endothelial cell injury due to copper-catalyzed hydrogen peroxide generation from homocysteine. J. Clin. Investig. 1986, 77, 1370–1376. [Google Scholar] [CrossRef] [PubMed]
- Blundell, G.; Jones, B.G.; Rose, F.A.; Tudball, N. Homocysteine mediated endothelial cell toxicity and its amelioration. Atherosclerosis 1996, 122, 163–172. [Google Scholar] [CrossRef]
- Tyagi, N.; Ovechkin, A.V.; Lominadze, D.; Moshal, K.S.; Tyagi, S.C. Mitochondrial mechanism of microvascular endothelial cells apoptosis in hyperhomocysteinemia. J. Cell. Biochem. 2006, 98, 1150–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Yoshizumi, M.; Lai, K.; Tsai, J.C.; Perrella, M.A.; Haber, E.; Lee, M.E. Inhibition of growth and p21ras methylation in vascular endothelial cells by homocysteine but not cysteine. J. Biol. Chem. 1997, 272, 25380–25385. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jia, J.; Ao, G.; Hu, L.; Liu, H.; Xiao, Y.; Du, H.; Alkayed, N.J.; Liu, C.F.; Cheng, J. Hydrogen sulfide protects blood-brain barrier integrity following cerebral ischemia. J. Neurochem. 2014, 129, 827–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, H.; Plate, K.H. Angiogenesis after cerebral ischemia. Acta Neuropathol. 2009, 117, 481–496. [Google Scholar] [CrossRef] [PubMed]
- Krupinski, J.; Kaluza, J.; Kumar, P.; Kumar, S.; Wang, J.M. Role of angiogenesis in patients with cerebral ischemic stroke. Stroke 1994, 25, 1794–1798. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, S.; Xiong, X.; Ling, L.; He, R.; Wang, M.; Deng, W.; Liu, Z.; Li, Y. Lipoprostaglandin e1 modifies cognitive impairment in rats with vascular cognitive impairment by promoting angiogenesis via the VEGF/VEGFR pathway. Mol. Med. Rep. 2017, 16, 3117–3124. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.P.; Liu, H.J.; Liu, X.F. Vegf promotes angiogenesis and functional recovery in stroke rats. J. Surg. Res. 2010, 23, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Papapetropoulos, A.; Pyriochou, A.; Altaany, Z.; Yang, G.; Marazioti, A.; Zhou, Z.; Jeschke, M.G.; Branski, L.K.; Herndon, D.N.; Wang, R.; et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 21972–21977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, W.J.; Wang, M.J.; Moore, P.K.; Jin, H.M.; Yao, T.; Zhu, Y.C. The novel proangiogenic effect of hydrogen sulfide is dependent on akt phosphorylation. Cardiovasc. Res. 2007, 76, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C.; Papapetropoulos, A. Hydrogen sulphide and angiogenesis: Mechanisms and applications. Br. J. Pharmacol. 2011, 164, 853–865. [Google Scholar] [CrossRef] [PubMed]
- Gupta, J.; Mitra, N.; Kanetsky, P.A.; Devaney, J.; Wing, M.R.; Reilly, M.; Shah, V.O.; Balakrishnan, V.S.; Guzman, N.J.; Girndt, M.; et al. Association between albuminuria, kidney function, and inflammatory biomarker profile in ckd in cric. Clin. J. Am. Soc. Nephrol. 2012, 7, 1938–1946. [Google Scholar] [CrossRef] [PubMed]
- Carrero, J.J.; Stenvinkel, P. Inflammation in end-stage renal disease--what have we learned in 10 years? Semin. Dial. 2010, 23, 498–509. [Google Scholar] [CrossRef] [PubMed]
- Gouroju, S.; Rao, P.; Bitla, A.R.; Vinapamula, K.S.; Manohar, S.M.; Vishnubhotla, S. Role of gut-derived uremic toxins on oxidative stress and inflammation in patients with chronic kidney disease. Indian J. Nephrol. 2017, 27, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Bologa, R.M.; Levine, D.M.; Parker, T.S.; Cheigh, J.S.; Serur, D.; Stenzel, K.H.; Rubin, A.L. Interleukin-6 predicts hypoalbuminemia, hypocholesterolemia, and mortality in hemodialysis patients. Am. J. Kidney Dis. 1998, 32, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Barreto, D.V.; Barreto, F.C.; Liabeuf, S.; Temmar, M.; Lemke, H.D.; Tribouilloy, C.; Choukroun, G.; Vanholder, R.; Massy, Z.A. Plasma interleukin-6 is independently associated with mortality in both hemodialysis and pre-dialysis patients with chronic kidney disease. Kidney Int. 2010, 77, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Krane, V.; Winkler, K.; Drechsler, C.; Lilienthal, J.; Marz, W.; Wanner, C. Effect of atorvastatin on inflammation and outcome in patients with type 2 diabetes mellitus on hemodialysis. Kidney Int. 2008, 74, 1461–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo-Rodriguez, E.; Pizarro-Sanchez, S.; Sanz, A.B.; Ramos, A.M.; Sanchez-Nino, M.D.; Martin-Cleary, C.; Fernandez-Fernandez, B.; Ortiz, A. Inflammatory cytokines as uremic toxins: “Ni son todos los que estan, ni estan todos los que son”. Toxins 2017, 9, 114. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Osaka, M.; Edamatsu, T.; Itoh, Y.; Yoshida, M. Crucial role of the aryl hydrocarbon receptor (AhR) in indoxyl sulfate-induced vascular inflammation. J. Atheroscler. Thromb. 2016, 23, 960–975. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Higuchi, Y.; Yagi, Y.; Nishijima, F.; Yamato, H.; Ishii, H.; Osaka, M.; Yoshida, M. Reduction of indoxyl sulfate by ast-120 attenuates monocyte inflammation related to chronic kidney disease. J. Leukoc. Biol. 2013, 93, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: A family of cell surface receptors. Cell 1987, 48, 549–554. [Google Scholar] [CrossRef]
- Sheppard, D. In vivo functions of integrins: Lessons from null mutations in mice. Matrix Biol. 2000, 19, 203–209. [Google Scholar] [CrossRef]
- Li, C.; Ding, X.Y.; Xiang, D.M.; Xu, J.; Huang, X.L.; Hou, F.F.; Zhou, Q.G. Enhanced M1 and impaired M2 macrophage polarization and reduced mitochondrial biogenesis via inhibition of AMP kinase in chronic kidney disease. Cell. Physiol. Biochem. 2015, 36, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, T.; Yamamoto, S.; Ito, T.; Sato, Y.; Matsuo, K.; Takahashi, Y.; Kaneko, Y.; Goto, S.; Kazama, J.J.; Gejyo, F.; et al. Indoxyl sulfate promotes macrophage IL-1β production by activating aryl hydrocarbon receptor/nf-kappa/mapk cascades, but the nlrp3 inflammasome was not activated. Toxins 2018, 10, 124. [Google Scholar] [CrossRef] [PubMed]
- Barisione, C.; Garibaldi, S.; Furfaro, A.L.; Nitti, M.; Palmieri, D.; Passalacqua, M.; Garuti, A.; Verzola, D.; Parodi, A.; Ameri, P.; et al. Moderate increase of indoxyl sulfate promotes monocyte transition into profibrotic macrophages. PloS ONE 2016, 11, e0149276. [Google Scholar] [CrossRef] [PubMed]
- Worthmann, H.; Chen, S.; Martens-Lobenhoffer, J.; Li, N.; Deb, M.; Tryc, A.B.; Goldbecker, A.; Dong, Q.; Kielstein, J.T.; Bode-Boger, S.M.; et al. High plasma dimethylarginine levels are associated with adverse clinical outcome after stroke. J. Atheroscler. Thromb. 2011, 18, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Brouns, R.; Marescau, B.; Possemiers, I.; Sheorajpanday, R.; De Deyn, P.P. Dimethylarginine levels in cerebrospinal fluid of hyperacute ischemic stroke patients are associated with stroke severity. Neurochem. Res. 2009, 34, 1642–1649. [Google Scholar] [CrossRef] [PubMed]
- Luneburg, N.; von Holten, R.A.; Topper, R.F.; Schwedhelm, E.; Maas, R.; Boger, R.H. Symmetric dimethylarginine is a marker of detrimental outcome in the acute phase after ischaemic stroke: Role of renal function. Clin. Sci. 2012, 122, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Schepers, E.; Barreto, D.V.; Liabeuf, S.; Glorieux, G.; Eloot, S.; Barreto, F.C.; Massy, Z.; Vanholder, R. Symmetric dimethylarginine as a proinflammatory agent in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 2374–2383. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Martens-Lobenhoffer, J.; Weissenborn, K.; Kielstein, J.T.; Lichtinghagen, R.; Deb, M.; Li, N.; Tryc, A.B.; Goldbecker, A.; Dong, Q.; et al. Association of dimethylarginines and mediators of inflammation after acute ischemic stroke. J. Neuroinflamm. 2012, 9, 251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glorieux, G.L.; Dhondt, A.W.; Jacobs, P.; Van Langeraert, J.; Lameire, N.H.; De Deyn, P.P.; Vanholder, R.C. In vitro study of the potential role of guanidines in leukocyte functions related to atherogenesis and infection. Kidney Int. 2004, 65, 2184–2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ting, K.K.; Brew, B.J.; Guillemin, G.J. Effect of quinolinic acid on human astrocytes morphology and functions: Implications in alzheimer's disease. J. Neuroinflamm. 2009, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argiles, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [PubMed]
- St'astny, F.; Skultetyova, I.; Pliss, L.; Jezova, D. Quinolinic acid enhances permeability of rat brain microvessels to plasma albumin. Brain Res. Bull. 2000, 53, 415–420. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Brew, B.J. Implications of the kynurenine pathway and quinolinic acid in alzheimer’s disease. Redox Rep. 2002, 7, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Brew, B.J.; Noonan, C.E.; Takikawa, O.; Cullen, K.M. Indoleamine 2,3 dioxygenase and quinolinic acid immunoreactivity in alzheimer's disease hippocampus. Neuropathol. Appl. Neurobiol. 2005, 31, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Guidetti, P.; Luthi-Carter, R.E.; Augood, S.J.; Schwarcz, R. Neostriatal and cortical quinolinate levels are increased in early grade huntington’s disease. Neurobiol. Dis. 2004, 17, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Raison, C.L.; Dantzer, R.; Kelley, K.W.; Lawson, M.A.; Woolwine, B.J.; Vogt, G.; Spivey, J.R.; Saito, K.; Miller, A.H. CSF concentrations of brain tryptophan and kynurenines during immune stimulation with IFN-α: Relationship to CNS immune responses and depression. Mol. Psychiatry 2010, 15, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Rassoulpour, A.; Wu, H.Q.; Medoff, D.; Tamminga, C.A.; Roberts, R.C. Increased cortical kynurenate content in schizophrenia. Biol. Psychiatry 2001, 50, 521–530. [Google Scholar] [CrossRef]
- Ogawa, T.; Matson, W.R.; Beal, M.F.; Myers, R.H.; Bird, E.D.; Milbury, P.; Saso, S. Kynurenine pathway abnormalities in parkinson’s disease. Neurology 1992, 42, 1702–1706. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Dai, J.; Remick, D.G.; Wang, X. Homocysteine mediated expression and secretion of monocyte chemoattractant protein-1 and interleukin-8 in human monocytes. Circ. Res. 2003, 93, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Antonelli, E.; Distrutti, E.; Rizzo, G.; Mencarelli, A.; Orlandi, S.; Zanardo, R.; Renga, B.; Di Sante, M.; Morelli, A.; et al. Inhibition of hydrogen sulfide generation contributes to gastric injury caused by anti-inflammatory nonsteroidal drugs. Gastroenterology 2005, 129, 1210–1224. [Google Scholar] [CrossRef] [PubMed]
- Zanardo, R.C.; Brancaleone, V.; Distrutti, E.; Fiorucci, S.; Cirino, G.; Wallace, J.L. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006, 20, 2118–2120. [Google Scholar] [CrossRef] [PubMed]
- Mariggio, M.A.; Minunno, V.; Riccardi, S.; Santacroce, R.; De Rinaldis, P.; Fumarulo, R. Sulfide enhancement of pmn apoptosis. Immunopharmacol. Immunotoxicol. 1998, 20, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Kanellis, J.; Watanabe, S.; Li, J.H.; Kang, D.H.; Li, P.; Nakagawa, T.; Wamsley, A.; Sheikh-Hamad, D.; Lan, H.Y.; Feng, L.; et al. Uric acid stimulates monocyte chemoattractant protein-1 production in vascular smooth muscle cells via mitogen-activated protein kinase and cyclooxygenase-2. Hypertension 2003, 41, 1287–1293. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G.; Raupachova, J.; Horl, W.H. The uraemic toxin phenylacetic acid contributes to inflammation by priming polymorphonuclear leucocytes. Nephrol. Dial. Transplant. 2013, 28, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, K.; Domaniewski, T.; Mysliwiec, M.; Pawlak, D. The kynurenines are associated with oxidative stress, inflammation and the prevalence of cardiovascular disease in patients with end-stage renal disease. Atherosclerosis 2009, 204, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Rist, P.M.; Jimenez, M.C.; Rexrode, K.M. Prospective association between β2-microglobulin levels and ischemic stroke risk among women. Neurology 2017, 88, 2176–2182. [Google Scholar] [CrossRef] [PubMed]
- Kuragano, T.; Kida, A.; Furuta, M.; Nanami, M.; Otaki, Y.; Hasuike, Y.; Nonoguchi, H.; Nakanishi, T. The impact of β2-microglobulin clearance on the risk factors of cardiovascular disease in hemodialysis patients. ASAIO J. 2010, 56, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Hosoya, K.; Tachikawa, M. Roles of organic anion/cation transporters at the blood-brain and blood-cerebrospinal fluid barriers involving uremic toxins. Clin. Exp. Nephrol. 2011, 15, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Sekine, T.; Watanabe, N.; Hosoyamada, M.; Kanai, Y.; Endou, H. Expression cloning and characterization of a novel multispecific organic anion transporter. J. Biol. Chem. 1997, 272, 18526–18529. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Takanaga, H.; Ohtsuki, S.; Deguchi, T.; Kang, Y.S.; Hosoya, K.; Terasaki, T. Rat organic anion transporter 3 (roat3) is responsible for brain-to-blood efflux of homovanillic acid at the abluminal membrane of brain capillary endothelial cells. J. Cereb. Blood Flow Metab. 2003, 23, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Sweet, D.H.; Miller, D.S.; Pritchard, J.B.; Fujiwara, Y.; Beier, D.R.; Nigam, S.K. Impaired organic anion transport in kidney and choroid plexus of organic anion transporter 3 (oat3 (slc22a8)) knockout mice. J. Biol. Chem. 2002, 277, 26934–26943. [Google Scholar] [CrossRef] [PubMed]
- Hayer-Zillgen, M.; Bruss, M.; Bonisch, H. Expression and pharmacological profile of the human organic cation transporters hoct1, hoct2 and hoct3. Br. J. Pharmacol. 2002, 136, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H. Organic cation transporters in intestine, kidney, liver, and brain. Annu. Rev. Physiol. 1998, 60, 243–266. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, S.; Asaba, H.; Takanaga, H.; Deguchi, T.; Hosoya, K.; Otagiri, M.; Terasaki, T. Role of blood-brain barrier organic anion transporter 3 (OAT3) in the efflux of indoxyl sulfate, a uremic toxin: Its involvement in neurotransmitter metabolite clearance from the brain. J. Neurochem. 2002, 83, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Tachikawa, M.; Kasai, Y.; Takahashi, M.; Fujinawa, J.; Kitaichi, K.; Terasaki, T.; Hosoya, K. The blood-cerebrospinal fluid barrier is a major pathway of cerebral creatinine clearance: Involvement of transporter-mediated process. J. Neurochem. 2008, 107, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef] [PubMed]
- David, S.; Kroner, A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat. Rev. Neurosci. 2011, 12, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, P.; Guo, Y.; Wang, H.; Leak, R.K.; Chen, S.; Gao, Y.; Chen, J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012, 43, 3063–3070. [Google Scholar] [CrossRef] [PubMed]
- Kigerl, K.A.; Gensel, J.C.; Ankeny, D.P.; Alexander, J.K.; Donnelly, D.J.; Popovich, P.G. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci. 2009, 29, 13435–13444. [Google Scholar] [CrossRef] [PubMed]
- Clayton, K.A.; Van Enoo, A.A.; Ikezu, T. Alzheimer’s disease: The role of microglia in brain homeostasis and proteopathy. Front. Neurosci. 2017, 11, 680. [Google Scholar] [CrossRef] [PubMed]
- Garrison, A.M.; Parrott, J.M.; Tunon, A.; Delgado, J.; Redus, L.; O’Connor, J.C. Kynurenine pathway metabolic balance influences microglia activity: Targeting kynurenine monooxygenase to dampen neuroinflammation. Psychoneuroendocrinology 2018, 94, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Parrott, J.M.; O’Connor, J.C. Kynurenine 3-monooxygenase: An influential mediator of neuropathology. Front. Psychiatry 2015, 6, 116. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilcock, D.M.; Vitek, M.P.; Colton, C.A. Vascular amyloid alters astrocytic water and potassium channels in mouse models and humans with alzheimer's disease. Neuroscience 2009, 159, 1055–1069. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Brambilla, L.; Valori, C.F.; Roncoroni, C.; Crugnola, A.; Yokota, T.; Bredesen, D.E.; Volterra, A. Focal degeneration of astrocytes in amyotrophic lateral sclerosis. Cell Death Differ. 2008, 15, 1691–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzocco, S.; Popolo, A.; Bianco, G.; Pinto, A.; Autore, G. Pro-apoptotic effect of methylguanidine on hydrogen peroxide-treated rat glioma cell line. Neurochem. Int. 2010, 57, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wu, Z.B.; Zhuge, Q.; Zheng, W.; Shao, B.; Wang, B.; Sun, F.; Jin, K. Glial scar formation occurs in the human brain after ischemic stroke. Int. J. Med. Sci. 2014, 11, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Grande, B.; Swana, M.; Nguyen, L.; Englezou, P.; Maysami, S.; Allan, S.M.; Rothwell, N.J.; Garlanda, C.; Denes, A.; Pinteaux, E. The acute-phase protein ptx3 is an essential mediator of glial scar formation and resolution of brain edema after ischemic injury. J. Cereb. Blood Flow Metab. 2014, 34, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Adesso, S.; Magnus, T.; Cuzzocrea, S.; Campolo, M.; Rissiek, B.; Paciello, O.; Autore, G.; Pinto, A.; Marzocco, S. Indoxyl sulfate affects glial function increasing oxidative stress and neuroinflammation in chronic kidney disease: Interaction between astrocytes and microglia. Front. Pharmacol. 2017, 8, 370. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Croitoru-Lamoury, J.; Dormont, D.; Armati, P.J.; Brew, B.J. Quinolinic acid upregulates chemokine production and chemokine receptor expression in astrocytes. GLIA 2003, 41, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S.; Abramov, A.Y. Mechanism of oxidative stress in neurodegeneration. Oxid. Med. Cell. Longev. 2012, 2012, 428010. [Google Scholar] [CrossRef] [PubMed]
- Gilgun-Sherki, Y.; Melamed, E.; Offen, D. Oxidative stress induced-neurodegenerative diseases: The need for antioxidants that penetrate the blood brain barrier. Neuropharmacology 2001, 40, 959–975. [Google Scholar] [CrossRef]
- Okada, A.; Nangaku, M.; Jao, T.M.; Maekawa, H.; Ishimono, Y.; Kawakami, T.; Inagi, R. D-serine, a novel uremic toxin, induces senescence in human renal tubular cells via gcn2 activation. Sci. Rep. 2017, 7, 11168. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, M.; Tatebe, J.; Watanabe, I.; Yamazaki, J.; Ikeda, T.; Morita, T. Aryl hydrocarbon receptor mediates indoxyl sulfate-induced cellular senescence in human umbilical vein endothelial cells. J. Atheroscler. Thromb. 2014, 21, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.J.; Small, D.M.; Ng, K.L.; Vesey, D.A.; Vitetta, L.; Francis, R.S.; Gobe, G.C.; Morais, C. Indoxyl sulfate induces apoptosis and hypertrophy in human kidney proximal tubular cells. Toxicol. Pathol. 2018, 46. [Google Scholar] [CrossRef] [PubMed]
- Hunkerler, Z.; Koken, T.; Koca, B.; Kahraman, A. Role of uremic toxins on apoptosis with varying periods of hemodialysis. Ther. Apher. Dial. 2017, 21, 38–42. [Google Scholar] [CrossRef] [PubMed]
- D’Hooge, R.; Van de Vijver, G.; Van Bogaert, P.P.; Marescau, B.; Vanholder, R.; De Deyn, P.P. Involvement of voltage- and ligand-gated Ca2+ channels in the neuroexcitatory and synergistic effects of putative uremic neurotoxins. Kidney Int. 2003, 63, 1764–1775. [Google Scholar] [CrossRef] [PubMed]
- De Deyn, P.P.; Vanholder, R.; D’Hooge, R. Nitric oxide in uremia: Effects of several potentially toxic guanidino compounds. Kidney Int. 2003, 63, S25–S28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipton, S.A.; Kim, W.K.; Choi, Y.B.; Kumar, S.; D'Emilia, D.M.; Rayudu, P.V.; Arnelle, D.R.; Stamler, J.S. Neurotoxicity associated with dual actions of homocysteine at the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 5923–5928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, R.P.; Swan, J.H.; Griffiths, T.; Meldrum, B.S. Blockade of n-methyl-d-aspartate receptors may protect against ischemic damage in the brain. Science 1984, 226, 850–852. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A.; Rosenberg, P.A. Excitatory amino acids as a final common pathway for neurologic disorders. N. Engl. J. Med. 1994, 330, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.K.; He, Y.; Park, J.S.; Bieri, G.; Snethlage, C.E.; Lin, K.; Gontier, G.; Wabl, R.; Plambeck, K.E.; Udeochu, J.; et al. β2-microglobulin is a systemic pro-aging factor that impairs cognitive function and neurogenesis. Nat. Med. 2015, 21, 932–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arvidsson, A.; Collin, T.; Kirik, D.; Kokaia, Z.; Lindvall, O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat. Med. 2002, 8, 963–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Xu, D.; Qi, H.; Yuan, Y.; Liu, H.; Yao, S.; Yuan, S.; Zhang, J. Enriched environment promotes post-stroke neurogenesis through nf-kappab-mediated secretion of IL-17a from astrocytes. Brain Res. 2018, 1687, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, S.; Hu, Z.; Zhu, G.; Zheng, G.; Wang, G. Enriched housing promotes post-stroke neurogenesis through calpain 1-stat3/hif-1alpha/vegf signaling. Brain Res. Bull. 2018, 139, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.F.; Wang, Y.C.; Zong, L.; Chen, Z.Y.; Li, Y. Elevating integrin-linked kinase expression has rescued hippocampal neurogenesis and memory deficits in an ad animal model. Brain Res. 2018, 1695, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Armstrong, J.S.; Chu, S.H.; Jia-Ling, S.; Wong, B.S.; Cheung, N.S.; Halliwell, B.; Moore, P.K. The novel neuromodulator hydrogen sulfide: An endogenous peroxynitrite ‘scavenger’? J. Neurochem. 2004, 90, 765–768. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H. Physiological role of hydrogen sulfide and polysulfide in the central nervous system. Neurochem. Int. 2013, 63, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Tsugane, M.; Oka, J.; Kimura, H. Hydrogen sulfide induces calcium waves in astrocytes. FASEB J. 2004, 18, 557–559. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Yang, X.; Zhao, S.; Wei, C.; Yin, Y.; Liu, T.; Jiang, S.; Xie, J.; Wan, X.; Mao, M.; et al. Hydrogen sulfide prevents ogd/r-induced apoptosis via improving mitochondrial dysfunction and suppressing an ros-mediated caspase-3 pathway in cortical neurons. Neurochem. Int. 2013, 63, 826–831. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Huang, Y.; Lin, W.; Gao, D.; Fei, Z. Protective effects of hydrogen sulfide in a rat model of traumatic brain injury via activation of mitochondrial adenosine triphosphate-sensitive potassium channels and reduction of oxidative stress. J. Surg. Res. 2013, 184, e27–e35. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Shan, H.; Chang, P.; Wang, T.; Dong, W.; Chen, X.; Tao, L. Hydrogen sulfide offers neuroprotection on traumatic brain injury in parallel with reduced apoptosis and autophagy in mice. PloS ONE 2014, 9, e87241. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Shan, H.; Wang, T.; Liu, W.; Wang, Y.; Wang, L.; Zhang, L.; Chang, P.; Dong, W.; Chen, X.; et al. Dynamic change of hydrogen sulfide after traumatic brain injury and its effect in mice. Neurochem. Res. 2013, 38, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Tu, C.; Zhao, J.; Ou, D.; Chen, G.; Liu, Y.; Xiao, X. Exogenous hydrogen sulfide protects against global cerebral ischemia/reperfusion injury via its anti-oxidative, anti-inflammatory and anti-apoptotic effects in rats. Brain Res. 2013, 1491, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Bolati, D.; Adijiang, A.; Enomoto, A.; Nishijima, F.; Dateki, M.; Niwa, T. Senescence and dysfunction of proximal tubular cells are associated with activated p53 expression by indoxyl sulfate. Am. J. Physiol. Cell. Physiol. 2010, 299, C1110–C1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.C.; Lu, Y.C.; Chiu, C.A.; Yu, T.H.; Hung, W.C.; Wang, C.P.; Lu, L.F.; Chung, F.M.; Lee, Y.J.; Tsai, I.T. Levels of indoxyl sulfate are associated with severity of coronary atherosclerosis. Clin. Investig. Med. 2013, 36, E42–E49. [Google Scholar] [CrossRef]
- Ryu, J.H.; Yu, M.; Lee, S.; Ryu, D.R.; Kim, S.J.; Kang, D.H.; Choi, K.B. Ast-120 improves microvascular endothelial dysfunction in end-stage renal disease patients receiving hemodialysis. Yonsei Med. J. 2016, 57, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Inami, Y.; Hamada, C.; Seto, T.; Hotta, Y.; Aruga, S.; Inuma, J.; Azuma, K.; Io, H.; Kaneko, K.; Watada, H.; et al. Effect of ast-120 on endothelial dysfunction in adenine-induced uremic rats. Int. J. Nephrol. 2014, 2014, 164125. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Tanaka, A.; Oyama, J.; Yamasaki, A.; Shimomura, M.; Hiwatashi, A.; Ueda, Y.; Amaha, M.; Nomura, M.; Matsumura, D.; et al. Long-term effects of ast-120 on the progression and prognosis of pre-dialysis chronic kidney disease: A 5-year retrospective study. Heart Vessels 2016, 31, 1625–1632. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Song, Y.S.; Chan, P.H. Inhibition of nadph oxidase is neuroprotective after ischemia-reperfusion. J. Cereb. Blood Flow Metab. 2009, 29, 1262–1272. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.M.; Pennypacker, K.R. Targeting antioxidant enzyme expression as a therapeutic strategy for ischemic stroke. Neurochem. Int. 2017, 107, 23–32. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Impact of uremic toxins-induced oxidative stress on the occurrence and severity of cerebrovascular diseases. RVLM: rostral ventrolateral medulla.
Figure 1.
Impact of uremic toxins-induced oxidative stress on the occurrence and severity of cerebrovascular diseases. RVLM: rostral ventrolateral medulla.

Figure 2.
Impact of uremic toxins on neurological damage: a mechanistic view based on a review of the recent literature. B2M: β-2-microglobulin, BBB: blood-brain barrier, DMA: dimethylarginines, ECs: endothelial cells, GFAP: glial fibrillary acidic protein, GP: guanidino compounds, iNOS: inducible nitric oxide synthase, IS: indoxyl sulfate, MLCK: myosin light chain kinase, MLC: myosin light chain, NO: nitric oxide, Pi: inorganic phosphate, QUIN: quinolinic acid, ROS: reactive oxygen species, SOD: superoxide dismutase. ICAM: intercellular adhesion molecule, VCAM: vascular cell adhesion molecule, GABA-A: γ-aminobutyric acid receptor A, NMDA: n-methyl-d-aspartic acid, MCP1: Monocyte chemoattractant protein 1, MAC-1: macrophage-1 antigen, TNF-α: tumour necrosis factor, CXCR4: C-X-C chemokine receptor type 4, CXCR5: C-X-C chemokine receptor type 5, CCR3: C-C chemokine receptor type 3, RANTES: Regulated on Activation, Normal T Expressed and Secreted
Figure 2.
Impact of uremic toxins on neurological damage: a mechanistic view based on a review of the recent literature. B2M: β-2-microglobulin, BBB: blood-brain barrier, DMA: dimethylarginines, ECs: endothelial cells, GFAP: glial fibrillary acidic protein, GP: guanidino compounds, iNOS: inducible nitric oxide synthase, IS: indoxyl sulfate, MLCK: myosin light chain kinase, MLC: myosin light chain, NO: nitric oxide, Pi: inorganic phosphate, QUIN: quinolinic acid, ROS: reactive oxygen species, SOD: superoxide dismutase. ICAM: intercellular adhesion molecule, VCAM: vascular cell adhesion molecule, GABA-A: γ-aminobutyric acid receptor A, NMDA: n-methyl-d-aspartic acid, MCP1: Monocyte chemoattractant protein 1, MAC-1: macrophage-1 antigen, TNF-α: tumour necrosis factor, CXCR4: C-X-C chemokine receptor type 4, CXCR5: C-X-C chemokine receptor type 5, CCR3: C-C chemokine receptor type 3, RANTES: Regulated on Activation, Normal T Expressed and Secreted

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Assem, M.; Lando, M.; Grissi, M.; Kamel, S.; Massy, Z.A.; Chillon, J.-M.; Hénaut, L. The Impact of Uremic Toxins on Cerebrovascular and Cognitive Disorders. Toxins 2018, 10, 303. https://doi.org/10.3390/toxins10070303
AMA Style
Assem M, Lando M, Grissi M, Kamel S, Massy ZA, Chillon J-M, Hénaut L. The Impact of Uremic Toxins on Cerebrovascular and Cognitive Disorders. Toxins. 2018; 10(7):303. https://doi.org/10.3390/toxins10070303
Chicago/Turabian StyleAssem, Maryam, Mathilde Lando, Maria Grissi, Saïd Kamel, Ziad A. Massy, Jean-Marc Chillon, and Lucie Hénaut. 2018. "The Impact of Uremic Toxins on Cerebrovascular and Cognitive Disorders" Toxins 10, no. 7: 303. https://doi.org/10.3390/toxins10070303
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.