Immunotoxins and Anticancer Drug Conjugate Assemblies: The Role of the Linkage between Components


Abstract
:1. Introduction
2. Immunotoxins





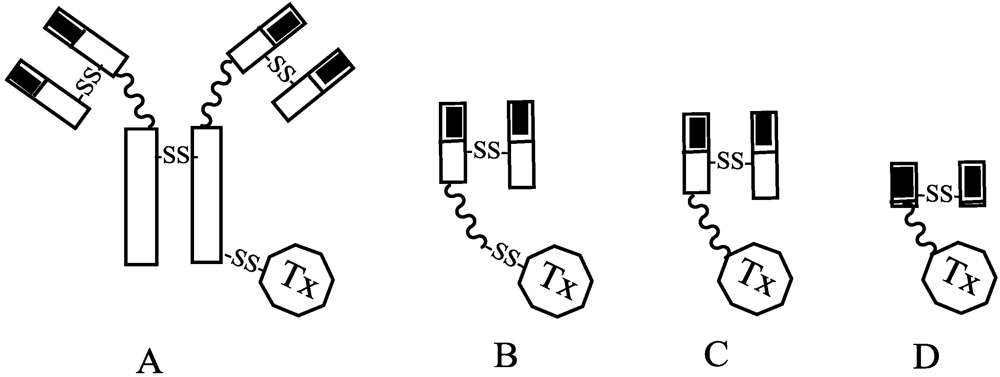{kind=link}
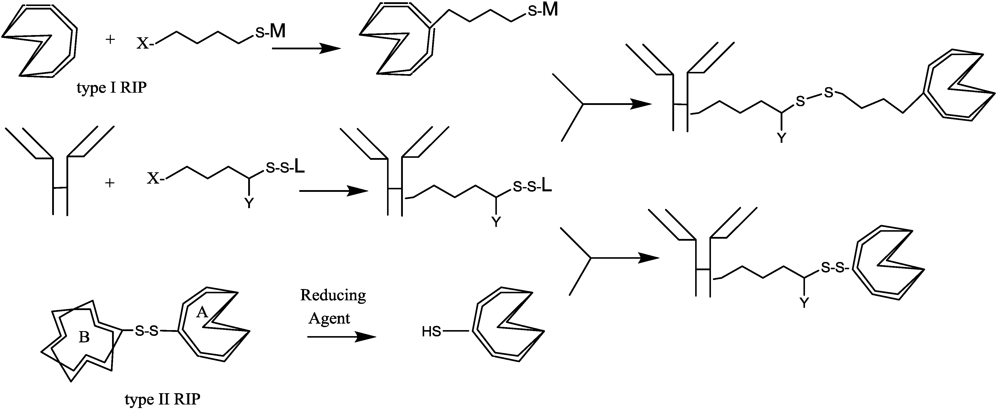{kind=link}
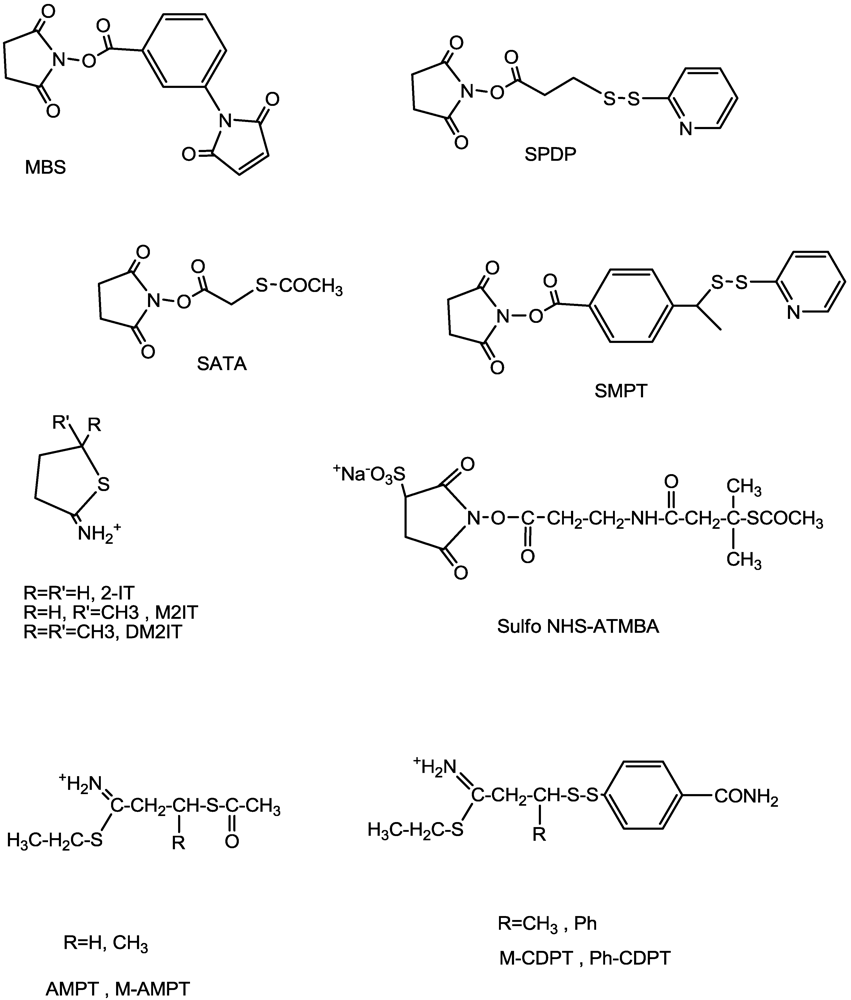{kind=link}
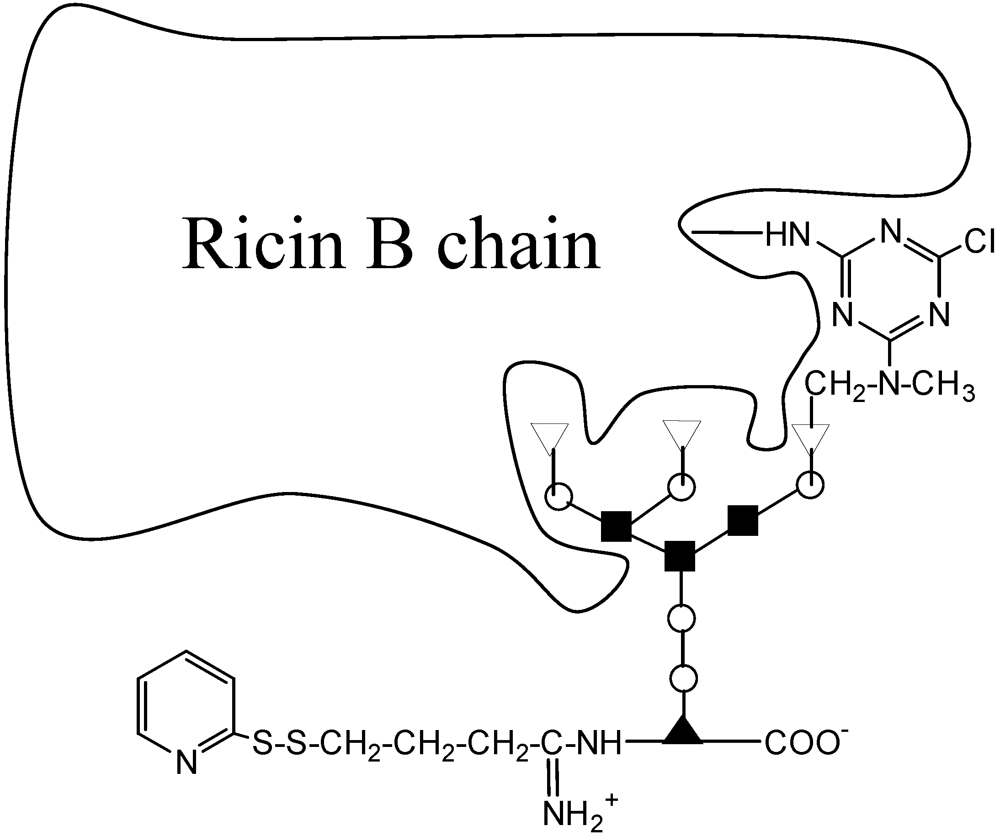{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Immunotoxin Agent | Antigen Target | Toxic Component | Diseases | Clinical Phase | Company |
|---|---|---|---|---|---|
| Ontak | IL-2R | DT | T-CLL, B-CLL, NHL | launched | Eisai |
| BL22 | CD22 | PE | Hairy Cell Leukemia, B-CLL, NHL | II | NCI |
| LMB-2 | CD25 | PE | NHL, leukemias | II | NCI |
| CAT-8015 | CD22 | PE | CLL, PLL, SLL | II | Medimmune |
| Combotox | CD19/CD22 | dgA | Leukemias | I | Abiogen |
| HuM-195/rGel | CD33 | r-Gelonin | Leukemias | I | Targa Ther. |
| MR1-1 | EGFRvIII | PE | Solid ca. | I | Ivax Corp. |
| SS1P; CAT-5001 | mesothelin | PE | Solid ca | II | NCI |
| Zemab | HER-2 | PE | Breast ca | I | Novartis |
| RFT-5.dgA | IL-2R | dgA | lymphomas | II | UTSMC |
| Cintredekin besudotox | IL13R | PE | Brain ca | III | Insys |
3. Antibody-Drug Conjugates


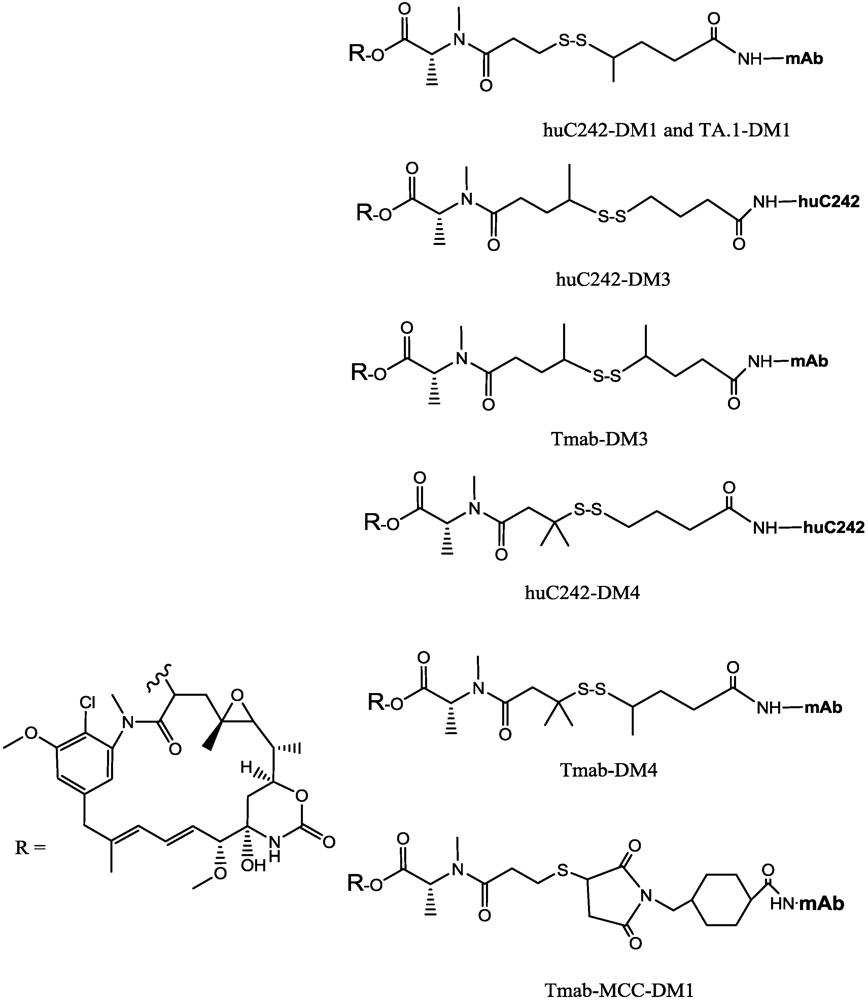
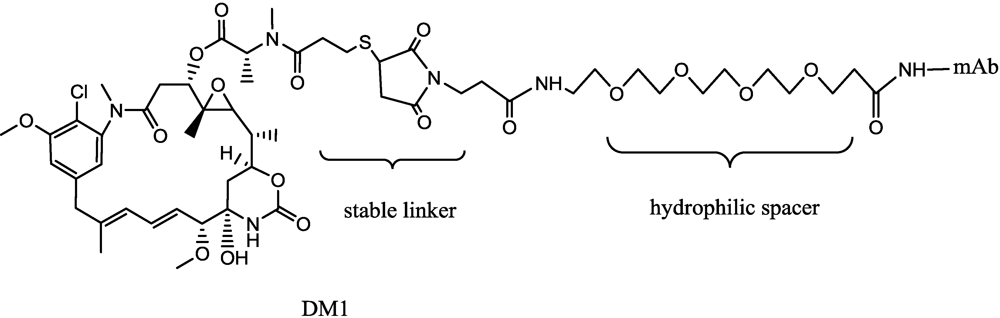
3.1. Maytansinoids





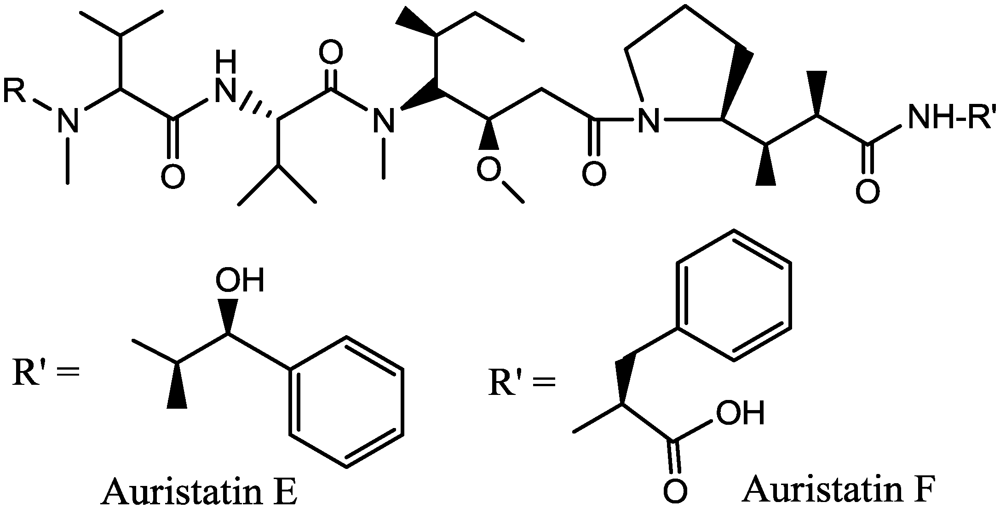
3.2. Dolastatins, Auristatin



3.3. Calicheamicins




4. Conclusions
Conflict of Interest
References
- Cancer facts and figures. Available online: http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-026238.pdf (accessed on 6 July 2011).
- Greish, K.; Fang, J.; Inutsuka, T.; Nagamitsu, A.; Maeda, H. Macromolecular therapeutics: Advantages and prospects with special emphasis on solid tumour targeting. Clin. Pharmacokinet. 2003, 42, 1089–1105. [Google Scholar]
- Maeda, H. Tumor-selective delivery of macromolecular drugs via the EPR effect: Background and future prospects. Bioconjugate Chem. 2010, 21, 797–802. [Google Scholar]
- Jain, R.K. Transport of molecules across tumor vasculature. Cancer Metastasis Rev. 1987, 6, 559–593. [Google Scholar]
- Roberts, W.G.; Palade, G.E. Neovasculature induced by vascular endothelial growth factor is fenestrated. Cancer Res. 1997, 57, 765–772. [Google Scholar]
- Padera, T.P.; Kadambi, A.; di Tomaso, E.; Carreira, C.M.; Brown, E.B.; Boucher, Y.; Choi, N.C.; Mathisen, D.; Wain, J.; Mark, E.J.; et al. Lymphatic metastasis in the absence of functional intratumor lymphatics. Science 2002, 296, 1883–1886. [Google Scholar] [PubMed]
- Leu, A.J.; Berk, D.A.; Lymboussaki, A.; Alitalo, K.; Jain, R.K. Absence of functional lymphatics within a murine sarcoma: A molecular and functional evaluation. Cancer Res. 2000, 60, 4324–4327. [Google Scholar]
- Cheng, Y.; Xu, T. The effect of dendrimers on the pharmacodynamic and pharmacokinetic behaviors of non-covalently or covalently attached drugs. Eur. J. Med. Chem. 2008, 43, 2291–2297. [Google Scholar]
- Li, J.; Zhu, Z. Research and development of next generation of antibody-based therapeutics. Acta Pharmacol. Sin. 2010, 31, 1198–1207. [Google Scholar]
- Reichert, J.M. Antibody-based therapeutics to watch in 2011. MAbs 2011, 3, 76–99. [Google Scholar]
- Reichert, J.M. Monoclonal antibodies as innovative therapeutics. Curr. Pharm. Biotechnol. 2008, 9, 423–430. [Google Scholar]
- Hughes, B. Antibody-drug conjugates for cancer: Poised to deliver? Nat. Rev. Drug Discov. 2010, 9, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Ducry, L.; Stump, B. Antibody-drug conjugates: Linking cytotoxic payloads to monoclonal antibodies. Bioconjugate Chem. 2009, 21, 5–13. [Google Scholar]
- Ulbrich, K.; Subr, V. Polymeric anticancer drugs with pH-controlled activation. Adv. Drug Delivery Rev. 2004, 56, 1023–1050. [Google Scholar]
- De Groot, F.M.H.; Damen, E.W.P.; Scheeren, H.W. Anticancer prodrugs for application in monotherapy: Targeting hypoxia, tumor-associated enzymes, and receptors. Curr. Med. Chem. 2001, 8, 1093–1122. [Google Scholar] [PubMed]
- Singh, Y.; Palombo, M.; Sinko, P.J. Recent trends in targeted anticancer prodrug and conjugate design. Curr. Med. Chem. 2008, 15, 1802–1826. [Google Scholar]
- Damen, E.W.P.; Nevalainen, T.J.; van den Bergh, T.J.M.; de Groot, F.M.H.; Scheeren, H.W. Synthesis of novel paclitaxel prodrugs designed for bioreductive activation in hypoxic tumour tissue. Bioorg. Med. Chem. 2002, 10, 71–77. [Google Scholar]
- Duncan, R. Polymer conjugates as anticancer nanomedicines. Nat. Rev. Cancer 2006, 6, 688–701. [Google Scholar]
- Stirpe, F.; Battelli, M.G. Ribosome-inactivating proteins: Progress and problems. Cell. Mol. Life Sci. 2006, 63, 1850–1866. [Google Scholar]
- Girbés, T.; Ferreras, J.M.; Arias, F.J.; Stirpe, F. Description, distribution, activity and phylogenetic relationship of ribosome-inactivating proteins in plants, fungi and bacteria. Mini-Rev. Med. Chem. 2004, 4, 461–476. [Google Scholar]
- De virgilio, M.; Lombardi, A.; Caliandro, R.; Fabbrini, M.S. Ribosome-Inactivating proteins: From plant defense to tumor attack. Toxins 2010, 2, 2699–2737. [Google Scholar]
- Ng, T.B.; Wong, J.H.; Wang, H. Recent progress in research on ribosome inactivating proteins. Curr. Protein Pept. Sci. 2010, 11, 37–53. [Google Scholar]
- Yamaizumi, M.; Mekada, E.; Uchida, T.; Okada, Y. One molecule of diphtheria toxin fragment A introduced into a cell can kill the cell. Cell 1978, 15, 245–250. [Google Scholar]
- Battelli, M.G. Cytotoxicity and toxicity to animals and humans of ribosome-inactivating proteins. Mini-Rev. Med. Chem. 2004, 4, 513–521. [Google Scholar] [PubMed]
- Bagaria, A.; Surendranath, K.; Ramagopal, U.A.; Ramakumar, S.; Karande, A.A. Structure-function analysis and insights into the reduced toxicity of Abrus precatorius agglutinin I in relation to abrin. J. Biol. Chem. 2006, 281, 34465–34474. [Google Scholar]
- Conde, F.P.; Fernandez-Puentes, C.; Montero, M.T.V.; Vazquez, D. Protein toxins that catalytically inactivate ribosomes from eukaryotic microorganisms Studies on the mode of action of alpha sarcin, mitogillin and restrictocin: Response to alpha sarcin antibodies. FEMS Microbiol. Lett. 1978, 4, 349–355. [Google Scholar]
- Ng, T.B.; Wang, H.X. Flammin and velin: New ribosome inactivating polypeptides from the mushroom Flammulina velutipes. Peptides 2004, 25, 929–933. [Google Scholar]
- Shapira, A.; Benhar, I. Toxin-Based Therapeutic Approaches. Toxins 2010, 2, 2519–2583. [Google Scholar]
- Bolognesi, A.; Polito, L. Immunotoxins and other conjugates: Pre-clinical studies. Mini-Rev. Med. Chem. 2004, 4, 563–583. [Google Scholar] [PubMed]
- Pasqualucci, L.; Flenghi, L.; Terenzi, A.; Bolognesi, A.; Stirpe, F.; Bigerna, B.; Falini, B. Immunotoxin therapy of hematological malignancies. Haematologica 1995, 80, 546–556. [Google Scholar]
- Fitzgerald, D.; Idziorek, T.; Batra, J.K.; Willingham, M.; Pastan, I. Antitumor activity of a thioether-linked immunotoxin: OVB3-PE. Bioconjugate Chem. 1990, 1, 264–268. [Google Scholar]
- Letvin, N.L.; Goldmacher, V.S.; Ritz, J. In vivo administration of lymphocyte-specific monoclonal antibodies in nonhuman primates. In vivo stability of disulfide-linked immunotoxin conjugates. J. Clin. Invest. 1986, 77, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Blakey, D.C.; Watson, G.J.; Knowles, P.P.; Thorpe, P.E. Effect of chemical deglycosylation of ricin A chain on the in vivo fate and cytotoxic activity of an immunotoxin composed of ricin A chain and anti-Thy 1.1 antibody. Cancer Res. 1987, 47, 947–952. [Google Scholar] [PubMed]
- Ebert, R.F.; Spryn, L.A. Immunotoxin construction with a ribosome-inactivating protein from barley. Bioconjugate Chem. 1990, 1, 331–336. [Google Scholar]
- Cazzola, M.; Bergamaschi, G.; Dezza, L.; D’Uva, R.; Ponchio, L.; Rosti, V.; Ascari, E. Cytotoxic activity of an anti-transferrin receptor immunotoxin on normal and leukemic human hematopoietic progenitors. Cancer Res. 1991, 51, 536–541. [Google Scholar]
- Scott, C.F., Jr.; Lambert, J.M.; Goldmacher, V.S. The pharmacokinetics and toxicity of murine monoclonal antibodies and of gelonin conjugates of these antibodies. Int. J. Immunopharmacol. 1987, 9, 211–225. [Google Scholar]
- Bjorn, M.J.; Ring, D.; Frankel, A. Evaluation of monoclonal antibodies for the development of breast cancer immunotoxins. Cancer Res. 1985, 45, 1214–1221. [Google Scholar]
- Thorpe, P.E.; Wallace, P.M.; Knowles, P.P.; Relf, M.G.; Brown, A.N.F.; Watson, G.J.; Knyba, R.E.; Wawrzynczak, E.J.; Blakey, D.C. New coupling agents for the synthesis of immunotoxins containing a hindered disulfide bond with improved stability in vivo. Cancer Res. 1987, 47, 5924–5931. [Google Scholar] [PubMed]
- Ghetie, V.; Till, M.A.; Ghetie, M.A.; Tucker, T.; Porter, J.; Patzer, E.J.; Richardson, J.A.; Uhr, J.W.; Vitetta, E.S. Preparation and characterization of conjugates of recombinant CD4 and deglycosylated ricin a chain using different cross-linkers. Bioconjugate Chem. 1990, 1, 24–31. [Google Scholar]
- Ghetie, V.; Vitetta, E.S. Chemical construction of immunotoxins. Appl. Biochem. Biotechnol.: Part B Mol. Biotechnol. 2001, 18, 251–268. [Google Scholar]
- Arpicco, S.; Dosio, F.; Brusa, P.; Crosasso, P.; Cattel, L. New coupling reagents for the preparation of disulfide cross-linked conjugates with increased stability. Bioconjugate Chem. 1997, 8, 327–337. [Google Scholar]
- Greenfield, L.; Bloch, W.; Moreland, M. Thiol-containing cross-linking agent with enhanced steric hindrance. Bioconjugate Chem. 1990, 1, 400–410. [Google Scholar]
- McIntyre, G.D.; Scott, C.F., Jr.; Ritz, J.; Blattler, W.A.; Lambert, J.M. Preparation and characterization of interleukin-2-gelonin conjugates made using different cross-linking reagents. Bioconjugate Chem. 1994, 5, 88–97. [Google Scholar]
- Thorpe, P.E.; Ross, W.C.J.; Brown, A.N.F. Blockade of the galactose-binding sites of ricin by its linkage to antibody. Specific cytotoxic effects of the conjugates. Eur. J. Biochem. 1984, 140, 63–71. [Google Scholar]
- Cattel, L.; Delprino, L.; Brusa, P.; Dosio, F.; Comoglio, P.M.; Prat, M. Comparison of blocked and non-blocked ricin-antibody immunotoxins against human gastric carcinoma and colorectal adenocarcinoma cell lines. Cancer Immunol. Immunother. 1988, 27, 233–240. [Google Scholar]
- Lambert, J.M. The galactose-binding sites of the cytotoxic lectin ricin can be chemically blocked in high yield with reactive ligands prepared by chemical modification of glycopeptides containing triantennary n-linked oligosaccharides. Biochemistry 1991, 30, 3234–3247. [Google Scholar] [CrossRef] [PubMed]
- Collinson, A.R.; Lambert, J.M.; Liu, Y.; O’Dea, C.; Shah, S.A.; Rasmussen, R.A.; Goldmacher, V.S. Anti-CD6-blocked ricin: An anti-pan T-cell immunotoxin. Int. J. Immunopharmacol. 1994, 16, 37–49. [Google Scholar]
- Grossbard, M.L.; Multani, P.S.; Freedman, A.S.; O’Day, S.; Gribben, J.G.; Rhuda, C.; Neuberg, D.; Nadler, L.M. A Phase II study of adjuvant therapy with anti-B4-blocked ricin after autologous bone marrow transplantation for patients with relapsed B-cell non- Hodgkin’s lymphoma. Clin. Cancer Res. 1999, 5, 2392–2398. [Google Scholar]
- Tsimberidou, A.M.; Giles, F.J.; Kantarjian, H.M.; Keating, M.J.; O’Brien, S.M. Anti-B4 blocked ricin post chemotherapy in patients with chronic lymphocytic leukemia—Long-term follow-up of a monoclonal antibody-based approach to residual disease. Leuk. Lymphoma 2003, 44, 1719–1725. [Google Scholar]
- Szatrowski, T.P.; Dodge, R.K.; Reynolds, C.; Westbrook, C.A.; Frankel, S.R.; Sklar, J.; Stewart, C.C.; Hurd, D.D.; Kolitz, J.E.; Velez-Garcia, E.; et al. Lineage specific treatment of adult patients with acute lymphoblastic leukemia in first remission with anti-B4-blocked ricin or high-dose cytarabine: Cancer and leukemia group B study 9311. Cancer 2003, 97, 1471–1480. [Google Scholar] [CrossRef] [PubMed]
- Blakey, D.C.; Thorpe, P.E. Effect of chemical deglycosylation on the in vivo fate of ricin A-chain. Cancer Drug Delivery 1986, 3, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, P.E.; Wallace, P.M.; Knowles, P.P.; Relf, M.G.; Brown, A.N.F.; Watson, G.J.; Blakey, D.C.; Newell, D.R. Improved antitumor effects of immunotoxins prepared with deglycosylated ricin A-chain and hindered disulfide linkages. Cancer Res. 1988, 48, 6396–6403. [Google Scholar]
- Ghetie, V.; Thorpe, P.; Ghetie, M.A.; Knowles, P.; Uhr, J.W.; Vitetta, E.S. The GLP large scale preparation of immunotoxins containing deglycosylated ricin A chain and a hindered disulfide bond. J. Immunol. Methods 1991, 142, 223–230. [Google Scholar]
- Bjorn, M.J.; Groetsema, G.; Scalapino, L. Antibody-Pseudomonas exotoxin A conjugates cytotoxic to human breast cancer cells in vitro. Cancer Res. 1986, 46, 3262–3267. [Google Scholar] [PubMed]
- Thorpe, P.E.; Ross, W.C.J. The preparation and cytotoxic properties of antibody-toxin conjugates. Immunol. Rev. 1982, 62, 119–158. [Google Scholar]
- Greenfield, L.; Johnson, V.G.; Youle, R.J. Mutations in diphtheria toxin separate binding from entry and amplify immunotoxin selectivity. Science 1987, 238, 536–539. [Google Scholar]
- Brinkmann, U.; Pai, L.H.; FitzGerald, D.J.; Willingham, M.; Pastan, I. B3(Fv)-PE38KDEL, a single-chain immunotoxin that causes complete regression of a human carcinoma in mice. Proc. Natl. Acad. Sci. 1991, 88, 8616–8620. [Google Scholar]
- Kreitman, R.J. Chimeric fusion proteins—Pseudomonas exotoxin-based. Curr. Opin. Invest. Drugs 2001, 2, 1282–1293. [Google Scholar]
- Van Oosterhout, Y.V.J.M.; van Emst, L.; Schattenberg, A.V.M.B.; Tax, W.J.M.; Ruiter, D.J.; Spits, H.; Nagengast, O.M.; Masereeuw, R.; Evers, S.; de Witte, T.; Preijers, F.W.M.B. A combination of anti-CD3 and anti-CD7 ricin A-immunotoxins for the in vivo treatment of acute graft versus host disease. Blood 2000, 95, 3693–3701. [Google Scholar] [PubMed]
- Martin, P.J.; Pei, J.; Gooley, T.; Anasetti, C.; Appelbaum, F.R.; Deeg, J.; Hansen, J.A.; Nash, R.A.; Petersdorf, E.W.; Storb, R.; et al. Evaluation of a CD25-specific immunotoxin for prevention of graft-versus-host disease after unrelated marrow transplantation. Biol. Blood Marrow Transplant. 2004, 10, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Schnell, R.; Borchmann, P.; Staak, J.O.; Schindler, J.; Ghetie, V.; Vitetta, E.S.; Engert, A. Clinical evaluation of ricin A-chain immunotoxins in patients with Hodgkin’s lymphoma. Ann. Oncol. 2003, 14, 729–736. [Google Scholar]
- Kreitman, R.J.; Wilson, W.H.; White, J.D.; Stetler-Stevenson, M.; Jaffe, E.S.; Giardina, S.; Waldmann, T.A.; Pastan, I. Phase I trial of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) in patients with hematologic malignancies. J. Clin. Oncol. 2000, 18, 1622–1636. [Google Scholar]
- Pastan, I.; Hassan, R.; FitzGerald, D.J.; Kreitman, R.J. Immunotoxin treatment of cancer. Annu. Rev. Med. 2007, 58, 221–237. [Google Scholar]
- Kreitman, R.J.; Pastan, I. Accumulation of a recombinant immunotoxin in a tumor in vivo: Fewer than 1000 molecules per cell are sufficient for complete responses. Cancer Res. 1998, 58, 968–975. [Google Scholar]
- Li, Y.M.; Hall, W.A. Targeted Toxins in Brain Tumor Therapy. Toxins 2010, 2, 2645–2662. [Google Scholar]
- Onda, M.; Beers, R.; Xiang, L.; Nagata, S.; Wang, Q.C.; Pastan, I. An immunotoxin with greatly reduced immunogenicity by identification and removal of B cell epitopes. Proc. Natl. Acad. Sci. USA 2008, 105, 11311–11316. [Google Scholar]
- Benhar, I.; Pastan, I. Identification of residues that stabilize the single-chain Fv of monoclonal antibodies B3. J. Biol. Chem. 1995, 270, 23373–23380. [Google Scholar]
- Keller, J.; Heisler, I.; Tauber, R.; Fuchs, H. Development of a novel molecular adapter for the optimization of immunotoxins. J. Control. Release 2001, 74, 259–261. [Google Scholar]
- Heisler, I.; Keller, J.; Tauber, R.; Sutherland, M.; Fuchs, H. A cleavable adapter to reduce nonspecific cytotoxicity of recombinant immunotoxins. Int. J. Cancer 2003, 103, 277–282. [Google Scholar]
- Dang, N.H.; Fayad, L.; McLaughlin, P.; Romaguara, J.E.; Hagemeister, F.; Goy, A.; Neelapu, S.; Samaniego, F.; Walker, P.L.; Wang, M.; et al. Phase II trial of the combination of denileukin diftitox and rituximab for relapsed/refractory B-cell non-Hodgkin lymphoma. Br. J. Haematol. 2007, 138, 502–505. [Google Scholar] [CrossRef] [PubMed]
- Gerena-Lewis, M.; Crawford, J.; Bonomi, P.; Maddox, A.M.; Hainsworth, J.; McCune, D.E.; Shukla, R.; Zeigler, H.; Hurtubise, P.; Chowdhury, T.R.; et al. A phase II trial of denileukin diftitox in patients with previously treated advanced non-small cell lung cancer. Am. J. Clin. Oncol.: Cancer Clin. Trials 2009, 32, 269–273. [Google Scholar]
- Kadin, M.E.; Vonderheid, E.C. Targeted therapies: Denileukin diftitox-a step towards a magic bullet’ for CTCL. Nat. Rev. Clin. Oncol. 2010, 7, 430–432. [Google Scholar]
- Chari, R.V.J. Targeted cancer therapy: Conferring specificity to cytotoxic drugs. Acc. Chem. Res. 2008, 41, 98–107. [Google Scholar]
- Chari, R.V.J. Targeted delivery of chemotherapeutics: Tumor-activated prodrug therapy. Adv. Drug Delivery Rev. 1998, 31, 89–104. [Google Scholar]
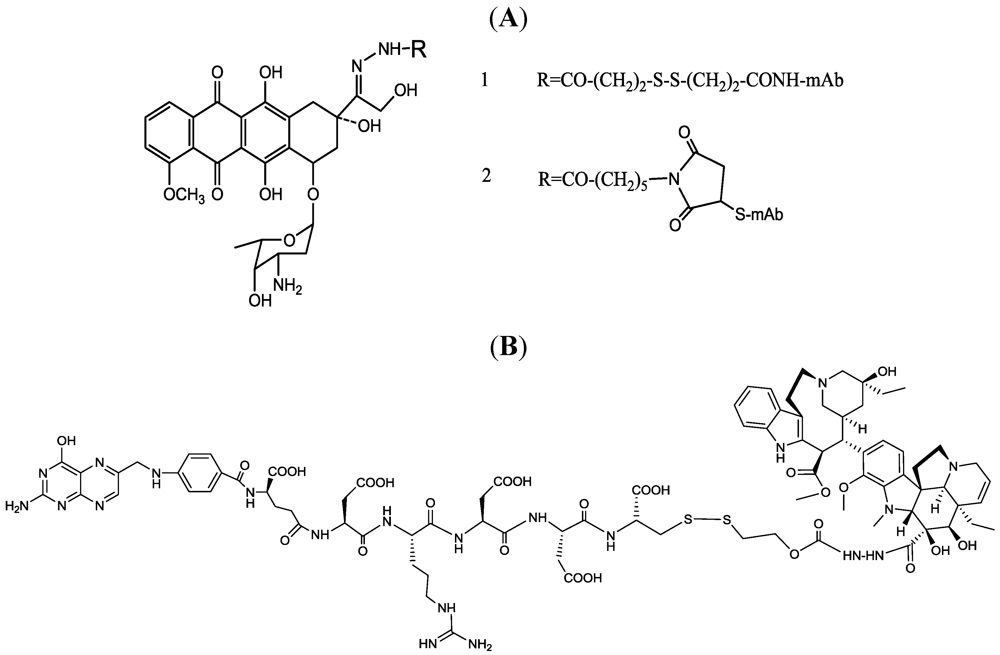
- Endo, N.; Takeda, Y.; Kishida, K.; Kato, Y.; Saito, M.; Umemoto, N.; Hara, T. Target-selective cytotoxicity of methotrexate conjugated with monoclonal anti-mm46 antibody. Cancer Immunol. Immunother. 1987, 25, 1–6. [Google Scholar] [PubMed]
- Pimm, M.V.; Paul, M.A.; Ogumuyiwa, Y.; Baldwin, R.W. Biodistribution and tumor-localization of a daunomycin monoclonal antibody conjugate in nude-mice with human-tumor xenografts. Cancer Immunol. Immunother. 1988, 27, 267–271. [Google Scholar]
- Spearman, M.E.; Goodwin, R.M.; Apelgren, L.D.; Bumol, T.F. Disposition of the monoclonal antibody-vinca alkaloid conjugate ks1/4-davlb (ly256787) and free 4-desacetylvinblastine in tumor-bearing nude-mice. J. Pharmacol. Exp. Ther. 1987, 241, 695–703. [Google Scholar]
- Kato, Y.; Tsukada, Y.; Hara, T.; Hirai, H. Enhanced antitumor activity of mitomycin C conjugated with anti-alpha-fetoprotein antibody by a novel method of conjugation. J. Appl. Biochem. 1983, 5, 313–319. [Google Scholar]
- Rowland, A.J.; Pietersz, G.A.; McKenzie, I.F.C. Preclinical investigation of the antitumor effects of anti-cd19-idarubicin immunoconjugates. Cancer Immunol. Immunother. 1993, 37, 195–202. [Google Scholar]
- Smyth, M.J.; Pietersz, G.A.; McKenzie, I.F.C. Selective enhancement of antitumor-activity of n-acetyl melphalan upon conjugation to monoclonal-antibodies. Cancer Res. 1987, 47, 62–69. [Google Scholar]
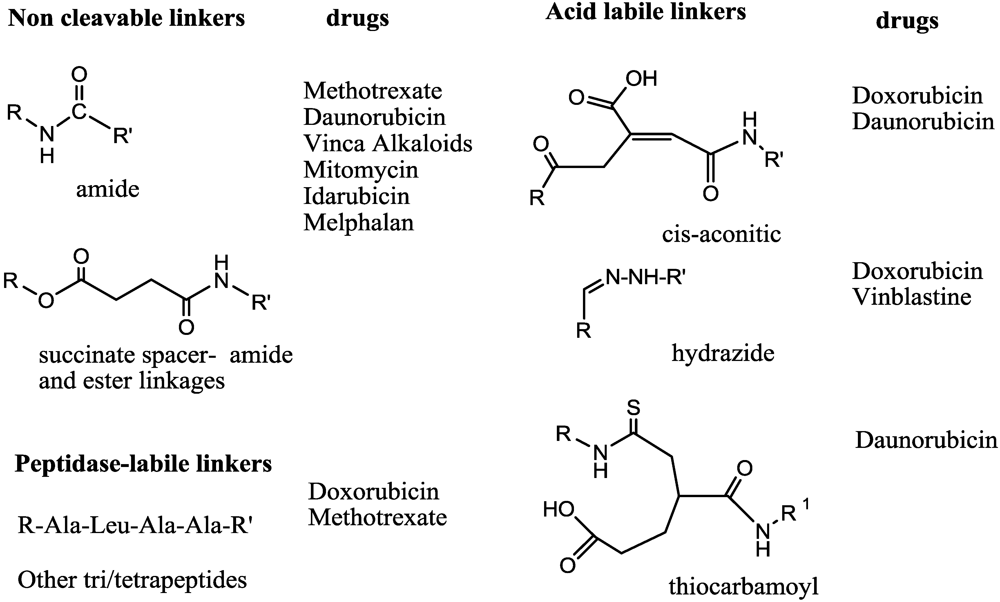
- Trouet, A.; Masquelier, M.; Baurain, R.; Deprez-De Campeneere, D. A covalent linkage between daunorubicin and proteins that is stable in serum and reversible by lysosomal hydrolases, as required for a lysosomotropic drug-carrier conjugate: in vitro and in vivo studies. Proc. Natl. Acad. Sci. USA 1982, 79, 626–629. [Google Scholar]
- Umemoto, N.; Kato, Y.; Endo, N.; Takeda, Y.; Hara, T. Preparation and in vitro cyto-toxicity of a methotrexate-anti-mm46 monoclonal-antibody conjugate via an oligopeptide spacer. Int. J. Cancer 1989, 43, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.C.; Ryser, H.J. cis-Aconityl spacer between daunomycin and macromolecular carriers: A model of pH-sensitive linkage releasing drug from a lysosomotropic conjugate. Biochem. Biophys. Res. Commun. 1981, 102, 1048–1054. [Google Scholar]
- Dillman, R.O.; Johnson, D.E.; Shawler, D.L.; Koziol, J.A. Superiority of an acid-labile daunorubicin monoclonal antibody immunoconjugate compared to free drug. Cancer Res. 1988, 48, 6097–6102. [Google Scholar] [PubMed]
- Greenfield, R.S.; Kaneko, T.; Daues, A.; Edson, M.A.; Fitzgerald, K.A.; Olech, L.J.; Grattan, J.A.; Spitalny, G.L.; Braslawsky, G.R. Evaluation invitro of adriamycin immunoconjugates synthesized using an acid-sensitive hydrazone linker. Cancer Res. 1990, 50, 6600–6607. [Google Scholar] [PubMed]
- Trail, P.A.; Willner, D.; Lasch, S.J.; Henderson, A.J.; Hofstead, S.; Casazza, A.M.; Firestone, R.A.; Hellstrom, I.; Hellstrom, K.E. Cure of xenografted human carcinomas by BR96-doxorubicin immunoconjugates. Science 1993, 261, 212–215. [Google Scholar]
- King, H.D.; Staab, A.J.; Pham-Kaplita, K.; Yurgaitis, D.; Firestone, R.A.; Lasch, S.J.; Trail, P.A. BR96 conjugates of highly potent anthracyclines. Bioorg. Med. Chem. Lett. 2003, 13, 2119–2122. [Google Scholar]
- Graeser, R.; Esser, N.; Unger, H.; Fichtner, I.; Zhu, A.; Unger, C.; Kratz, F. INNO-206, the (6-maleimidocaproyl hydrazone derivative of doxorubicin), shows superior antitumor efficacy compared to doxorubicin in different tumor xenograft models and in an orthotopic pancreas carcinoma model. Invest. New Drugs 2010, 28, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Schneck, D.; Butler, F.; Dugan, W.; Littrell, D.; Petersen, B.; Bowsher, R.; DeLong, A.; Dorrbecker, S. Disposition of a murine monoclonal antibody vinca conjugate (KS1/4-DAVLB) in patients with adenocarcinomas. Clin. Pharmacol. Ther. 1990, 47, 36–41. [Google Scholar]
- Laguzza, B.C.; Nichols, C.L.; Briggs, S.L.; Cullinan, G.J.; Johnson, D.A.; Starling, J.J.; Baker, A.L.; Bumol, T.F.; Corvalan, J.R.F. New antitumor monoclonal-antibody vinca conjugates LY203725 and related-compounds—design, preparation, and representative in vivo activity. J. Med. Chem. 1989, 32, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Dosio, F.; Milla, P.; Cattel, L. EC-145, a folate-targeted Vinca alkaloid conjugate for the potential treatment of folate receptor-expressing cancers. Curr. Opin. Invest. Drugs 2010, 11, 1424–1433. [Google Scholar]
- Sun, M.M.C.; Beam, K.S.; Cerveny, C.G.; Hamblett, K.J.; Blackmore, R.S.; Torgov, M.Y.; Handley, F.G.M.; Ihle, N.C.; Senter, P.D.; Alley, S.C. Reduction-alkylation strategies for the modification of specific monoclonal antibody disulfides. Bioconjugate Chem. 2005, 16, 1282–1290. [Google Scholar]
- McDonagh, C.F.; Turcott, E.; Westendorf, L.; Webster, J.B.; Alley, S.C.; Kim, K.; Andreyka, J.; Stone, I.; Hamblett, K.J.; Francisco, J.A.; Carter, P. Engineered antibody-drug conjugates with defined sites and stoichiometries of drug attachment. Protein Eng., Des. Sel. 2006, 19, 299–307. [Google Scholar] [CrossRef]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.C.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [PubMed]
- Issell, B.F.; Crooke, S.T. Maytansine. Cancer Treat. Rev. 1978, 5, 199–207. [Google Scholar]
- Okamoto, K.; Harada, K.; Ikeyama, S.; Iwasa, S. Therapeutic effect of ansamitocin targeted to tumor by a bispecific monoclonal-antibody. Jpn. J. Cancer Res. 1992, 83, 761–768. [Google Scholar]
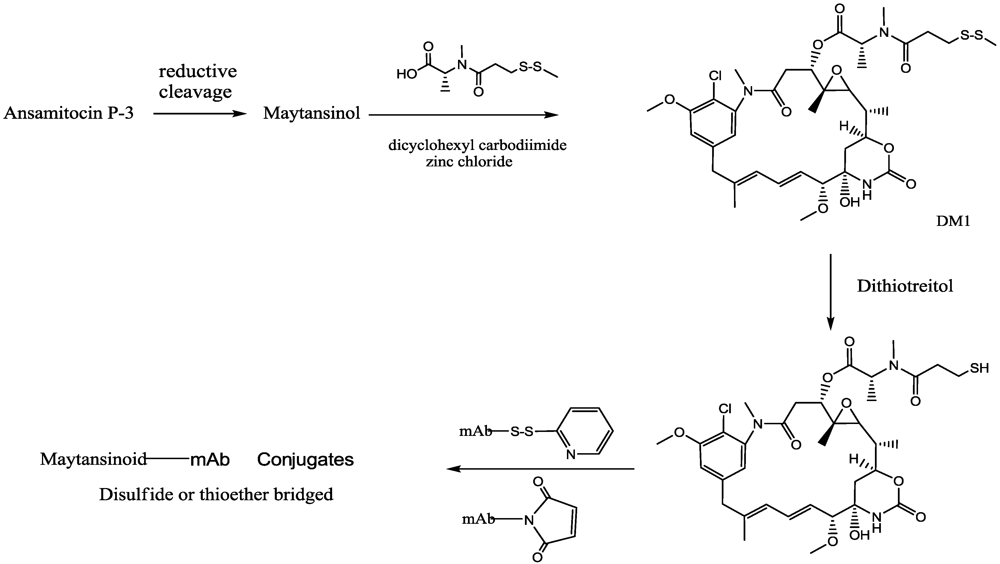
- Chari, R.V.J.; Martell, B.A.; Gross, J.L.; Cook, S.B.; Shah, S.A.; Blattler, W.A.; McKenzie, S.J.; Goldmacher, V.S. Immunoconjugates containing novel maytansinoids: Promising anticancer drugs. Cancer Res. 1992, 52, 127–131. [Google Scholar]
- Liu, C.N.; Tadayoni, B.M.; Bourret, L.A.; Mattocks, K.M.; Derr, S.M.; Widdison, W.C.; Kedersha, N.L.; Ariniello, P.D.; Goldmacher, V.S.; Lambert, J.M.; et al. Eradication of large colon tumor xenografts by targeted delivery of maytansinoids. Proc. Natl. Acad. Sci. USA 1996, 93, 8618–8623. [Google Scholar]
- Cassady, J.M.; Chan, K.K.; Floss, H.G.; Leistner, E. Recent developments in the maytansinoid antitumor agents. Chem. Pharm. Bull. 2004, 52, 1–26. [Google Scholar]
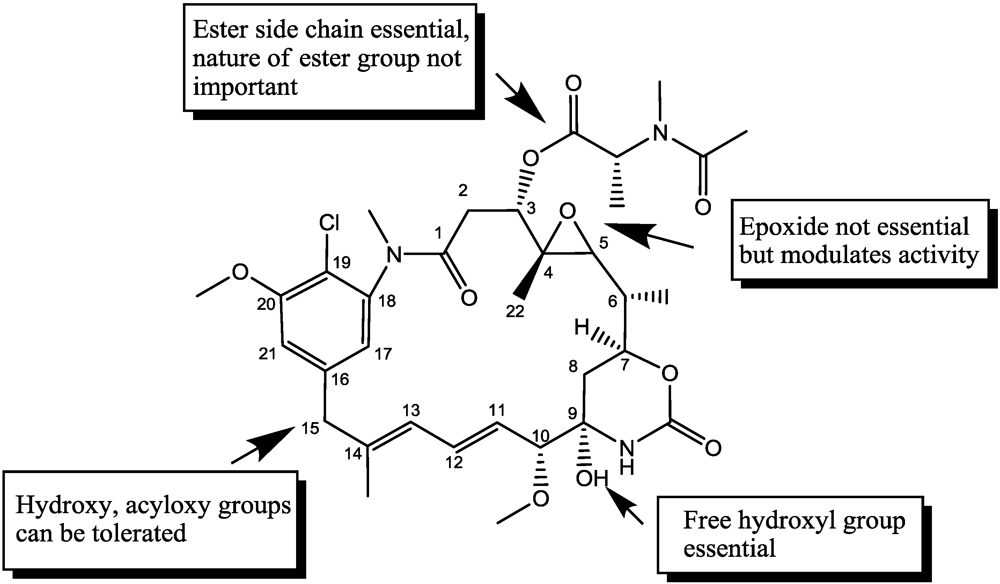
- Kupchan, S.M.; Sneden, A.T.; Branfman, A.R.; Howie, G.A.; Rebhun, L.I.; McIvor, W.E.; Wang, R.W.; Schnaitman, T.C. Structural requirements for antileukemic activity among the naturally occurring and semisynthetic maytansinoids. J. Med. Chem. 1978, 21, 31–37. [Google Scholar]
- Widdison, W.C.; Wilhelm, S.D.; Cavanagh, E.E.; Whiteman, K.R.; Leece, B.A.; Kovtun, Y.; Goldmacher, V.S.; Xie, H.S.; Steeves, R.M.; Lutz, R.J.; et al. Semisynthetic maytansine analogues for the targeted treatment of cancer. J. Med. Chem. 2006, 49, 4392–4408. [Google Scholar] [PubMed]
- Tijink, B.M.; Buter, J.; de Bree, R.; Giaccone, G.; Lang, M.S.; Staab, A.; Leemans, C.R.; van Dongen, G. A phase I dose escalation study with anti-CD44v6 bivatuzumab mertansine in patients with incurable squamous cell carcinoma of the head and neck or esophagus. Clin. Cancer Res. 2006, 12, 6064–6072. [Google Scholar]
- Krop, I.E.; Beeram, M.; Modi, S.; Jones, S.F.; Holden, S.N.; Yu, W.; Girish, S.; Tibbitts, J.; Yi, J.H.; Sliwkowski, M.X.; et al. Phase I Study of Trastuzumab-DM1, an HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-Positive metastatic breast cancer. J. Clin. Oncol. 2010, 28, 2698–2704. [Google Scholar] [PubMed]
- Ross, S.; Spencer, S.D.; Holcomb, I.; Tan, C.; Hongo, J.; Devaux, B.; Rangell, L.; Keller, G.A.; Schow, P.; Steeves, R.M.; et al. Prostate stem cell antigen as therapy target: Tissue expression and in vivo efficacy of an immunoconjugate. Cancer Res. 2002, 62, 2546–2553. [Google Scholar] [PubMed]
- Helft, P.R.; Schilsky, R.L.; Hoke, F.J.; Williams, D.; Kindler, H.L.; Sprague, E.; DeWitte, M.; Martino, H.K.; Erickson, J.; Pandite, L.; et al. A phase I study of cantuzumab mertansine administered as a single intravenous infusion once weekly in patients with advanced solid tumors. Clin. Cancer Res. 2004, 10, 4363–4368. [Google Scholar] [PubMed]
- Russo, A.; Degraff, W.; Friedman, N.; Mitchell, J.B. Selective modulation of glutathione levels in human normal versus tumor-cells and subsequent differential response to chemotherapy drugs. Cancer Res. 1986, 46, 2845–2848. [Google Scholar] [PubMed]
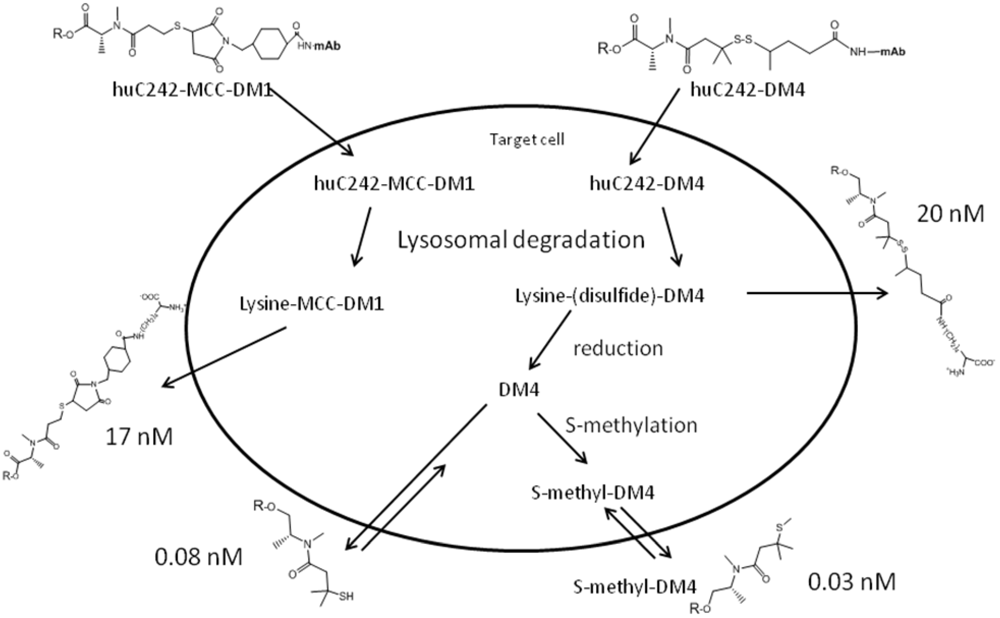
- Erickson, H.K.; Park, P.U.; Widdison, W.C.; Kovtun, Y.V.; Garrett, L.M.; Hoffman, K.; Lutz, R.J.; Goldmacher, V.S.; Blättler, W.A. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res. 2006, 66, 4426–4433. [Google Scholar]
- Phillips, G.D.L.; Li, G.M.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blattler, W.A.; Lambert, J.M.; Chari, R.V.J.; Lutz, R.J.; et al. Trgeting HER2-Positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [PubMed]
- Erickson, H.K.; Widdison, W.C.; Mayo, M.F.; Whiteman, K.; Audette, C.; Wilhelm, S.D.; Singh, R. Tumor delivery and in vivo processing of disulfide-linked and thioether-linked antibody-maytansinoid conjugates. Bioconjugate Chem. 2010, 21, 84–92. [Google Scholar] [CrossRef]
- Polson, A.G.; Calemine-Fenaux, J.; Chan, P.; Chang, W.; Christensen, E.; Clark, S.; de Sauvage, F.J.; Eaton, D.; Elkins, K.; Michael Elliott, J.; et al. Antibody-drug conjugates for the treatment of non-Hodgkin’s lymphoma: Target and linker-drug selection. Cancer Res. 2009, 69, 2358–2364. [Google Scholar] [PubMed]
- Lopus, M.; Oroudjev, E.; Wilson, L.; Wilhelm, S.; Widdison, W.; Chari, R.; Jordan, M.A. Maytansine and Cellular Metabolites of Antibody-Maytansinoid Conjugates Strongly Suppress Microtubule Dynamics by Binding to Microtubules. Mol. Cancer Ther. 2010, 9, 2689–2699. [Google Scholar]
- Loo, T.W.; Clarke, D.M. Recent progress in understanding the mechanism of P-glycoprotein-mediated drug efflux. J. Membr. Biol. 2005, 206, 173–185. [Google Scholar]
- Kovtun, Y.V.; Audette, C.A.; Mayo, M.F.; Jones, G.E.; Doherty, H.; Maloney, E.K.; Erickson, H.K.; Sun, X.; Wilhelm, S.; Ab, O.; et al. Antibody-maytansinoid conjugates designed to bypass multidrug resistance. Cancer Res. 2010, 70, 2528–2537. [Google Scholar] [PubMed]
- Ikeda, H.; Hideshima, T.; Fulciniti, M.; Lutz, R.J.; Yasui, H.; Okawa, Y.; Kiziltepe, T.; Vallet, S.; Pozzi, S.; Santo, L.; et al. The monoclonal antibody nBT062 conjugated to cytotoxic maytansinoids has selective cytotoxicity against CD138-positive multiple myeloma cells in vitro and in vivo. Clin. Cancer Res. 2009, 15, 4028–4037. [Google Scholar] [PubMed]
- Thompson, D.S.; Patnaik, A.; Bendell, J.C.; Papadopoulos, K.; Infante, J.R.; Mastico, R.A.; Johnson, D.; Qin, A.; O’Leary, J.J.; Tolcher, A.W. A phase I dose-escalation study of IMGN388 in patients with solid tumors. J. Clin. Oncol. 2010, 28, 3058. [Google Scholar]
- Al-Katib, A.M.; Aboukameel, A.; Mohammad, R.; Bissery, M.-C.; Zuany-Amorim, C. Superior antitumor activity of SAR3419 to rituximab in xenograft models for Non-Hodgkin’s lymphoma. Clin. Cancer Res. 2009, 15, 4038–4045. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R. The dolastatins. In Fortschritte der Chemie Organischer Naturstoffe Progress in the Chemistry of Organic Natural Products Progrèss Dans La Chimie des Substances Organiques Naturelles; Springer: Berlin, Germany, 1997; Volume 70, pp. 1–79. [Google Scholar]
- Mohammad, R.M.; Varterasian, M.L.; Almatchy, V.P.; Hannoudi, G.N.; Pettit, G.R.; Al-Katib, A. Successful treatment of human chronic lymphocytic leukemia xenografts with combination biological agents auristatin PE and bryostatin 1. Clin. Cancer Res. 1998, 4, 1337–1343. [Google Scholar]
- Pettit, G.R.; Flahive, E.J.; Boyd, M.R.; Bai, R.; Hamel, E.; Pettit, R.K.; Schmidt, J.M. Antineoplastic agents 360. Synthesis and cancer cell growth inhibitory studies of dolastatin 15 structural modifications. Anti-Cancer Drug Des. 1998, 13, 47–66. [Google Scholar]
- Turner, T.; Jackson, W.H.; Pettit, G.R.; Wells, A.; Kraft, A.S. Treatment of human prostate cancer cells with dolastatin 10, a peptide isolated from a marine shell-less mollusc. Prostate 1998, 34, 175–181. [Google Scholar]
- Bai, R.; Pettit, G.R.; Hamel, E. Binding of dolastatin-10 to tubulin at a distinct site for peptide antimitotic agents near the exchangeable nucleotide and vinca alkaloid sites. J. Biol. Chem. 1990, 265, 17141–17149. [Google Scholar] [PubMed]
- Kalemkerian, G.P.; Ou, X.; Adil, M.R.; Rosati, R.; Khoulani, M.M.; Madan, S.K.; Pettit, G.R. Activity of dolastatin 10 against small-cell lung cancer in vitro and in vivo: Induction of apoptosis and bcl-2 modification. Cancer Chemother. Pharmacol. 1999, 43, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Krug, L.M.; Miller, V.A.; Kalemkerian, G.P.; Kraut, M.J.; Ng, K.K.; Heelan, R.T.; Pizzo, B.A.; Perez, W.; McClean, N.; Kris, M.G. Phase II study of dolastatin-10 in patients with advanced non-small-cell lung cancer. Ann. Oncol. 2000, 11, 227–228. [Google Scholar]
- Miyazaki, K.; Kobayashi, M.; Natsume, T.; Gondo, M.; Mikami, T.; Sakakibara, K.; Tsukagoshi, S. Synthesis and antitumor activity of novel dolastatin 10 analogs. Chem. Pharm. Bull. 1995, 43, 1706–1718. [Google Scholar]
- Pettit, G.R.; Srirangam, J.K.; Barkoczy, J.; Williams, M.D.; Durkin, K.P.M.; Boyd, M.R.; Bai, R.; Hamel, E.; Schmidt, J.M.; Chapuis, J.C. Antineoplastic agents 337. Synthesis of dolastatin 10 structural modifications. Anti-Cancer Drug Des. 1995, 10, 529–544. [Google Scholar]
- Pettit, G.R.; Srirangam, J.K.; Barkoczy, J.; Williams, M.D.; Boyd, M.R.; Hamel, E.; Pettit, R.K.; Hogan, F.; Bai, R.L.; Chapuis, J.C.; et al. Antineoplastic agents 365. Dolastatin 10 SAR probes. Anti-Cancer Drug Des. 1998, 13, 243–277. [Google Scholar]
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B.A.; Cerveny, C.G.; Chace, D.F.; DeBlanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B.; et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 2003, 21, 778–784. [Google Scholar] [PubMed]
- Dubowchik, G.M.; Firestone, R.A.; Padilla, L.; Willner, D.; Hofstead, S.J.; Mosure, K.; Knipe, J.O.; Lasch, S.J.; Trail, P.A. Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: Model studies of enzymatic drug release and antigen-specific in vitro anticancer activity. Bioconjugate Chem. 2002, 13, 855–869. [Google Scholar] [CrossRef]
- Doronina, S.O.; Bovee, T.D.; Meyer, D.W.; Miyamoto, J.B.; Anderson, M.E.; Morris-Tilden, C.A.; Senter, P.D. Novel peptide linkers for highly potent antibody-auristatin conjugate. Bioconjugate Chem. 2008, 19, 1960–1963. [Google Scholar]
- Younes, A.; Bartlett, N.L.; Leonard, J.P.; Kennedy, D.A.; Lynch, C.M.; Sievers, E.L.; Forero-Torres, A. Brentuximab Vedotin (SGN-35) for Relapsed CD30-Positive Lymphomas. N. Engl. J. Med. 2010, 363, 1812–1821. [Google Scholar]
- Ma, D.; Hopf, C.E.; Malewicz, A.D.; Donovan, G.P.; Senter, P.D.; Goeckeler, W.F.; Maddon, P.J.; Olson, W.C. Potent antitumor activity of an auristatin-conjugated, fully human monoclonal antibody to prostate-specific membrane antigen. Clin. Cancer Res. 2006, 12, 2591–2596. [Google Scholar]
- Alley, S.C.; Okeley, N.M.; Senter, P.D. Antibody-drug conjugates: Targeted drug delivery for cancer. Curr. Opin. Chem. Biol. 2010, 14, 529–537. [Google Scholar]
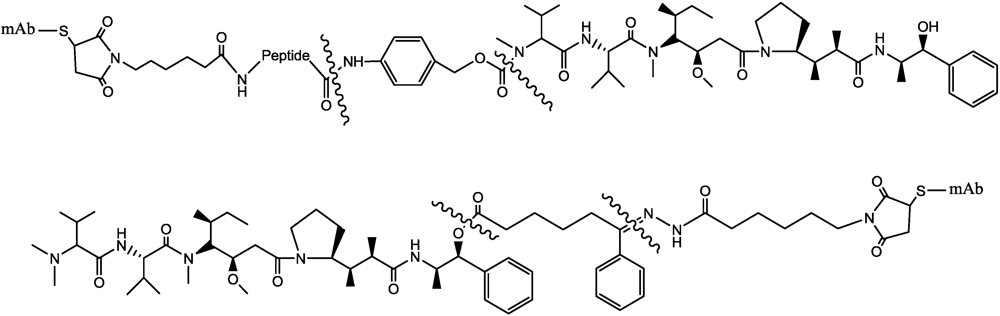
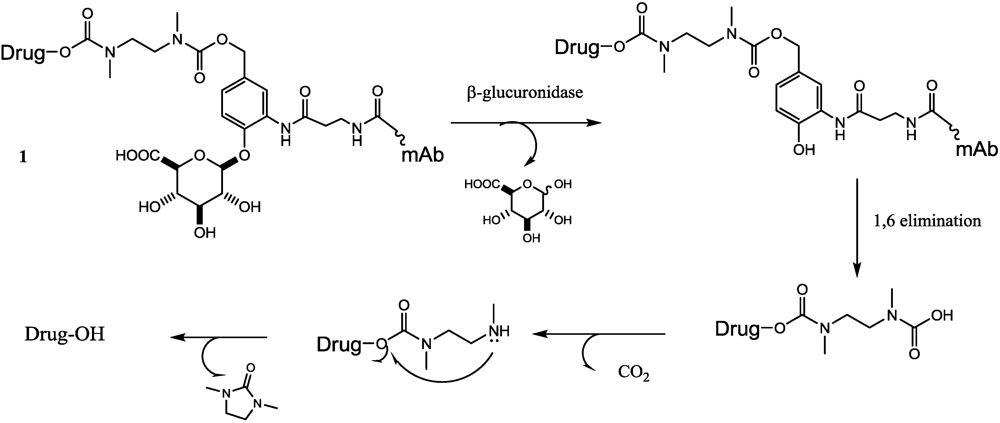
- Jeffrey, S.C.; Andreyka, J.B.; Bernhardt, S.X.; Kissler, K.M.; Kline, T.; Lenox, J.S.; Moser, R.F.; Nguyen, M.T.; Okeley, N.M.; Stone, I.J.; et al. Development and properties of beta-glucuronide linkers for monoclonal antibody-drug conjugates. Bioconjugate Chem. 2006, 17, 831–840. [Google Scholar] [CrossRef]
- Jeffrey, S.C.; Nguyen, M.T.; Moser, R.F.; Meyer, D.L.; Miyamoto, J.B.; Senter, P.D. Minor groove binder antibody conjugates employing a water soluble beta-glucuronide linker. Bioorg. Med. Chem. Lett. 2007, 17, 2278–2280. [Google Scholar]
- Jiang, X.; Garcia-Fortanet, J.; de Brabander, J.K. Synthesis and complete stereochemical assignment of psymberin/irciniastatin A. J. Am. Chem. Soc. 2005, 127, 11254–11255. [Google Scholar]
- De Graaf, M.; Boven, E.; Scheeren, H.W.; Haisma, H.J.; Pinedo, H.M. Beta-glucuronidase-mediated drug release. Curr. Pharm. Des. 2002, 8, 1391–1403. [Google Scholar]
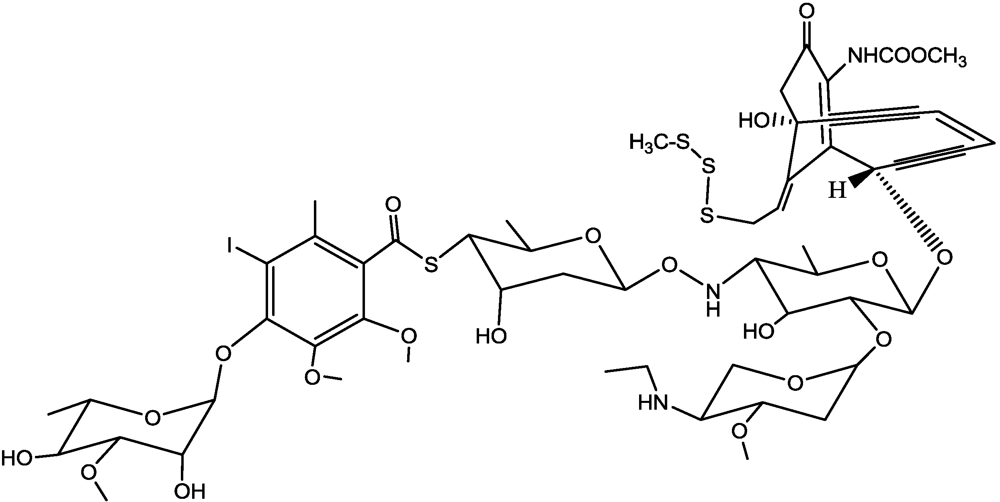
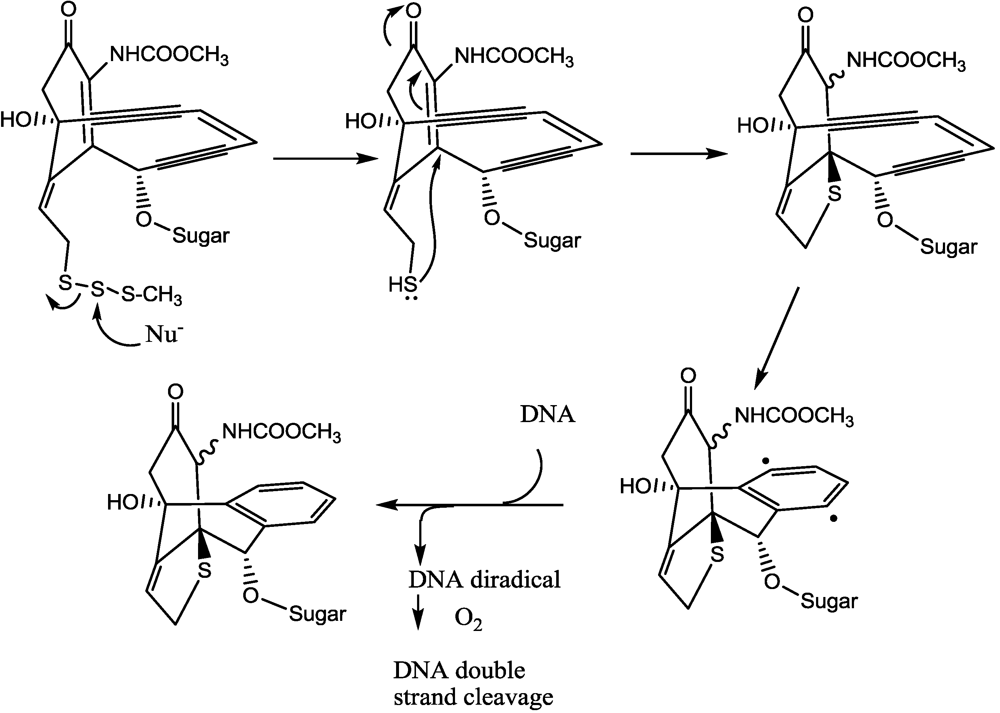
- Lee, M.D.; Dunne, T.S.; Chang, C.C.; Ellestad, G.A.; Siegel, M.M.; Morton, G.O.; McGahren, W.J.; Borders, D.B. Calichemicins, a novel family of antitumor antibiotics. 2. Chemistry and structure of calichemicin.gamma.1I. J. Am. Chem. Soc. 1987, 109, 3466–3468. [Google Scholar]
- Lee, M.D.; Dunne, T.S.; Siegel, M.M.; Chang, C.C.; Morton, G.O.; Borders, D.B. Calichemicins, a novel family of antitumor antibiotics. 1. Chemistry and partial structure of calichemicin.gamma.1I. J. Am. Chem. Soc. 1987, 109, 3464–3466. [Google Scholar]
- Smith, A.L.; Nicolaou, K.C. The enediyne antibiotics. J. Med. Chem. 1996, 39, 2103–2117. [Google Scholar]
- Hinman, L.M.; Hamann, P.R.; Wallace, R.; Menendez, A.T.; Durr, F.E.; Upeslacis, J. Preparation and characterization of monoclonal-antibody conjugates of the calicheamicins: A novel and potent family of antitumor antibiotics. Cancer Res. 1993, 53, 3336–3342. [Google Scholar]
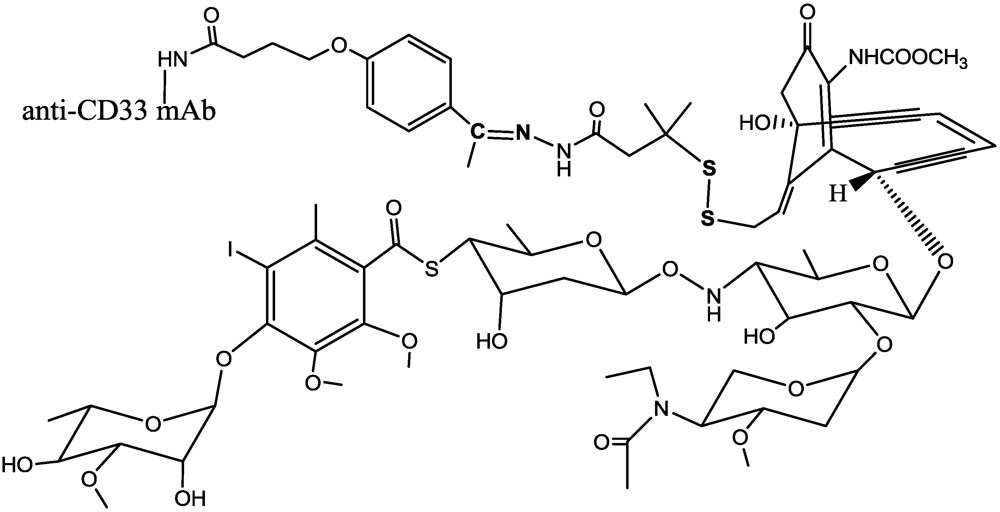
- Hamann, P.R.; Hinman, L.M.; Beyer, C.F.; Lindh, D.; Upeslacis, J.; Flowers, D.A.; Bernstein, I. An anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Choice of linker. Bioconjugate Chem. 2002, 13, 40–46. [Google Scholar] [CrossRef]
- Hamann, P.R.; Hinman, L.M.; Hollander, I.; Beyer, C.F.; Lindh, D.; Holcomb, R.; Hallett, W.; Tsou, H.R.; Upeslacis, J.; Shochat, D.; et al. Gemtuzumab ozogamicin, a potent and selective anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Bioconjugate Chem. 2002, 13, 47–58. [Google Scholar] [CrossRef]
- Van der Velden, V.H.J.; te Mervelde, J.G.; Hoogeveen, P.G.; Bernstein, I.D.; Houtsmuller, A.B.; Berger, M.S.; van Dongen, J.J.M. Targeting of the CD33-calicheamicin immunoconjugate Mylotarg (CMA-676) in acute myeloid leukemia: In vivo and in vitro saturation and internalization by leukemic and normal myeloid cells. Blood 2001, 97, 3197–3204. [Google Scholar] [CrossRef] [PubMed]
- DiJoseph, J.F.; Dougher, M.M.; Kalyandrug, L.B.; Armellino, D.C.; Boghaert, E.R.; Hamann, P.R.; Moran, J.K.; Damle, N.K. Antitumor efficacy of a combination of CMC-544 (inotuzumab ozogamicin), a CD22-targeted cytotoxic immunoconjugate of calicheamicin, and rituximab against non-Hodgkin’s B-cell lymphoma. Clin. Cancer Res. 2006, 12, 242–249. [Google Scholar] [PubMed]
- Wong, B.Y.; Dang, N.H. Inotuzumab ozogamicin as novel therapy in lymphomas. Exp. Opin. Biol. Ther. 2010, 10, 1251–1258. [Google Scholar]
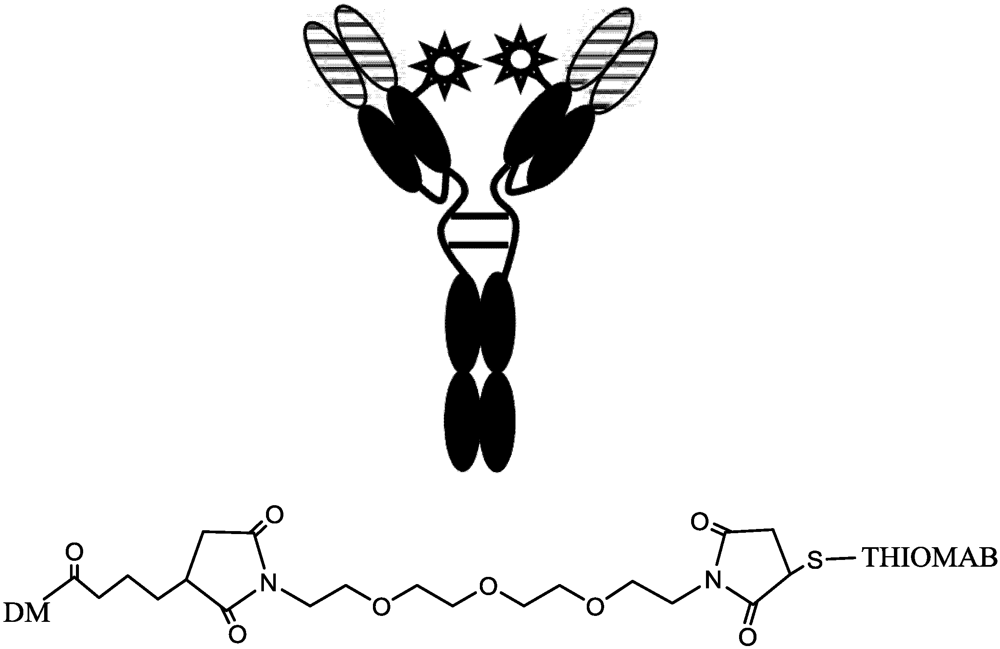
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [PubMed]
- Junutula, J.R.; Flagella, K.M.; Graham, R.A.; Parsons, K.L.; Ha, E.; Raab, H.; Bhakta, S.; Nguyen, T.; Dugger, D.L.; Li, G.M.; et al. Engineered Thio-Trastuzumab-DM1 Conjugate with an Improved Therapeutic Index to Target Human Epidermal Growth Factor Receptor 2-Positive Breast Cancer. Clin. Cancer Res. 2010, 16, 4769–4778. [Google Scholar] [PubMed]
- Puri, S.; Mahapatra, A.K.; Hussain, E.; Sarkar, C.; Sinha, S.; Joshi, B.H. A review of studies on targeting interleukin 4 receptor for central nervous system malignancy. Curr. Mol. Med. 2009, 9, 732–739. [Google Scholar]
- Brown, J.; Rasamoelisolo, M.; Spearman, M.; Bosc, D.; Cizeau, J.; Entwistle, J.; MacDonald, G.C. Preclinical assessment of an anti-EpCAM immunotoxin: Locoregional delivery provides a safer alternative to systemic administration. Cancer Biother. Radiopharm. 2009, 24, 477–487. [Google Scholar]
- Polson, A.G.; Ho, W.Y.; Ramakrishnan, V. Investigational antibody-drug conjugates for hematological malignancies. Exp. Opin. Invest. Drugs 2011, 20, 75–85. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dosio, F.; Brusa, P.; Cattel, L. Immunotoxins and Anticancer Drug Conjugate Assemblies: The Role of the Linkage between Components. Toxins 2011, 3, 848-883. https://doi.org/10.3390/toxins3070848
Dosio F, Brusa P, Cattel L. Immunotoxins and Anticancer Drug Conjugate Assemblies: The Role of the Linkage between Components. Toxins. 2011; 3(7):848-883. https://doi.org/10.3390/toxins3070848
Chicago/Turabian StyleDosio, Franco, Paola Brusa, and Luigi Cattel. 2011. "Immunotoxins and Anticancer Drug Conjugate Assemblies: The Role of the Linkage between Components" Toxins 3, no. 7: 848-883. https://doi.org/10.3390/toxins3070848