The Interactions of Human Neutrophils with Shiga Toxins and Related Plant Toxins: Danger or Safety?

Abstract
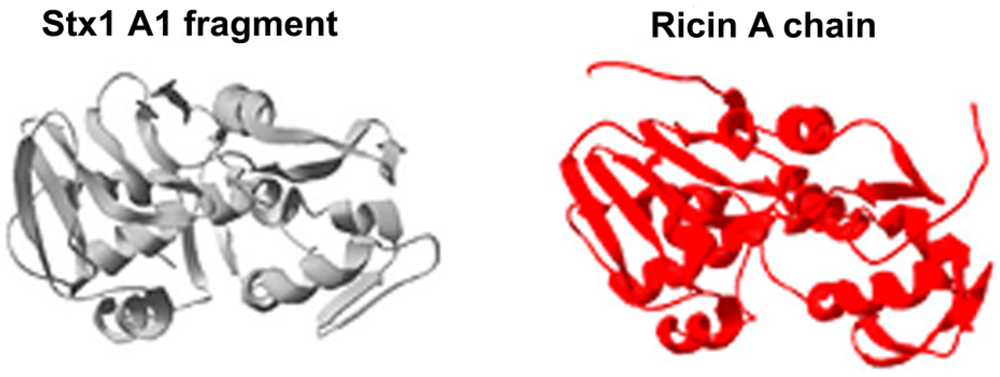
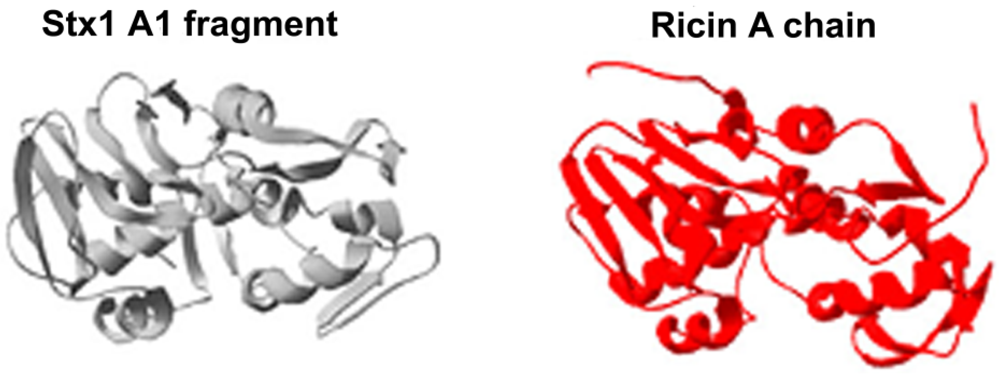
:1. Shiga Toxins and Ricin Are Analogous Toxins
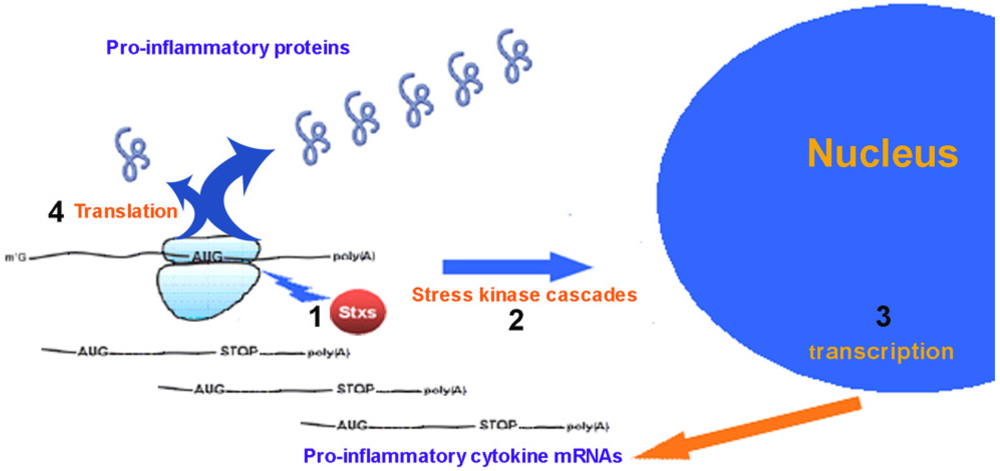
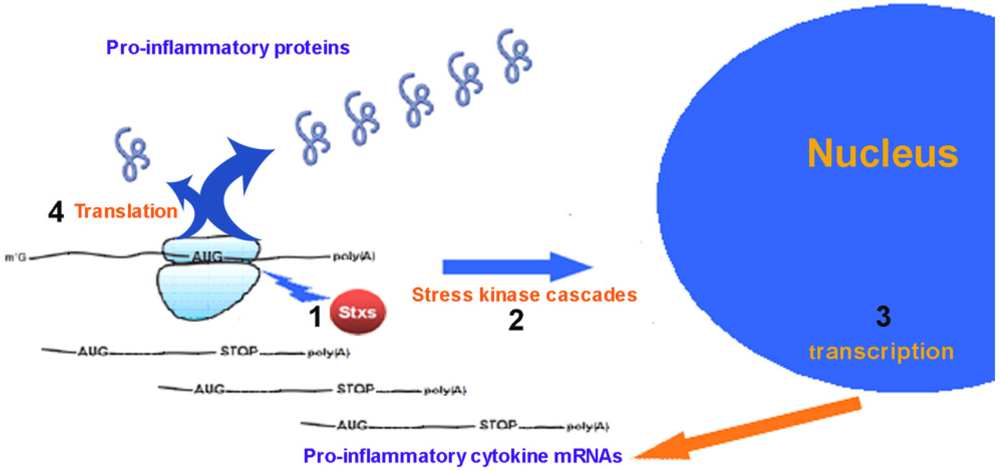
2. Pro-Inflammatory Cytokines Produced by Eukaryotic Cells in Response to Ricin and Stx
3. Role of Stx in the Pathogenesis of HUS
4. Transport of Stx in Blood and the Role of Stx/PMN Interactions

{kind=link}
{kind=link}
| Paper | Toxin | Purification scheme a | LPS b pg/µg | Purification method |
|---|---|---|---|---|
| Positive results | ||||
| Brigotti et al., [112,114] | Stx1 | AGl | <3 | [126] |
| Stx2 | AS, AE, AP1 | <3 | [127] c | |
| Griener et al., [110] | Stx1 | ASy | <0.077 | [128] |
| Stx2 | ASy | <0.077 | [128] | |
| King et al., [116] | Stx1 | AS, AP1 | <3 | [129] |
| Liu et al., [119] | Stx2 | CE, HPLC | n.a. | [130] d |
| rStx2 | Dr. Gondaira (Denka Seiken, Tokyo) | <0.001 | n.d. | |
| Stahl et al., [83] | Stx2 | AP1e | n.a. | [131] |
| Te Loo et al., [99] | Stx1 | Dr. Karmali (Health, Toronto, Canada) | n.d. | n.d. |
| Negative results | ||||
| Aoki et al., [118] | Stx1 | AS, AE, Ch, HPLC | <2500 | [132] |
| Stx2 | AS, AE, Ch, HPLC | <2500 | [133] | |
| Fernandez et al., [105] | Stx1 | Dr. Juniki (Denka Seiken, Nigata, Japan) | <40 | n.d. |
| Stx2 | Dr. Juniki (Denka Seiken, Nigata, Japan) | <40 | n.d. | |
| Flagler et al., [115] | Stx1 | AS, AE, HA, AG, GF | <11 | [115] |
| Stx2 | AS, AE, AP, AG, PS | <11 | [115] | |
| Geelen et al., [109] | Stx1 | Dr. Karmali (Health, Toronto, Canada) | n.d. | n.d. |
| Stx2 | Toxin Technology, Sarasota FL, USA | n.d. | n.d. | |
| Holle et al., [117] | Stx1 | Prof. Lord (Warwick University, UK) AGl f | n.d. | [126] |
| Stx2 | Toxin Technology, Sarasota FL, USA | n.d. | n.d. |
5. PMN Recognize the A Chain of Stx and Related Plant Toxins
6. Transfer of Stx from PMN to Gb3Cer-Expressing Cells
7. Where Do PMN Bind Stx During the Natural Course of Disease?
| Total toxin (nM) | Stx1 a | Ricin A chain a | ||||
|---|---|---|---|---|---|---|
| PMN receptor saturation (%) | Free toxin (%) | Bound toxin (%) | PMN receptor saturation (%) | Free toxin (%) | Bound toxin (%) | |
| 100 | 90.83 | 99.09 | 0.91 | - | - | - |
| 10 | 48.75 | 95.12 | 4.88 | 90.10 | 90.99 | 9.01 |
| 1 | 8.39 | 91.61 | 8.39 | 38.20 | 61.80 | 38.20 |
| 0.1 | 0.90 | 90.98 | 9.02 | 4.88 | 51.25 | 48.75 |
| 0.01 | 0.09 | 90.92 | 9.08 | 0.50 | 50.12 | 49.88 |
| 0.001 | 0.01 | 90.91 | 9.09 | 0.05 | 50.01 | 49.99 |
8. Binding of Stx to PMN in Patients with HUS
| High % Stx-positive PMN (mean ± SD, n = 4) | Low % Stx-positive PMN (mean ± SD, n = 3) | t Test | |
|---|---|---|---|
| Creatinine (µM) | 229 ± 124 | 627 ± 219 | p < 0.05 |
| Hb concentration (M) | 5.0 ± 1.3 | 4.2 ± 1.8 | p = 0.52 |
| Platelets × 109/L | 36.3 ± 14.6 | 64.7 ± 28.2 | p = 0.14 |
9. The Nature of the Stx-Recognizing Receptors on PMN
10. Conclusions
Acknowledgments
Conflict of Interest
References
- Stillmark, H. Ueber ricin, ein giftiges ferment aus den samen von ricinus communis l. Und einigen anderen euphorbiaceen. 1988. [Google Scholar]
- Olsnes, S.; Pihl, A. Different biological properties of the two constituent peptide chains of ricin, a toxic protein inhibiting protein synthesis. Biochemistry 1973, 12, 3121–3126. [Google Scholar] [CrossRef]
- Lin, J.Y.; Liu, K.; Chen, C.C.; Tung, T.C. Effect of crystalline ricin on the biosynthesis of protein, rna, and DNA in experimental tumor cell. Cancer Res. 1971, 31, 921–924. [Google Scholar]
- Olsnes, S.; Pihl, A. Ricin—a potent inhibitor of protein synthesis. FEBS Lett. 1972, 20, 327–329. [Google Scholar] [CrossRef]
- Olsnes, S.; Pihl, A. Treatment of abrin and ricin with -mercaptoethanol opposite effects on their toxicity in mice and their ability to inhibit protein synthesis in a cell-free system. FEBS Lett. 1972, 28, 48–50. [Google Scholar] [CrossRef]
- Montanaro, L.; Sperti, S.; Stirpe, F. Inhibition by ricin of protein synthesis in vitro. Ribosomes as the target of the toxin. Biochem. J. 1973, 136, 677–683. [Google Scholar]
- Sperti, S.; Montanaro, L.; Mattioli, A.; Stirpe, F. Inhibition by ricin of protein synthesis in vitro: 60 s ribosomal subunit as the target of the toxin. Biochem. J. 1973, 136, 813–815. [Google Scholar]
- Carrasco, L.; Fernandez-Puentes, C.; Vazquez, D. Effects of ricin on the ribosomal sites involved in the interaction of the elongation factors. Eur. J. Biochem. 1975, 54, 499–503. [Google Scholar] [CrossRef]
- Montanaro, L.; Sperti, S.; Mattioli, A.; Testoni, G.; Stirpe, F. Inhibition by ricin of protein synthesis in vitro. Inhibition of the binding of elongation factor 2 and of adenosine diphosphate-ribosylated elongation factor 2 to ribosomes. Biochem. J. 1975, 146, 127–131. [Google Scholar]
- Olsnes, S.; Fernandez-Puentes, C.; Carrasco, L.; Vazquez, D. Ribosome inactivation by the toxic lectins abrin and ricin. Kinetics of the enzymic activity of the toxin a-chains. Eur. J. Biochem. 1975, 60, 281–288. [Google Scholar] [CrossRef]
- Endo, Y.; Mitsui, K.; Motizuki, M.; Tsurugi, K. The mechanism of action of ricin and related toxic lectins on eukaryotic ribosomes. The site and the characteristics of the modification in 28 S ribosomal rna caused by the toxins. J. Biol. Chem. 1987, 262, 5908–5912. [Google Scholar]
- Endo, Y.; Tsurugi, K. Rna n-glycosidase activity of ricin a-chain. Mechanism of action of the toxic lectin ricin on eukaryotic ribosomes. J. Biol. Chem. 1987, 262, 8128–8130. [Google Scholar]
- Chan, Y.L.; Endo, Y.; Wool, I.G. The sequence of the nucleotides at the alpha-sarcin cleavage site in rat 28 S ribosomal ribonucleic acid. J. Biol. Chem. 1983, 258, 12768–12770. [Google Scholar]
- Moazed, D.; Robertson, J.M.; Noller, H.F. Interaction of elongation factors ef-g and ef-tu with a conserved loop in 23S RNA. Nature 1988, 334, 362–364. [Google Scholar]
- Stirpe, F.; Battelli, M.G. Ribosome-inactivating proteins: Progress and problems. Cell. Mol. Life. Sci. 2006, 63, 1850–1866. [Google Scholar] [CrossRef]
- McIver, J.; Grady, G.F.; Keusch, G.T. Production and characterization of exotoxin(s) of shigella dysenteriae type 1. J. Infect. Dis. 1975, 131, 559–566. [Google Scholar] [CrossRef]
- Brown, J.E.; Rothman, S.W.; Doctor, B.P. Inhibition of protein synthesis in intact hela cells by shigella dysenteriae 1 toxin. Infect. Immun. 1980, 29, 98–107. [Google Scholar]
- Thompson, M.R.; Steinberg, M.S.; Gemski, P.; Formal, S.B.; Doctor, B.P. Inhibition of in vitro protein synthesis by shigella dysenteriae 1 toxin. Biochem. Biophys. Res. Commun. 1976, 71, 783–788. [Google Scholar] [CrossRef]
- Brown, J.E.; Ussery, M.A.; Leppla, S.H.; Rothman, S.W. Inhibition of protein synthesis by shiga toxin: Activation of the toxin and inhibition of peptide elongation. FEBS Lett. 1980, 117, 84–88. [Google Scholar] [CrossRef]
- Olsnes, S.; Eiklid, K. Isolation and characterization of shigella shigae cytotoxin. J. Biol. Chem. 1980, 255, 284–289. [Google Scholar]
- Reisbig, R.; Olsnes, S.; Eiklid, K. The cytotoxic activity of shigella toxin. Evidence for catalytic inactivation of the 60 s ribosomal subunit. J. Biol. Chem. 1981, 256, 8739–8744. [Google Scholar]
- Endo, Y.; Tsurugi, K.; Yutsudo, T.; Takeda, Y.; Ogasawara, T.; Igarashi, K. Site of action of a vero toxin (vt2) from Escherichia coli o157:H7 and of shiga toxin on eukaryotic ribosomes. RNA n-glycosidase activity of the toxins. Eur. J. Biochem. 1988, 171, 45–50. [Google Scholar] [CrossRef]
- Barbieri, L.; Valbonesi, P.; Brigotti, M.; Montanaro, L.; Stirpe, F.; Sperti, S. Shiga-like toxin i is a polynucleotide:Adenosine glycosidase. Mol. Microbiol. 1998, 29, 661–662. [Google Scholar] [CrossRef]
- Barbieri, L.; Valbonesi, P.; Bonora, E.; Gorini, P.; Bolognesi, A.; Stirpe, F. Polynucleotide: Adenosine glycosidase activity of ribosome-inactivating proteins: Effect on DNA, rna and poly(a). Nucleic. Acids. Res. 1997, 25, 518–522. [Google Scholar]
- Barbieri, L.; Gorini, P.; Valbonesi, P.; Castiglioni, P.; Stirpe, F. Unexpected activity of saporins. Nature 1994, 372, 624. [Google Scholar]
- Brigotti, M.; Alfieri, R.; Sestili, P.; Bonelli, M.; Petronini, P.G.; Guidarelli, A.; Barbieri, L.; Stirpe, F.; Sperti, S. Damage to nuclear DNA induced by shiga toxin 1 and ricin in human endothelial cells. FASEB J. 2002, 16, 365–372. [Google Scholar]
- Brigotti, M.; Carnicelli, D.; Ravanelli, E.; Vara, A.G.; Martinelli, C.; Alfieri, R.R.; Petronini, P.G.; Sestili, P. Molecular damage and induction of proinflammatory cytokines in human endothelial cells exposed to shiga toxin 1, shiga toxin 2, and alpha-sarcin. Infect. Immun. 2007, 75, 2201–2207. [Google Scholar]
- Fraser, M.E.; Chernaia, M.M.; Kozlov, Y.V.; James, M.N. Crystal structure of the holotoxin from shigella dysenteriae at 2.5 a resolution. Nat. Struct. Biol. 1994, 1, 59–64. [Google Scholar] [CrossRef]
- Katzin, B.J.; Collins, E.J.; Robertus, J.D. Structure of ricin a-chain at 2.5 a. Proteins 1991, 10, 251–259. [Google Scholar] [CrossRef]
- McCluskey, A.J.; Poon, G.M.; Bolewska-Pedyczak, E.; Srikumar, T.; Jeram, S.M.; Raught, B.; Gariepy, J. The catalytic subunit of shiga-like toxin 1 interacts with ribosomal stalk proteins and is inhibited by their conserved c-terminal domain. J. Mol. Biol. 2008, 378, 375–386. [Google Scholar]
- Monzingo, A.F.; Robertus, J.D. X-ray analysis of substrate analogs in the ricin a-chain active site. J. Mol. Biol. 1992, 227, 1136–1145. [Google Scholar]
- Fraser, M.E.; Cherney, M.M.; Marcato, P.; Mulvey, G.L.; Armstrong, G.D.; James, M.N. Binding of adenine to stx2, the protein toxin from Escherichia coli o157:H7. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006, 62, 627–630. [Google Scholar] [CrossRef]
- Yan, X.; Day, P.; Hollis, T.; Monzingo, A.F.; Schelp, E.; Robertus, J.D.; Milne, G.W.; Wang, S. Recognition and interaction of small rings with the ricin a-chain binding site. Proteins 1998, 31, 33–41. [Google Scholar]
- Licastro, F.; Morini, M.C.; Bolognesi, A.; Stirpe, F. Ricin induces the production of tumour necrosis factor-alpha and interleukin-1 beta by human peripheral-blood mononuclear cells. Biochem. J. 1993, 294, 517–520. [Google Scholar]
- Thorpe, C.M.; Hurley, B.P.; Lincicome, L.L.; Jacewicz, M.S.; Keusch, G.T.; Acheson, D.W. Shiga toxins stimulate secretion of interleukin-8 from intestinal epithelial cells. Infect. Immun. 1999, 67, 5985–5993. [Google Scholar]
- Gonzalez, T.V.; Farrant, S.A.; Mantis, N.J. Ricin induces il-8 secretion from human monocyte/macrophages by activating the p38 map kinase pathway. Mol. Immunol. 2006, 43, 1920–1923. [Google Scholar] [CrossRef]
- Yamasaki, C.; Nishikawa, K.; Zeng, X.T.; Katayama, Y.; Natori, Y.; Komatsu, N.; Oda, T. Induction of cytokines by toxins that have an identical rna n-glycosidase activity: Shiga toxin, ricin, and modeccin. Biochim. Biophys. Acta 1671, 44–50. [Google Scholar]
- Iordanov, M.S.; Pribnow, D.; Magun, J.L.; Dinh, T.H.; Pearson, J.A.; Magun, B.E. Ultraviolet radiation triggers the ribotoxic stress response in mammalian cells. J. Biol. Chem. 1998, 273, 15794–15803. [Google Scholar]
- Iordanov, M.S.; Pribnow, D.; Magun, J.L.; Dinh, T.H.; Pearson, J.A.; Chen, S.L.; Magun, B.E. Ribotoxic stress response: Activation of the stress-activated protein kinase jnk1 by inhibitors of the peptidyl transferase reaction and by sequence-specific rna damage to the alpha-sarcin/ricin loop in the 28s rRNA. Mol. Cell. Biol. 1997, 17, 3373–3381. [Google Scholar]
- Korcheva, V.; Wong, J.; Corless, C.; Iordanov, M.; Magun, B. Administration of ricin induces a severe inflammatory response via nonredundant stimulation of erk, jnk, and p38 mapk and provides a mouse model of hemolytic uremic syndrom. Am. J. Pathol. 2005, 166, 323–339. [Google Scholar] [CrossRef]
- Van Setten, P.A.; Monnens, L.A.; Verstraten, R.G.; Van den Heuvel, L.P.; Van Hinsbergh, V.W. Effects of verocytotoxin-1 on nonadherent human monocytes: Binding characteristics, protein synthesis, and induction of cytokine releas. Blood 1996, 88, 174–183. [Google Scholar]
- Zoja, C.; Angioletti, S.; Donadelli, R.; Zanchi, C.; Tomasoni, S.; Binda, E.; Imberti, B.; Te Loo, M.; Monnens, L.; Remuzzi, G.; et al. Shiga toxin-2 triggers endothelial leukocyte adhesion and transmigration via nf-kappab dependent up-regulation of il-8 and mcp-1. Kidney Int. 2002, 62, 846–856. [Google Scholar] [CrossRef]
- Ramegowda, B.; Tesh, V.L. Differentiation-associated toxin receptor modulation, cytokine production, and sensitivity to shiga-like toxins in human monocytes and monocytic cell line. Infect. Immun. 1996, 64, 1173–1180. [Google Scholar]
- Noris, M.; Remuzzi, G. Hemolytic uremic syndrome. J. Am. Soc. Nephrol. 2005, 16, 1035–1050. [Google Scholar] [CrossRef]
- Morigi, M.; Galbusera, M.; Binda, E.; Imberti, B.; Gastoldi, S.; Remuzzi, A.; Zoja, C.; Remuzzi, G. Verotoxin-1-induced up-regulation of adhesive molecules renders microvascular endothelial cells thrombogenic at high shear stress. Blood 2001, 98, 1828–1835. [Google Scholar]
- Matussek, A.; Lauber, J.; Bergau, A.; Hansen, W.; Rohde, M.; Dittmar, K.E.; Gunzer, M.; Mengel, M.; Gatzlaff, P.; Hartmann, M.; et al. Molecular and functional analysis of shiga toxin-induced response patterns in human vascular endothelial cells. Blood 2003, 102, 1323–1332. [Google Scholar]
- Persson, S.; Olsen, K.E.; Ethelberg, S.; Scheutz, F. Subtyping method for Escherichia coli shiga toxin (verocytotoxin) 2 variants and correlations to clinical manifestations. J. Clin. Microbiol. 2007, 45, 2020–2024. [Google Scholar] [CrossRef]
- Friedrich, A.W.; Bielaszewska, M.; Zhang, W.L.; Pulz, M.; Kuczius, T.; Ammon, A.; Karch, H. Escherichia coli harboring shiga toxin 2 gene variants: Frequency and association with clinical symptoms. J. Infect. Dis. 2002, 185, 74–84. [Google Scholar] [CrossRef]
- Yamasaki, C.; Natori, Y.; Zeng, X.T.; Ohmura, M.; Yamasaki, S.; Takeda, Y. Induction of cytokines in a human colon epithelial cell line by shiga toxin 1 (stx1) and stx2 but not by non-toxic mutant stx1 which lacks n-glycosidase activity. FEBS Lett. 1999, 442, 231–234. [Google Scholar] [CrossRef]
- Sakiri, R.; Ramegowda, B.; Tesh, V.L. Shiga toxin type 1 activates tumor necrosis factor-alpha gene transcription and nuclear translocation of the transcriptional activators nuclear factor-kappab and activator protein-1. Blood 1998, 92, 558–566. [Google Scholar]
- Smith, W.E.; Kane, A.V.; Campbell, S.T.; Acheson, D.W.; Cochran, B.H.; Thorpe, C.M. Shiga toxin 1 triggers a ribotoxic stress response leading to p38 and jnk activation and induction of apoptosis in intestinal epithelial cells. Infect. Immun. 2003, 71, 1497–1504. [Google Scholar]
- Karmali, M.A.; Petric, M.; Lim, C.; Fleming, P.C.; Arbus, G.S.; Lior, H. The association between idiopathic hemolytic uremic syndrome and infection by verotoxin-producing Escherichia coli. J. Infect. Dis. 1985, 151, 775–782. [Google Scholar]
- Griffin, P.M.; Tauxe, R.V. The epidemiology of infections caused by Escherichia coli o157:H7, other enterohemorrhagic E. coli, and the associated hemolytic uremic syndrome. Epidemiol. Rev. 1991, 13, 60–98. [Google Scholar]
- Trompeter, R.S.; Schwartz, R.; Chantler, C.; Dillon, M.J.; Haycock, G.B.; Kay, R.; Barratt, T.M. Haemolytic-uraemic syndrome: An analysis of prognostic features. Arch. Dis. Child. 1983, 58, 101–105. [Google Scholar] [CrossRef]
- Tozzi, A.E.; Caprioli, A.; Minelli, F.; Gianviti, A.; De Petris, L.; Edefonti, A.; Montini, G.; Ferretti, A.; De Palo, T.; Gaido, M.; et al. Shiga toxin-producing Escherichia coli infections associated with hemolytic uremic syndrome, Italy, 1988-2000. Emerg. Infect. Dis. 2003, 9, 106–108. [Google Scholar]
- Paton, J.C.; Paton, A.W. Pathogenesis and diagnosis of shiga toxin-producing Escherichia coli infections. Clin. Microbiol. Rev. 1998, 11, 450–479. [Google Scholar]
- Tarr, P.I.; Gordon, C.A.; Chandler, W.L. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 2005, 365, 1073–1086. [Google Scholar]
- O'Loughlin, E.V.; Robins-Browne, R.M. Effect of shiga toxin and shiga-like toxins on eukaryotic cells. Microbes. Infect. 2001, 3, 493–507. [Google Scholar]
- Strockbine, N.A.; Jackson, M.P.; Sung, L.M.; Holmes, R.K.; O’Brien, A.D. Cloning and sequencing of the genes for shiga toxin from shigella dysenteriae type 1. J. Bacteriol. 1988, 170, 1116–1122. [Google Scholar]
- Zoja, C.; Buelli, S.; Morigi, M. Shiga toxin-associated hemolytic uremic syndrome: Pathophysiology of endothelial dysfunction. Pediatr. Nephrol. 2010, 25, 2231–2240. [Google Scholar]
- Bielaszewska, M.; Karch, H. Consequences of enterohaemorrhagic Escherichia coli infection for the vascular endothelium. Thromb. Haemost. 2005, 94, 312–318. [Google Scholar]
- Nakao, H.; Takeda, T. Escherichia coli shiga toxin. J. Nat. Toxins 2000, 9, 299–313. [Google Scholar]
- Tesh, V.L. Induction of apoptosis by shiga toxins. Future Microbiol. 2010, 5, 431–453. [Google Scholar]
- Tesh, V.L. The induction of apoptosis by shiga toxins and ricin. Curr. Top. Microbiol. Immunol. 2012, 357, 137–178. [Google Scholar]
- Nataro, J.P.; Kaper, J.B. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 1998, 11, 142–201. [Google Scholar]
- Frankel, G.; Phillips, A.D. Attaching effacing Escherichia coli and paradigms of tir-triggered actin polymerization: Getting off the pedestal. Cell Microbiol. 2008, 10, 549–556. [Google Scholar]
- Karmali, M.A. Host and pathogen determinants of verocytotoxin-producing Escherichia coli-associated hemolytic uremic syndrome. Kidney Int. Suppl. 2009, 75, S4–S7. [Google Scholar]
- Los, J.M.; Los, M.; Wegrzyn, G. Bacteriophages carrying shiga toxin genes: Genomic variations, detection and potential treatment of pathogenic bacteria. Future Microbiol. 2011, 6, 909–924. [Google Scholar]
- Zumbrun, S.D.; Hanson, L.; Sinclair, J.F.; Freedy, J.; Melton-Celsa, A.R.; Rodriguez-Canales, J.; Hanson, J.C.; O’Brien, A.D. Human intestinal tissue and cultured colonic cells contain globotriaosylceramide synthase mrna and the alternate shiga toxin receptor globotetraosylceramide. Infect. Immun. 2010, 78, 4488–4499. [Google Scholar]
- Schuller, S. Shiga toxin interaction with human intestinal epithelium. Toxins 2011, 3, 626–639. [Google Scholar]
- Engedal, N.; Skotland, T.; Torgersen, M.L.; Sandvig, K. Shiga toxin and its use in targeted cancer therapy and imaging. Microb. Biotechnol. 2011, 4, 32–46. [Google Scholar]
- Distler, U.; Souady, J.; Hulsewig, M.; Drmic-Hofman, I.; Haier, J.; Friedrich, A.W.; Karch, H.; Senninger, N.; Dreisewerd, K.; Berkenkamp, S.; et al. Shiga toxin receptor gb3cer/cd77: Tumor-association and promising therapeutic target in pancreas and colon cancer. PLoS One 2009, 4, E6813. [Google Scholar]
- Muthing, J.; Schweppe, C.H.; Karch, H.; Friedrich, A.W. Shiga toxins, glycosphingolipid diversity, and endothelial cell injury. Thromb. Haemost. 2009, 101, 252–264. [Google Scholar]
- Malyukova, I.; Murray, K.F.; Zhu, C.; Boedeker, E.; Kane, A.; Patterson, K.; Peterson, J.R.; Donowitz, M.; Kovbasnjuk, O. Macropinocytosis in shiga toxin 1 uptake by human intestinal epithelial cells and transcellular transcytosis. Am. J. Physiol. Gastrointest. Liver. Physiol. 2009, 296, G78–G92. [Google Scholar]
- Acheson, D.W.; Moore, R.; De Breucker, S.; Lincicome, L.; Jacewicz, M.; Skutelsky, E.; Keusch, G.T. Translocation of shiga toxin across polarized intestinal cells in tissue culture. Infect. Immun. 1996, 64, 3294–3300. [Google Scholar]
- Hurley, B.P.; Thorpe, C.M.; Acheson, D.W. Shiga toxin translocation across intestinal epithelial cells is enhanced by neutrophil transmigration. Infect. Immun. 2001, 69, 6148–6155. [Google Scholar]
- Richardson, S.E.; Karmali, M.A.; Becker, L.E.; Smith, C.R. The histopathology of the hemolytic uremic syndrome associated with verocytotoxin-producing Escherichia coli infections. Hum. Pathol. 1988, 19, 1102–1108. [Google Scholar]
- Ray, P.E.; Liu, X.H. Pathogenesis of shiga toxin-induced hemolytic uremic syndrome. Pediatr. Nephrol. 2001, 16, 823–839. [Google Scholar]
- Inward, C.D.; Howie, A.J.; Fitzpatrick, M.M.; Rafaat, F.; Milford, D.V.; Taylor, C.M. Renal histopathology in fatal cases of diarrhoea-associated haemolytic uraemic syndrome. British association for paediatric nephrology. Pediatr. Nephrol. 1997, 11, 556–559. [Google Scholar] [CrossRef]
- Proulx, F.; Seidman, E.G.; Karpman, D. Pathogenesis of shiga toxin-associated hemolytic uremic syndrome. Pediatr. Res. 2001, 50, 163–171. [Google Scholar]
- Ghosh, S.A.; Polanowska-Grabowska, R.K.; Fujii, J.; Obrig, T.; Gear, A.R. Shiga toxin binds to activated platelets. J. Thromb. Haemost. 2004, 2, 499–506. [Google Scholar]
- Karpman, D.; Papadopoulou, D.; Nilsson, K.; Sjogren, A.C.; Mikaelsson, C.; Lethagen, S. Platelet activation by shiga toxin and circulatory factors as a pathogenetic mechanism in the hemolytic uremic syndrome. Blood 2001, 97, 3100–3108. [Google Scholar]
- Stahl, A.L.; Sartz, L.; Nelsson, A.; Bekassy, Z.D.; Karpman, D. Shiga toxin and lipopolysaccharide induce platelet-leukocyte aggregates and tissue factor release, a thrombotic mechanism in hemolytic uremic syndrome. PLoS One 2009, 4, e6990. [Google Scholar]
- Stahl, A.L.; Sartz, L.; Karpman, D. Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia coli-induced hemolytic uremic syndrome. Blood 2011, 117, 5503–5513. [Google Scholar]
- Caprioli, A.; Morabito, S.; Scheutz, F.; Chart, H.; Oswald, E.; Brigotti, M.; Monnens, L.; Aspan, A.; La Ragione, R.; Low, C.; Newell, D. Pathogenesis of verocytotoxin/shiga toxins-producing Escherichia coli infection. In Proceedings of Emerging Infectious Diseases, Rome, Italy, December 2006.
- Taylor, C.M.; Williams, J.M.; Lote, C.J.; Howie, A.J.; Thewles, A.; Wood, J.A.; Milford, D.V.; Raafat, F.; Chant, I.; Rose, P.E. A laboratory model of toxin-induced hemolytic uremic syndrome. Kidney Int. 1999, 55, 1367–1374. [Google Scholar]
- Fu, X.J.; Iijima, K.; Nozu, K.; Hamahira, K.; Tanaka, R.; Oda, T.; Yoshikawa, N.; Matsuo, M. Role of p38 map kinase pathway in a toxin-induced model of hemolytic uremic syndrome. Pediatr. Nephrol. 2004, 19, 844–852. [Google Scholar]
- Messmann, R.A.; Vitetta, E.S.; Headlee, D.; Senderowicz, A.M.; Figg, W.D.; Schindler, J.; Michiel, D.F.; Creekmore, S.; Steinberg, S.M.; Kohler, D.; et al. A phase i study of combination therapy with immunotoxins igg-hd37-deglycosylated ricin a chain (dga) and igg-rfb4-dga (combotox) in patients with refractory cd19(+), cd22(+) b cell lymphoma. Clin. Cancer. Res. 2000, 6, 1302–1313. [Google Scholar]
- Caprioli, A.; Luzzi, I.; Rosmini, F.; Pasquini, P.; Cirrincione, R.; Gianviti, A.; Matteucci, M.C.; Rizzoni, G. Hemolytic-uremic syndrome and vero cytotoxin-producing Escherichia coli infection in italy. The hus italian study group. J. Infect. Dis. 1992, 166, 154–158. [Google Scholar]
- Brigotti, M.; Tazzari, P.L.; Ravanelli, E.; Carnicelli, D.; Rocchi, L.; Arfilli, V.; Scavia, G.; Minelli, F.; Ricci, F.; Pagliaro, P.; et al. Clinical relevance of shiga toxin concentrations in the blood of patients with hemolytic uremic syndrome. Pediatr. Infect. Dis. J. 2011, 30, 486–490. [Google Scholar]
- Bitzan, M.; Richardson, S.; Huang, C.; Boyd, B.; Petric, M.; Karmali, M.A. Evidence that verotoxins (shiga-like toxins) from Escherichia coli bind to p blood group antigens of human erythrocytes in vitro. Infect. Immun. 1994, 62, 3337–3347. [Google Scholar]
- Geelen, J.M.; Van der Velden, T.J.; Van den Heuvel, L.P.; Monnens, L.A. Interactions of shiga-like toxin with human peripheral blood monocytes. Pediatr. Nephrol. 2007, 22, 1181–1187. [Google Scholar]
- Cooling, L.L.; Walker, K.E.; Gille, T.; Koerner, T.A. Shiga toxin binds human platelets via globotriaosylceramide (pk antigen) and a novel platelet glycosphingolipid. Infect. Immun. 1998, 66, 4355–4366. [Google Scholar]
- Cohen, A.; Madrid-Marina, V.; Estrov, Z.; Freedman, M.H.; Lingwood, C.A.; Dosch, H.M. Expression of glycolipid receptors to shiga-like toxin on human b lymphocytes: A mechanism for the failure of long-lived antibody response to dysenteric disease. Int. Immunol. 1990, 2, 1–8. [Google Scholar]
- Obrig, T.G.; Louise, C.B.; Lingwood, C.A.; Boyd, B.; Barley-Maloney, L.; Daniel, T.O. Endothelial heterogeneity in shiga toxin receptors and responses. J. Biol. Chem. 1993, 268, 15484–15488. [Google Scholar]
- Louise, C.B.; Obrig, T.G. Shiga toxin-associated hemolytic-uremic syndrome: Combined cytotoxic effects of shiga toxin, interleukin-1 beta, and tumor necrosis factor alpha on human vascular endothelial cells in vitro. Infect. Immun. 1991, 59, 4173–4179. [Google Scholar]
- Van de Kar, N.C.; Monnens, L.A.; Karmali, M.A.; Van Hinsbergh, V.W. Tumor necrosis factor and interleukin-1 induce expression of the verocytotoxin receptor globotriaosylceramide on human endothelial cells: Implications for the pathogenesis of the hemolytic uremic syndrome. Blood 1992, 80, 2755–2764. [Google Scholar]
- Gariepy, J. The use of shiga-like toxin 1 in cancer therapy. Crit. Rev. Oncol. Hematol. 2001, 39, 99–106. [Google Scholar]
- Te Loo, D.M.; Monnens, L.A.; Van Der Velden, T.J.; Vermeer, M.A.; Preyers, F.; Demacker, P.N.; Van Den Heuvel, L.P.; Van Hinsbergh, V.W. Binding and transfer of verocytotoxin by polymorphonuclear leukocytes in hemolytic uremic syndrome. Blood 2000, 95, 3396–3402. [Google Scholar]
- Walters, M.D.; Matthei, I.U.; Kay, R.; Dillon, M.J.; Barratt, T.M. The polymorphonuclear leucocyte count in childhood haemolytic uraemic syndrome. Pediatr. Nephrol. 1989, 3, 130–134. [Google Scholar]
- Coad, N.A.; Marshall, T.; Rowe, B.; Taylor, C.M. Changes in the postenteropathic form of the hemolytic uremic syndrome in children. Clin. Nephrol. 1991, 35, 10–16. [Google Scholar]
- Milford, D.V.; Taylor, C.M.; Guttridge, B.; Hall, S.M.; Rowe, B.; Kleanthous, H. Haemolytic uraemic syndromes in the british isles 1985-8: Association with verocytotoxin producing Escherichia coli. Part 1: Clinical and epidemiological aspects. Arch. Dis. Child. 1990, 65, 716–721. [Google Scholar] [CrossRef]
- Fitzpatrick, M.M.; Shah, V.; Filler, G.; Dillon, M.J.; Barratt, T.M. Neutrophil activation in the haemolytic uraemic syndrome: Free and complexed elastase in plasma. Pediatr. Nephrol. 1992, 6, 50–53. [Google Scholar]
- Fernandez, G.C.; Gomez, S.A.; Ramos, M.V.; Bentancor, L.V.; Fernandez-Brando, R.J.; Landoni, V.I.; Lopez, L.; Ramirez, F.; Diaz, M.; Alduncin, M.; et al. The functional state of neutrophils correlates with the severity of renal dysfunction in children with hemolytic uremic syndrome. Pediatr. Res. 2007, 61, 123–128. [Google Scholar]
- Fernandez, G.C.; Gomez, S.A.; Rubel, C.J.; Bentancor, L.V.; Barrionuevo, P.; Alduncin, M.; Grimoldi, I.; Exeni, R.; Isturiz, M.A.; Palermo, M.S. Impaired neutrophils in children with the typical form of hemolytic uremic syndrome. Pediatr. Nephrol. 2005, 20, 1306–1314. [Google Scholar]
- Fernandez, G.C.; Rubel, C.; Barrionuevo, P.; Lopez, L.; Ramirez, F.; Diaz, M.; Isturiz, M.A.; Palermo, M.S. Phenotype markers and function of neutrophils in children with hemolytic uremic syndrome. Pediatr. Nephrol. 2002, 17, 337–344. [Google Scholar]
- Fitzpatrick, M.M.; Shah, V.; Trompeter, R.S.; Dillon, M.J.; Barratt, T.M. Interleukin-8 and polymorphoneutrophil leucocyte activation in hemolytic uremic syndrome of childhood. Kidney Int. 1992, 42, 951–956. [Google Scholar]
- Forsyth, K.D.; Simpson, A.C.; Fitzpatrick, M.M.; Barratt, T.M.; Levinsky, R.J. Neutrophil-mediated endothelial injury in haemolytic uraemic syndrome. Lancet 1989, 2, 411–414. [Google Scholar]
- Geelen, J.M.; Van der Velden, T.J.; Te Loo, D.M.; Boerman, O.C.; Van den Heuvel, L.P.; Monnens, L.A. Lack of specific binding of shiga-like toxin (verocytotoxin) and non-specific interaction of shiga-like toxin 2 antibody with human polymorphonuclear leucocytes. Nephrol. Dial. Transplant. 2007, 22, 749–755. [Google Scholar]
- Griener, T.P.; Mulvey, G.L.; Marcato, P.; Armstrong, G.D. Differential binding of shiga toxin 2 to human and murine neutrophils. J. Med. Microbiol. 2007, 56, 1423–1430. [Google Scholar]
- Tazzari, P.L.; Ricci, F.; Carnicelli, D.; Caprioli, A.; Tozzi, A.E.; Rizzoni, G.; Conte, R.; Brigotti, M. Flow cytometry detection of shiga toxins in the blood from children with hemolytic uremic syndrome. Cytometry. B. Clin. Cytom. 2004, 61, 40–44. [Google Scholar]
- Brigotti, M.; Carnicelli, D.; Ravanelli, E.; Barbieri, S.; Ricci, F.; Bontadini, A.; Tozzi, A.E.; Scavia, G.; Caprioli, A.; Tazzari, P.L. Interactions between shiga toxins and human polymorphonuclear leukocytes. J. Leukoc. Biol. 2008, 84, 1019–1027. [Google Scholar]
- Arfilli, V.; Carnicelli, D.; Rocchi, L.; Ricci, F.; Pagliaro, P.; Tazzari, P.L.; Brigotti, M. Shiga toxin 1 and ricin a chain bind to human polymorphonuclear leucocytes through a common receptor. Biochem. J. 2010, 432, 173–180. [Google Scholar]
- Brigotti, M.; Tazzari, P.L.; Ravanelli, E.; Carnicelli, D.; Barbieri, S.; Rocchi, L.; Arfilli, V.; Scavia, G.; Ricci, F.; Bontadini, A.; et al. Endothelial damage induced by shiga toxins delivered by neutrophils during transmigration. J. Leukoc. Biol. 2010, 88, 201–210. [Google Scholar]
- Flagler, M.J.; Strasser, J.E.; Chalk, C.L.; Weiss, A.A. Comparative analysis of the abilities of shiga toxins 1 and 2 to bind to and influence neutrophil apoptosis. Infect. Immun. 2007, 75, 760–765. [Google Scholar]
- King, A.J.; Sundaram, S.; Cendoroglo, M.; Acheson, D.W.; Keusch, G.T. Shiga toxin induces superoxide production in polymorphonuclear cells with subsequent impairment of phagocytosis and responsiveness to phorbol esters. J. Infect. Dis. 1999, 179, 503–507. [Google Scholar]
- Holle, J.U.; Williams, J.M.; Harper, L.; Savage, C.O.; Taylor, C.M. Effect of verocytotoxins (shiga-like toxins) on human neutrophils in vitro. Pediatr. Nephrol. 2005, 20, 1237–1244. [Google Scholar]
- Aoki, Y.; Takeda, T. Effect of shiga toxins on granulocyte function. Microb. Pathog. 2002, 32, 279–285. [Google Scholar]
- Liu, J.; Akahoshi, T.; Sasahana, T.; Kitasato, H.; Namai, R.; Sasaki, T.; Inoue, M.; Kondo, H. Inhibition of neutrophil apoptosis by verotoxin 2 derived from Escherichia coli o157:H7. Infect. Immun. 1999, 67, 6203–6205. [Google Scholar]
- Te Loo, D.M.; Van Hinsbergh, V.W.; Van den Heuvel, L.P.; Monnens, L.A. Detection of verocytotoxin bound to circulating polymorphonuclear leukocytes of patients with hemolytic uremic syndrome. J. Am. Soc. Nephrol. 2001, 12, 800–806. [Google Scholar]
- Brigotti, M.; Caprioli, A.; Tozzi, A.E.; Tazzari, P.L.; Ricci, F.; Conte, R.; Carnicelli, D.; Procaccino, M.A.; Minelli, F.; Ferretti, A.V.; et al. Shiga toxins present in the gut and in the polymorphonuclear leukocytes circulating in the blood of children with hemolytic-uremic syndrome. J. Clin. Microbiol. 2006, 44, 313–317. [Google Scholar]
- Athens, J.W. Granulocyte kinetics in health and disease. Natl. Cancer Inst. Monogr. 1969, 30, 135–155. [Google Scholar]
- Ivan, E.; Colovai, A.I. Human fc receptors: Critical targets in the treatment of autoimmune diseases and transplant rejections. Hum. Immunol. 2006, 67, 479–491. [Google Scholar]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar]
- Van der Poel, C.E.; Spaapen, R.M.; Van de Winkel, J.G.; Leusen, J.H. Functional characteristics of the high affinity igg receptor, fcgammari. J. Immunol. 2011, 186, 2699–2704. [Google Scholar]
- Ryd, M.; Alfredsson, H.; Blomberg, L.; Andersson, A.; Lindberg, A.A. Purification of shiga toxin by alpha-D-galactose-(1→4)-beta-D-galactose-(1→4)-beta-D-glucose-(1→) receptor ligand-based chromatography. FEBS Lett. 1989, 258, 320–322. [Google Scholar]
- Downes, F.P.; Barrett, T.J.; Green, J.H.; Aloisio, C.H.; Spika, J.S.; Strockbine, N.A.; Wachsmuth, I.K. Affinity purification and characterization of shiga-like toxin ii and production of toxin-specific monoclonal antibodies. Infect. Immun. 1988, 56, 1926–1933. [Google Scholar]
- Mulvey, G.; Vanmaele, R.; Mrazek, M.; Cahill, M.; Armstrong, G.D. Affinity purification of shiga-like toxin i and shiga-like toxin ii. J. Microbiol.Meth. 1998, 32, 247–252. [Google Scholar]
- Donohue-Rolfe, A.; Acheson, D.W.; Kane, A.V.; Keusch, G.T. Purification of shiga toxin and shiga-like toxins i and ii by receptor analog affinity chromatography with immobilized p1 glycoprotein and production of cross-reactive monoclonal antibodies. Infect. Immun. 1989, 57, 3888–3893. [Google Scholar]
- MacLeod, D.L.; Gyles, C.L. Purification and characterization of an Escherichia coli shiga-like toxin ii variant. Infect. Immun. 1990, 58, 1232–1239. [Google Scholar]
- Acheson, D.W.; Jacewicz, M.; Kane, A.V.; Donohue-Rolfe, A.; Keusch, G.T. One step high yield affinity purification of shiga-like toxin ii variants and quantitation using enzyme linked immunosorbent assays. Microb. Pathog. 1993, 14, 57–66. [Google Scholar]
- Noda, M.; Yutsudo, T.; Nakabayashi, N.; Hirayama, T.; Takeda, Y. Purification and some properties of shiga-like toxin from Escherichia coli o157:H7 that is immunologically identical to shiga toxin. Microb. Pathog. 1987, 2, 339–349. [Google Scholar]
- Yutsudo, T.; Nakabayashi, N.; Hirayama, T.; Takeda, Y. Purification and some properties of a vero toxin from Escherichia coli o157:H7 that is immunologically unrelated to shiga toxin. Microb. Pathog. 1987, 3, 21–30. [Google Scholar]
- Brigotti, M.; Carnicelli, D.; Arfilli, V.; Rocchi, L.; Ricci, F.; Pagliaro, P.; Tazzari, P.L.; Vara, A.G.; Amelia, M.; Manoli, F.; et al. Change in conformation with reduction of alpha-helix content causes loss of neutrophil binding activity in fully cytotoxic shiga toxin 1. J. Biol. Chem. 2011, 286, 34514–34521. [Google Scholar]
- Mulvey, G.L.; Marcato, P.; Kitov, P.I.; Sadowska, J.; Bundle, D.R.; Armstrong, G.D. Assessment in mice of the therapeutic potential of tailored, multivalent shiga toxin carbohydrate ligands. J. Infect. Dis. 2003, 187, 640–649. [Google Scholar]
- Marcato, P.; Vander Helm, K.; Mulvey, G.L.; Armstrong, G.D. Serum amyloid p component binding to shiga toxin 2 requires both a subunit and b pentamer. Infect. Immun. 2003, 71, 6075–6078. [Google Scholar]
- Kimura, T.; Tani, S.; Matsumoto Yi, Y.; Takeda, T. Serum amyloid p component is the shiga toxin 2-neutralizing factor in human blood. J. Biol. Chem. 2001, 276, 41576–41579. [Google Scholar]
- Armstrong, G.D.; Mulvey, G.L.; Marcato, P.; Griener, T.P.; Kahan, M.C.; Tennent, G.A.; Sabin, C.A.; Chart, H.; Pepys, M.B. Human serum amyloid p component protects against Escherichia coli o157:H7 shiga toxin 2 in vivo: Therapeutic implications for hemolytic-uremic syndrome. J. Infect. Dis. 2006, 193, 1120–1124. [Google Scholar] [CrossRef]
- Winter, K.R.; Stoffregen, W.C.; Dean-Nystrom, E.A. Shiga toxin binding to isolated porcine tissues and peripheral blood leukocytes. Infect. Immun. 2004, 72, 6680–6684. [Google Scholar]
- Menge, C.; Eisenberg, T.; Stamm, I.; Baljer, G. Comparison of binding and effects of Escherichia coli shiga toxin 1 on bovine and ovine granulocytes. Vet. Immunol. Immunopathol. 2006, 113, 392–403. [Google Scholar]
- Macher, B.A.; Klock, J.C. Isolation and chemical characterization of neutral glycosphingolipids of human neutrophils. J. Biol. Chem. 1980, 255, 2092–2096. [Google Scholar]
- Collins, S.J. The hl-60 promyelocytic leukemia cell line: Proliferation, differentiation, and cellular oncogene expression. Blood 1987, 70, 1233–1244. [Google Scholar]
- Olins, A.L.; Olins, D.E. The mechanism of granulocyte nuclear shape determination: Possible involvement of the centrosome. Eur. J. Cell. Biol. 2005, 84, 181–188. [Google Scholar]
- Kiguchi, K.; Henning-Chubb, C.; Huberman, E. Glycosphingolipid patterns in human promyelocytic hl-60 leukemia cells susceptible or resistant to differentiation induction by phorbol 12-myristate 13-acetate. Biochim. Biophys. Acta 1993, 1176, 27–36. [Google Scholar]
- Nakamura, M.; Tsunoda, A.; Sakoe, K.; Gu, J.; Nishikawa, A.; Taniguchi, N.; Saito, M. Total metabolic flow of glycosphingolipid biosynthesis is regulated by udp-glcnac:Lactosylceramide beta 1→3n-acetylglucosaminyltransferase and cmp-neuac:Lactosylceramide alpha 2→3 sialyltransferase in human hematopoietic cell line hl-60 during differentiation. J. Biol. Chem. 1992, 267, 23507–23514. [Google Scholar]
- Nojiri, H.; Takaku, F.; Tetsuka, T.; Motoyoshi, K.; Miura, Y.; Saito, M. Characteristic expression of glycosphingolipid profiles in the bipotential cell differentiation of human promyelocytic leukemia cell line hl-60. Blood 1984, 64, 534–541. [Google Scholar]
- Keepers, T.R.; Psotka, M.A.; Gross, L.K.; Obrig, T.G. A murine model of hus: Shiga toxin with lipopolysaccharide mimics the renal damage and physiologic response of human disease. J. Am. Soc. Nephrol. 2006, 17, 3404–3414. [Google Scholar]
- Ramos, M.V.; Auvynet, C.; Poupel, L.; Rodero, M.; Mejias, M.P.; Panek, C.A.; Vanzulli, S.; Combadiere, C.; Palermo, M. Chemokine receptor ccr1 disruption limits renal damage in a murine model of hemolytic uremic syndrome. Am. J. Pathol. 2011, 180, 1040–1048. [Google Scholar]
- Sauter, K.A.; Melton-Celsa, A.R.; Larkin, K.; Troxell, M.L.; O’Brien, A.D.; Magun, B.E. Mouse model of hemolytic-uremic syndrome caused by endotoxin-free shiga toxin 2 (stx2) and protection from lethal outcome by anti-stx2 antibody. Infect. Immun. 2008, 76, 4469–4478. [Google Scholar]
- Kuligowski, M.P.; Kitching, A.R.; Hickey, M.J. Leukocyte recruitment to the inflamed glomerulus: A critical role for platelet-derived p-selectin in the absence of rolling. J. Immunol. 2006, 176, 6991–6999. [Google Scholar]
- Kelly, J.; Oryshak, A.; Wenetsek, M.; Grabiec, J.; Handy, S. The colonic pathology of Escherichia coli o157:H7 infection. Am. J. Surg.Pathol. 1990, 14, 87–92. [Google Scholar]
- Kelly, J.K.; Pai, C.H.; Jadusingh, I.H.; Macinnis, M.L.; Shaffer, E.A.; Hershfield, N.B. The histopathology of rectosigmoid biopsies from adults with bloody diarrhea due to verotoxin-producing Escherichia coli. Am. J. Clin. Pathol. 1987, 88, 78–82. [Google Scholar]
- Griffin, P.M.; Olmstead, L.C.; Petras, R.E. Escherichia coli o157:H7-associated colitis. A clinical and histological study of 11 cases. Gastroenterology 1990, 99, 142–149. [Google Scholar]
- Taylor, F.B., Jr.; Tesh, V.L.; DeBault, L.; Li, A.; Chang, A.C.; Kosanke, S.D.; Pysher, T.J.; Siegler, R.L. Characterization of the baboon responses to shiga-like toxin: Descriptive study of a new primate model of toxic responses to stx-1. Am. J. Pathol. 1999, 154, 1285–1299. [Google Scholar]
- Te Loo, D.M.; Heuvelink, A.E.; de Boer, E.; Nauta, J.; Van der Walle, J.; Schroder, C.; van Hinsbergh, V.W.; Chart, H.; Van de Kar, N.C.; Van den Heuvel, L.P. Vero cytotoxin binding to polymorphonuclear leukocytes among households with children with hemolytic uremic syndrome. J. Infect. Dis. 2001, 184, 446–450. [Google Scholar]
- Inward, C.D.; Varagunam, M.; Adu, D.; Milford, D.V.; Taylor, C.M. Cytokines in haemolytic uraemic syndrome associated with verocytotoxin-producing Escherichia coli infection. Arch. Dis. Child. 1997, 77, 145–147. [Google Scholar]
- Van Setten, P.A.; Van Hinsbergh, V.W.; Van den Heuvel, L.P.; Preyers, F.; Dijkman, H.B.; Assmann, K.J.; Van der Velden, T.J.; Monnens, L.A. Monocyte chemoattractant protein-1 and interleukin-8 levels in urine and serum of patents with hemolytic uremic syndrome. Pediatr. Res. 1998, 43, 759–767. [Google Scholar]
- Smith, Q.R.; Rapoport, S.I. Cerebrovascular permeability coefficients to sodium, potassium, and chlorid. J. Neurochem. 1986, 46, 1732–1742. [Google Scholar]
- Pardridge, W. Introduction to the Blood-Brain Barrier. Methodology, Biology and Pathology. Cambridge University Press: Cambridge, UK,, 1998; pp. 1–8. [Google Scholar]
- Fujii, J.; Kinoshita, Y.; Kita, T.; Higure, A.; Takeda, T.; Tanaka, N.; Yoshida, S. Magnetic resonance imaging and histopathological study of brain lesions in rabbits given intravenous verotoxin 2. Infect. Immun. 1996, 64, 5053–5060. [Google Scholar]
- Takahashi, K.; Funata, N.; Ikuta, F.; Sato, S. Neuronal apoptosis and inflammatory responses in the central nervous system of a rabbit treated with shiga toxin-2. J. Neuroinflammation 2008, 5, 11. [Google Scholar]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar]
- Creagh, E.M.; O’Neill, L.A. Tlrs, nlrs and rlrs: A trinity of pathogen sensors that co-operate in innate immunity. Trends. Immunol. 2006, 27, 352–357. [Google Scholar]
- Torgersen, M.L.; Engedal, N.; Pedersen, A.M.; Husebye, H.; Espevik, T.; Sandvig, K. Toll-like receptor 4 facilitates binding of shiga toxin to colon carcinoma and primary umbilical vein endothelial cells. FEMS Immunol. Med. Microbiol. 2011, 61, 63–75. [Google Scholar]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar]
- Lopez, E.L.; Contrini, M.M.; Glatstein, E.; Gonzalez Ayala, S.; Santoro, R.; Ezcurra, G.; Teplitz, E.; Matsumoto, Y.; Sato, H.; Sakai, K.; et al. An epidemiologic surveillance of shiga-like toxin-producing Escherichia coli infection in argentinean children: Risk factors and serum shiga-like toxin 2 values. Pediatr. Infect. Dis. J. 2012, in press. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Brigotti, M. The Interactions of Human Neutrophils with Shiga Toxins and Related Plant Toxins: Danger or Safety? Toxins 2012, 4, 157-190. https://doi.org/10.3390/toxins4030157
Brigotti M. The Interactions of Human Neutrophils with Shiga Toxins and Related Plant Toxins: Danger or Safety? Toxins. 2012; 4(3):157-190. https://doi.org/10.3390/toxins4030157
Chicago/Turabian StyleBrigotti, Maurizio. 2012. "The Interactions of Human Neutrophils with Shiga Toxins and Related Plant Toxins: Danger or Safety?" Toxins 4, no. 3: 157-190. https://doi.org/10.3390/toxins4030157