Anthrax Lethal Toxin-Induced Gene Expression Changes in Mouse Lung

Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Lethal Toxin and Mutant Toxin
2.3. Injections and Sampling
2.4. RNA Isolation and evaluation of Global Gene Expression
2.5. Data Analysis
3. Results
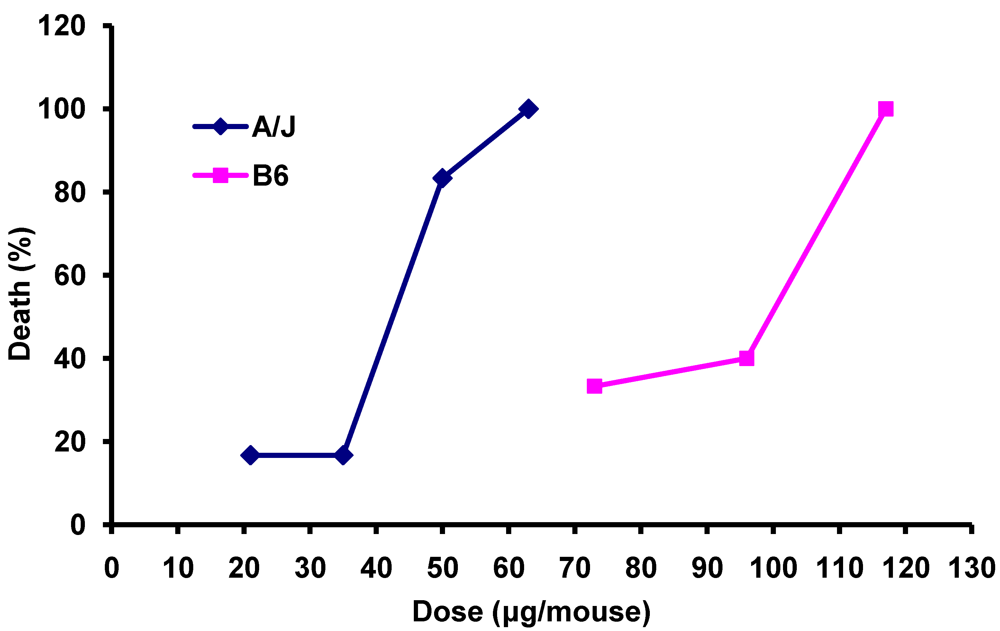
3.1. Differential Susceptibility of A/J and C57BL/6 Mice to Death Induced by Anthrax LeTx

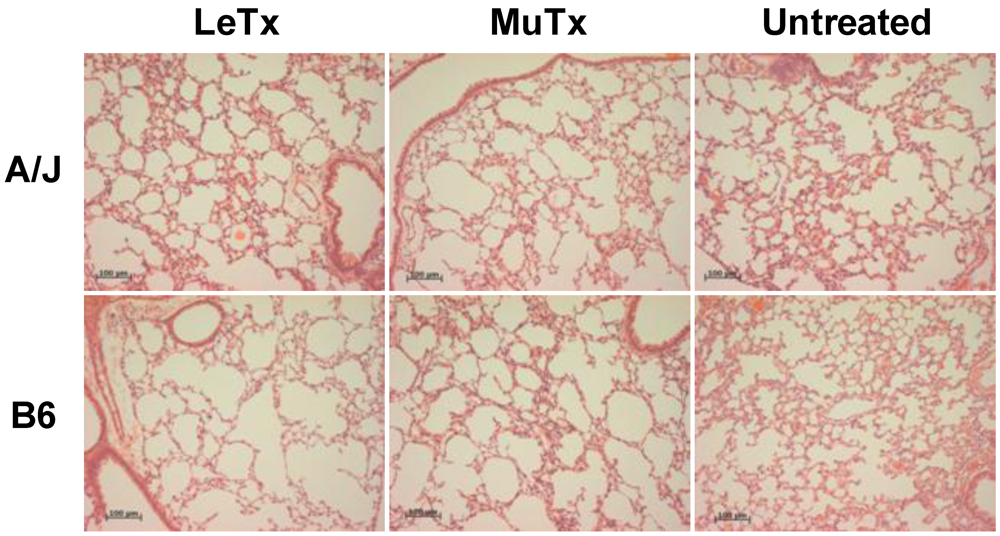
3.2. Lack of Obvious Pulmonary Pathology in Mice Receiving Systemic Exposure to Anthrax LeTx

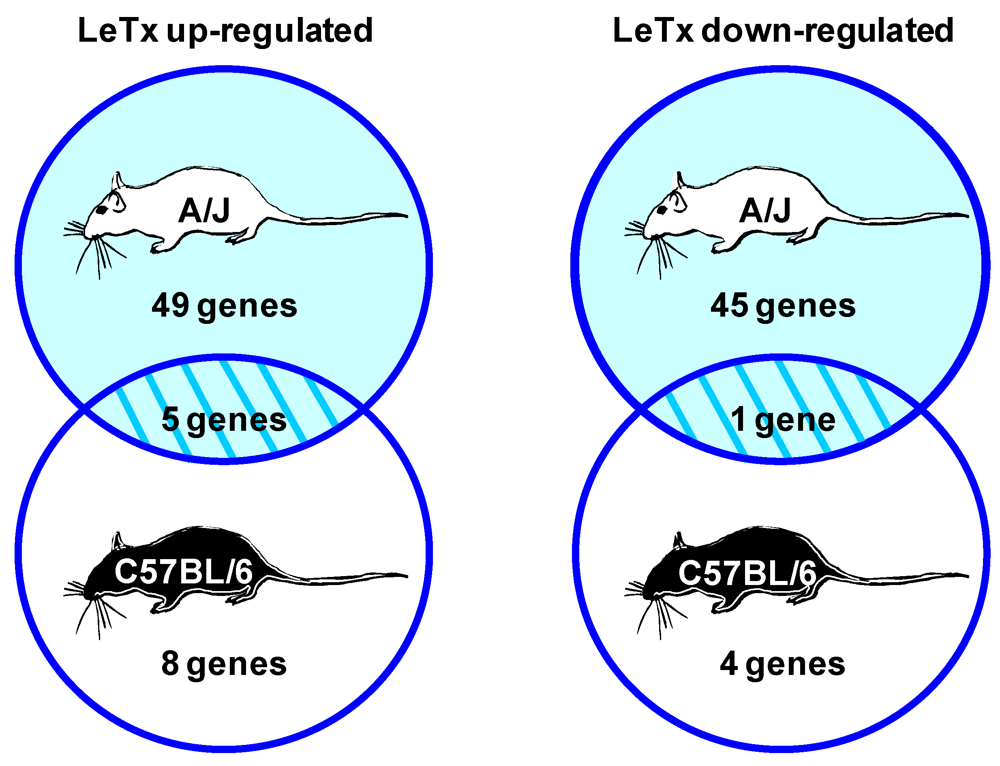
3.3. LeTx-Specific Gene Expression Changes in Mouse Lung Include Genes that are Candidates for Mediating or Promoting Vascular Leakage and Pulmonary Edema

{kind=link}
{kind=link}
{kind=link}
 |
 |

| Chemotaxis of Leukocytes (p = 2.47 × 10−12) | Quantity of Lymphocytes (p = 2.91 × 10−6) | Cellular Movement (p = 8.77 × 10−11) | Cellular Apoptosis (p = 1.31 ×10−5) | |
| Up-Regulated | Ccr7 | Ccr7 | Ccr7 | Ccr7 |
| Eln | Cd3d | Cd3e | Cd3e | |
| Plec1 | Cd3e | Cd69 | Cd69 | |
| Prkcb | Cd3g | Eln | Gadd45b | |
| Cd69 | Lck | Lck | ||
| Lck | Plec1 | Msx1 | ||
| Rasgrp1 | Prkcb | Prkcb | ||
| Satb1 | Rasgrp1 | Rasgrp1 | ||
| Siglecg | Tubb2b | Satb1 | ||
| Wisp2 | Sox17 | |||
| Down-Regulated | Ccbp2 | Bcl2a1b | Adrb2 | Adrb2 |
| Ccl11 | Notch4 | Ccbp2 | Bcl2a1b | |
| Chi3l4 | Ccl11 | Ccl11 | ||
| Csf2ra | Ccl6 | Csf2ra | ||
| Ear2 | Chi3l4 | Lcn2 | ||
| Plaur | Csf2ra | Plaur | ||
| S100a8 | Ear2 | Serpina3n | ||
| S100a9 | Plaur | Tlr4 | ||
| Serpina3n | S100a8 | |||
| Tlr4 | S100a9 | |||
| Serpina3n | ||||
| Stc1 | ||||
| Tlr4 | ||||
| Activation of Lymphocytes (p = 3.11 × 10−6) | Uptake of Arachidonic Acid (p = 1.06 × 10−4) | Orthostatic Hypotension (p = 7.29 × 10−4) | ||
| Up-Regulated | Cd3d | Ddc | ||
| Cd3e | ||||
| Cd3g | ||||
| Igkv1-117 | ||||
| Lck | ||||
| Nlrc3 | ||||
| Prkcb | ||||
| Satb1 | ||||
| Down-regulated | Ccl11 | S100a8 | Adrb2 | |
| Tlr4 | S100a9 |
3.4. Gene Expression Differences that May Participate in Differential Susceptibility of A/J and B6 Strains to Death by LeTx
 |
4. Discussion
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Jernigan, J.A.; Stephens, D.S.; Ashford, D.A.; Omenaca, C.; Topiel, M.S.; Galbraith, M.; Tapper, M.; Fisk, T.L.; Zaki, S.; Popovic, T.; et al. Bioterrorism-related inhalational anthrax: The first 10 cases reported in the united states. Emerg. Infect. Dis. 2001, 7, 933–944. [Google Scholar]
- Collier, R.J.; Young, J.A. Anthrax toxin. Annu. Rev. Cell Dev. Biol. 2003, 19, 45–70. [Google Scholar]
- van der Goot, G.; Young, J.A. Receptors of anthrax toxin and cell entry. Mol. Aspects Med. 2009, 30, 406–412. [Google Scholar]
- Duesbery, N.S.; Webb, C.P.; Leppla, S.H.; Gordon, V.M.; Klimpel, K.R.; Copeland, T.D.; Ahn, N.G.; Oskarsson, M.K.; Fukasawa, K.; Paull, K.D.; et al. Proteolytic inactivation of map-kinase-kinase by anthrax lethal factor. Science 1998, 280, 734–737. [Google Scholar]
- Culley, N.C.; Pinson, D.M.; Chakrabarty, A.; Mayo, M.S.; Levine, S.M. Pathophysiological manifestations in mice exposed to anthrax lethal toxin. Infect. Immunol. 2005, 73, 7006–7010. [Google Scholar]
- Cui, X.; Moayeri, M.; Li, Y.; Li, X.; Haley, M.; Fitz, Y.; Correa-Araujo, R.; Banks, S.M.; Leppla, S.H.; Eichacker, P.Q. Lethality during continuous anthrax lethal toxin infusion is associated with circulatory shock but not inflammatory cytokine or nitric oxide release in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R699–R709. [Google Scholar]
- Moayeri, M.; Haines, D.; Young, H.A.; Leppla, S.H. Bacillus anthracis lethal toxin induces tnf-alpha-independent hypoxia-mediated toxicity in mice. J. Clin. Invest. 2003, 112, 670–682. [Google Scholar]
- Gill, D.M. Bacterial toxins: A table of lethal amounts. Microbiol. Rev. 1982, 46, 86–94. [Google Scholar]
- Dixon, T.C.; Meselson, M.; Guillemin, J.; Hanna, P.C. Anthrax. N. Engl. J. Med. 1999, 341, 815–826. [Google Scholar]
- Moayeri, M.; Leppla, S.H. Cellular and systemic effects of anthrax lethal toxin and edema toxin. Mol. Aspects Med. 2009, 30, 439–455. [Google Scholar]
- Fang, H.; Xu, L.; Chen, T.Y.; Cyr, J.M.; Frucht, D.M. Anthrax lethal toxin has direct and potent inhibitory effects on b cell proliferation and immunoglobulin production. J. Immunol. 2006, 176, 6155–6161. [Google Scholar]
- Comer, J.E.; Chopra, A.K.; Peterson, J.W.; Konig, R. Direct inhibition of t-lymphocyte activation by anthrax toxins in vivo. Infect. Immunol. 2005, 73, 8275–8281. [Google Scholar]
- During, R.L.; Li, W.; Hao, B.; Koenig, J.M.; Stephens, D.S.; Quinn, C.P.; Southwick, F.S. Anthrax lethal toxin paralyzes neutrophil actin-based motility. J. Infect. Dis. 2005, 192, 837–845. [Google Scholar]
- Warfel, J.M.; Steele, A.D.; D’Agnillo, F. Anthrax lethal toxin induces endothelial barrier dysfunction. Am. J. Pathol. 2005, 166, 1871–1881. [Google Scholar]
- Jackson laboratory phenome database. Available online: http://www.jax.org (accessed on 6 September 2011).
- Salles, I.I.; Voth, D.E.; Ward, S.C.; Averette, K.M.; Tweten, R.K.; Bradley, K.A.; Ballard, J.D. Cytotoxic activity of bacillus anthracis protective antigen observed in a macrophage cell line overexpressing antxr1. Cell. Microbiol. 2006, 8, 1272–1281. [Google Scholar]
- Voth, D.E.; Hamm, E.E.; Nguyen, L.G.; Tucker, A.E.; Salles, I.I.; Ortiz-Leduc, W.; Ballard, J.D. Bacillus anthracis oedema toxin as a cause of tissue necrosis and cell type-specific cytotoxicity. Cell. Microbiol. 2005, 7, 1139–1149. [Google Scholar]
- Dozmorov, I.; Centola, M. An associative analysis of gene expression array data. Bioinformatics 2003, 19, 204–211. [Google Scholar]
- Knowlton, N.; Dozmorov, I.M.; Centola, M. Microarray data analysis toolbox (mdat): For normalization, adjustment and analysis of gene expression data. Bioinformatics 2004, 20, 3687–3690. [Google Scholar]
- Dozmorov, I.; Lefkovits, I. Internal standard-based analysis of microarray data. Part 1: Analysis of differential gene expressions. Nucleic Acids Res. 2009, 37, 6323–6339. [Google Scholar]
- Dozmorov, M.G.; Guthridge, J.M.; Hurst, R.E.; Dozmorov, I.M. A comprehensive and universal method for assessing the performance of differential gene expression analyses. PLoS One 2010, 5, e12657. [Google Scholar]
- Benbrook, D.M.; Lightfoot, S.; Ranger-Moore, J.; Liu, T.; Chengedza, S.; Berry, W.L.; Dozmorov, I. Gene expression analysis of biological systems driving an organotypic model of endometrial carcinogenesis and chemoprevention. Gene Regul. Syst. Biol. 2008, 2, 21–42. [Google Scholar]
- Torgerson, T.R.; Genin, A.; Chen, C.; Zhang, M.; Zhou, B.; Anover-Sombke, S.; Frank, M.B.; Dozmorov, I.; Ocheltree, E.; Kulmala, P.; et al. Foxp3 inhibits activation-induced nfat2 expression in t cells thereby limiting effector cytokine expression. J. Immunol. 2009, 183, 907–915. [Google Scholar]
- Xu, Z.; Potula, H.H.; Vallurupalli, A.; Perry, D.; Baker, H.; Croker, B.P.; Dozmorov, I.; Morel, L. Cyclin-dependent kinase inhibitor cdkn2c regulates b cell homeostasis and function in the nzm2410-derived murine lupus susceptibility locus sle2c1. J. Immunol. 2011, 186, 6673–6682. [Google Scholar]
- Bioconductor diffgeneanalysis. Available online: http://www.bioconductor.org/packages/2.5/bioc/html/diffGeneAnalysis.html (accessed on 6 September).
- Moayeri, M.; Martinez, N.W.; Wiggins, J.; Young, H.A.; Leppla, S.H. Mouse susceptibility to anthrax lethal toxin is influenced by genetic factors in addition to those controlling macrophage sensitivity. Infect. Immun. 2004, 72, 4439–4447. [Google Scholar]
- Boyden, E.D.; Dietrich, W.F. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat. Genet. 2006, 38, 240–244. [Google Scholar]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene expression omnibus: Ncbi gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar]
- Ncbi gene expression omnibus. Available online: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE28554 (accessed on 7 September 2011).
- Pai, J.T.; Ruoslahti, E. Identification of endothelial genes up-regulated in vivo. Gene 2005, 347, 21–33. [Google Scholar]
- Favre, C.J.; Mancuso, M.; Maas, K.; McLean, J.W.; Baluk, P.; McDonald, D.M. Expression of genes involved in vascular development and angiogenesis in endothelial cells of adult lung. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1917–H1938. [Google Scholar]
- Rouleau, C.; Menon, K.; Boutin, P.; Guyre, C.; Yoshida, H.; Kataoka, S.; Perricone, M.; Shankara, S.; Frankel, A.E.; Duesbery, N.S.; et al. The systemic administration of lethal toxin achieves a growth delay of human melanoma and neuroblastoma xenografts: Assessment of receptor contribution. Int. J. Oncol. 2008, 32, 739–748. [Google Scholar]
- Kitamura, K.; Kangawa, K.; Kawamoto, M.; Ichiki, Y.; Nakamura, S.; Matsuo, H.; Eto, T. Adrenomedullin: A novel hypotensive peptide isolated from human pheochromocytoma. Biochem. Biophys. Res. Commun. 1993, 192, 553–560. [Google Scholar]
- Mutlu, G.M.; Dumasius, V.; Burhop, J.; McShane, P.J.; Meng, F.J.; Welch, L.; Dumasius, A.; Mohebahmadi, N.; Thakuria, G.; Hardiman, K.; et al. Upregulation of alveolar epithelial active na+ transport is dependent on beta2-adrenergic receptor signaling. Circ. Res. 2004, 94, 1091–1100. [Google Scholar]
- Factor, P.; Adir, Y.; Mutlu, G.M.; Burhop, J.; Dumasius, V. Effects of beta2-adrenergic receptor overexpression on alveolar epithelial active transport. J. Allergy Clin. Immunol. 2002, 110, S242–S246. [Google Scholar]
- Tang, K.M.; Wang, G.R.; Lu, P.; Karas, R.H.; Aronovitz, M.; Heximer, S.P.; Kaltenbronn, K.M.; Blumer, K.J.; Siderovski, D.P.; Zhu, Y.; et al. Regulator of g-protein signaling-2 mediates vascular smooth muscle relaxation and blood pressure. Nat. Med. 2003, 9, 1506–1512. [Google Scholar]
- Kirby, J.E. Anthrax lethal toxin induces human endothelial cell apoptosis. Infect. Immun. 2004, 72, 430–439. [Google Scholar]
- Gozes, Y.; Moayeri, M.; Wiggins, J.F.; Leppla, S.H. Anthrax lethal toxin induces ketotifen-sensitive intradermal vascular leakage in certain inbred mice. Infect. Immun. 2006, 74, 1266–1272. [Google Scholar]
- Okuda, J.; Arikawa, Y.; Takeuchi, Y.; Mahmoud, M.M.; Suzaki, E.; Kataoka, K.; Suzuki, T.; Okinaka, Y.; Nakai, T. Intracellular replication of edwardsiella tarda in murine macrophage is dependent on the type iii secretion system and induces an up-regulation of anti-apoptotic nf-kappab target genes protecting the macrophage from staurosporine-induced apoptosis. Microb. Pathog. 2006, 41, 226–240. [Google Scholar]
- van Hinsbergh, V.W.; van Nieuw Amerongen, G.P. Intracellular signalling involved in modulating human endothelial barrier function. J. Anat. 2002, 200, 549–560. [Google Scholar]
- Arora, N.; Leppla, S.H. Residues 1-254 of anthrax toxin lethal factor are sufficient to cause cellular uptake of fused polypeptides. J. Biol. Chem. 1993, 268, 3334–3341. [Google Scholar]
- Duesbery, N.S.; Resau, J.; Webb, C.P.; Koochekpour, S.; Koo, H.M.; Leppla, S.H.; Vande Woude, G.F. Suppression of ras-mediated transformation and inhibition of tumor growth and angiogenesis by anthrax lethal factor, a proteolytic inhibitor of multiple mek pathways. Proc. Natl. Acad. Sci. USA 2001, 98, 4089–4094. [Google Scholar]
- Alfano, R.W.; Leppla, S.H.; Liu, S.; Bugge, T.H.; Ortiz, J.M.; Lairmore, T.C.; Duesbery, N.S.; Mitchell, I.C.; Nwariaku, F.; Frankel, A.E. Inhibition of tumor angiogenesis by the matrix metalloproteinase-activated anthrax lethal toxin in an orthotopic model of anaplastic thyroid carcinoma. Mol. Cancer Ther. 2010, 9, 190–201. [Google Scholar]
- Pliyev, B.K.; Antonova, O.A.; Menshikov, M. Participation of the urokinase-type plasminogen activator receptor (upar) in neutrophil transendothelial migration. Mol. Immunol. 2011, 48, 1168–1177. [Google Scholar]
- Rijneveld, A.W.; Levi, M.; Florquin, S.; Speelman, P.; Carmeliet, P.; van Der Poll, T. Urokinase receptor is necessary for adequate host defense against pneumococcal pneumonia. J. Immunol. 2002, 168, 3507–3511. [Google Scholar]
- Gyetko, M.R.; Sud, S.; Kendall, T.; Fuller, J.A.; Newstead, M.W.; Standiford, T.J. Urokinase receptor-deficient mice have impaired neutrophil recruitment in response to pulmonary pseudomonas aeruginosa infection. J. Immunol. 2000, 165, 1513–1519. [Google Scholar]
- Ridger, V.C.; Wagner, B.E.; Wallace, W.A.; Hellewell, P.G. Differential effects of cd18, cd29, and cd49 integrin subunit inhibition on neutrophil migration in pulmonary inflammation. J. Immunol. 2001, 166, 3484–3490. [Google Scholar]
- Moen, S.T.; Yeager, L.A.; Lawrence, W.S.; Ponce, C.; Galindo, C.L.; Garner, H.R.; Baze, W.B.; Suarez, G.; Peterson, J.W.; Chopra, A.K. Transcriptional profiling of murine organ genes in response to infection with bacillus anthracis ames spores. Microb. Pathog. 2008, 44, 293–310. [Google Scholar]
- Heximer, S.P.; Knutsen, R.H.; Sun, X.; Kaltenbronn, K.M.; Rhee, M.H.; Peng, N.; Oliveira-dos-Santos, A.; Penninger, J.M.; Muslin, A.J.; Steinberg, T.H.; et al. Hypertension and prolonged vasoconstrictor signaling in rgs2-deficient mice. J. Clin. Invest. 2003, 111, 445–452. [Google Scholar]
- Cui, X.; Li, Y.; Moayeri, M.; Choi, G.H.; Subramanian, G.M.; Li, X.; Haley, M.; Fitz, Y.; Feng, J.; Banks, S.M.; et al. Late treatment with a protective antigen-directed monoclonal antibody improves hemodynamic function and survival in a lethal toxin-infused rat model of anthrax sepsis. J. Infect. Dis. 2005, 191, 422–434. [Google Scholar]
- Siderovski, D.P.; Heximer, S.P.; Forsdyke, D.R. A human gene encoding a putative basic helix-loop-helix phosphoprotein whose mrna increases rapidly in cycloheximide-treated blood mononuclear cells. DNA Cell Biol. 1994, 13, 125–147. [Google Scholar]
- Kato, H.; Shichiri, M.; Marumo, F.; Hirata, Y. Adrenomedullin as an autocrine/paracrine apoptosis survival factor for rat endothelial cells. Endocrinology 1997, 138, 2615–2620. [Google Scholar]
- Di Paola, R.; Talero, E.; Galuppo, M.; Mazzon, E.; Bramanti, P.; Motilva, V.; Cuzzocrea, S. Adrenomedullin in inflammatory process associated with experimental pulmonary fibrosis. Respir. Res. 2011, 12, 41. [Google Scholar]
- Kennedy, S.P.; Sun, D.; Oleynek, J.J.; Hoth, C.F.; Kong, J.; Hill, R.J. Expression of the rat adrenomedullin receptor or a putative human adrenomedullin receptor does not correlate with adrenomedullin binding or functional response. Biochem. Biophys. Res. Commun. 1998, 244, 832–837. [Google Scholar]
- Ramachandran, V.; Arumugam, T.; Langley, R.; Hwang, R.F.; Vivas-Mejia, P.; Sood, A.K.; Lopez-Berestein, G.; Logsdon, C.D. The admr receptor mediates the effects of adrenomedullin on pancreatic cancer cells and on cells of the tumor microenvironment. PLoS One 2009, 4, e7502. [Google Scholar]
- Crandall, E.D.; Heming, T.A.; Palombo, R.L.; Goodman, B.E. Effects of terbutaline on sodium transport in isolated perfused rat lung. J. Appl. Physiol. 1986, 60, 289–294. [Google Scholar]
- Frank, J.A.; Wang, Y.; Osorio, O.; Matthay, M.A. Beta-adrenergic agonist therapy accelerates the resolution of hydrostatic pulmonary edema in sheep and rats. J. Appl. Physiol. 2000, 89, 1255–1265. [Google Scholar]
- Bogatcheva, N.V.; Verin, A.D. The role of cytoskeleton in the regulation of vascular endothelial barrier function. Microvasc. Res. 2008, 76, 202–207. [Google Scholar]
- Friedlander, A.M. Macrophages are sensitive to anthrax lethal toxin through an acid-dependent process. J. Biol. Chem. 1986, 261, 7123–7126. [Google Scholar]
- Friedlander, A.M.; Bhatnagar, R.; Leppla, S.H.; Johnson, L.; Singh, Y. Characterization of macrophage sensitivity and resistance to anthrax lethal toxin. Infect. Immun. 1993, 61, 245–252. [Google Scholar]
- Liu, S.; Miller-Randolph, S.; Crown, D.; Moayeri, M.; Sastalla, I.; Okugawa, S.; Leppla, S.H. Anthrax toxin targeting of myeloid cells through the cmg2 receptor is essential for establishment of bacillus anthracis infections in mice. Cell Host Microbe 2010, 8, 455–462. [Google Scholar]
- Barson, H.V.; Mollenkopf, H.; Kaufmann, S.H.; Rijpkema, S. Anthrax lethal toxin suppresses chemokine production in human neutrophil nb-4 cells. Biochem. Biophys. Res. Commun. 2008, 374, 288–293. [Google Scholar]
- St Croix, B.; Rago, C.; Velculescu, V.; Traverso, G.; Romans, K.E.; Montgomery, E.; Lal, A.; Riggins, G.J.; Lengauer, C.; Vogelstein, B.; et al. Genes expressed in human tumor endothelium. Science 2000, 289, 1197–1202. [Google Scholar]
- Lyubchenko, T.; Woodward, H.; Veo, K.D.; Burns, N.; Nijmeh, H.; Liubchenko, G.A.; Stenmark, K.R.; Gerasimovskaya, E.V. P2y1 and p2y13 purinergic receptors mediate Ca2+ signaling and proliferative responses in pulmonary artery vasa vasorum endothelial cells. Am. J. Physiol. Cell Physiol. 2011, 300, C266–C275. [Google Scholar]
- Malla, R.R.; Gopinath, S.; Gondi, C.S.; Alapati, K.; Dinh, D.H.; Gujrati, M.; Rao, J.S. Cathepsin b and upar knockdown inhibits tumor-induced angiogenesis by modulating vegf expression in glioma. Cancer Gene Ther. 2011, 18, 419–434. [Google Scholar]
- Paccani, S.R.; Tonello, F.; Ghittoni, R.; Natale, M.; Muraro, L.; D’Elios, M.M.; Tang, W.J.; Montecucco, C.; Baldari, C.T. Anthrax toxins suppress t lymphocyte activation by disrupting antigen receptor signaling. J. Exp. Med. 2005, 201, 325–331. [Google Scholar]
- Hirsh, M.I.; Manov, I.; Cohen-Kaplan, V.; Iancu, T.C. Ultrastructural features of lymphocyte suppression induced by anthrax lethal toxin and treated with chloroquine. Lab. Invest. 2007, 87, 182–188. [Google Scholar]
- Guidi-Rontani, C.; Duflot, E.; Mock, M. Anthrax lethal toxin-induced mitogenic response of human t-cells. FEMS Microbiol. Lett. 1997, 157, 285–289. [Google Scholar]
- Rosenstein, M.; Ettinghausen, S.E.; Rosenberg, S.A. Extravasation of intravascular fluid mediated by the systemic administration of recombinant interleukin 2. J. Immunol. 1986, 137, 1735–1742. [Google Scholar]
- Puri, R.K.; Rosenberg, S.A. Combined effects of interferon alpha and interleukin 2 on the induction of a vascular leak syndrome in mice. Cancer Immunol. Immunother. 1989, 28, 267–274. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dumas, E.K.; Cox, P.M.; Fullenwider, C.O.; Nguyen, M.; Centola, M.; Frank, M.B.; Dozmorov, I.; James, J.A.; Farris, A.D. Anthrax Lethal Toxin-Induced Gene Expression Changes in Mouse Lung. Toxins 2011, 3, 1111-1130. https://doi.org/10.3390/toxins3091111
Dumas EK, Cox PM, Fullenwider CO, Nguyen M, Centola M, Frank MB, Dozmorov I, James JA, Farris AD. Anthrax Lethal Toxin-Induced Gene Expression Changes in Mouse Lung. Toxins. 2011; 3(9):1111-1130. https://doi.org/10.3390/toxins3091111
Chicago/Turabian StyleDumas, Eric K., Philip M. Cox, Charles O’Connor Fullenwider, Melissa Nguyen, Michael Centola, Mark Barton Frank, Igor Dozmorov, Judith A. James, and A. Darise Farris. 2011. "Anthrax Lethal Toxin-Induced Gene Expression Changes in Mouse Lung" Toxins 3, no. 9: 1111-1130. https://doi.org/10.3390/toxins3091111