Antibodies against Anthrax: Mechanisms of Action and Clinical Applications

{kind=link}
{kind=link}
{kind=link}
Abstract
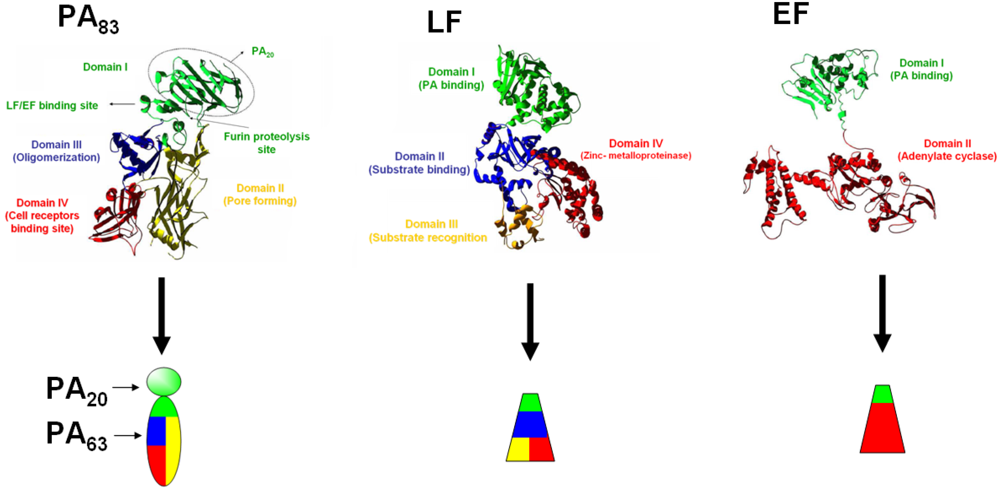
:1. Introduction


2. Antibodies Neutralizing the Anthrax Toxins
2.1. Antibodies Inhibiting PA/Receptors Interaction

2.2. Antibodies Inhibiting PA Cleavage by Furin
2.3. Antibodies Inhibiting PA Heptamerization
2.4. Antibodies Inhibiting PA-LF/EF Interactions
2.5. Antibodies Inhibiting Endocytosis and Translocation
3. Antibodies Directed Against Bacillus anthracis Capsule
4. The Particular Case of Antibody Enhancing Toxin Activity
5. Discussion and Conclusion
Acknowledgments
Disclaimer
References
- Inglesby, T.V.; O’Toole, T.; Henderson, D.A.; Bartlett, J.G.; Ascher, M.S.; Eitzen, E.; Friedlander, A.M.; Gerberding, J.; Hauer, J.; Hughes, J.; et al. Anthrax as a biological weapon, 2002: Updated recommendations for management. J. Am. Med. Assoc. 2002, 287, 2236–2252. [Google Scholar]
- Green, B.D.; Battisti, L.; Koehler, T.M.; Thorne, C.B.; Ivins, B.E. Demonstration of a capsule plasmid in Bacillus anthracis. Infect. Immun. 1985, 49, 291–297. [Google Scholar]
- Friedlander, A.M. Macrophages are sensitive to anthrax lethal toxin through an acid-dependent process. J. Biol. Chem. 1986, 261, 7123–7126. [Google Scholar]
- Barth, H.; Aktories, K.; Popoff, M.R.; Stiles, B.G. Binary bacterial toxins: Biochemistry, biology, and applications of common clostridium and bacillus protein. Microbiol. Mol. Biol. Rev. 2004, 68, 373–402. [Google Scholar]
- Collier, R.J.; Young, J.A. Anthrax toxin. Annu. Rev. Cell Dev. Biol. 2003, 19, 45–70. [Google Scholar]
- Petosa, C.; Collier, R.J.; Klimpel, K.R.; Leppla, S.H.; Liddington, R.C. Crystal structure of the anthrax toxin protective antigen. Nature 1997, 385, 833–838. [Google Scholar]
- Pannifer, A.D.; Wong, T.Y.; Schwarzenbacher, R.; Renatus, M.; Petosa, C.; Bienkowska, J.; Lacy, D.B.; Collier, R.J.; Park, S.; Leppla, S.H.; et al. Crystal structure of the anthrax lethal factor. Nature 2001, 414, 229–233. [Google Scholar] [PubMed]
- Drum, C.L.; Yan, S.Z.; Bard, J.; Shen, Y.Q.; Lu, D.; Soelaiman, S.; Grabarek, Z.; Bohm, A.; Tang, W.J. Structural basis for the activation of anthrax adenylyl cyclase exotoxin by calmodulin. Nature 2002, 415, 396–402. [Google Scholar]
- Lacy, D.B.; Wigelsworth, D.J.; Melnyk, R.A.; Harrison, S.C.; Collier, R.J. Structure of heptameric protective antigen bound to an anthrax toxin receptor: A role for receptor in pH-dependent pore formation. Proc. Natl. Acad. Sci. USA 2004, 101, 13147–13151. [Google Scholar]
- Santelli, E.; Bankston, L.A.; Leppla, S.H.; Liddington, R.C. Crystal structure of a complex between anthrax toxin and its host cell receptor. Nature 2004, 430, 905–908. [Google Scholar]
- Mogridge, J.; Mourez, M.; Collier, R.J. Involvement of domain 3 in oligomerization by the protective antigen moiety of anthrax toxin. J. Bacteriol. 2001, 183, 2111–2116. [Google Scholar]
- Little, S.F.; Lowe, J.R. Location of receptor-binding region of protective antigen from Bacillus anthracis. Biochem. Biophys. Res. Commun. 1991, 180, 531–537. [Google Scholar]
- Rosovitz, M.J.; Schuck, P.; Varughese, M.; Chopra, A.P.; Mehra, V.; Singh, Y.; McGinnis, L.M.; Leppla, S.H. Alanine-scanning mutations in domain 4 of anthrax toxin protective antigen reveal residues important for binding to the cellular receptor and to a neutralizing monoclonal antibody. J. Biol. Chem. 2003, 278, 30936–30944. [Google Scholar]
- Bradley, K.A.; Mogridge, J.; Mourez, M.; Collier, R.J.; Young, J.A. Identification of the cellular receptor for anthrax toxin. Nature 2001, 414, 225–229. [Google Scholar]
- Scobie, H.M.; Rainey, G.J.; Bradley, K.A.; Young, J.A. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 5170–5174. [Google Scholar]
- Vitale, G.; Bernardi, L.; Napolitani, G.; Mock, M.; Montecucco, C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem. J. 2000, 352, 739–745. [Google Scholar]
- Duesbery, N.S.; Webb, C.P.; Leppla, S.H.; Gordon, V.M.; Klimpel, K.R.; Copeland, T.D.; Ahn, N.G.; Oskarsson, M.K.; Fukasawa, K.; Paull, K.D.; et al. Proteolytic inactivation of map-kinase-kinase by anthrax lethal factor. Science 1998, 280, 734–737. [Google Scholar] [PubMed]
- Leppla, S.H. Anthrax toxin edema factor: A bacterial adenylate cyclase that increases cyclic amp concentrations of eukaryotic cells. Proc. Natl. Acad. Sci. USA 1982, 79, 3162–3166. [Google Scholar]
- Leppla, S.H.; Arora, N.; Varughese, M. Anthrax toxin fusion proteins for intracellular delivery of macromolecules. J. Appl. Microbiol. 1999, 87, 284. [Google Scholar]
- Quinn, C.P.; Singh, Y.; Klimpel, K.R.; Leppla, S.H. Functional mapping of anthrax toxin lethal factor by in-frame insertion mutagenesis. J. Biol. Chem. 1991, 266, 20124–20130. [Google Scholar]
- Legge, T.M. The milroy lectures on industrial anthrax: Delivered before the royal college of physicians of london. Br. Med. J. 1905, 1, 641–643. [Google Scholar]
- Meyerhoff, A.; Albrecht, R.; Meyer, J.M.; Dionne, P.; Higgins, K.; Murphy, D. US food and drug administration approval of ciprofloxacin hydrochloride for management of postexposure inhalational anthrax. Clin. Infect. Dis. 2004, 39, 303–308. [Google Scholar]
- FDA. Prescription drug product: Doxycycline and penicillin g procaine administration for inhalational anthrax (post-exposure). Fed. Regist. 2001, 55679–55682.
- Shepard, C.W.; Soriano-Gabarro, M.; Zell, E.R.; Hayslett, J.; Lukacs, S.; Goldstein, S.; Factor, S.; Jones, J.; Ridzon, R.; Williams, I.; et al. ntimicrobial postexposure prophylaxis for anthrax: Adverse events and adherence. Emerg. Infect. Dis. 2002, 8, 1124–1132. [Google Scholar]
- CDC. Update: Investigation of bioterrorism-related anthrax and adverse events from antimicrobial prophylaxis.DOI: PubMed:. MMWR Morb. Mortal. Wkly. Rep. 2001, 50, 973–976. [PubMed]
- Beharry, Z.; Chen, H.; Gadhachanda, V.R.; Buynak, J.D.; Palzkill, T. Evaluation of penicillin-based inhibitors of the class a and b beta-lactamases from Bacillus anthracis. Biochem. Biophys. Res. Commun. 2004, 313, 541–545. [Google Scholar]
- Chen, Y.; Tenover, F.C.; Koehler, T.M. Beta-lactamase gene expression in a penicillin-resistant Bacillus anthracis strain. Antimicrob. Agents Chemother. 2004, 48, 4873–4877. [Google Scholar]
- Athamna, A.; Athamna, M.; Abu-Rashed, N.; Medlej, B.; Bast, D.J.; Rubinstein, E. Selection of Bacillus anthracis isolates resistant to antibiotics. J. Antimicrob. Chemother. 2004, 54, 424–428. [Google Scholar]
- Price, L.B.; Vogler, A.; Pearson, T.; Busch, J.D.; Schupp, J.M.; Keim, P. In vitro selection and characterization of Bacillus anthracis mutants with high-level resistance to ciprofloxacin. Antimicrob. Agents Chemother. 2003, 47, 2362–2365. [Google Scholar]
- Choe, C.H.; Bouhaouala, S.S.; Brook, I.; Elliot, T.B.; Knudson, G.B. In vitro development of resistance to ofloxacin and doxycycline in Bacillus anthracis sterne. Antimicrob.DOI: PubMed:. Agents Chemother. 2000, 44. [Google Scholar]
- Albrecht, M.T.; Li, H.; Williamson, E.D.; LeButt, C.S.; Flick-Smith, H.C.; Quinn, C.P.; Westra, H.; Galloway, D.; Mateczun, A.; Goldman, S.; et al. Human monoclonal antibodies against anthrax lethal factor and protective antigen act independently to protect against Bacillus anthracis infection and enhance endogenous immunity to anthrax. Infect. Immun. 2007, 75, 5425–5433. [Google Scholar] [PubMed]
- Wigelsworth, D.J.; Krantz, B.A.; Christensen, K.A.; Lacy, D.B.; Juris, S.J.; Collier, R.J. Binding stoichiometry and kinetics of the interaction of a human anthrax toxin receptor, cmg2, with protective antige. J. Biol. Chem. 2004, 279, 23349–23356. [Google Scholar]
- Lacy, D.B.; Wigelsworth, D.J.; Scobie, H.M.; Young, J.A.; Collier, R.J. Crystal structure of the von willebrand factor a domain of human capillary morphogenesis protein 2: An anthrax toxin receptor. Proc. Natl. Acad. Sci. USA 2004, 101, 6367–6372. [Google Scholar]
- Bradley, K.A.; Mogridge, J.; Jonah, G.; Rainey, A.; Batty, S.; Young, J.A. Binding of anthrax toxin to its receptor is similar to alpha integrin-ligand interactions. J. Biol. Chem. 2003, 278, 49342–49347. [Google Scholar]
- Little, S.F.; Leppla, S.H.; Cora, E. Production and characterization of monoclonal antibodies to the protective antigen component of Bacillus anthracis toxin. Infect. Immun. 1988, 56, 1807–1813. [Google Scholar] [PubMed]
- Laffly, E.; Danjou, L.; Condemine, F.; Vidal, D.; Drouet, E.; Lefranc, M.P.; Bottex, C.; Thullier, P. Selection of a macaque fab with framework regions like those in humans, high affinity, and ability to neutralize the protective antigen (pa) of Bacillus anthracis by binding to the segment of pa between residues 686 and 69. Antimicrob. Agents Chemother. 2005, 49, 3414–3420. [Google Scholar] [PubMed]
- Kelly-Cirino, C.D.; Mantis, N.J. Neutralizing monoclonal antibodies directed against defined linear epitopes on domain 4 of anthrax protective antigen. Infect. Immun. 2009, 77, 4859–4867. [Google Scholar]
- Chen, Z.; Moayeri, M.; Zhou, Y.H.; Leppla, S.; Emerson, S.; Sebrell, A.; Yu, F.; Svitel, J.; Schuck, P.; St Claire, M.; et al. Efficient neutralization of anthrax toxin by chimpanzee monoclonal antibodies against protective antigen. J. Infect. Dis. 2006, 193, 625–633. [Google Scholar] [PubMed]
- Brossier, F.; Levy, M.; Landier, A.; Lafaye, P.; Mock, M. Functional analysis of Bacillus anthracis protective antigen by using neutralizing monoclonal antibodies. Infect. Immun. 2004, 72, 6313–6317. [Google Scholar] [PubMed]
- Zhou, B.; Carney, C.; Janda, K.D. Selection and characterization of human antibodies neutralizing Bacillus anthracis toxin. Bioorg. Med. Chem. 2008, 16, 1903–1913. [Google Scholar] [PubMed]
- Mazumdar, S. Raxibacumab. MAbs 2009, 1, 531–538. [Google Scholar]
- Wild, M.A.; Xin, H.; Maruyama, T.; Nolan, M.J.; Calveley, P.M.; Malone, J.D.; Wallace, M.R.; Bowdish, K.S. Human antibodies from immunized donors are protective against anthrax toxin in vivo.DOI: PubMed:. Nat, Biotechnol. 2003, 21, 1305–1306. [Google Scholar]
- Migone, T.S.; Subramanian, G.M.; Zhong, J.; Healey, L.M.; Corey, A.; Devalaraja, M.; Lo, L.; Ullrich, S.; Zimmerman, J.; Chen, A.; et al. Raxibacumab for the treatment of inhalational anthrax. N. Engl. J. Med. 2009, 361, 135–144. [Google Scholar] [PubMed]
- Antoniu, S.A. Raxibacumab for inhalational anthrax: An effective specific therapeutic approach? Expert Opin. Investig. Drugs 2010, 19, 909–911. [Google Scholar]
- Mohamed, N.; Clagett, M.; Li, J.; Jones, S.; Pincus, S.; D’Alia, G.; Nardone, L.; Babin, M.; Spitalny, G.; Casey, L. A high-affinity monoclonal antibody to anthrax protective antigen passively protects rabbits before and after aerosolized Bacillus anthracis spore challenge. Infect. Immun. 2005, 73, 795–802. [Google Scholar] [PubMed]
- Klimpel, K.R.; Molloy, S.S.; Thomas, G.; Leppla, S.H. Anthrax toxin protective antigen is activated by a cell surface protease with the sequence specificity and catalytic properties of furin. Proc. Natl. Acad. Sci. USA 1992, 89, 10277–10281. [Google Scholar]
- Singh, Y.; Chaudhary, V.K.; Leppla, S.H. A deleted variant of Bacillus anthracis protective antigen is non-toxic and blocks anthrax toxin action in vivo. J. Biol. Chem. 1989, 264, 19103–19107. [Google Scholar] [PubMed]
- Christensen, K.A.; Krantz, B.A.; Melnyk, R.A.; Collier, R.J. Interaction of the 20 kda and 63 kda fragments of anthrax protective antigen: Kinetics and thermodynamics. Biochemistry 2005, 44, 1047–1053. [Google Scholar]
- Rivera, J.; Nakouzi, A.; Abboud, N.; Revskaya, E.; Goldman, D.; Collier, R.J.; Dadachova, E.; Casadevall, A. A monoclonal antibody to Bacillus anthracis protective antigen defines a neutralizing epitope in domain 1. Infect. Immun. 2006, 74, 4149–4156. [Google Scholar] [PubMed]
- Wang, F.; Ruther, P.; Jiang, I.; Sawada-Hirai, R.; Sun, S.M.; Nedellec, R.; Morrow, P.R.; Kang, A.S. Human monoclonal antibodies that neutralize anthrax toxin by inhibiting heptamer assembly. Hum. Antib. 2004, 13, 105–110. [Google Scholar]
- Milne, J.C.; Furlong, D.; Hanna, P.C.; Wall, J.S.; Collier, R.J. Anthrax protective antigen forms oligomers during intoxication of mammalian cells. J. Biol. Chem. 1994, 269, 20607–20612. [Google Scholar]
- Little, S.F.; Novak, J.M.; Lowe, J.R.; Leppla, S.H.; Singh, Y.; Klimpel, K.R.; Lidgerding, B.C.; Friedlander, A.M. Characterization of lethal factor binding and cell receptor binding domains of protective antigen of Bacillus anthracis using monoclonal antibodies. Microbiology 1996, 142, 707–715. [Google Scholar] [PubMed]
- Peterson, J.W.; Comer, J.E.; Baze, W.B.; Noffsinger, D.M.; Wenglikowski, A.; Walberg, K.G.; Hardcastle, J.; Pawlik, J.; Bush, K.; Taormina, J.; et al. Human monoclonal antibody avp-21d9 to protective antigen reduces dissemination of the Bacillus anthracis ames strain from the lungs in a rabbit model. Infect. Immun. 2007, 75, 3414–3424. [Google Scholar] [PubMed]
- Vitale, L.; Blanset, D.; Lowy, I.; O’Neill, T.; Goldstein, J.; Little, S.F.; Andrews, G.P.; Dorough, G.; Taylor, R.K.; Keler, T. Prophylaxis and therapy of inhalational anthrax by a novel monoclonal antibody to protective antigen that mimics vaccine-induced immunity. Infect. Immun. 2006, 74, 5840–5847. [Google Scholar]
- Radjainia, M.; Hyun, J.K.; Leysath, C.E.; Leppla, S.H.; Mitra, A.K. Anthrax toxin-neutralizing antibody reconfigures the protective antigen heptamer into a supercomplex. Proc. Natl. Acad. Sci. USA 2010, 107, 14070–14074. [Google Scholar]
- Elliott, J.L.; Mogridge, J.; Collier, R.J. A quantitative study of the interactions of Bacillus anthracis edema factor and lethal factor with activated protective antigen. Biochemistry 2000, 39, 6706–6713. [Google Scholar] [PubMed]
- Feld, G.K.; Thoren, K.L.; Kintzer, A.F.; Sterling, H.J.; Tang, I.I.; Greenberg, S.G.; Williams, E.R.; Krantz, B.A. Structural basis for the unfolding of anthrax lethal factor by protective antigen oligomers. Nat. Struct. Mol. Biol. 2010, 17, 1383–1390. [Google Scholar]
- Kintzer, A.F.; Thoren, K.L.; Sterling, H.J.; Dong, K.C.; Feld, G.K.; Tang, I.I.; Zhang, T.T.; Williams, E.R.; Berger, J.M.; Krantz, B.A. The protective antigen component of anthrax toxin forms functional octameric complexes. J. Mol. Biol. 2009, 392, 614–629. [Google Scholar] [PubMed]
- Cunningham, K.; Lacy, D.B.; Mogridge, J.; Collier, R.J. Mapping the lethal factor and edema factor binding sites on oligomeric anthrax protective antigen. Proc. Natl. Acad. Sci. USA 2002, 99, 7049–7053. [Google Scholar]
- Kumar, P.; Ahuja, N.; Bhatnagar, R. Purification of anthrax edema factor from Escherichia coli and identification of residues required for binding to anthrax protective antigen. Infect. Immun. 2001, 69, 6532–6536. [Google Scholar] [CrossRef] [PubMed]
- Lacy, D.B.; Lin, H.C.; Melnyk, R.A.; Schueler-Furman, O.; Reither, L.; Cunningham, K.; Baker, D.; Collier, R.J. A model of anthrax toxin lethal factor bound to protective antigen. Proc. Natl. Acad. Sci. USA 2005, 102, 16409–16414. [Google Scholar]
- Lacy, D.B.; Mourez, M.; Fouassier, A.; Collier, R.J. Mapping the anthrax protective antigen binding site on the lethal and edema factors. J. Biol. Chem. 2002, 277, 3006–3010. [Google Scholar]
- Melnyk, R.A.; Hewitt, K.M.; Lacy, D.B.; Lin, H.C.; Gessner, C.R.; Li, S.; Woods, V.L., Jr.; Collier, R.J. Structural determinants for the binding of anthrax lethal factor to oligomeric protective antigen. J. Biol. Chem. 2006, 281, 1630–1635. [Google Scholar]
- Steiniger, S.C.; Altobell, L.J., III; Zhou, B.; Janda, K.D. Selection of human antibodies against cell surface-associated oligomeric anthrax protective antigen. Mol. Immunol. 2007, 44, 2749–2755. [Google Scholar] [PubMed]
- Price, B.M.; Liner, A.L.; Park, S.; Leppla, S.H.; Mateczun, A.; Galloway, D.R. Protection against anthrax lethal toxin challenge by genetic immunization with a plasmid encoding the lethal factor protein. Infect. Immun. 2001, 69, 4509–4515. [Google Scholar]
- Zhao, P.; Liang, X.; Kalbfleisch, J.; Koo, H.M.; Cao, B. Neutralizing monoclonal antibody against anthrax lethal factor inhibits intoxication in a mouse model. Hum. Antib. 2003, 12, 129–135. [Google Scholar]
- Nguyen, M.L.; Crowe, S.R.; Kurella, S.; Teryzan, S.; Cao, B.; Ballard, J.D.; James, J.A.; Farris, A.D. Sequential b-cell epitopes of Bacillus anthracis lethal factor bind lethal toxin-neutralizing antibodies. Infect. Immun. 2009, 77, 162–169. [Google Scholar] [PubMed]
- Little, S.F.; Leppla, S.H.; Friedlander, A.M. Production and characterization of monoclonal antibodies against the lethal factor component of Bacillus anthracis lethal toxin. Infect. Immun. 1990, 58, 1606–1613. [Google Scholar] [PubMed]
- Lim, N.K.; Kim, J.H.; Oh, M.S.; Lee, S.; Kim, S.Y.; Kim, K.S.; Kang, H.J.; Hong, H.J.; Inn, K.S. An anthrax lethal factor-neutralizing monoclonal antibody protects rats before and after challenge with anthrax toxin. Infect. Immun. 2005, 73, 6547–6551. [Google Scholar]
- Pelat, T.; Hust, M.; Laffly, E.; Condemine, F.; Bottex, C.; Vidal, D.; Lefranc, M.P.; Dubel, S.; Thullier, P. High-affinity, human antibody-like antibody fragment (single-chain variable fragment) neutralizing the lethal factor (lf) of Bacillus anthracis by inhibiting protective antigen-lf complex formation. Antimicrob. Agents Chemother. 2007, 51, 2758–2764. [Google Scholar] [PubMed]
- Kulshreshtha, P.; Bhatnagar, R. Inhibition of anthrax toxins with a bispecific monoclonal antibody that cross reacts with edema factor as well as lethal factor of Bacillus anthracis. Mol. Immunol. 2011. [Google Scholar]
- Little, S.F.; Leppla, S.H.; Burnett, J.W.; Friedlander, A.M. Structure-function analysis of Bacillus anthracis edema factor by using monoclonal antibodies. Biochem. Biophys. Res. Commun. 1994, 199, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Abrami, L.; Liu, S.; Cosson, P.; Leppla, S.H.; van der Goot, F.G. Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J. Cell Biol. 2003, 160, 321–328. [Google Scholar]
- Abrami, L.; Lindsay, M.; Parton, R.G.; Leppla, S.H.; van der Goot, F.G. Membrane insertion of anthrax protective antigen and cytoplasmic delivery of lethal factor occur at different stages of the endocytic pathway. J. Cell Biol. 2004, 166, 645–651. [Google Scholar] [Green Version]
- Chen, Z.; Moayeri, M.; Crown, D.; Emerson, S.; Gorshkova, I.; Schuck, P.; Leppla, S.H.; Purcell, R.H. Novel chimpanzee/human monoclonal antibodies that neutralize anthrax lethal factor, and evidence for possible synergy with anti-protective antigen antibody. Infect. Immun. 2009, 77, 3902–3908. [Google Scholar]
- Chen, Z.; Moayeri, M.; Zhao, H.; Crown, D.; Leppla, S.H.; Purcell, R.H. Potent neutralization of anthrax edema toxin by a humanized monoclonal antibody that competes with calmodulin for edema factor binding. Proc. Natl. Acad. Sci. USA 2009, 106, 13487–13492. [Google Scholar]
- Ivins, B.E.; Welkos, S.L. Cloning and expression of the Bacillus anthracis protective antigen gene in bacillus subtilis. Infect. Immun. 1986, 54, 537–542. [Google Scholar] [PubMed]
- Drysdale, M.; Heninger, S.; Hutt, J.; Chen, Y.; Lyons, C.R.; Koehler, T.M. Capsule synthesis by Bacillus anthracis is required for dissemination in murine inhalation anthrax. Embo J. 2005, 24, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.T.; Fellows, P.F.; Leighton, T.J.; Lucas, A.H. Induction of opsonic antibodies to the gamma-d-glutamic acid capsule of Bacillus anthracis by immunization with a synthetic peptide-carrier protein conjugate. FEMS Immunol. Med. Microbiol. 2004, 40, 231–237. [Google Scholar] [PubMed]
- Kozel, T.R.; Murphy, W.J.; Brandt, S.; Blazar, B.R.; Lovchik, J.A.; Thorkildson, P.; Percival, A.; Lyons, C.R. Mabs to Bacillus anthracis capsular antigen for immunoprotection in anthrax and detection of antigenemia. Proc. Natl. Acad. Sci. USA 2004, 101, 5042–5047. [Google Scholar]
- Kozel, T.R.; Thorkildson, P.; Brandt, S.; Welch, W.H.; Lovchik, J.A.; AuCoin, D.P.; Vilai, J.; Lyons, C.R. Protective and immunochemical activities of monoclonal antibodies reactive with the Bacillus anthracis polypeptide capsule. Infect. Immun. 2007, 75, 152–163. [Google Scholar] [PubMed]
- Chen, Z.; Schneerson, R.; Lovchik, J.; Lyons, C.R.; Zhao, H.; Dai, Z.; Kubler-Kielb, J.; Leppla, S.H.; Purcell, R.H. Pre- and postexposure protection against virulent anthrax infection in mice by humanized monoclonal antibodies to Bacillus anthracis capsule. Proc. Natl. Acad. Sci. USA 2011, 108, 739–744. [Google Scholar]
- Tirado, S.M.; Yoon, K.J. Antibody-dependent enhancement of virus infection and disease. Viral Immunol. 2003, 16, 69–86. [Google Scholar]
- Mohamed, N.; Li, J.; Ferreira, C.S.; Little, S.F.; Friedlander, A.M.; Spitalny, G.L.; Casey, L.S. Enhancement of anthrax lethal toxin cytotoxicity: A subset of monoclonal antibodies against protective antigen increases lethal toxin-mediated killing of murine macrophages. Infect. Immun. 2004, 72, 3276–3283. [Google Scholar]
- Little, S.F.; Webster, W.M.; Fisher, D.E. Monoclonal antibodies directed against protective antigen of Bacillus anthracis enhance lethal toxin activity in vivo. FEMS Immunol. Med. Microbiol. 2011, 62, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Pelat, T.; Hüst, M.; Laffly, E.; Condemine, F.; Bottex, C.; Vidal, D.; Lefranc, M.P.; Dübel, S.; Thullier, P. High-affinity, human antibody-like antibody fragment (single-chain variable fragment) neutralizing the lethal factor (lf) of Bacillus anthracis by inhibiting protective antigen-lf complex formation. Antimicrob. Agents Chemother. 2007, 51, 2758–2764. [Google Scholar] [PubMed]
- Adekar, S.P.; Takahashi, T.; Jones, R.M.; Al-Saleem, F.H.; Ancharski, D.M.; Root, M.J.; Kapadnis, B.P.; Simpson, L.L.; Dessain, S.K. Neutralization of botulinum neurotoxin by a human monoclonal antibody specific for the catalytic light chain. PLoS One 2008, 3. [Google Scholar] [CrossRef]
- Baillie, L.W. Past, imminent and future human medical countermeasures for anthrax. J. Appl. Microbiol. 2006, 101, 594–606. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Froude II, J.W.; Thullier, P.; Pelat, T. Antibodies against Anthrax: Mechanisms of Action and Clinical Applications. Toxins 2011, 3, 1433-1452. https://doi.org/10.3390/toxins3111433
Froude II JW, Thullier P, Pelat T. Antibodies against Anthrax: Mechanisms of Action and Clinical Applications. Toxins. 2011; 3(11):1433-1452. https://doi.org/10.3390/toxins3111433
Chicago/Turabian StyleFroude II, Jeffrey W., Philippe Thullier, and Thibaut Pelat. 2011. "Antibodies against Anthrax: Mechanisms of Action and Clinical Applications" Toxins 3, no. 11: 1433-1452. https://doi.org/10.3390/toxins3111433