Isolation and Characterization of [D-Leu1]microcystin-LY from Microcystis aeruginosa CPCC-464

 , and
, and
Abstract
:1. Introduction
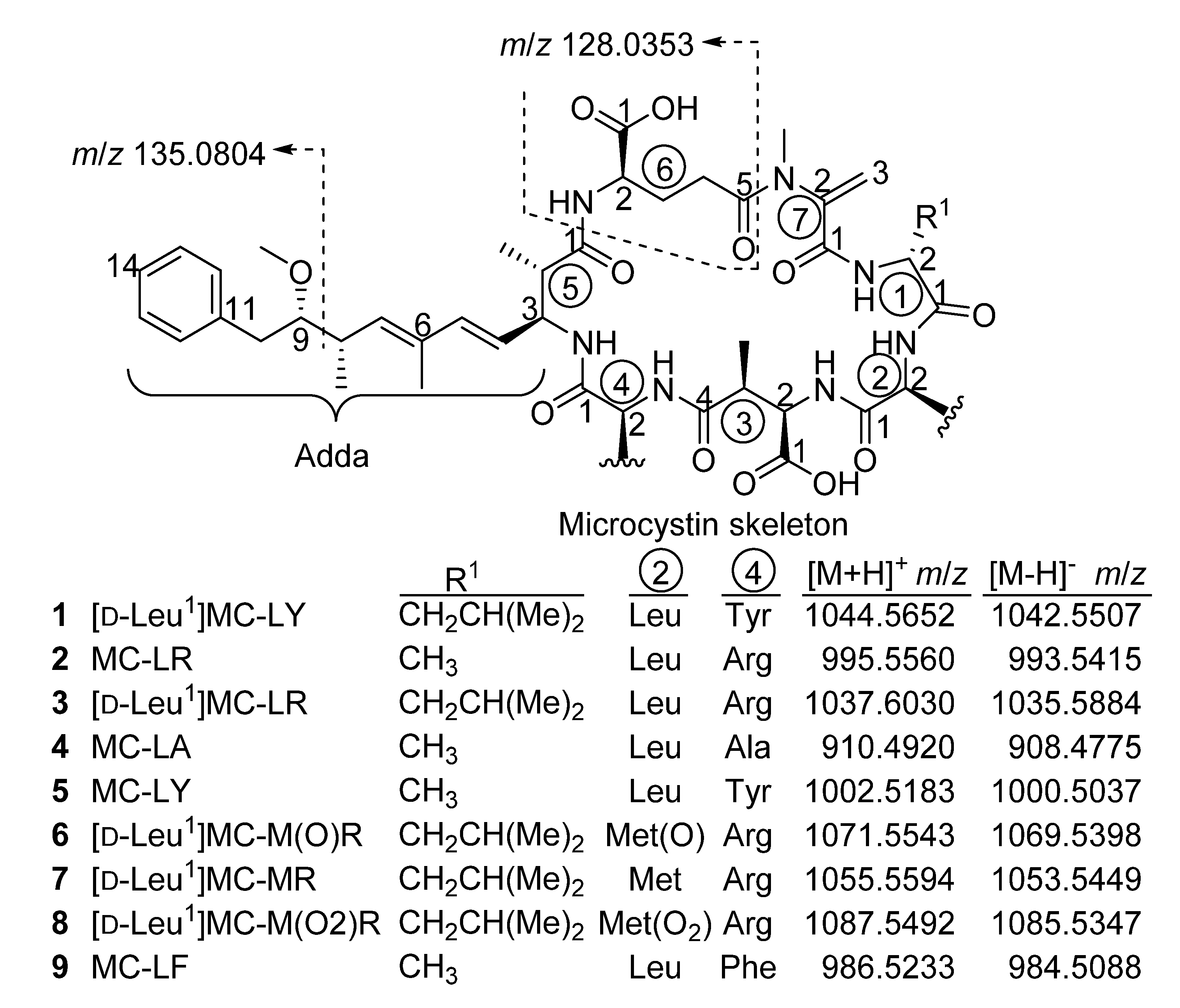
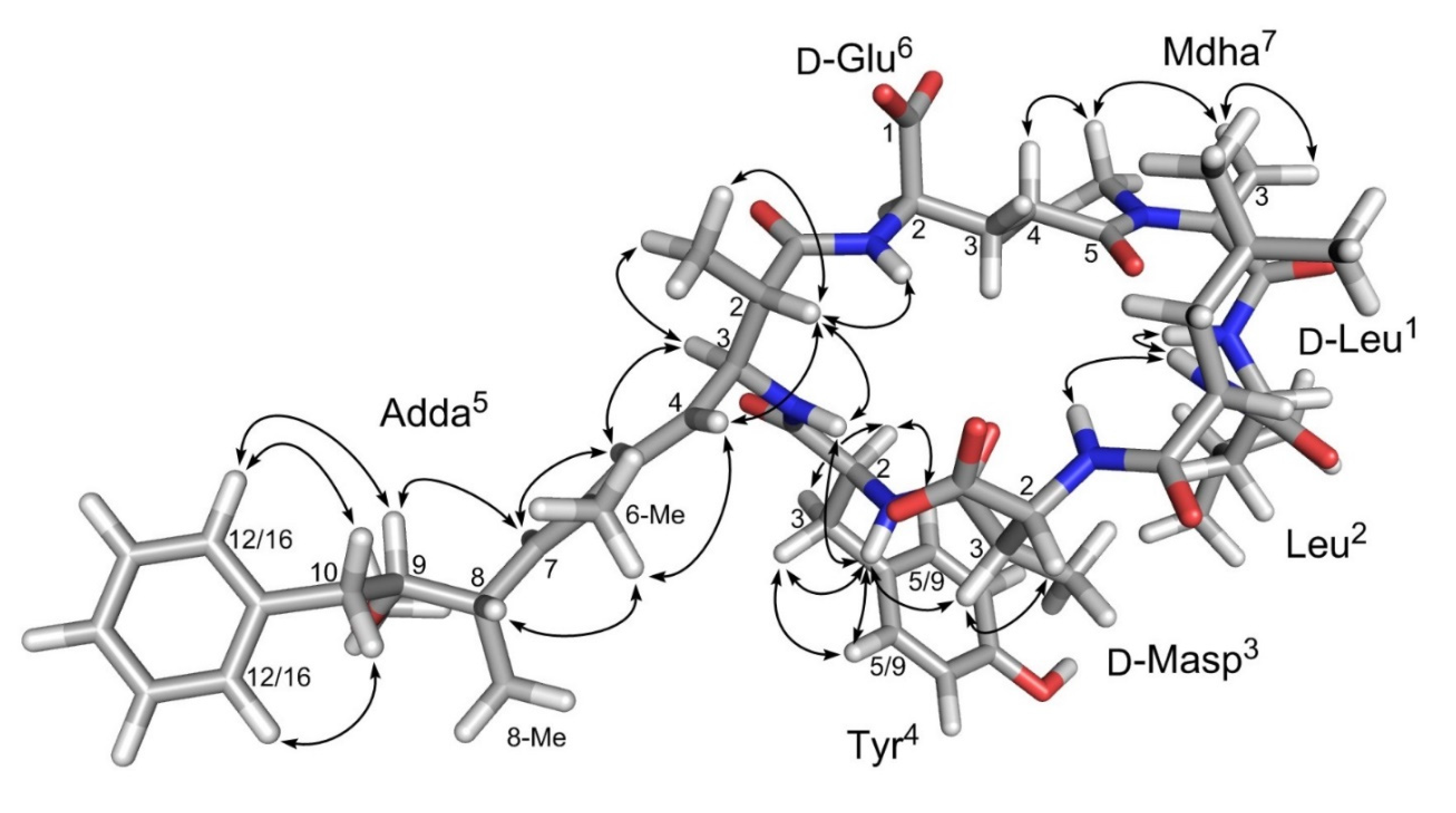
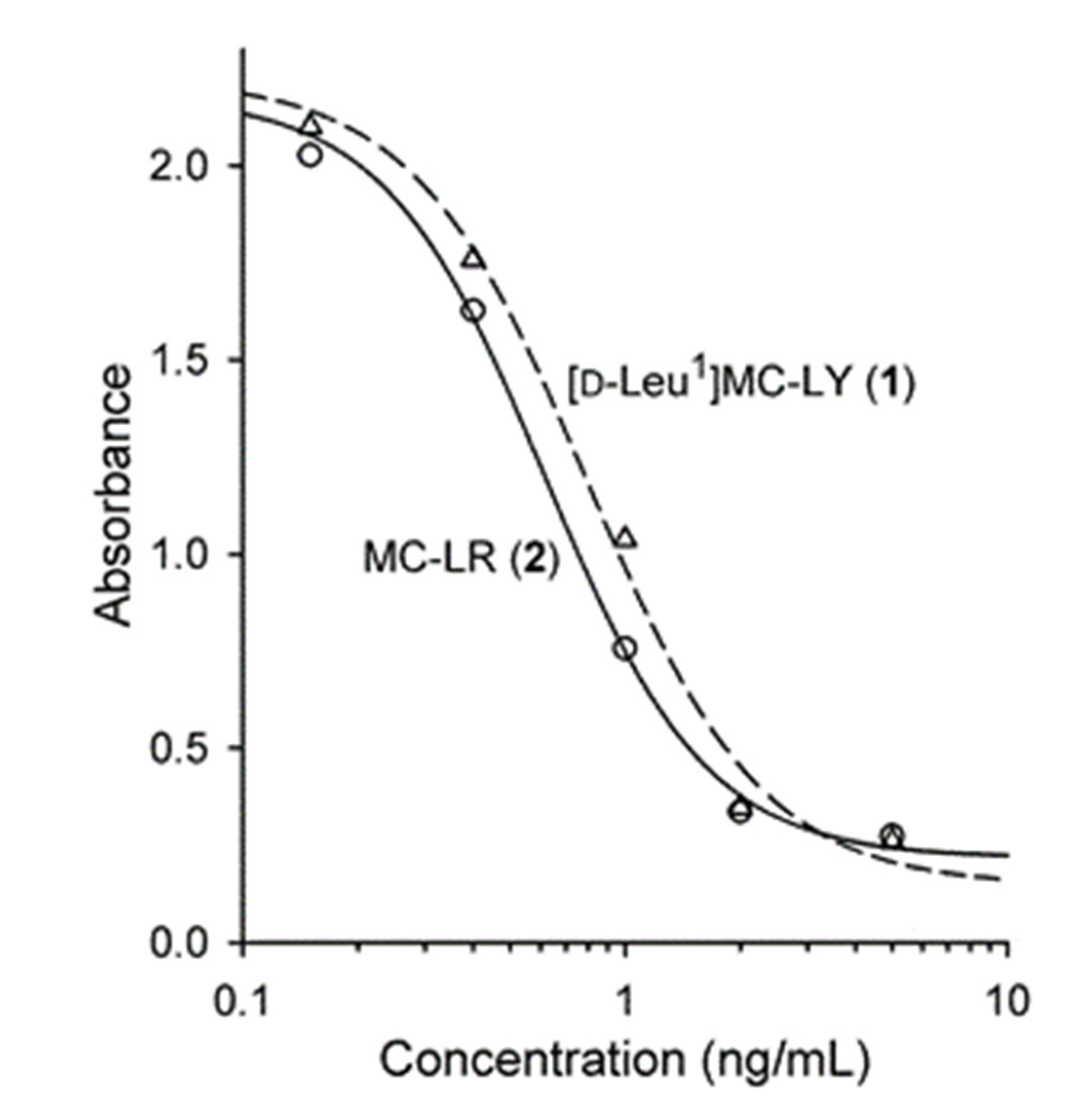
2. Results and Discussion
3. Conclusions
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Toxins and Other Materials
4.3. Biological Material
4.4. Toxin Isolation from Culture Biomass
4.5. Preparation of Reference Material
4.6. Protein Phosphatase Inhibition Assay
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Catherine, A.; Bernard, C.; Spoof, L.; Bruno, M. Microcystins and nodularins. In Handbook of Cyanobacterial Monitoring and Cyanobacterial Analysis, 1st ed.; Meriluoto, J., Spoof, L., Codd, G.A., Eds.; John Wiley & Sons: Chichester, UK, 2017; p. 109. [Google Scholar]
- Gulledge, B.M.; Aggen, J.B.; Eng, H.; Sweimeh, K.; Chamberlin, A.R. Microcystin analogues comprised only of adda and a single additional amino acid retain moderate activity as PP1/PP2A inhibitors. Bioorg. Med. Chem. Lett. 2003, 13, 2907–2911. [Google Scholar] [CrossRef]
- Gulledge, B.M.; Aggen, J.B.; Chamberlin, A.R. Linearized and truncated microcystin analogues as inhibitors of protein phosphatases 1 and 2A. Bioorg. Med. Chem. Lett. 2003, 13, 2903–2906. [Google Scholar] [CrossRef]
- Spoof, L.; Catherine, A. Appendix 3. Tables of microcystins and nodularins. In Handbook of Cyanobacterial Monitoring and Cyanobacterial Analysis; Meriluoto, J., Spoof, L., Codd, G.A., Eds.; John Wiley & Sons: Chichester, UK, 2017; pp. 526–537. [Google Scholar]
- Hollingdale, C.; Thomas, K.; Lewis, N.; Bekri, K.; McCarron, P.; Quilliam, M.A. Feasibility study on production of a matrix reference material for cyanobacterial toxins. Anal. Bioanal. Chem. 2015, 407, 5353–5363. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Namikoshi, M.; Brittain, S.M.; Carmichael, W.W.; Murphy, T. [D-Leu1] microcystin-LR, a new microcystin isolated from waterbloom in a Canadian prairie lake. Toxicon 2001, 39, 855–862. [Google Scholar] [CrossRef]
- Matthiensen, A.; Beattie, K.A.; Yunes, J.S.; Kaya, K.; Codd, G.A. [D-Leu1] Microcystin-LR, from the cyanobacterium Microcystis RST 9501 and from a Microcystis bloom in the Patos Lagoon estuary, Brazil. Phytochemistry 2000, 55, 383–387. [Google Scholar] [CrossRef]
- Schripsema, J.; Dagino, D. Complete assignment of the NMR spectra of [D-Leu1]-microcystin-LR and analysis of its solution structure. Mag. Res. Chem. 2002, 40, 614–617. [Google Scholar] [CrossRef]
- Ortiz, X.; Korenkova, E.; Jobst, K.J.; MacPherson, K.A.; Reiner, E.J. A high throughput targeted and non-targeted method for the analysis of microcystins and anatoxin-A using on-line solid phase extraction coupled to liquid chromatography-quadrupole time-of-flight high resolution mass spectrometry. Anal. Bioanal. Chem. 2017, 409, 4959–4969. [Google Scholar] [CrossRef]
- Caixach, J.; Flores, C.; Spoof, L.; Meriluoto, J.; Schmidt, W.; Mazur-Marzec, H.; Hiskia, A.; Kaloudis, T.; Furey, A. Liquid chromatography–mass spectrometry. In Handbook of Cyanobacterial Monitoring and Cyanobacterial Analysis, 1st ed.; Meriluoto, J., Spoof, L., Codd, G.A., Eds.; John Wiley & Sons: Chichester, UK, 2017; pp. 218–257. [Google Scholar]
- Foss, A.J.; Miles, C.O.; Samdal, I.A.; Løvberg, K.E.; Wilkins, A.L.; Rise, F.; Jaabæk, J.A.H.; McGowan, P.C.; Aubel, M.T. Analysis of free and metabolized microcystins in samples following a bird mortality event. Harmful Algae 2018, 80, 117–129. [Google Scholar] [CrossRef]
- Trogen, G.; Edlund, U.; Larsson, G.; Sethson, I. The solution NMR structure of a blue-green algae hepatotoxin, microcystin-RR. A comparison with the structure of microcystin-LR. Eur. J. Biochem. 1998, 258, 301–312. [Google Scholar] [CrossRef] [Green Version]
- Stewart, A.K.; Strangman, W.K.; Percy, A.; Wright, J.L.C. The biosynthesis of (15)N-labeled microcystins and the comparative MS/MS fragmentation of natural abundance and their (15)N-labeled congeners using LC-MS/MS. Toxicon 2018, 144, 91–102. [Google Scholar] [CrossRef]
- Bortoli, S.; Volmer, D. Account: Characterization and identification of microcystins by mass spectrometry. Eur. J. Mass Spectrom. 2014, 20, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Miles, C.O.; Sandvik, M.; Nonga, H.E.; Rundberget, T.; Wilkins, A.L.; Rise, F.; Ballot, A. Identification of microcystins in a Lake Victoria cyanobacterial bloom using LC–MS with thiol derivatization. Toxicon 2013, 70, 21–31. [Google Scholar] [CrossRef]
- Mayumi, T.; Kato, H.; Imanishi, S.; Kawasaki, Y.; Hasegawa, M.; Harada, K. Structural characterization of microcystins by LC/MS/MS under ion trap conditions. J. Antibiot. 2006, 59, 710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorr, F.A.; Oliveira-Silva, D.; Lopes, N.P.; Iglesias, J.; Volmer, D.A.; Pinto, E. Dissociation of deprotonated microcystin variants by collision-induced dissociation following electrospray ionization. Rapid. Commun. Mass Spectrom. 2011, 25, 1981–1992. [Google Scholar] [CrossRef] [PubMed]
- Burton, I.W.; Quilliam, M.A.; Walter, J.A. Quantitative 1H NMR with external standards: Use in preparation of calibration solutions for algal toxins and other natural products. Anal. Chem. 2005, 77, 3123–3131. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.; Crain, S.; Quilliam, M.A.; Chen, Y.M.; Wechsler, D. Analysis of natural toxins by liquid chromatography-chemiluminescence nitrogen detection and application to the preparation of certified reference materials. J. AOAC Int. 2016, 99, 1173–1184. [Google Scholar] [CrossRef]
- Gulledge, B.M.; Aggen, J.B.; Huang, H.; Nairn, A.C.; Chamberlin, A.R. The Microcystins and Nodularins: Cyclic polypeptide inhibitors of PP1 and PP2A. Curr. Med. Chem. 2002, 9, 1991–2003. [Google Scholar] [CrossRef]
- Ikehara, T.; Imamura, S.; Sano, T.; Nakashima, J.; Kuniyoshi, K.; Oshiro, N.; Yoshimoto, M.; Yasumoto, T. The effect of structural variation in 21 microcystins on their inhibition of PP2A and the effect of replacing cys269 with glycine. Toxicon 2009, 54, 539–544. [Google Scholar] [CrossRef]
- Miles, C.O.; Melanson, J.E.; Ballot, A. Sulfide oxidations for LC-MS analysis of methionine-containing microcystins in Dolichospermum flos-aquae NIVA-CYA 656. Environ. Sci. Technol. 2014, 48, 13307–13315. [Google Scholar] [CrossRef]
- Pick, F.R. Blooming algae: A Canadian perspective on the rise of toxic cyanobacteria. Can. J. Fish. Aquat. Sci. 2016, 73, 1149–1158. [Google Scholar] [CrossRef] [Green Version]
- Mantzouki, E.; Lürling, M.; Fastner, J.; De Senerpont Domis, L.; Wilk-Woźniak, E.; Koreivienė, J.; Seelen, L.; Teurlincx, S.; Verstijnen, Y.; Krztoń, W.; et al. Temperature effects explain continental scale distribution of cyanobacterial toxins. Toxins 2018, 10, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shishido, T.; Kaasalainen, U.; Fewer, D.; Rouhiainen, L.; Jokela, J.; Wahlsten, M.; Fiore, M.; Yunes, J.; Rikkinen, J.; Sivonen, K. Convergent evolution of [D-Leucine1] microcystin-LR in taxonomically disparate cyanobacteria. BMC Evol. Biol. 2013, 13, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Y.; Rosso, L.; Sedan, D.; Giannuzzi, L.; Andrinolo, D.; Volmer, D.A. Seven new microcystin variants discovered from a native Microcystis aeruginosa strain—Unambiguous assignment of product ions by tandem mass spectrometry. Rapid. Commun. Mass Spectrom. 2015, 29, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Kaasalainen, U.; Fewer, D.P.; Jokela, J.; Wahlsten, M.; Sivonen, K.; Rikkinen, J. Cyanobacteria produce a high variety of hepatotoxic peptides in lichen symbiosis. Proc. Natl. Acad. Sci. USA 2012, 109, 5886–5891. [Google Scholar] [CrossRef] [Green Version]
- Samdal, I.A.; Ballot, A.; Lovberg, K.E.; Miles, C.O. Multihapten approach leading to a sensitive ELISA with broad cross-reactivity to microcystins and nodularin. Environ. Sci. Technol. 2014, 48, 8035–8043. [Google Scholar] [CrossRef] [Green Version]
- Fischer, W.J.; Garthwaite, I.; Miles, C.O.; Ross, K.M.; Aggen, J.B.; Chamberlin, A.R.; Towers, N.R.; Dietrich, D.R. Congener-independent immunoassay for microcystins and nodularins. Environ. Sci. Technol. 2001, 35, 4849–4856. [Google Scholar] [CrossRef] [Green Version]
- Rippka, R.; Deruelles, J.; Waterbury, J.B.; Herdman, M.; Stanier, R.Y. Generic assignments, strain histories and properties of pure cultures of cyanobacteria. Microbiology 1979, 1, 111. [Google Scholar] [CrossRef] [Green Version]
- Allen, M.M.; Stanier, R.Y. Growth and division of some unicellular blue-green algae. J. Gen. Microbiol. 1968, 51, 199–202. [Google Scholar] [CrossRef] [Green Version]
- Abraxis Microcystins/Nodularins PP2A, Microtiter Plate: Test for the Detection of Microcystins and Nodularins in Water. Available online: https://www.abraxiskits.com/wp-content/uploads/2017/03/Microcystins-PP2A-Plate.pdf (accessed on 14 February 2019).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Unit | Position | δc, Type | δH, Multiplicity (J in Hz) | HMBC |
|---|---|---|---|---|
| D-Leu1 | 1 | ND | ||
| 2 | 52.0, CH | 4.44, m | ||
| 2-NH | 7.89, d (7.7) | |||
| 3a | 39.2, CH2 | 1.62, m | 4 | |
| 3b | 1.33, m | |||
| 4 | 24.8, CH | 1.57, m | ||
| 4-Me | 19.9, CH3 | 0.83, d (6.3) | ||
| 5 | 22.6, CH3 | 0.83, d (6.3) | ||
| Leu2 | 1 | 174.5, C | ||
| 2 | 53.8, CH | 4.23, ddd (10.4, 6.6, 4.0) | ||
| 2-NH | 8.28, d (6.5) | 1 | ||
| 3a | 39.8, CH2 | 1.92, ddd (13.7, 12.0, 4.0) | 2,5/6 | |
| 3b | 1.51, ddd (13.7, 10.4, 3.8) | |||
| 4 | 24.5, CH | 1.76, m | ||
| 4-Me | 20.0, CH3 | 0.88, d (6.6) | ||
| 5 | 22.8, CH3 | 0.88, d (6.6) | ||
| D-Masp3 | 1 | 175.6, C | ||
| 2 | 54.6, CH | 4.60, dd (9.0, 3.8) | 1,4 | |
| 2-NH | 7.72, d (9.0) | Leu2-1 | ||
| 3 | 40.7, CH | 3.04, dq (7.2, 3.8) | ||
| 3-Me | 13.8, CH3 | 0.86, d (7.2) | 2,4 | |
| 4 | 177.4, C | |||
| Tyr4 | 1 | 170.6, C | ||
| 2 | 54.1, CH | 4.35, ddd (11.7, 9.3, 3.3) | 1 | |
| 2-NH | 8.89, d (9.3) | |||
| 3a | 36.3, CH2 | 3.29, m | 1 | |
| 3b | 2.54, dd (14.1, 11.7) | 2,4,5/9 | ||
| 4 | 128.3, C | |||
| 5/9 | 130.2, CH | 6.99, d (8.5) | 3,7,5/9 | |
| 6/8 | 115.0, CH | 6.62, d (8.5) | 4,7,6/8 | |
| 7 | 156.4, C | |||
| Adda5 | 1 | 176.0, C | ||
| 2 | 44.1, CH | 2.76, m | ||
| 2-Me | 14.8, CH3 | 1.08, d (6.9) | 1,2,3 | |
| 3 | 55.5, CH | 4.70, ~q (9.7) | 1,2,4,5 | |
| 3-NH | 7.36, d (9.2) | |||
| 4 | 125.2, CH | 5.46, dd (15.5, 9.0) | 6 | |
| 5 | 138.0, CH | 6.32, d (15.5) | 3,7,6-Me | |
| 6 | 132.8, C | |||
| 6-Me | 11.8, CH3 | 1.63, s | 5,6,7 | |
| 7 | 136.2, CH | 5.49, d (10.1) | 5,6,8,9,6-Me | |
| 8 | 36.5, CH | 2.61, m | ||
| 8-Me | 15.5, CH3 | 1.03, d (6.7) | 7,8,9 | |
| 9 | 87.2, CH | 3.27, m | ||
| 9-OMe | 57.6, CH3 | 3.24, s | 9 | |
| 10a | 37.7, CH2 | 2.83, dd (13.9, 4.7) | 12/16 | |
| 10b | 2.68, dd (13.9, 7.4) | 9,11,12/16 | ||
| 11 | 139.4, C | |||
| 12/16 | 129.5, CH | 7.19, m | 10,14,12/16 | |
| 13/15 | 128.1, CH | 7.25, t (7.6) | 11,13/15 | |
| 14 | 125.9, CH | 7.17, m | ||
| D-Glu6 | 1 | 174.8, C | ||
| 2 | 53.3, CH | 4.31, ~q (7.2) | 1,3 | |
| 2-NH | 7.55, brs | |||
| 3a | 27.0, CH2 | 2.10, m | ||
| 3b | 1.79, m | |||
| 4a | 32.0, CH2 | 2.72, m | ||
| 4b | 2.59, m | |||
| 5 | 175.2, C | |||
| Mdha7 | 1 | 165.5, C | ||
| 2 | 145.1, C | |||
| 2-NMe | 37.3, CH3 | 3.36, s | 2, D-Glu-5 | |
| 3E | 113.3, CH2 | 5.84, s | 1 | |
| 3Z | 5.43, s | 1,2 |
| AA Origin | Reported for MC-LA (4) | MC-LA (4) | MC-LY (5) | [D-Leu1]MC-LY (1) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | Calc. m/z | Formula | m/z | Diff. | m/z | Diff. | m/z | Diff. |
| 44.0495 | C2H6N+ | ND | N/A | 136.0757 | 92.0262 | 136.0756 | 92.0261 | |||||||
| 56.0495 | C3H6N+ | 56.0497 | 0.0002 | 56.0497 | 0.0002 | 56.0497 | 0.0002 | |||||||
| 84.0444 | C4H6NO+ | 84.0443 | -0.0001 | 84.0444 | 0.0000 | 84.0444 | 0.0000 | |||||||
| 86.0964 | C5H12N+ | 86.0964 | 0.0000 | 86.0964 | 0.0000 | 86.0964 | 0.0000 | |||||||
| 103.0542 | C8H7+ | 103.0541 | -0.0001 | 103.0542 | 0.0000 | 103.0542 | 0.0000 | |||||||
| 107.0855 | C8H11+ | 107.0854 | -0.0001 | 107.0855 | 0.0000 | 107.0855 | 0.0000 | |||||||
| 127.0866 | C6H11N2O+ | 127.0865 | -0.0001 | 127.0866 | 0.0000 | 169.1334 | 42.0468 | |||||||
| 135.0804 | C9H11O+ | 135.0803 | -0.0001 | 135.0804 | 0.0000 | 135.0803 | -0.0001 | |||||||
| 135.1168 | C10H15+ | 135.1166 | -0.0002 | 135.1167 | -0.0001 | 135.1166 | -0.0002 | |||||||
| 155.0815 | C7H11N2O2+ | 155.0813 | -0.0002 | 155.0813 | -0.0002 | 197.1283 | 42.0468 | |||||||
| 163.1117 | C11H15O+ | 163.1115 | -0.0002 | 163.1117 | 0.0000 | 163.1115 | -0.0002 | |||||||
| 173.0921 | C7H13N2O3+ | 173.0917 | -0.0004 | 265.1180 | 92.0259 | 265.1179 | 92.0258 | |||||||
| 195.0764 | C9H11N2O3+ | 195.0763 | -0.0001 | 195.0764 | 0.0000 | 195.0763 | -0.0001 | |||||||
| 213.0870 | C9H13N2O4+ | 213.0868 | -0.0002 | 213.0869 | -0.0001 | 213.0868 | -0.0002 | |||||||
| 218.1135 | C8H16N3O4+ | 218.1133 | -0.0002 | 310.1393 | 92.0258 | 310.1392 | 92.0257 | |||||||
| 265.1587 | C19H21O+ | 265.1581 | -0.0006 | 265.1583 | -0.0004 | 265.1581 | -0.0006 | |||||||
| 268.1656 | C13H22N3O3+ | 268.1650 | -0.0006 | 268.1652 | -0.0004 | 310.2119 | 42.0463 | |||||||
| 292.1543 | C16H22NO4+ | 292.1538 | -0.0005 | 292.1541 | -0.0002 | 292.1540 | -0.0003 | |||||||
| 314.1710 | C14H24N3O5+ | 314.1705 | -0.0005 | 406.1967 | 92.0257 | 406.1965 | 92.0255 | |||||||
| 331.1976 | C14H27N4O5+ | 331.1970 | -0.0006 | 423.2230 | 92.0254 | 423.2229 | 92.0253 | |||||||
| 375.1914 | C20H27N2O5+ | 375.1906 | -0.0008 | 375.1912 | -0.0002 | 375.1907 | -0.0007 | |||||||
| 385.2082 | C17H29N4O6+ | 385.2073 | -0.0009 | 477.2359 | 92.0277 | 519.2807 | 134.0725 | |||||||
| 402.2347 | C17H32N5O6+ | 402.2342 | -0.0005 | 494.2602 | 92.0255 | 536.3072 | 134.0725 | |||||||
| 446.2286 | C23H32N3O6+ | 446.2276 | -0.0010 | 446.2283 | -0.0003 | 488.2745 | 42.0459 | |||||||
| 468.2453 | C21H34N5O7+ | 468.2443 | -0.0010 | 560.2707 | 92.0254 | 602.3180 | 134.0727 | |||||||
| 485.2718 | C21H37N6O7+ | 485.2708 | -0.0010 | 577.2976 | 92.0258 | 619.3447 | 134.0729 | |||||||
| 509.2646 | C29H37N2O6+ | 509.2637 | -0.0009 | 509.2640 | -0.0006 | 509.2640 | -0.0006 | |||||||
| 559.3126 | C29H43N4O7+ | 559.3117 | -0.0009 | 559.3157 | 0.0031 | 601.3610 | 42.0484 | |||||||
| 580.3017 | C32H42N3O7+ | 580.3008 | -0.0009 | 580.3010 | -0.0007 | 622.3479 | 42.0462 | |||||||
| 597.2879 | C26H41N6O10+ | 597.2872 | -0.0007 | 689.3134 | 92.0255 | 731.3605 | 134.0726 | |||||||
| 693.3858 | C38H53O8N4+ | 693.3854 | -0.0004 | 693.3854 | -0.0004 | 735.4322 | 42.0464 | |||||||
| 758.4083 | C37H56N7O10+ | 758.4076 | -0.0007 | 850.4350 | 92.0267 | 892.4804 | 134.0721 | |||||||
| 759.3923 | C37H55N6O11+ | 759.3917 | -0.0006 | 851.4190 | 92.0267 | 893.4652 | 134.0729 | |||||||
| 776.4189 | C37H58N7O11+ | 776.4187 | -0.0002 | 868.4445 | 92.0256 | 910.4906 | 134.0717 | |||||||
| 910.4920 | C46H68N7O12+ | 910.4908 | -0.0012 | 1002.5175 | 92.0255 | 1044.5634 | 134.0714 | |||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
LeBlanc, P.; Merkley, N.; Thomas, K.; Lewis, N.I.; Békri, K.; Renaud, S.L.; Pick, F.R.; McCarron, P.; Miles, C.O.; Quilliam, M.A. Isolation and Characterization of [D-Leu1]microcystin-LY from Microcystis aeruginosa CPCC-464. Toxins 2020, 12, 77. https://doi.org/10.3390/toxins12020077
LeBlanc P, Merkley N, Thomas K, Lewis NI, Békri K, Renaud SL, Pick FR, McCarron P, Miles CO, Quilliam MA. Isolation and Characterization of [D-Leu1]microcystin-LY from Microcystis aeruginosa CPCC-464. Toxins. 2020; 12(2):77. https://doi.org/10.3390/toxins12020077
Chicago/Turabian StyleLeBlanc, Patricia, Nadine Merkley, Krista Thomas, Nancy I. Lewis, Khalida Békri, Susan LeBlanc Renaud, Frances R. Pick, Pearse McCarron, Christopher O. Miles, and Michael A. Quilliam. 2020. "Isolation and Characterization of [D-Leu1]microcystin-LY from Microcystis aeruginosa CPCC-464" Toxins 12, no. 2: 77. https://doi.org/10.3390/toxins12020077