On the Interaction of Clostridium perfringens Enterotoxin with Claudins


Abstract
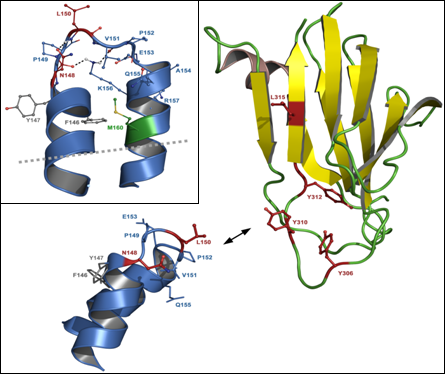
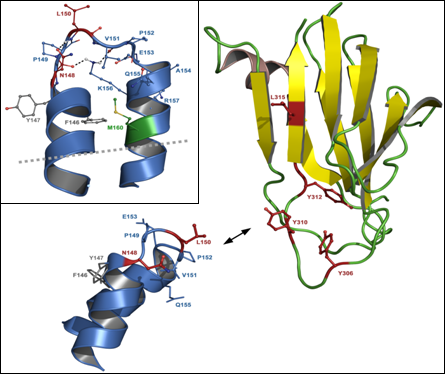
:
1. Introduction
2. Clostridium perfringens and Its Toxins
3. Clostridium perfringens Enterotoxin (CPE)
3.1. Functional domains of CPE
3.2. C-terminal claudin-binding domain of CPE
4. The CPE-Receptors: Claudins
5. The Interaction between Clostridium perfringens Enterotoxin and Claudins
5.1. Methods used to investigate interaction between CPE and claudins
5.1.1. Use of peptides in arrays and surface plasmon resonance spectroscopy
5.1.2. Pull-down assays
5.1.3. Cellular binding assays
5.1.3.1. Assay of CPE cytotoxicity
5.1.3.2. Competition binding assay
5.1.4. Methods for investigation of cCPE effects on paracellular permeability
5.2. Residues in CPE involved in binding to claudins
5.3. Residues in claudins involved in interaction with CPE

{kind=link}
{kind=link}
 : Assays based on peptides of claudins or CPE;
: Assays based on peptides of claudins or CPE;  : assays based on detergent-solubilized claudins;
: assays based on detergent-solubilized claudins;  : cellular-binding assays. The color shade corresponds to the following categories of binding capacity: 0–10% (-), 10–25% (low), 25–85% (medium) and >85% (high) of bound CPE relative to Cld3wt or Cld4wt. Entries marked with an asterisk were derived by cytotoxicity assays and the categories are defined as: highly sensitive (high), EC50 < 1 µg/mL; slightly sensitive (low), 1 µg/mL < EC50 < 30 µg/mL; and insensitive, 30 µg/mL < EC50 [43]. In some cases, no further quantification of the amount of bound CPE was available (+). All constructs without prefixes are derived from mouse sequences, the other constructs are derived from human (hu-); monkey (mk-); mouse/human chimaeras (muh-). monkey/human chimeras (mkh-). (p) indicates data obtained with CPE290–319, (L) data obtained with CPE116–319; all other data were obtained with full length CPE or constructs similar to CPE194–319.
: cellular-binding assays. The color shade corresponds to the following categories of binding capacity: 0–10% (-), 10–25% (low), 25–85% (medium) and >85% (high) of bound CPE relative to Cld3wt or Cld4wt. Entries marked with an asterisk were derived by cytotoxicity assays and the categories are defined as: highly sensitive (high), EC50 < 1 µg/mL; slightly sensitive (low), 1 µg/mL < EC50 < 30 µg/mL; and insensitive, 30 µg/mL < EC50 [43]. In some cases, no further quantification of the amount of bound CPE was available (+). All constructs without prefixes are derived from mouse sequences, the other constructs are derived from human (hu-); monkey (mk-); mouse/human chimaeras (muh-). monkey/human chimeras (mkh-). (p) indicates data obtained with CPE290–319, (L) data obtained with CPE116–319; all other data were obtained with full length CPE or constructs similar to CPE194–319.
 |
6. Potential Pharmacological Use of Clostridium perfringens Enterotoxin
6.1. cCPE as a TJ-Modulator
6.2. Treatment of tumors overexpressing claudins by cCPE-constructs
7. Conclusions and Remarks

References
- McClane, B.A. The complex interactions between Clostridium perfringens enterotoxin and epithelial tight junctions. Toxicon 2001, 39, 1781–1791. [Google Scholar]
- Smedley, J.G., III; McClane, B.A. Fine mapping of the N-terminal cytotoxicity region of Clostridium perfringens enterotoxin by site-directed mutagenesis. Infect. Immun. 2004, 72, 6914–6923. [Google Scholar] [CrossRef] [PubMed]
- Miyakawa, M.E.F.; Creydt, V.P.; Uzal, F.A.; McClane, B.A.; Ibarra, C. Clostridium perfringens enterotoxin damages the human intestine in vitro. Infect. Immun. 2005, 73, 8407–8410. [Google Scholar]
- Sonoda, N.; Furuse, M.; Sasaki, H.; Yonemura, S.; Katahira, J.; Horiguchi, Y.; Tsukita, S. Clostridium perfringens enterotoxin fragment removes specific claudins from tight junction strands: Evidence for direct involvement of claudins in tight junction barrier. J. Cell Biol. 1999, 147, 195–204. [Google Scholar]
- Kondoh, M.; Masuyama, A.; Takahashi, A.; Asano, N.; Mizuguchi, H.; Koizumi, N.; Fujii, M.; Hayakawa, T.; Horiguchi, Y.; Watanbe, Y. A novel strategy for the enhancement of drug absorption using a claudin modulator. Mol. Pharmacol. 2005, 67, 749–756. [Google Scholar]
- Rood, J.I. Virulence genes of Clostridium perfringens. Annu. Rev. Microbiol. 1998, 52, 333–360. [Google Scholar]
- Sakurai, J.; Nagahama, M.; Oda, M. Clostridium perfringens alpha-toxin: Characterization and mode of action. J. Biochem. 2004, 136, 569–574. [Google Scholar]
- Awad, M.M.; Ellemor, D.M.; Boyd, R.L.; Emmins, J.J.; Rood, J.I. Synergistic effects of alpha-toxin and perfringolysin O in Clostridium perfringens-mediated gas gangrene. Infect. Immun. 2001, 69, 7904–7910. [Google Scholar]
- Nagahama, M.; Hayashi, S.; Morimitsu, S.; Sakurai, J. Biological activities and pore formation of Clostridium perfringens beta toxin in HL 60 cells. J. Biol. Chem. 2003, 278, 36934–36941. [Google Scholar]
- Fisher, D.J.; Fernandez-Miyakawa, M.E.; Sayeed, S.; Poon, R.; Adams, V.; Rood, J.I.; Uzal, F.A.; McClane, B.A. Dissecting the contributions of Clostridium perfringens type C toxins to lethality in the mouse intravenous injection model. Infect. Immun. 2006, 74, 5200–5210. [Google Scholar]
- Miyata, S.; Minami, J.; Tamai, E.; Matsushita, O.; Shimamoto, S.; Okabe, A. Clostridium perfringens epsilon-toxin forms a heptameric pore within the detergent-insoluble microdomains of Madin-Darby canine kidney cells and rat synaptosomes. J. Biol. Chem. 2002, 277, 39463–39468. [Google Scholar]
- Petit, L.; Gibert, M.; Gourch, A.; Bens, M.; Vandewalle, A.; Popoff, M.R. Clostridium perfringens epsilon toxin rapidly decreases membrane barrier permeability of polarized MDCK cells. Cell. Microbiol. 2003, 5, 155–164. [Google Scholar]
- Petit, L.; Maier, E.; Gibert, M.; Popoff, M.R.; Benz, R. Clostridium perfringens epsilon toxin induces a rapid change of cell membrane permeability to ions and forms channels in artificial lipid bilayers. J. Biol. Chem. 2001, 276, 15736–15740. [Google Scholar]
- Loffler, A.; Labbe, R.G. Isolation of An Inclusion Body from Sporulating, Enterotoxin-Positive Clostridium-Perfringens. FEMS Microbiol. Lett. 1985, 27, 143–147. [Google Scholar]
- Loffler, A.; Labbe, R. Characterization of A Parasporal Inclusion Body from Sporulating, Enterotoxin-Positive Clostridium-Perfringens Type-A. J. Bacteriol. 1986, 165, 542–548. [Google Scholar]
- Duncan, C.L.; Sugiyama, H.; Strong, D.H. Rabbit Ileal Loop Response to Strains of Clostridium perfringens. J. Bacteriol. 1968, 95, 1560–1566. [Google Scholar]
- Strong, D.H.; Duncan, C.L.; Perna, G. Clostridium-Perfringens Type-A Food Poisoning. 2. Response of Rabbit Ileum As An Indication of Enteropathogenicity of Strains of Clostridium-Perfringens in Human Beings. Infect. Immun. 1971, 3, 171–178. [Google Scholar] [PubMed]
- Niilo, L. Fluid Secretory Response of Bovine Thiry Jejunal Fistula to Enterotoxin of Clostridium-Perfringens. Infect. Immun. 1973, 7, 1–4. [Google Scholar]
- Skjelkvale, R.; Uemura, T. Experimental Diarrhea in Human Volunteers Following Oral-Administration of Clostridium-Perfringens Enterotoxin. J. Appl. Bacteriol. 1977, 43, 281–286. [Google Scholar]
- Sarker, R.; Carman, R.J.; McClane, B.A. Inactivation of the gene (cpe) encoding Clostridium perfringens enterotoxin eliminates the ability of two cpe-positive C-perfringens type A human gastrointestinal disease isolates to affect rabbit ileal loops. Mol. Microbiol. 1999, 33, 946–958. [Google Scholar]
- Czeczulin, J.R.; Hanna, P.C.; McClane, B.A. Cloning, Nucleotide Sequencing, and Expression of the Clostridium-Perfringens Enterotoxin Gene in Escherichia-Coli. Infect. Immun. 1993, 61, 3429–3439. [Google Scholar]
- Fujinaga, Y.; Inoue, K.; Shimazaki, S.; Tomochika, K.; Tsuzuki, K.; Fujii, N.; Watanabe, T.; Ohyama, T.; Takeshi, K.; Inoue, K.; Oguma, K. Molecular Construction of Clostridium-Botulinum Type-C Progenitor Toxin and Its Gene Organization. Biochem. Biophys. Res. Commun. 1994, 205, 1291–1298. [Google Scholar]
- Sherman, S.; Klein, E.; McClane, B.A. Clostridium-Perfringens Type-A Enterotoxin Induces Tissue-Damage and Fluid Accumulation in Rabbit Ileum. J. Diarrhoeal Dis. Res. 1994, 12, 200–207. [Google Scholar]
- McDonel, J.L.; Chang, L.W.; Pounds, J.G.; Duncan, C.L. Effects of Clostridium-Perfringens Enterotoxin on Rat and Rabbit Ileum - Electron-Microscopic Study. Lab. Invest. 1978, 39, 210–218. [Google Scholar]
- Singh, U.; Mitic, L.L.; Wieckowski, E.U.; Anderson, J.M.; McClane, B.A. Comparative biochemical and immunocytochemical studies reveal differences in the effects of Clostridium perfringens enterotoxin on polarized CaCo-2 cells versus Vero cells. J. Biol. Chem. 2001, 276, 33402–33412. [Google Scholar]
- Matsuda, M.; Sugimoto, N. Calcium-Independent and Dependent Steps in Action of Clostridium-Perfringens Enterotoxin on Hela and Vero Cells. Biochem. Biophys. Res. Commun. 1979, 91, 629–636. [Google Scholar]
- McClane, B.A.; McDonel, J.L. The effects of Clostridium perfringens enterotoxin on morphology, viability, and macromolecular synthesis in Vero cells. J. Cell Physiol. 1979, 99, 191–200. [Google Scholar]
- Katahira, J.; Sugiyama, H.; Inoue, N.; Horiguchi, Y.; Matsuda, M.; Sugimoto, N. Clostridium perfringens enterotoxin utilizes two structurally related membrane proteins as functional receptors in vivo. J. Biol. Chem. 1997, 272, 26652–26658. [Google Scholar] [PubMed]
- Robertson, S.L.; Smedley, J.G., III.; Singh, U.; Chakrabarti, G.; Van Itallie, C.M.; Anderson, J.M.; McClane, B.A. Compositional and stoichiometric analysis of Clostridium perfringens enterotoxin complexes in Caco-2 cells and claudin 4 fibroblast transfectants. Cell Microbiol. 2007, 9, 2734–2755. [Google Scholar]
- Wieckowski, E.U.; Wnek, A.P.; McClane, B.A. Evidence That An Approximate-To-50-Kda Mammalian Plasma-Membrane Protein with Receptor-Like Properties Mediates the Amphiphilicity of Specifically Bound Clostridium-Perfringens Enterotoxin. J. Biol. Chem. 1994, 269, 10838–10848. [Google Scholar]
- Wieckowski, E.U.; Kokai-Kun, J.F.; McClane, B.A. Characterization of membrane-associated Clostridium perfringens enterotoxin following pronase treatment. Infect. Immun. 1998, 66, 5897–5905. [Google Scholar]
- Smedley, J.G.; Uzal, F.A.; McClane, B.A. Identification of a prepore large-complex stage in the mechanism of action of Clostridium perfringens enterotoxin. Infect. Immun. 2007, 75, 2381–2390. [Google Scholar]
- Chakrabarti, G.; McClane, B.A. The importance of calcium influx, calpain and calmodulin for the activation of CaCo-2 cell death pathways by Clostridium perfringens enterotoxin. Cell. Microbiol. 2005, 7, 129–146. [Google Scholar]
- Kokai-Kun, J.F.; McClane, B.A. Deletion analysis of the Clostridium perfringens enterotoxin. Infect. Immun. 1997, 65, 1014–1022. [Google Scholar]
- Kokai-Kun, J.F.; Benton, K.; Wieckowski, E.U.; McClane, B.A. Identification of a Clostridium perfringens enterotoxin region required for large complex formation and cytotoxicity by random mutagenesis. Infect. Immun. 1999, 67, 5634–5641. [Google Scholar] [PubMed]
- Hanna, P.C.; Mietzner, T.A.; Schoolnik, G.K.; McClane, B.A. Localization of the receptor-binding region of Clostridium perfringens enterotoxin utilizing cloned toxin fragments and synthetic peptides. The 30 C-terminal amino acids define a functional binding region. J. Biol. Chem. 1991, 266, 11037–11043. [Google Scholar] [PubMed]
- Winkler, L.; Gehring, C.; Wenzel, A.; Muller, S.L.; Piehl, C.; Krause, G.; Blasig, I.E.; Piontek, J. Molecular Determinants of the Interaction between Clostridium perfringens Enterotoxin Fragments and Claudin-3. J. Biol. Chem. 2009, 284, 18863–18872. [Google Scholar]
- Van Itallie, C.M.; Betts, L.; Smedley, J.G.; McClane, B.A.; Anderson, J.M. Structure of the claudin-binding domain of Clostridium perfringens enterotoxin. J. Biol. Chem. 2008, 283, 268–274. [Google Scholar]
- Wilson, J.J.; Matsushita, O.; Okabe, A.; Sakon, J. A bacterial collagen-binding domain with novel calcium-binding motif controls domain orientation. EMBO J. 2003, 22, 1743–1752. [Google Scholar]
- Boonserm, P.; Davis, P.; Ellar, D.J.; Li, J. Crystal structure of the mosquito-iarvicidal toxin Cry4Ba and its biological implications. J. Mol. Biol. 2005, 348, 363–382. [Google Scholar]
- Katahira, J.; Inoue, N.; Horiguchi, Y.; Matsuda, M.; Sugimoto, N. Molecular cloning and functional characterization of the receptor for Clostridium perfringens enterotoxin. J. Cell Biol. 1997, 136, 1239–1247. [Google Scholar]
- Morita, K.; Furuse, M.; Fujimoto, K.; Tsukita, S. Claudin multigene family encoding four-transmembrane domain protein components of tight junction strands. Proc. Natl. Acad. Sci. USA 1999, 96, 511–516. [Google Scholar]
- Fujita, K.; Katahira, J.; Horiguchi, Y.; Sonoda, N.; Furuse, M.; Tsukita, S. Clostridium perfringens enterotoxin binds to the second extracellular loop of claudin-3, a tight junction integral membrane protein. FEBS Lett. 2000, 476, 258–261. [Google Scholar]
- Kimura, J.; Abe, H.; Kamitani, S.; Toshima, H.; Fukui, A.; Miyake, M.; Kamata, Y.; Sugita-Konishi, Y.; Yamamoto, S.; Horiguchi, Y. Clostridium perfringens Enterotoxin Interacts with Claudins via Electrostatic Attraction. J. Biol. Chem. 2010, 285, 401–408. [Google Scholar] [PubMed]
- Krause, G.; Winkler, L.; Mueller, S.L.; Haseloff, R.F.; Piontek, J.; Blasig, I.E. Structure and function of claudins. Biochim. Biophys. Acta 2008, 1778, 631–645. [Google Scholar]
- Staehelin, L.A. Structure and function of intercellular junctions. Int. Rev. Cytol. 1974, 39, 191–283. [Google Scholar]
- Furuse, M.; Hirase, T.; Itoh, M.; Nagafuchi, A.; Yonemura, S.; Tsukita, S.; Tsukita, S. Occludin: a novel integral membrane protein localizing at tight junctions. J. Cell Biol. 1993, 123, 1777–1788. [Google Scholar]
- Ikenouchi, J.; Furuse, M.; Furuse, K.; Sasaki, H.; Tsukita, S.; Tsukita, S. Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J. Cell Biol. 2005, 171, 939–945. [Google Scholar]
- Steed, E.; Rodrigues, N.T.; Balda, M.S.; Matter, K. Identification of MarvelD3 as a tight junction-associated transmembrane protein of the occludin family. BMC Cell Biol. 2009, 10, 95. [Google Scholar]
- Gonzalez-Mariscal, L.; Betanzos, A.; Nava, P.; Jaramillo, B.E. Tight junction proteins. Prog. Biophys. Mol. Biol. 2003, 81, 1–44. [Google Scholar]
- Umeda, K.; Ikenouchi, J.; Katahira-Tayama, S.; Furuse, K.; Sasaki, H.; Nakayama, M.; Matsui, T.; Tsukita, S.; Furuse, M.; Tsukita, S. ZO-1 and ZO-2 independently determine where claudins are polymerized in tight-junction strand formation. Cell 2006, 126, 741–754. [Google Scholar]
- Yu, A.S.; Cheng, M.H.; Angelow, S.; Gunzel, D.; Kanzawa, S.A.; Schneeberger, E.E.; Fromm, M.; Coalson, R.D. Molecular basis for cation selectivity in claudin-2-based paracellular pores: identification of an electrostatic interaction site. J. Gen. Physiol. 2009, 133, 111–127. [Google Scholar]
- Daugherty, B.L.; Ward, C.; Smith, T.; Ritzenthaler, J.D.; Koval, M. Regulation of heterotypic claudin compatibility. J. Biol. Chem. 2007, 282, 30005–30013. [Google Scholar]
- Piontek, J.; Winkler, L.; Wolburg, H.; Muller, S.L.; Zuleger, N.; Piehl, C.; Wiesner, B.; Krause, G.; Blasig, I.E. Formation of tight junction: determinants of homophilic interaction between classic claudins. FASEB J. 2008, 22, 146–158. [Google Scholar]
- Zhang, J.; Piontek, J.; Wolburg, H.; Piehl, C.; Liss, M.; Otten, C.; Christ, A.; Willnow, T.E.; Blasig, I.E.; Abdelilah-Seyfried, S. Establishment of a neuroepithelial barrier by Claudin5a is essential for zebrafish brain ventricular lumen expansion. Proc. Natl. Acad. Sci. USA 2010, 107, 1425–1430. [Google Scholar]
- Piehl, C.; Piontek, J.; Cording, J.; Wolburg, H.; Blasig, I.E. Participation of the second extracellular loop of claudin-5 in paracellular tightening against ions, small and large molecules. Cell Mol. Life Sci. 2010. [Google Scholar]
- Krause, G.; Winkler, L.; Piehl, C.; Blasig, I.; Piontek, J.; Muller, S.L. Structure and Function of Extracellular Claudin Domains. Ann. N.Y. Acad. Sci. 2009, 1165, 34–43. [Google Scholar]
- Ling, J.; Liao, H.; Clark, R.; Wong, M.S.; Lo, D.D. Structural constraints for the binding of short peptides to claudin-4 revealed by surface plasmon resonance. J. Biol. Chem. 2008, 283, 30585–30595. [Google Scholar]
- Harada, M.; Kondoh, M.; Ebihara, C.; Takahashi, A.; Komiya, E.; Fujii, M.; Mizuguchi, H.; Tsunoda, S.; Horiguchi, Y.; Yagi, K.; Watanabe, Y. Role of tyrosine residues in modulation of claudin-4 by the C-terminal fragment of Clostridium perfringens enterotoxin. Biochem. Pharmacol. 2007, 73, 206–214. [Google Scholar]
- Takahashi, A.; Komiya, E.; Kakutani, H.; Yoshida, T.; Fujii, M.; Horiguchi, Y.; Mizuquchi, H.; Tsutsumi, Y.; Tsunoda, S.I.; Koizumi, N.; Isoda, K.; Yagi, K.; Watanabe, Y.; Kondoh, M. Domain mapping of a claudin-4 modulator, the C-terminal region of C-terminal fragment of Clostridium perfringens enterotoxin, by site-directed mutagenesis. Biochem. Pharmacol. 2008, 75, 1639–1648. [Google Scholar]
- Lohrberg, D.; Krause, E.; Schumann, M.; Piontek, J.; Winkler, L.; Blasig, I.E.; Haseloff, R.F. A strategy for enrichment of claudins based on their affinity to Clostridium perfringens enterotoxin. BMC Cell Biol. 2009, 10, 61. [Google Scholar]
- Robertson, S.L.; Smedley, J.G.; McClane, B.A. Identification of a Claudin-4 Residue Important for Mediating the Host Cell Binding and Action of Clostridium perfringens Enterotoxin. Infect. Immun. 2010, 78, 505–517. [Google Scholar]
- Ebihara, C.; Kondoh, M.; Hasuike, N.; Harada, M.; Mizuguchi, H.; Horiguchi, Y.; Fujii, M.; Watanabe, Y. Preparation of a claudin-targeting molecule using a C-terminal fragment of Clostridium perfringens enterotoxin. J. Pharmacol. Exp. Ther. 2006, 316, 255–260. [Google Scholar]
- Swisshelm, K.; Machl, A.; Planitzer, S.; Robertson, R.; Kubbies, M.; Hosier, S. SEMP1, a senescence-associated cDNA isolated from human mammary epithelial cells, is a member of an epithelial membrane protein superfamily. Gene 1999, 226, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Uchida, H.; Kondoh, M.; Hanada, T.; Takahashi, A.; Hamakubo, T.; Yagi, K. A claudin-4 modulator enhances the mucosal absorption of a biologically active peptide. Biochem. Pharmacol. 2010, 79, 1437–1444. [Google Scholar]
- Ebihara, C.; Kondoh, M.; Harada, M.; Fujii, M.; Mizuguchi, H.; Tsunoda, S.; Horiguchi, Y.; Yagi, K.; Watanabe, Y. Role of Tyr306 in the C-terminal fragment of Clostridium perfringens enterotoxin for modulation of tight junction. Biochem. Pharmacol. 2007, 73, 824–830. [Google Scholar]
- Takahashi, A.; Kondoh, M.; Masuyama, A.; Fujii, M.; Mizuguchi, H.; Horiguchi, Y.; Watanabe, Y. Role of C-terminal regions of the C-terminal fragment of Clostridium perfringens enterotoxin in its interaction with claudin-4. J. Control. Release 2005, 108, 56–62. [Google Scholar]
- Mizuno, N.; Niwa, T.; Yotsumoto, Y.; Sugiyama, Y. Impact of drug transporter studies on drug discovery and development. Pharmacol. Rev. 2003, 55, 425–461. [Google Scholar]
- Aungst, B.J. Intestinal permeation enhancers. J. Pharm. Sci. 2000, 89, 429–442. [Google Scholar]
- Salama, N.N.; Eddington, N.D.; Fasano, A. Tight junction modulation and its relationship to drug delivery. Adv. Drug Delivery Rev. 2006, 58, 15–28. [Google Scholar]
- Hochman, J.; Artursson, P. Mechanisms of Absorption Enhancement and Tight Junction Regulation. J. Controlled Release 1994, 29, 253–267. [Google Scholar]
- Yamamoto, A.; Uchiyama, T.; Nishikawa, R.; Fujita, T.; Muranishi, S. Effectiveness and toxicity screening of various absorption enhancers in the rat small intestine: Effects of absorption enhancers on the intestinal absorption of phenol red and the release of protein and phospholipids from the intestinal membrane. J. Pharm. Pharmacol. 1996, 48, 1285–1289. [Google Scholar]
- Kondoh, M.; Takahashi, A.; Fujii, M.; Yagi, K.; Watanabe, Y. A novel strategy for a drug delivery system using a claudin modulator. Biol. Pharm. Bull. 2006, 29, 1783–1789. [Google Scholar]
- Furuse, M.; Hata, M.; Furuse, K.; Yoshida, Y.; Haratake, A.; Sugitani, Y.; Noda, T.; Kubo, A.; Tsukita, S. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1-deficient mice. J. Cell Biol. 2002, 156, 1099–1111. [Google Scholar]
- Nitta, T.; Hata, M.; Gotoh, S.; Seo, Y.; Sasaki, H.; Hashimoto, N.; Furuse, M.; Tsukita, S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J. Cell Biol. 2003, 161, 653–660. [Google Scholar]
- Kominsky, S.L.; Tyler, B.; Sosnowski, J.; Brady, K.; Doucet, M.; Nell, D.; Smedley, J.G., III; McClane, B.; Brem, H.; Sukumar, S. Clostridium perfringens enterotoxin as a novel-targeted therapeutic for brain metastasis. Cancer Res. 2007, 67, 7977–7982. [Google Scholar] [PubMed]
- Santin, A.D.; Bellone, S.; Marizzoni, M.; Palmieri, M.; Siegel, E.R.; McKenney, J.K.; Hennings, L.; Comper, F.; Bandiera, E.; Pecorelli, S. Overexpression of claudin-3 and claudin-4 receptors in uterine serous papillary carcinoma - Novel targets for a type-specific therapy using Clostridium perfringens enterotoxin (CPE). Cancer 2007, 109, 1312–1322. [Google Scholar]
- Morin, P.J. Claudin proteins in ovarian cancer. Dis. Markers 2007, 23, 453–457. [Google Scholar]
- Morin, P.J. Claudin proteins in human cancer: Promising new targets for diagnosis and therapy. Cancer Res. 2005, 65, 9603–9606. [Google Scholar]
- Potala, S.; Sahoo, S.K.; Verma, R.S. Targeted therapy of cancer using diphtheria toxin-derived immunotoxins. Drug Discov. Today 2008, 13, 807–815. [Google Scholar]
- Kreitman, R.J.; Pastan, I. Immunotoxins in the treatment of refractory hairy cell leukemia. Hematol. Oncol. Clin. N. Am. 2006, 20, 1137–1151. [Google Scholar]
- Kakutani, H.; Kondoh, M.; Saeki, R.; Fujii, M.; Watanabe, Y.; Mizuguchi, H.; Yagi, K. Claudin-4-targeting of diphtheria toxin fragment A using a C-terminal fragment of Clostridium perfringens enterotoxin. Eur. J. Pharm. Biopharm. 2010, 75, 213–217. [Google Scholar]
- Yuan, X.Q.; Lin, X.J.; Manorek, G.; Kanatani, I.; Cheung, L.H.; Rosenblum, M.G.; Howell, S.B. Recombinant CPE fused to tumor necrosis factor targets human ovarian cancer cells expressing the claudin-3 and claudin-4 receptors. Mol. Cancer Ther. 2009, 8, 1906–1915. [Google Scholar]
- Michl, P.; Buchholz, M.; Rolke, M.; Kunsch, S.; Lohr, M.; McClane, B.; Tsukita, S.; Leder, G.; Adler, G.; Gress, T.M. Claudin-4: A new target for pancreatic cancer treatment using Clostridium perfringens enterotoxin. Gastroenterology 2001, 121, 678–684. [Google Scholar]
- Santin, A.D.; Cane, S.; Bellone, S.; Palmieri, M.; Siegel, E.R.; Thomas, M.; Roman, J.J.; Burnett, A.; Cannon, M.J.; Pecorelli, S. Treatment of chemotherapy-resistant human ovarian cancer xenografts in C.B-17/SCID mice by intraperitoneal administration of Clostridium perfringens enterotoxin. Cancer Res. 2005, 65, 4334–4342. [Google Scholar] [PubMed]
- Meertens, L.; Bertaux, C.; Cukierman, L.; Cormier, E.; Lavillette, D.; Cosset, F.L.; Dragic, T. The tight junction proteins claudin-1, -6, and -9 are entry cofactors for hepatitis C virus. J. Virol. 2008, 82, 3555–3560. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Veshnyakova, A.; Protze, J.; Rossa, J.; Blasig, I.E.; Krause, G.; Piontek, J. On the Interaction of Clostridium perfringens Enterotoxin with Claudins. Toxins 2010, 2, 1336-1356. https://doi.org/10.3390/toxins2061336
Veshnyakova A, Protze J, Rossa J, Blasig IE, Krause G, Piontek J. On the Interaction of Clostridium perfringens Enterotoxin with Claudins. Toxins. 2010; 2(6):1336-1356. https://doi.org/10.3390/toxins2061336
Chicago/Turabian StyleVeshnyakova, Anna, Jonas Protze, Jan Rossa, Ingolf E. Blasig, Gerd Krause, and Joerg Piontek. 2010. "On the Interaction of Clostridium perfringens Enterotoxin with Claudins" Toxins 2, no. 6: 1336-1356. https://doi.org/10.3390/toxins2061336