Cloning and Characterization of a Hybridoma Secreting a 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)-Specific Monoclonal Antibody and Recombinant F(ab)

Abstract
:1. Introduction
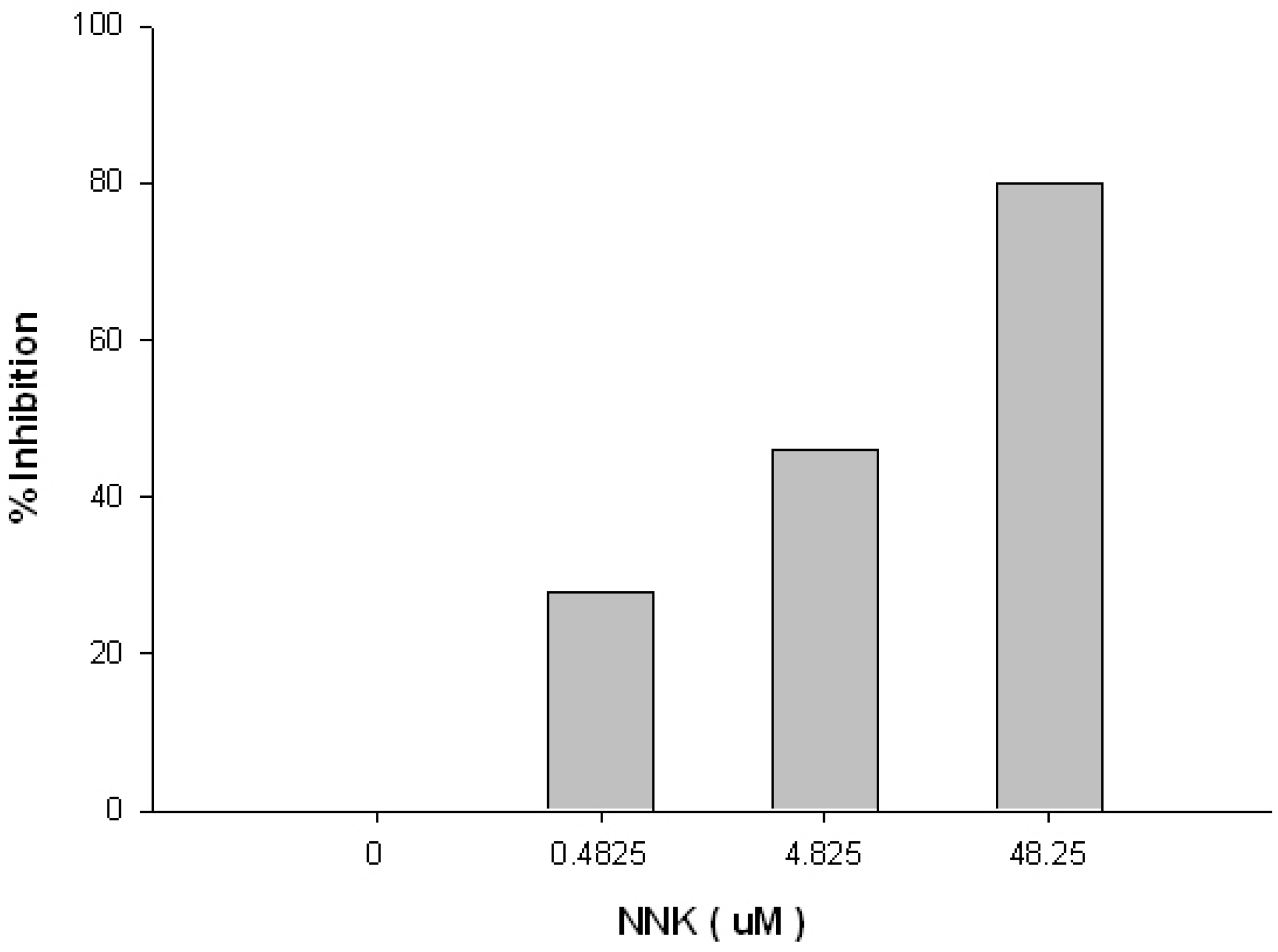
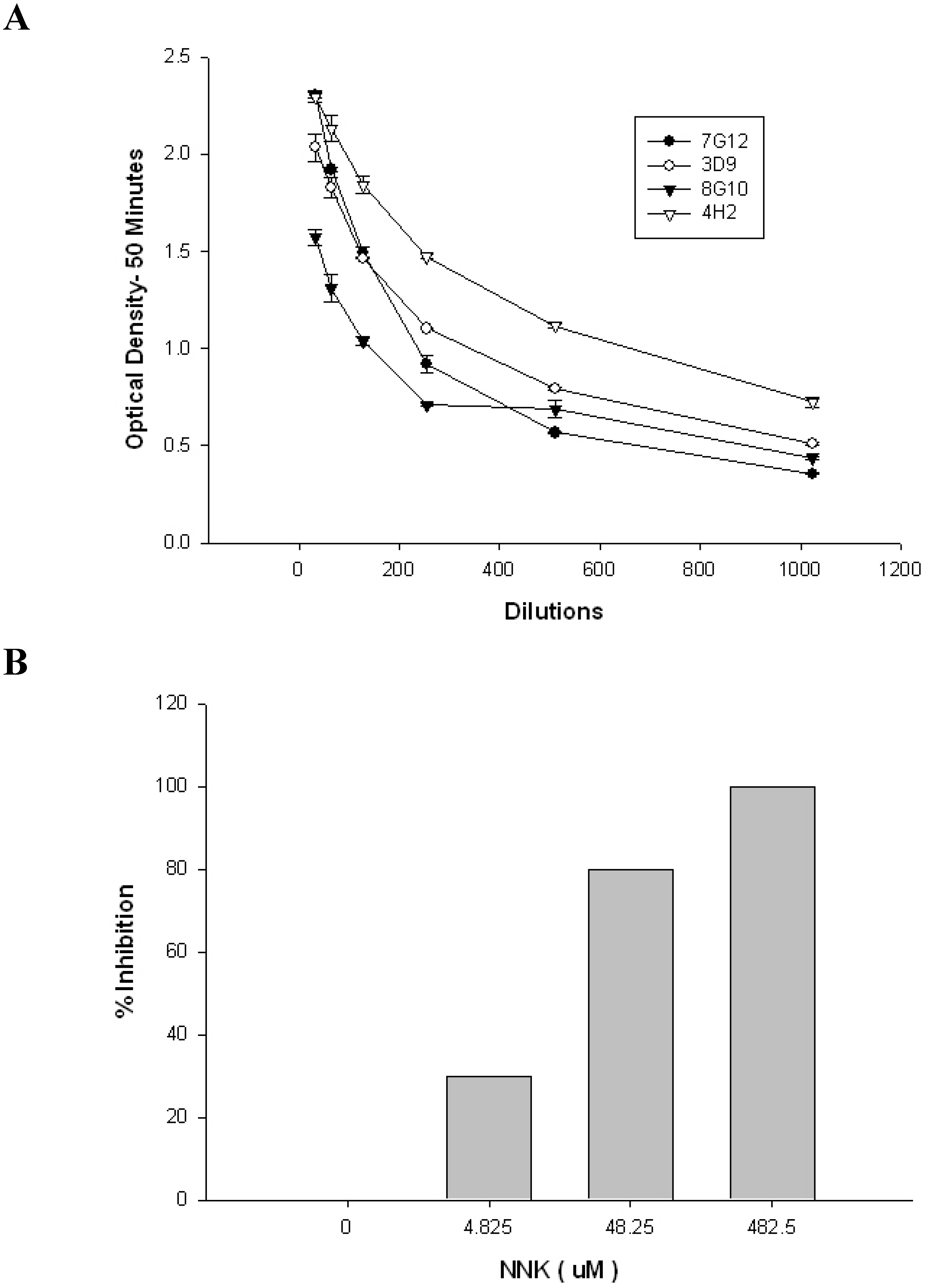
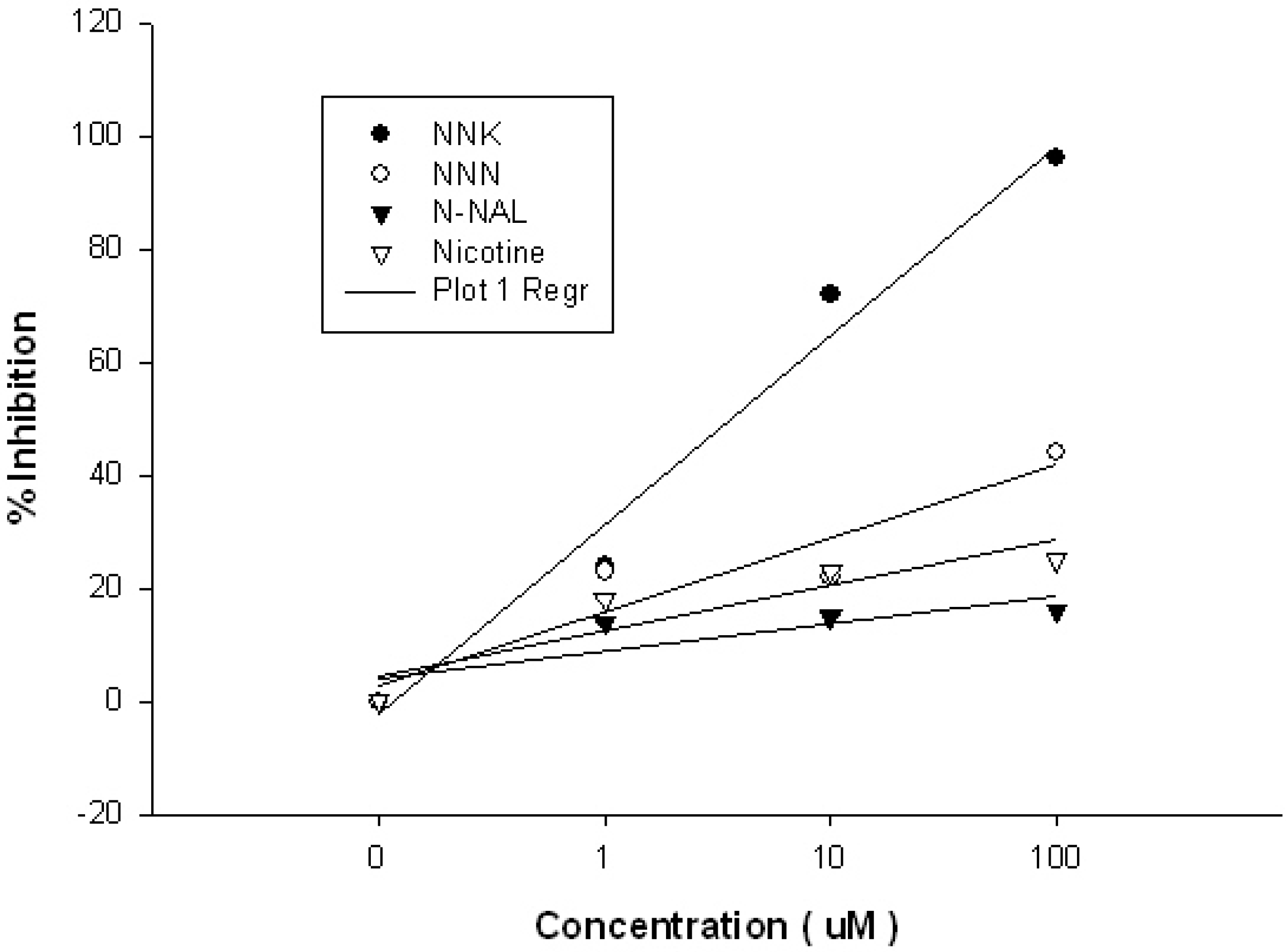
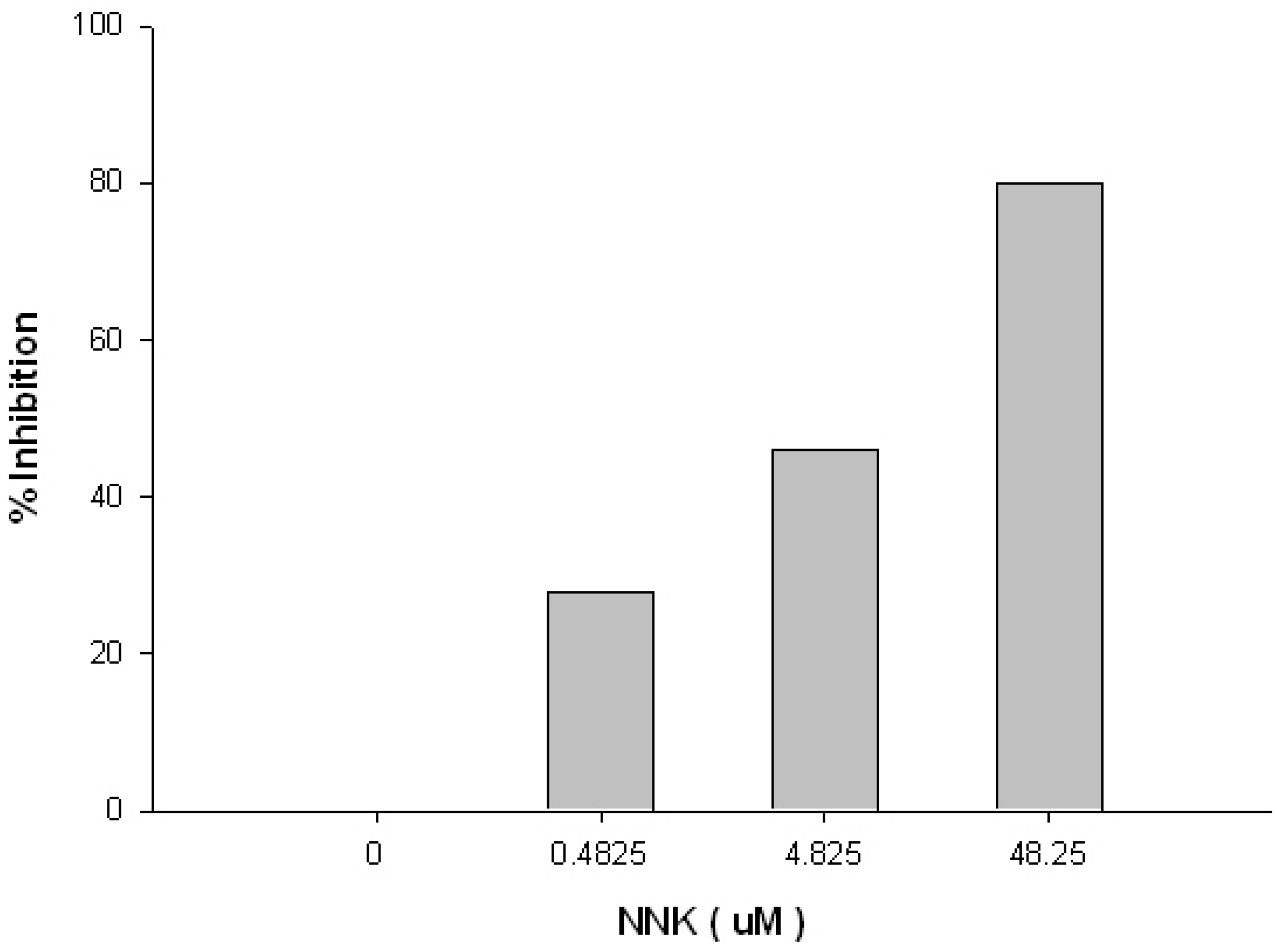
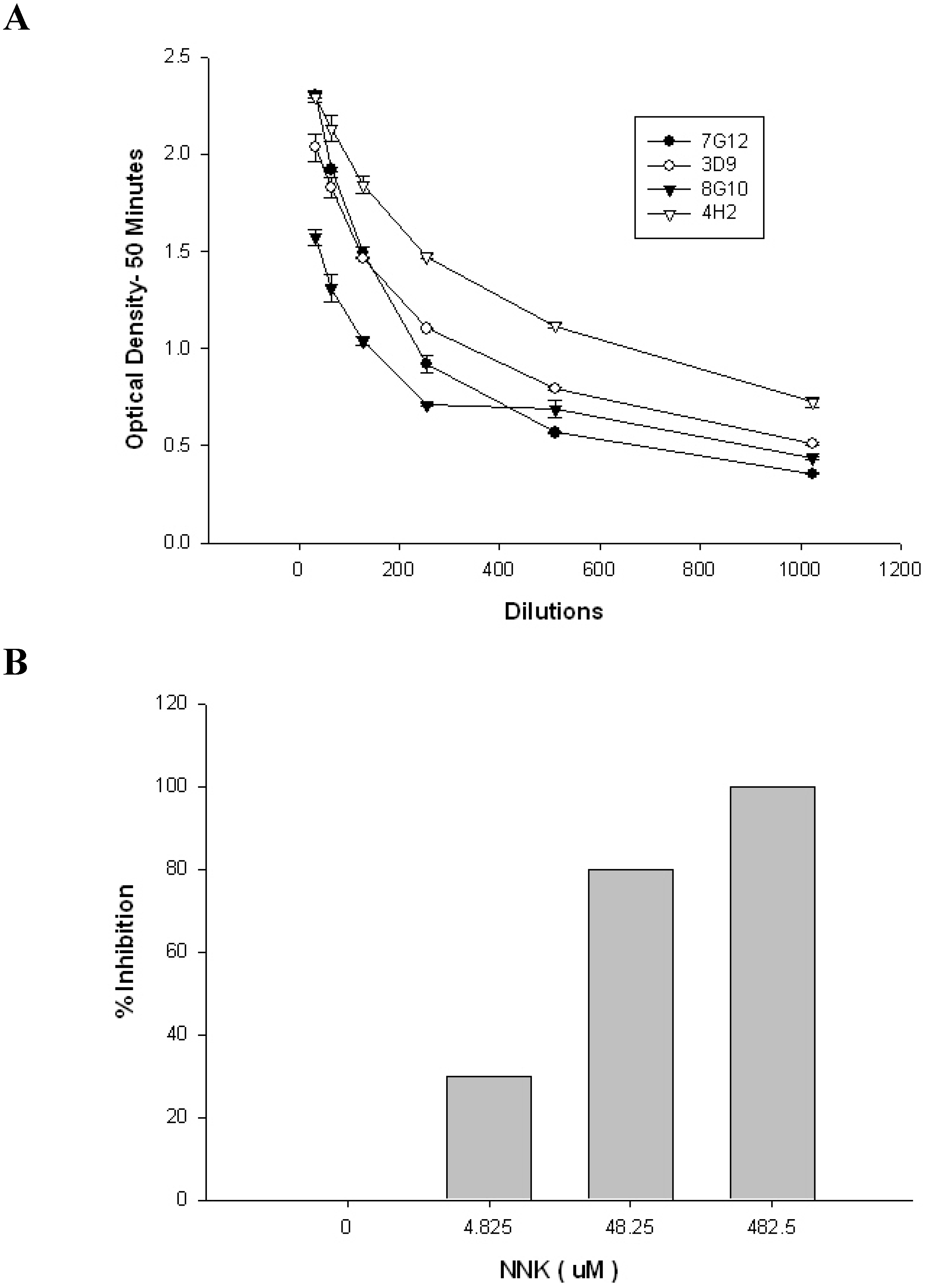
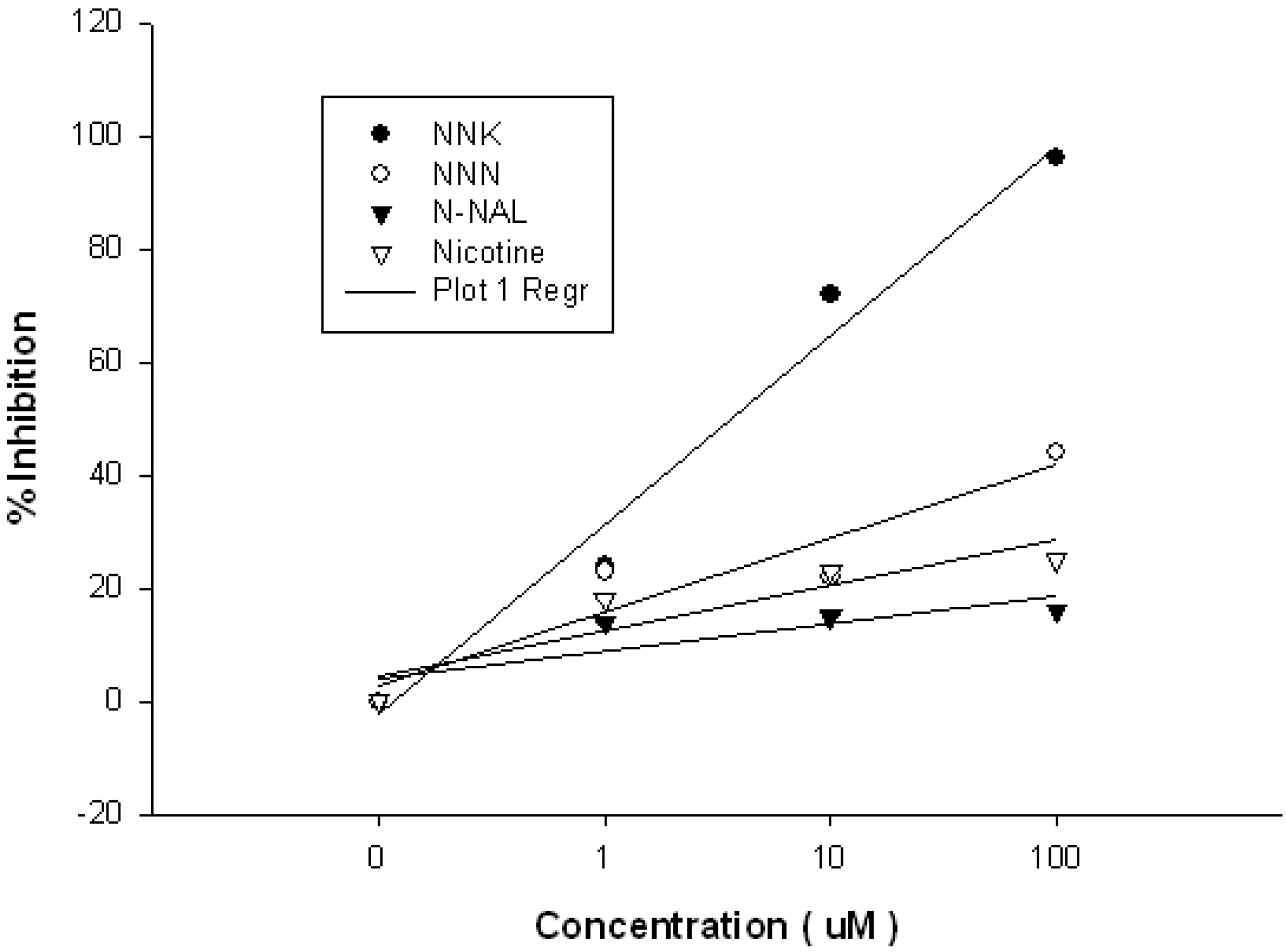
2. Results

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
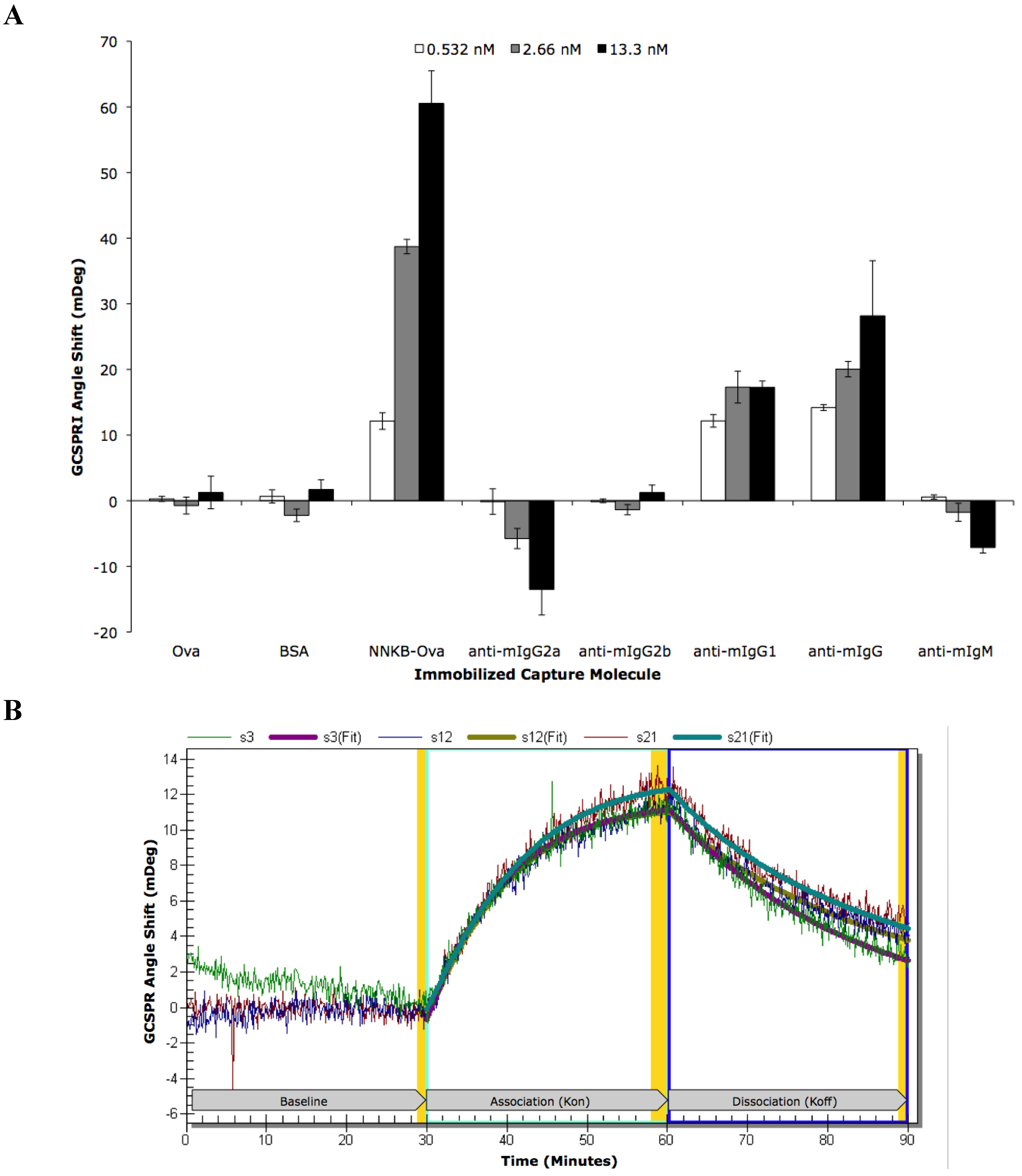
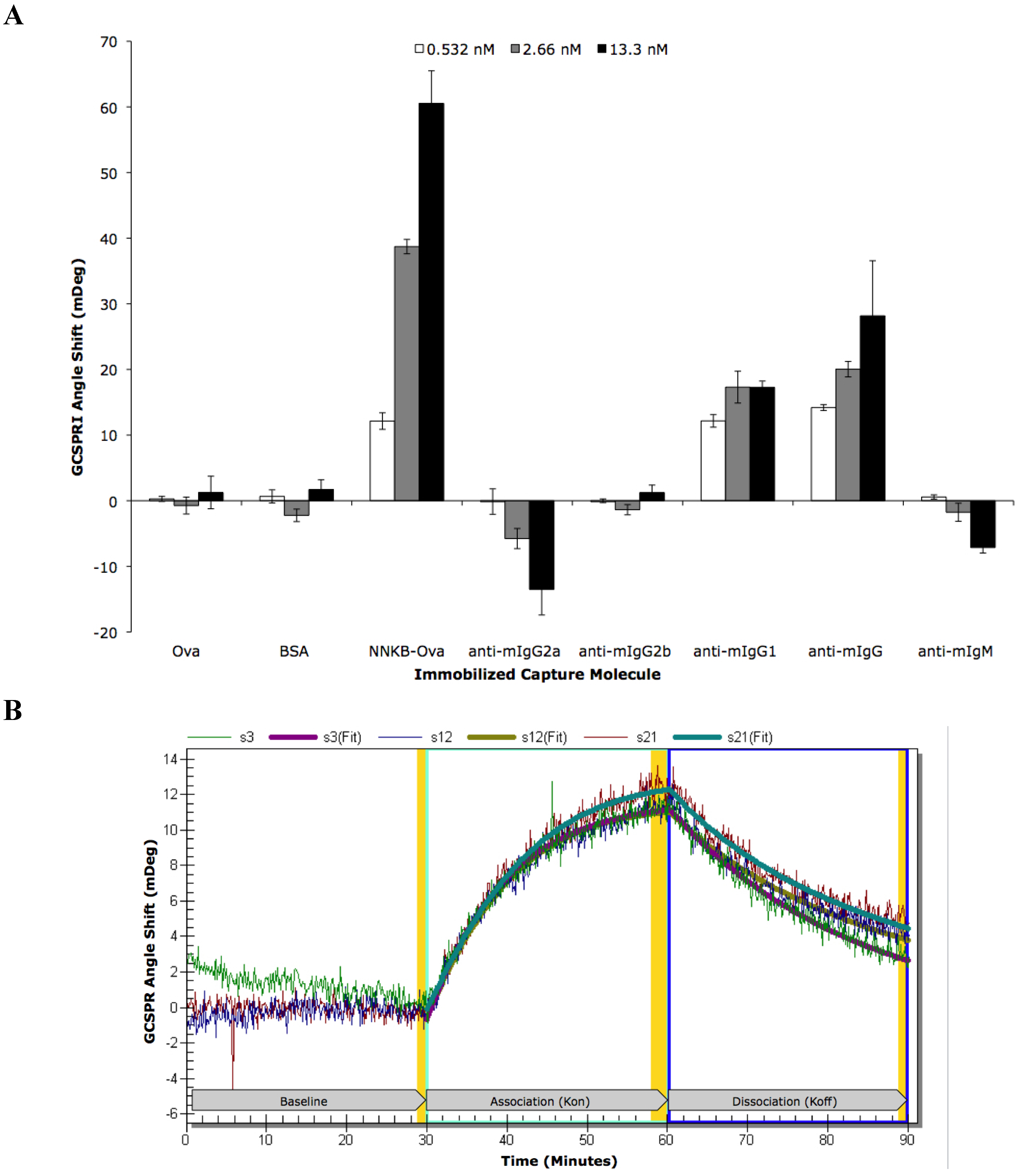
| ROI | ROI Content | Concentration | Kinetic Analysis | Kon | Koff | Ka (kon/koff) | Kd (koff/kon) |
|---|---|---|---|---|---|---|---|
| s3 | NNKB-Ova | 500 μg/mL | Local | 1.94 × 106 | 7.38 × 10−4 | 2.63 × 109 | 3.80 × 10−10 |
| s12 | NNKB-Ova | 500 μg/mL | Local | 3.60 × 106 | 8.18 × 10−4 | 4.40 × 109 | 2.27 × 10−10 |
| s21 | NNKB-Ova | 500 μg/mL | Local | 3.76 × 106 | 1.01 × 10−3 | 3.63 × 109 | 2.75 × 10−10 |
| All 3 | NNKB-Ova | 500 μg/mL | Global | 2.87 × 106 | 8.41 × 10−4 | 3.41 × 109 | 2.93 × 10−10 |
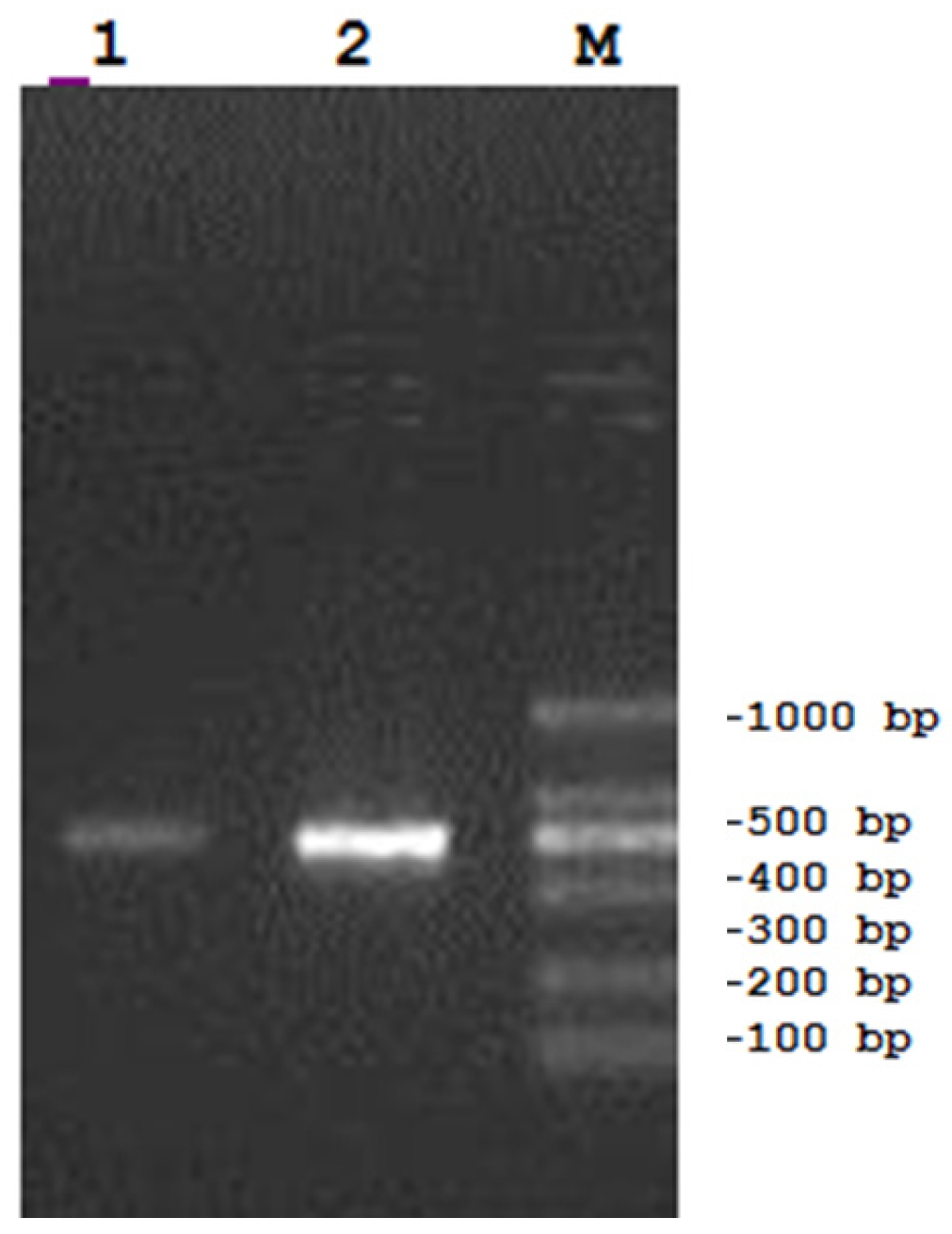
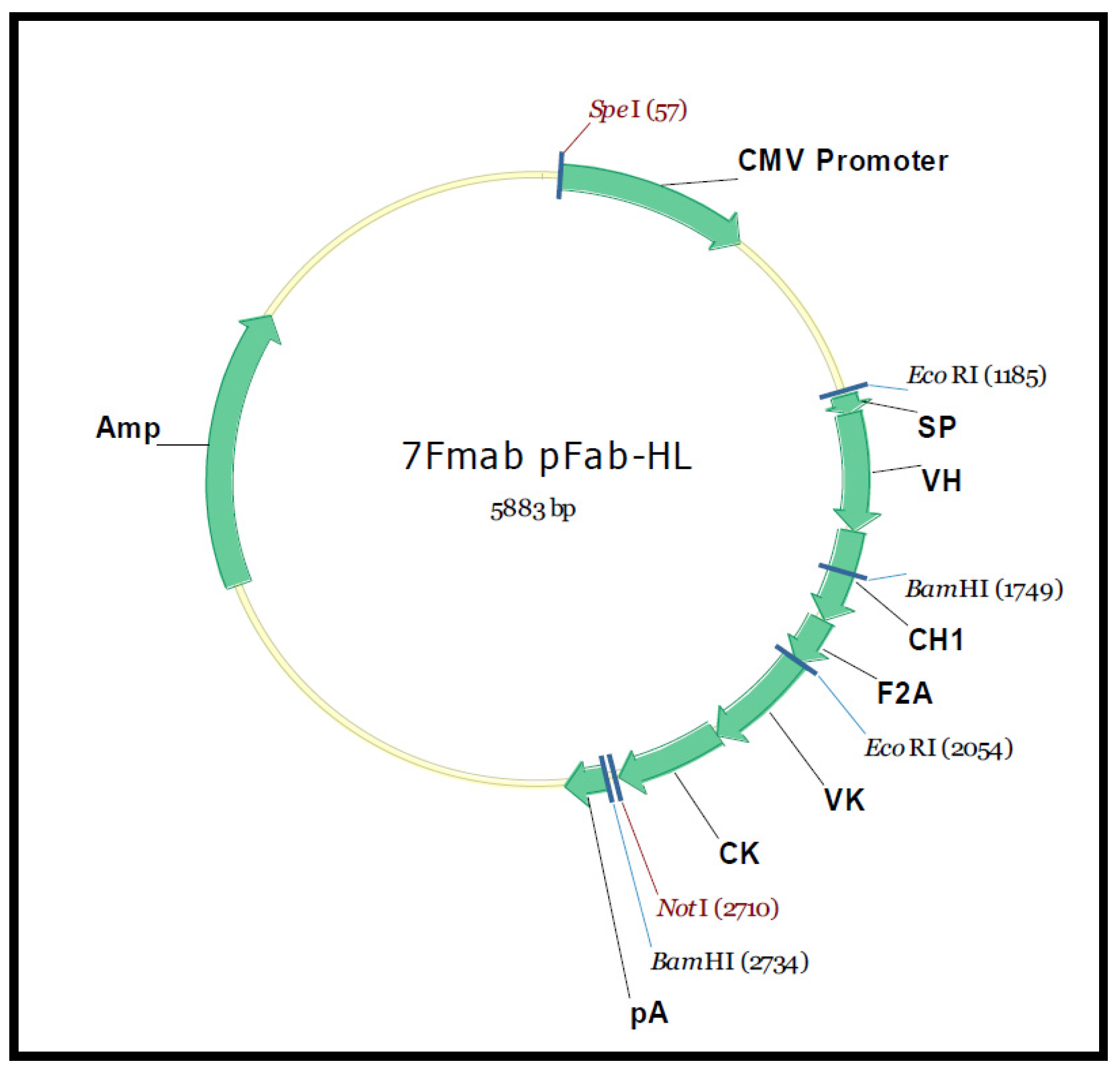

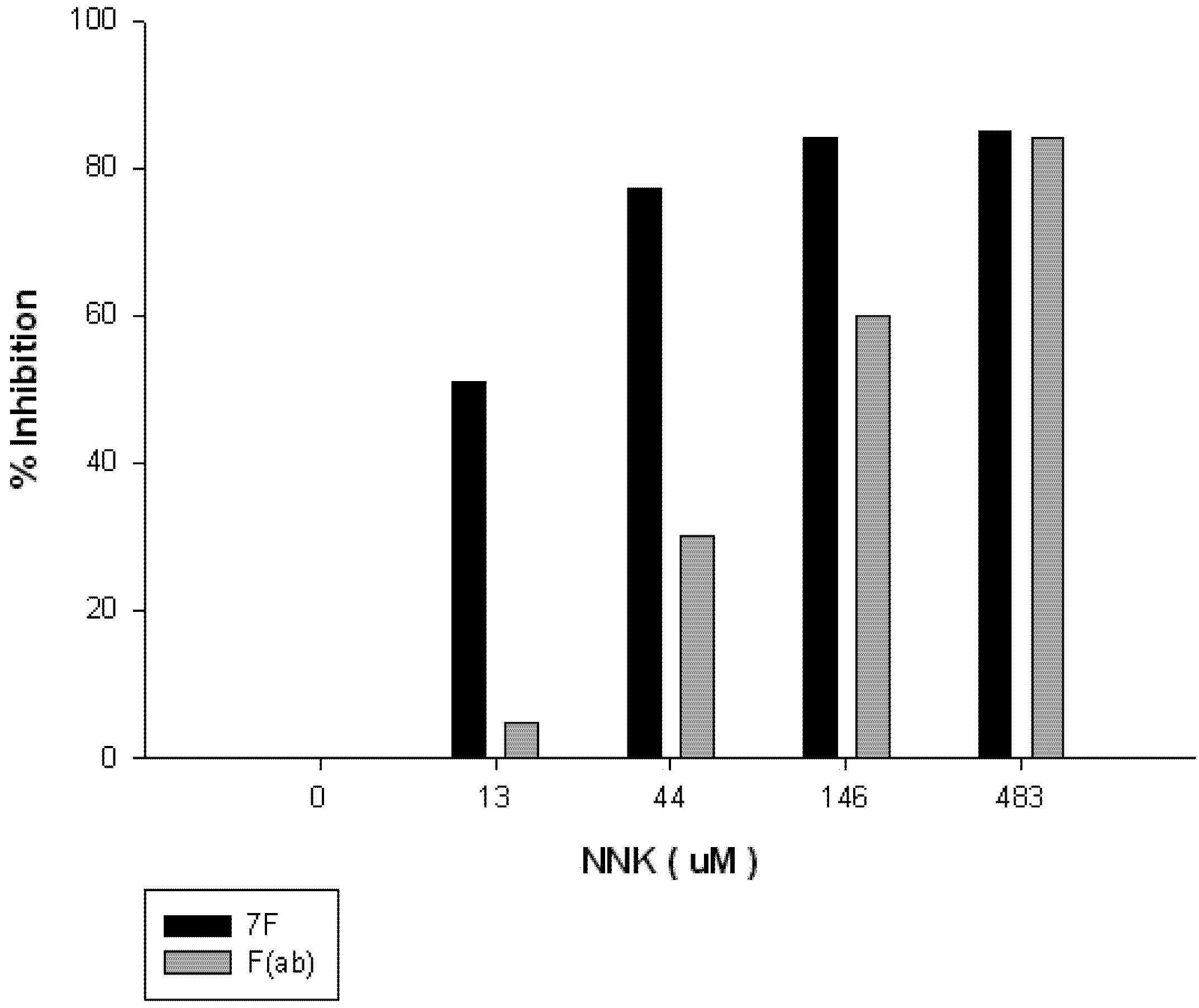
3. Materials and Methods
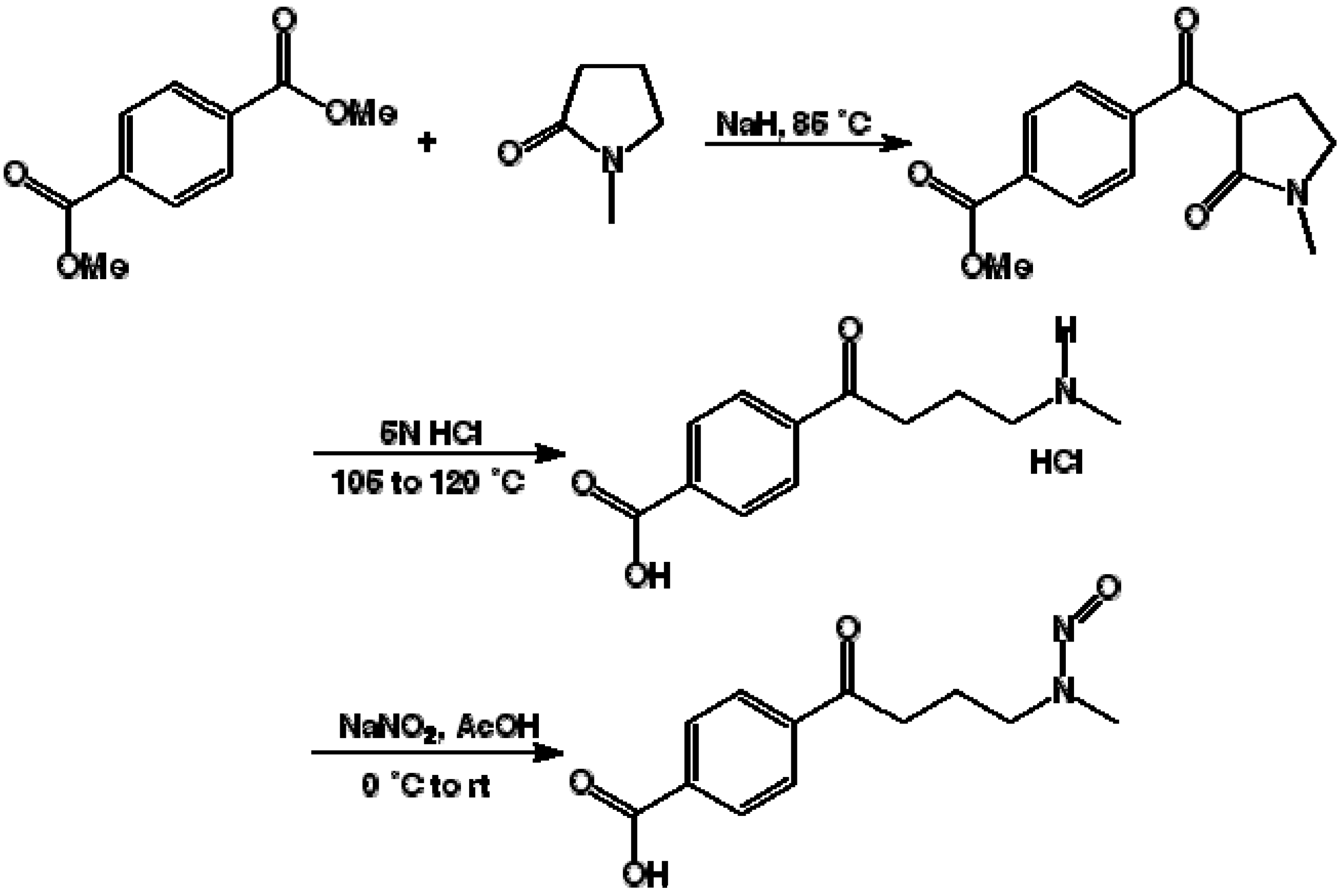
3.1. Synthesis of 4-(4-Methylnitrosoamino-1-oxobutyryl)benzoic Acid (NNKB)
3.2. Chemical Coupling of NNKB to Carrier Proteins
3.3. Antibody Generation in Mice
3.4. Enzyme Linked Immunosorbent Assays (ELISAs)
3.5. ELISA of F(ab)
3.6. Competitive ELISA
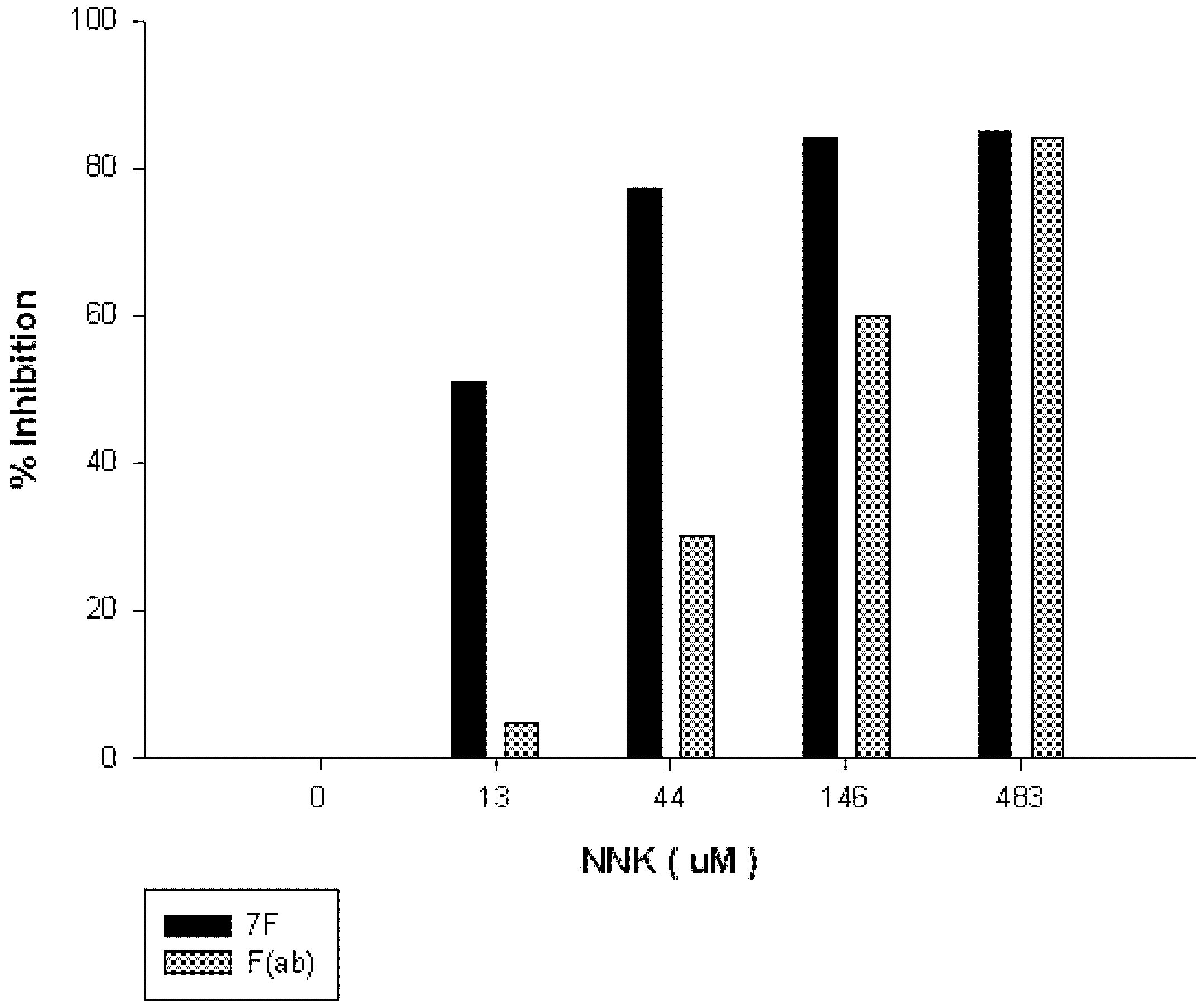
3.7. Competitive ELISA of F(ab)
3.8. Monoclonal Antibody Production
3.9. GCSPRI Analysis of Monoclonal Antibody Characterization
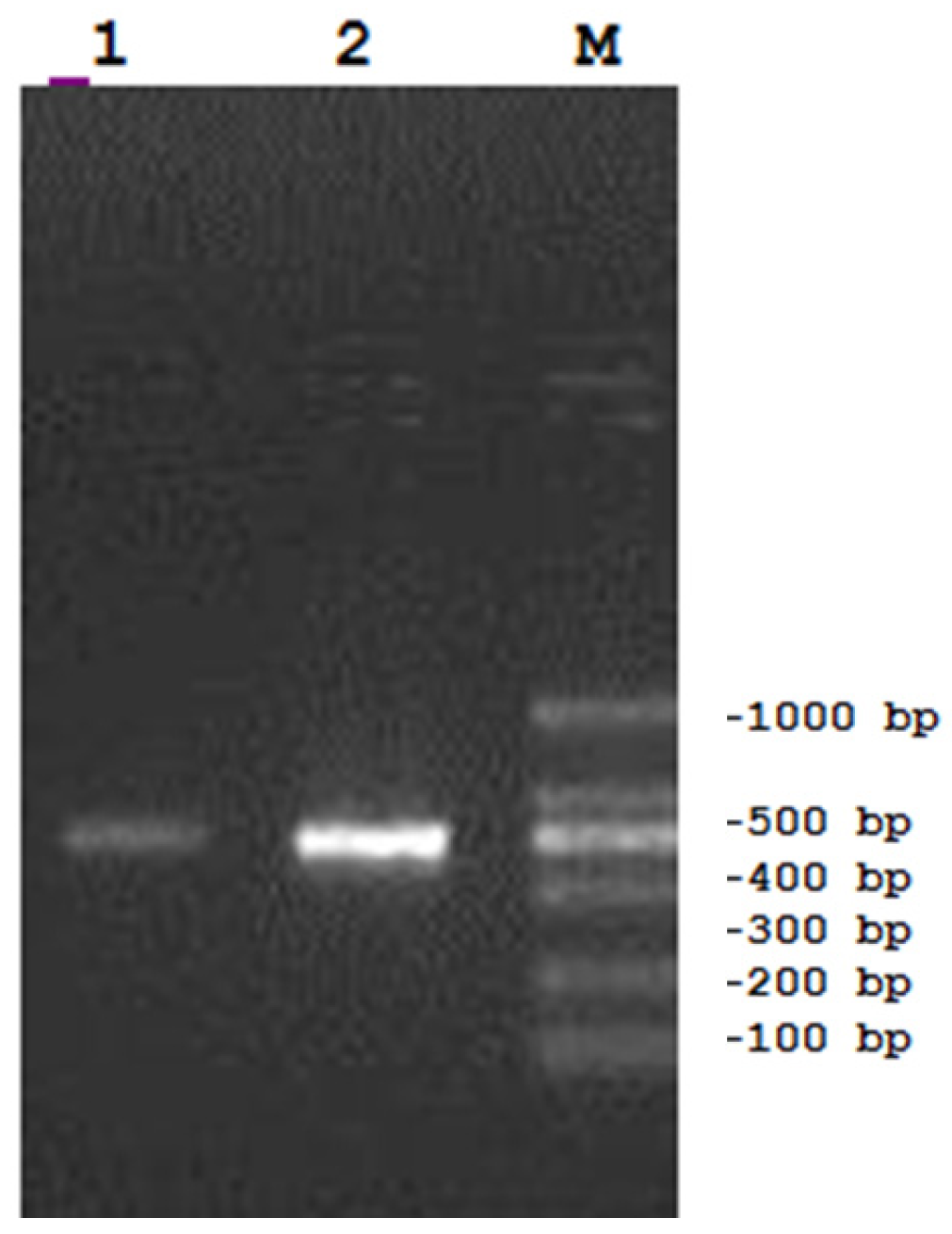
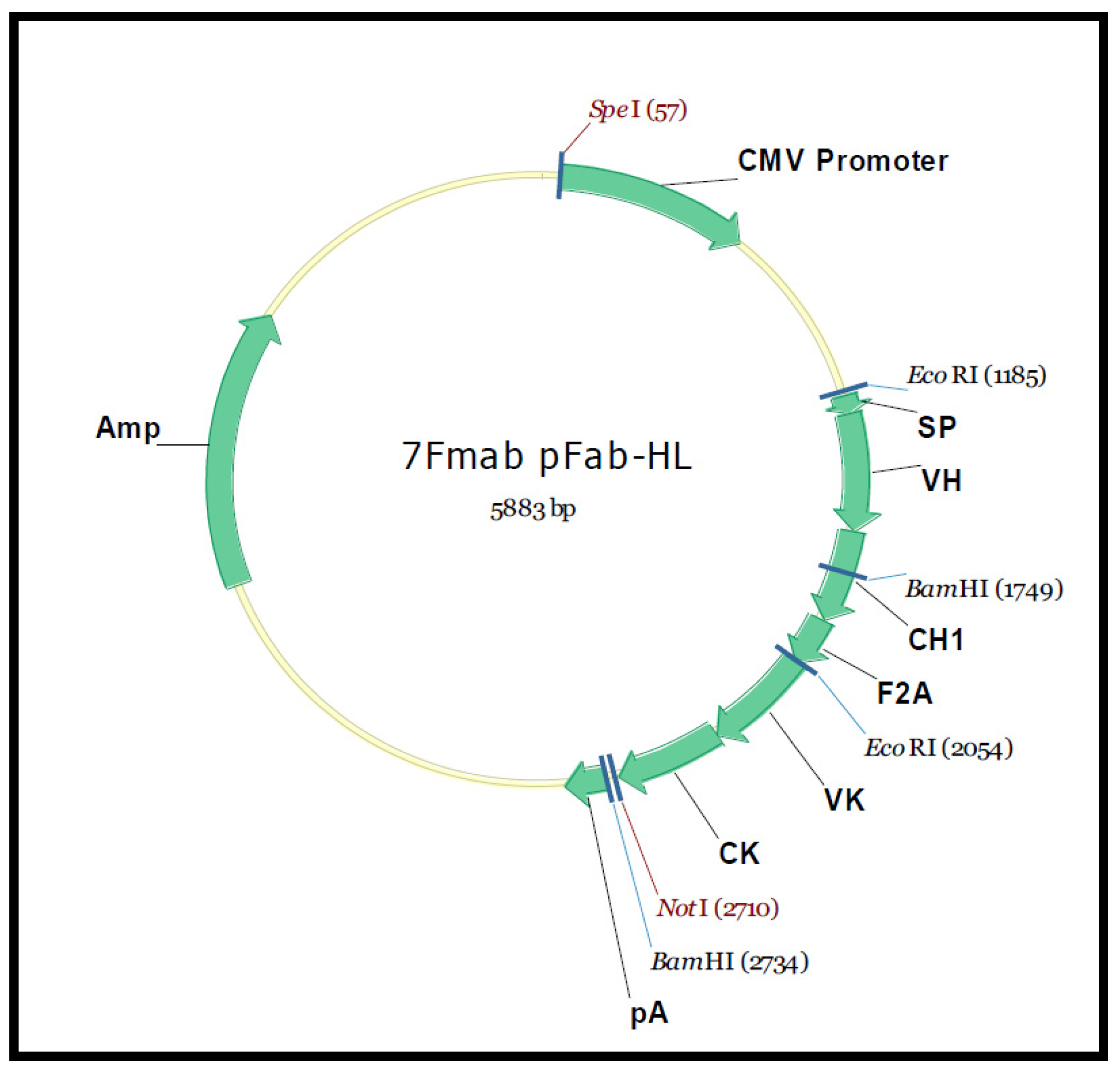
3.10. Cloning and Characterization of a F(ab)-Generating Single Chain Antibody
3.11. Statistical Analysis
4. Conclusions
Acknowledgements
References
- World Health Organization. Overall evaluations of carcinogenicity: An updating of IARC Monographs volumes 1 to 42. IARC Monogr. Eval. Carcinog. Risks Hum. Suppl. 1987, 7, 1–440.
- Rodu, B.; Jansson, C. Smokeless tobacco and oral cancer: A review of the risks and determinants. Crit. Rev. Oral Biol. Med. 2004, 15, 252–263. [Google Scholar] [CrossRef]
- Chiang, H.C.; Huang, Y.K.; Chen, P.F.; Chang, C.C.; Wang, C.J.; Lin, P.; Lee, H.L. 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone is correlated with 8-hydroxy-2'-deoxyguanosine in humans after exposure to environmental tobacco smoke. Sci. Total Environ. 2012, 414, 134–139. [Google Scholar] [CrossRef]
- Nilsson, R. The molecular basis for induction of human cancers by tobacco specific nitrosamines. Regul. Toxicol. Pharmacol. 2011, 60, 268–280. [Google Scholar] [CrossRef]
- Hecht, S.S. Tobacco smoke carcinogens and lung cancer. J. Natl. Cancer Inst. 1999, 91, 1194–1210. [Google Scholar] [CrossRef]
- Schuller, H.M. Cell type specific, receptor-mediated modulation of growth kinetics in human lung cancer cell lines by nicotine and tobacco-related nitrosamines. Biochem. Pharmacol. 1989, 38, 3439–3442. [Google Scholar] [CrossRef]
- Schuller, H.M. Nitrosamines as nicotinic receptor ligands. Life Sci. 2007, 80, 2274–2280. [Google Scholar] [CrossRef]
- Arredondo, J.; Chernyavsky, A.I.; Grando, S.A. Nicotinic receptors mediate tumorigenic action of tobacco-derived nitrosamines on immortalized oral epithelial cells. Cancer Biol. Ther. 2006, 5, 511–517. [Google Scholar] [CrossRef]
- Arredondo, J.; Chernyavsky, A.I.; Jolkovsky, D.L.; Pinkerton, K.E.; Grando, S.A. Receptor-mediated tobacco toxicity: Cooperation of the Ras/Raf-1/MEK1/ERK and JAK-2/STAT-3 pathways downstream of alpha7 nicotinic receptor in oral keratinocytes. Faseb. J. 2006, 20, 2093–2101. [Google Scholar] [CrossRef]
- Arredondo, J.; Chernyavsky, A.I.; Grando, S.A. The nicotinic receptor antagonists abolish pathobiologic effects of tobacco-derived nitrosamines on BEP2D cells. J. Cancer Res. Clin. Oncol. 2006, 132, 653–663. [Google Scholar] [CrossRef]
- Rodu, B.; Cole, P. Tobacco-related mortality. Nature 1994, 370, 184. [Google Scholar] [CrossRef]
- Hatsukami, D.K.; Lemmonds, C.; Tomar, S.L. Smokeless tobacco use: Harm reduction or induction approach? Prev. Med. 2004, 38, 309–317. [Google Scholar] [CrossRef]
- Rodu, B.; Phillips, C.V. Switching to smokeless tobacco as a smoking cessation method: Evidence from the 2000 National Health Interview Survey. Harm Reduct. J. 2008, 5, 18. [Google Scholar] [CrossRef]
- Silbart, L.K.; Rasmussen, M.V.; Oliver, A.R. Immunoprophylactic intervention in chemical toxicity and carcinogenicity. Vet. Hum. Toxicol. 1997, 39, 37–43. [Google Scholar]
- Kosten, T.; Owens, S.M. Immunotherapy for the treatment of drug abuse. Pharmacol. Ther. 2005, 108, 76–85. [Google Scholar] [CrossRef]
- Rasmussen, M.V.; Silbart, L.K. Peroral administration of specific antibody enhances carcinogen excretion. J. Immunother. 1998, 21, 418–426. [Google Scholar] [CrossRef]
- Silbart, L.K.; Keren, D.F. Reduction of intestinal carcinogen absorption by carcinogen-specific secretory immunity. Science 1989, 243, 1462–1464. [Google Scholar]
- Baud, F.J.; Borron, S.W.; Bismuth, C. Modifying toxicokinetics with antidotes. Toxicol. Lett. 1995, 82–83, 785–793. [Google Scholar]
- Moolten, F.; Capparell, N.; Boger, E. Reduction of respiratory tract binding of benzo[a]pyrene in mice by immunization. J. Natl. Cancer Inst. 1978, 61, 1347–1349. [Google Scholar]
- Moolten, F.L.; Schreiber, B.; Rizzone, A.; Weiss, A.J.; Boger, E. Protection of mice against 7,12-dimethylbenz(a)anthracene-induced skin tumors by immunization with a fluorinated analog of the carcinogen. Cancer Res. 1981, 41, 425–429. [Google Scholar]
- Adair, C.D.; Buckalew, V.M.; Graves, S.W.; Lam, G.K.; Johnson, D.D.; Saade, G.; Lewis, D.F.; Robinson, C.; Danoff, T.M.; Chauhan, N.; et al. Digoxin immune fab treatment for severe preeclampsia. Am. J. Perinatol. 2010, 27, 655–662. [Google Scholar] [CrossRef]
- Daniels, J.R.; Wessinger, W.D.; Hardwick, W.C.; Li, M.; Gunnell, M.G.; Hall, C.J.; Owens, S.M.; McMillan, D.E. Effects of anti-phencyclidine and anti-(+)-methamphetamine monoclonal antibodies alone and in combination on the discrimination of phencyclidine and (+)-methamphetamine by pigeons. Psychopharmacology 2006, 185, 36–44. [Google Scholar] [CrossRef]
- Hicks, M.J.; De, B.P.; Rosenberg, J.B.; Davidson, J.T.; Moreno, A.Y.; Janda, K.D.; Wee, S.; Koob, G.F.; Hackett, N.R.; Kaminsky, S.M.; et al. Cocaine analog coupled to disrupted adenovirus: A vaccine strategy to evoke high-titer immunity against addictive drugs. Mol. Ther. 2011, 19, 612–619. [Google Scholar] [CrossRef]
- Deng, W.; Yang, D.; Zhao, B.; Ouyang, Z.; Song, J.; Fan, N.; Liu, Z.; Zhao, Y.; Wu, Q.; Nashun, B.; et al. Use of the 2A peptide for generation of multi-transgenic pigs through a single round of nuclear transfer. PLoS One 2011, 6, e19986. [Google Scholar]
- Chan, H.Y.; V, S.; Xing, X.; Kraus, P.; Yap, S.P.; Ng, P.; Lim, S.L.; Lufkin, T. Comparison of IRES and F2A-based locus-specific multicistronic expression in stable mouse lines. PLoS One 2011, 6, e28885. [Google Scholar]
- Verrier, J.D.; Madorsky, I.; Coggin, W.E.; Geesey, M.; Hochman, M.; Walling, E.; Daroszewski, D.; Eccles, K.S.; Ludlow, R.; Semple-Rowland, S.L. Bicistronic lentiviruses containing a viral 2A cleavage sequence reliably co-express two proteins and restore vision to an animal model of LCA1. PLoS One 2011, 6, e20553. [Google Scholar] [CrossRef]
- De Felipe, P. Skipping the co-expression problem: The new 2A “CHYSEL” technology. Genet. Vaccines Ther. 2004, 2, 13. [Google Scholar] [CrossRef] [Green Version]
- Reed, C.D.; Rast, H.; Hu, W.G.; Mah, D.; Nagata, L.; Masri, S.A. Expression of furin-linked Fab fragments against anthrax toxin in a single mammalian expression vector. Protein Expr. Purif. 2007, 54, 261–266. [Google Scholar] [CrossRef]
- Tan, Y.; Liang, H.; Chen, A.; Guo, X. Coexpression of double or triple copies of the rabies virus glycoprotein gene using a “self-cleaving” 2A peptide-based replication-defective human adenovirus serotype 5 vector. Biologicals 2010, 38, 586–593. [Google Scholar] [CrossRef]
- Luke, G.; Escuin, H.; de Felipe, P.; Ryan, M. 2A to the fore—Research, technology and applications. Biotechnol. Genet. Eng. Rev. 2010, 26, 223–260. [Google Scholar]
- Hagemeyer, C.E.; von Zur Muhlen, C.; von Elverfeldt, D.; Peter, K. Single-chain antibodies as diagnostic tools and therapeutic agents. Thromb. Haemost. 2009, 101, 1012–1019. [Google Scholar]
- Pathak, T.; Thomas, N.F.; Akhtar, M.; Gani, D. Synthesis of [4–2H2]-, (4R)[4–2H1]- and (4S)[4–2H1]-4-(methylnitrosamino)-1-(3'-pyridyl)-1-butanone, C-4 deuterated isotopomers of the procarcinogen NNK. Tetrahedron 1990, 46, 1733–1744. [Google Scholar] [CrossRef]
- Harris, J.F.; Hawley, R.G.; Hawley, T.S.; Crawford-Sharpe, G.C. Increased frequency of both total and specific monoclonal antibody producing hybridomas using a fusion partner that constitutively expresses recombinant IL-6. J. Immunol. Methods 1992, 148, 199–207. [Google Scholar] [CrossRef]
- Goding, J.W. Antibody production by hybridomas. J. Immunol. Methods 1980, 39, 285–308. [Google Scholar] [CrossRef]
- Marusov, G.; Sweatt, A.; Pietrosimone, K.; Benson, D.; Geary, S.J.; Silbart, L.K.; Challa, S.; Lagoy, J.; Lawrence, D.A.; Lynes, M.A. A microarray biosensor for multiplexed detection of microbes using grating-coupled surface plasmon resonance imaging. Environ. Sci. Technol. 2012, 46, 348–359. [Google Scholar] [CrossRef]
- LaemmLi, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Rouan, S.K.; Otterness, I.G.; Cunningham, A.C.; Holden, H.E.; Rhodes, C.T. Reversal of colchicine-induced mitotic arrest in Chinese hamster cells with a colchicine-specific monoclonal antibody. Am. J. Pathol. 1990, 137, 779–787. [Google Scholar]
- Safadi, R.; Levy, I.; Amitai, Y.; Caraco, Y. Beneficial effect of digoxin-specific Fab antibody fragments in oleander intoxication. Arch. Intern. Med. 1995, 155, 2121–2125. [Google Scholar] [CrossRef]
- Shen, X.Y.; Orson, F.M.; Kosten, T.R. Vaccines against drug abuse. Clin. Pharmacol. Ther. 2012, 91, 60–70. [Google Scholar] [CrossRef]
- Treweek, J.B.; Janda, K.D. An antidote for acute cocaine toxicity. Mol. Pharm. 2012, 9, 969–978. [Google Scholar] [CrossRef]
- De Buck, S.S.; Schellenberger, M.T.; Ensch, C.; Muller, C.P. Effects of antibodies induced by a conjugate vaccine on 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone absorptive transport, metabolism, and proliferation of human lung cells. Int. J. Cancer 2009, 127, 513–520. [Google Scholar]
- Pentel, P.R.; Dufek, M.B.; Roiko, S.A.; Lesage, M.G.; Keyler, D.E. Differential effects of passive immunization with nicotine-specific antibodies on the acute and chronic distribution of nicotine to brain in rats. J. Pharmacol. Exp. Ther. 2006, 317, 660–666. [Google Scholar] [CrossRef]
- Gentry, W.B.; Ruedi-Bettschen, D.; Owens, S.M. Anti-(+)-methamphetamine monoclonal antibody antagonists designed to prevent the progression of human diseases of addiction. Clin. Pharmacol. Ther. 2010, 88, 390–393. [Google Scholar] [CrossRef]
- Mizuguchi, H.; Xu, Z.; Ishii-Watabe, A.; Uchida, E.; Hayakawa, T. IRES-dependent second gene expression is significantly lower than cap-dependent first gene expression in a bicistronic vector. Mol. Ther. 2000, 1, 376–382. [Google Scholar] [CrossRef]
- Donnelly, M.L.; Hughes, L.E.; Luke, G.; Mendoza, H.; ten Dam, E.; Gani, D.; Ryan, M.D. The “cleavage” activities of foot-and-mouth disease virus 2A site-directed mutants and naturally occurring “2A-like” sequences. J. Gen. Virol. 2001, 82, 1027–1041. [Google Scholar]
- Donnelly, M.L.; Gani, D.; Flint, M.; Monaghan, S.; Ryan, M.D. The cleavage activities of aphthovirus and cardiovirus 2A proteins. J. Gen. Virol. 1997, 78, 13–21. [Google Scholar]
- Groot Bramel-Verheije, M.H.; Rottier, P.J.; Meulenberg, J.J. Expression of a foreign epitope by porcine reproductive and respiratory syndrome virus. Virology 2000, 278, 380–289. [Google Scholar] [CrossRef]
- Schillberg, S.; Fischer, R.; Emans, N. Molecular farming” of antibodies in plants. Naturwissenschaften 2003, 90, 145–155. [Google Scholar]
- Kathuria, S.; Sriraman, R.; Nath, R.; Sack, M.; Pal, R.; Artsaenko, O.; Talwar, G.P.; Fischer, R.; Finnern, R. Efficacy of plant-produced recombinant antibodies against HCG. Hum. Reprod. 2002, 17, 2054–2061. [Google Scholar] [CrossRef]
- Barbi, T.; Drake, P.M.; Drever, M.; van Dolleweerd, C.J.; Porter, A.R.; Ma, J.K. Generation of transgenic plants expressing plasma membrane-bound antibodies to the environmental pollutant microcystin-LR. Transgenic Res. 2011, 20, 701–707. [Google Scholar] [CrossRef]
- Makvandi-Nejad, S.; McLean, M.D.; Hirama, T.; Almquist, K.C.; Mackenzie, C.R.; Hall, J.C. Transgenic tobacco plants expressing a dimeric single-chain variable fragment (scfv) antibody against Salmonella enterica serotype Paratyphi B. Transgenic Res. 2005, 14, 785–792. [Google Scholar] [CrossRef]
- Ramirez, N.; Rodriguez, M.; Ayala, M.; Cremata, J.; Perez, M.; Martinez, A.; Linares, M.; Hevia, Y.; Paez, R.; Valdes, R.; et al. Expression and characterization of an anti-(hepatitis B surface antigen) glycosylated mouse antibody in transgenic tobacco (Nicotiana tabacum) plants and its use in the immunopurification of its target antigen. Biotechnol. Appl. Biochem. 2003, 38, 223–230. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wanczyk, H.; Barker, T.; Rood, D.; Zapata, D.I.; Howell, A.R.; Richardson, S.K.; Zinckgraf, J.; Marusov, G.P.; Lynes, M.A.; Silbart, L.K. Cloning and Characterization of a Hybridoma Secreting a 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)-Specific Monoclonal Antibody and Recombinant F(ab). Toxins 2013, 5, 568-589. https://doi.org/10.3390/toxins5030568
Wanczyk H, Barker T, Rood D, Zapata DI, Howell AR, Richardson SK, Zinckgraf J, Marusov GP, Lynes MA, Silbart LK. Cloning and Characterization of a Hybridoma Secreting a 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)-Specific Monoclonal Antibody and Recombinant F(ab). Toxins. 2013; 5(3):568-589. https://doi.org/10.3390/toxins5030568
Chicago/Turabian StyleWanczyk, Heather, Tolga Barker, Debra Rood, Daniel I. Zapata, Amy R. Howell, Stewart K. Richardson, John Zinckgraf, Gregory P. Marusov, Michael A. Lynes, and Lawrence K. Silbart. 2013. "Cloning and Characterization of a Hybridoma Secreting a 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)-Specific Monoclonal Antibody and Recombinant F(ab)" Toxins 5, no. 3: 568-589. https://doi.org/10.3390/toxins5030568