The Cytotoxic Necrotizing Factor 1 from E. Coli: A Janus Toxin Playing with Cancer Regulators



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The CNF1 Protein: Structure and Activity
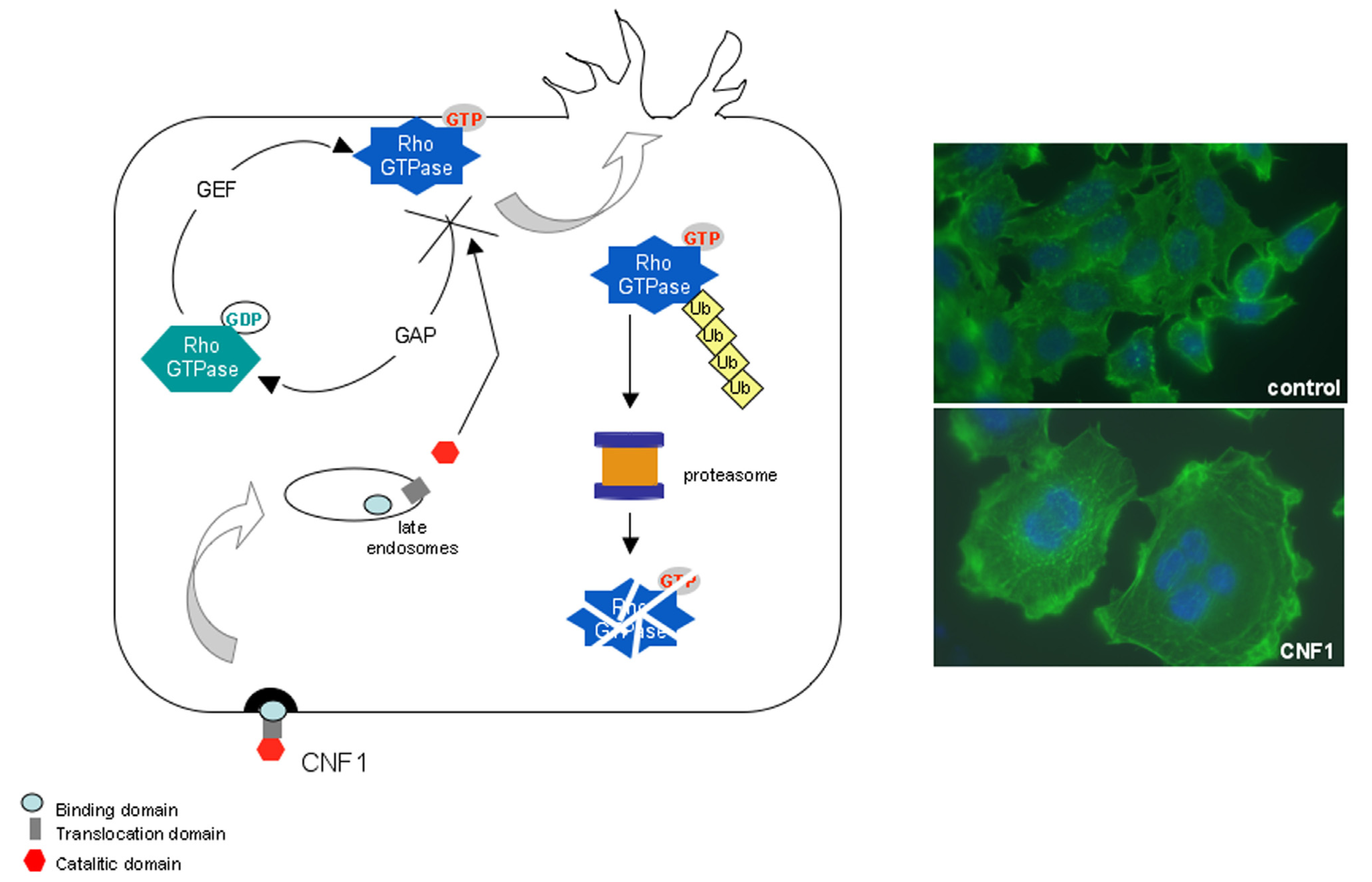
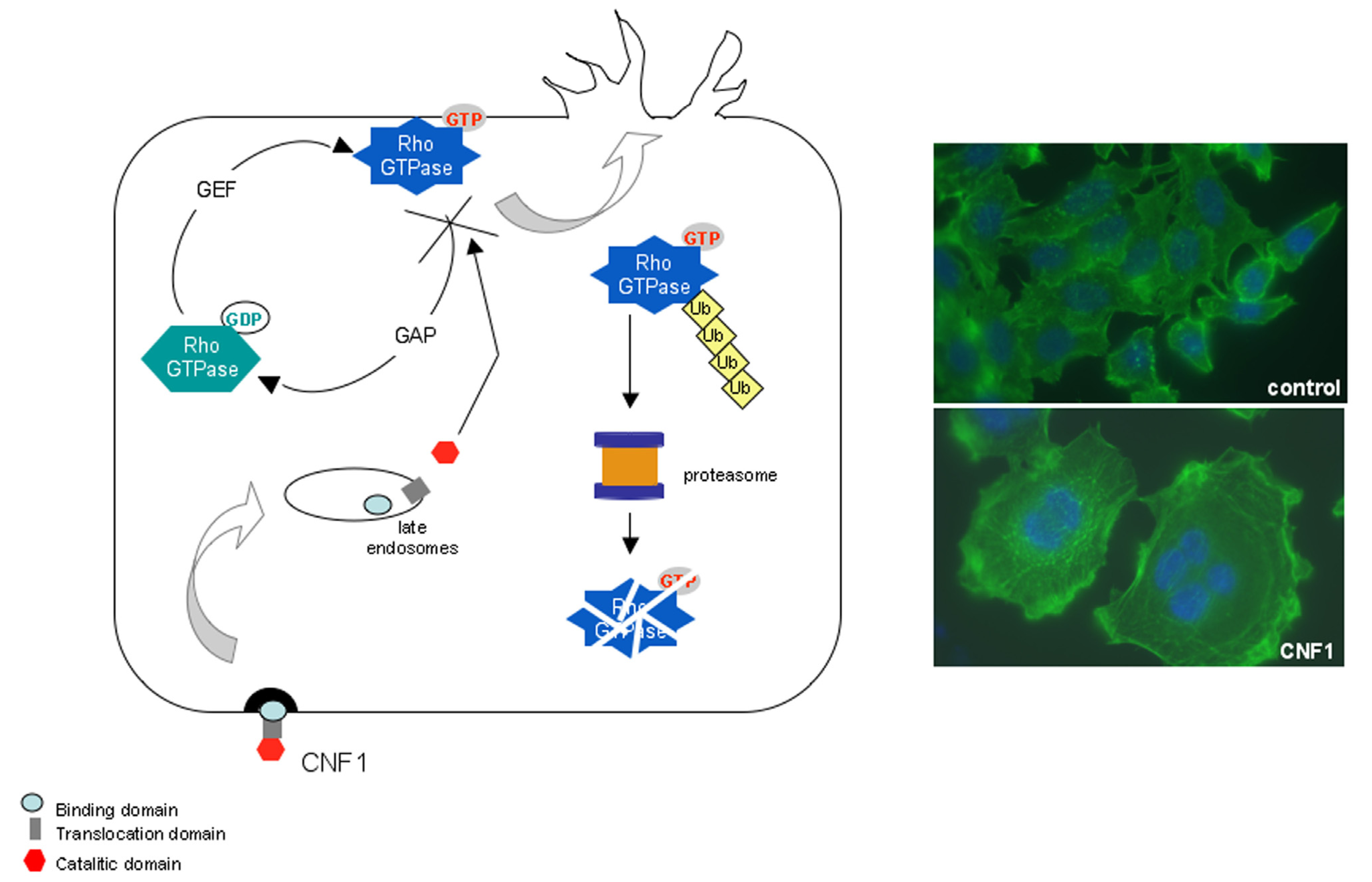
3. CNF1 Provokes Rho GTPase-Dependent Cellular Effects
4. CNF1 Modifies Mitochondrial Architecture and Hinders Apoptosis via the Pro-Inflammatory Akt/IKK/NF-kB Pathway
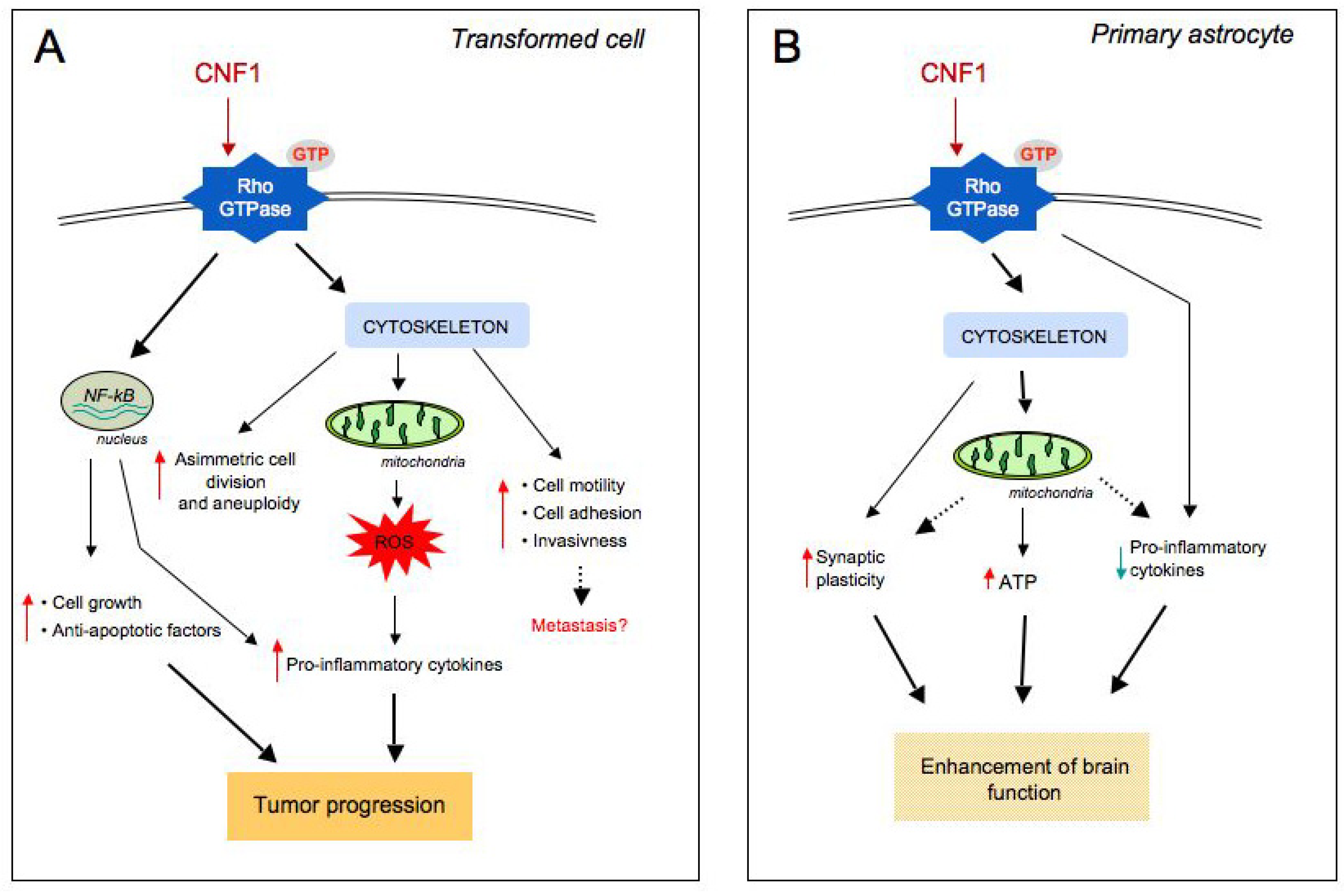
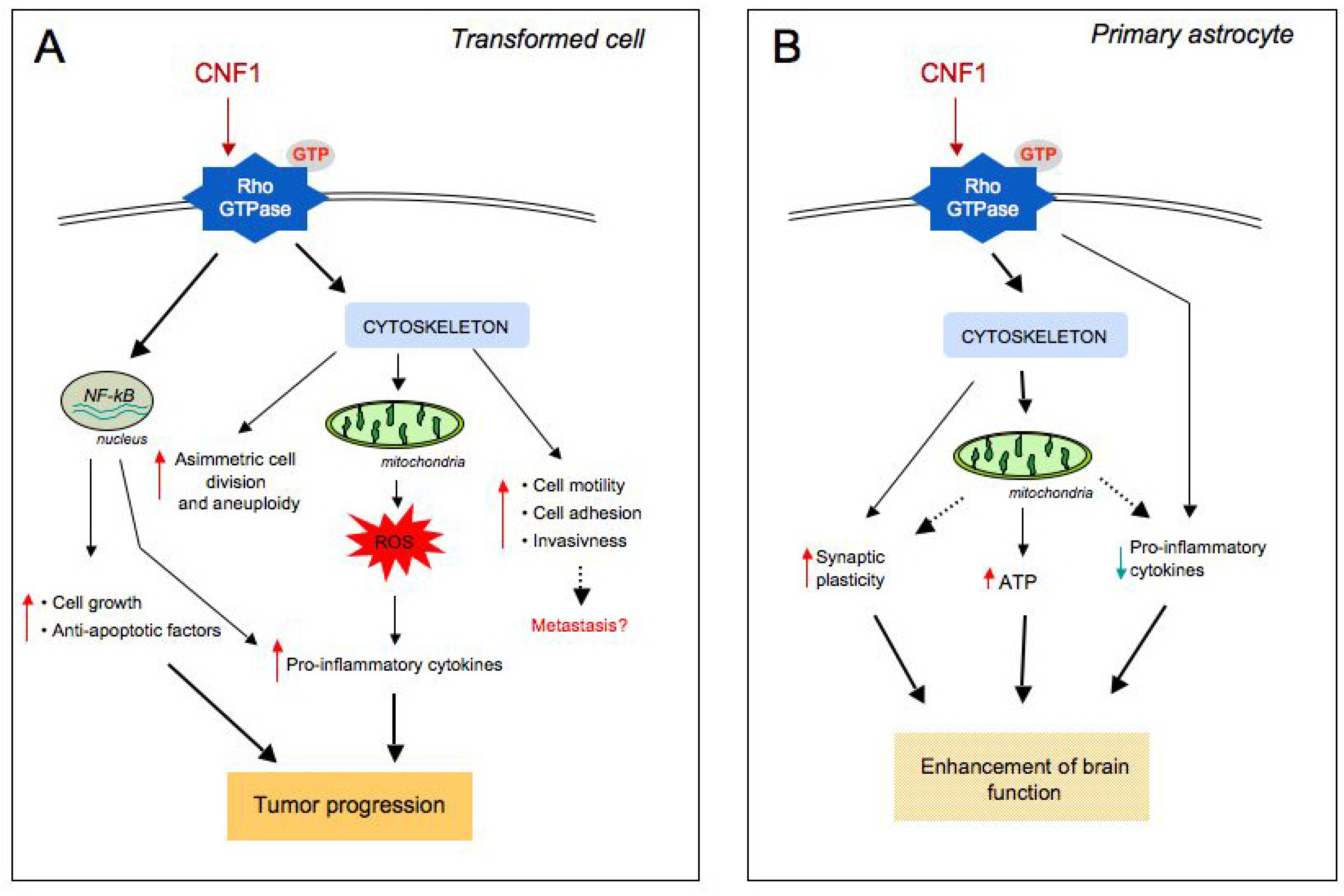
5. Evidence of a Link between CNF1 and Cancer in Humans
6. Conclusions: CNF1, a Janus Toxin Playing with Cell Regulation
Acknowledgments
Conflicts of Interest
References
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef]
- Ley, R.E.; Hamady, M.; Lozupone, C.; Turnbaugh, P.J.; Ramey, R.R.; Bircher, J.S.; Schlegel, M.L.; Tucker, T.A.; Schrenzel, M.D.; Knight, R.; Gordon, J.I. Evolution of mammals and their gut microbes. Science 2008, 320, 1647–1651. [Google Scholar] [CrossRef]
- Pop, M.; Deboy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic analysis of the human distal gut microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef]
- Plottel, C.S.; Blaser, M.J. Microbiome and malignancy. Cell Host Microbe 2011, 10, 324–335. [Google Scholar] [CrossRef]
- Sobhani, I.; Tap, J.; Roudot-Thoraval, F.; Roperch, J.P.; Letulle, S.; Langella, P.; Corthier, G.; Tran Van Nhieu, J.; Furet, J.P. Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS One 2011, 6, e16393. [Google Scholar] [CrossRef]
- Shanahan, F. The host-microbe interface within the gut. Best Pract. Res. Clin. Gastroenterol. 2002, 16, 915–931. [Google Scholar] [CrossRef]
- Lax, A.J.; Thomas, W. How bacteria could cause cancer: One step at a time. Trends Microbiol. 2002, 10, 293–299. [Google Scholar] [CrossRef]
- Karin, M.; Greten, F.R. NF-B: Linking inflammation and immunity to cancer development and progression. Nat. Rev. 2005, 5, 749–759. [Google Scholar] [CrossRef]
- Farinati, F.; Cardin, R.; Degan, P.; Rugge, M.; Di Mario, F.; Bonvicini, P.; Naccarato, R. Oxidative DNA damage accumulation in gastric carcinogenesis. Gut 1998, 42, 351–356. [Google Scholar] [CrossRef]
- Obst, B.; Wagner, S.; Sewing, K.; Beil, W. Helicobacter pylori causes DNA damage in gastric epithelial cells. Carcinogenesis 2000, 21, 1111–1115. [Google Scholar] [CrossRef]
- Hofseth, L.J.; Wargovich, M.J. Inflammation, cancer, and targets of ginseng. J. Nutr. 2007, 137, 183S–185S. [Google Scholar]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Morrison, W.B. Inflammation and cancer: A comparative view. J. Vet. Intern. Med. 2012, 26, 18–31. [Google Scholar] [CrossRef]
- Kaper, J.B.; Nataro, J.P.; Mobley, H.L. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2004, 2, 123–140. [Google Scholar] [CrossRef]
- Buc, E.; Dubois, D.; Sauvanet, P.; Raisch, J.; Delmas, J.; Darfeuille-Michaud, A.; Pezet, D.; Bonnet, R. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLoS One 2013, 8, e56964. [Google Scholar] [CrossRef]
- Boquet, P. The cytotoxic necrotizing factor 1 (CNF1) from Escherichia coli. Toxicon 2001, 39, 1673–1680. [Google Scholar] [CrossRef]
- Petkovsek, Z.; Elersic, K.; Guaina, M.; Zgur-Bertok, D.; Starcic Erjavec, M. Virulence potential of Escherichia coli isolates from skin and soft tissue infections. J. Clin Microbiol. 2009, 47, 1811–1817. [Google Scholar] [CrossRef]
- Caprioli, A.; Falbo, V.; Roda, L.G.; Ruggeri, F.M.; Zona, C. Partial purification and characterization of an Escherichia coli toxic factor that induces morphological cell alterations. Infect Immun. 1983, 39, 1300–1306. [Google Scholar]
- Caprioli, A.; Donelli, G.; Falbo, V.; Possenti, R.; Roda, L.G.; Roscetti, G.; Ruggeri, F.M. A cell division-active protein from E. coli. Biochem. Biophys. Res. Commun. 1984, 118, 587–593. [Google Scholar] [CrossRef]
- Lemichez, E.; Flatau, G.; Bruzzone, M.; Boquet, P.; Gauthier, M. Molecular localization of the Escherichia coli cytotoxic necrotizing factor CNF1 cell-binding and catalytic domains. Mol. Microbiol. 1997, 24, 1061–1070. [Google Scholar]
- Fabbri, A.; Gauthier, M.; Boquet, P. The 5' region of cnf1 harbours a translational regulatory mechanism for CNF1 synthesis and encodes the cell-binding domain of the toxin. Mol. Microbiol. 1999, 33, 108–118. [Google Scholar] [CrossRef]
- Falbo, V.; Pace, T.; Picci, L.; Pizzi, E.; Caprioli, A. Isolation and nucleotide sequence of the gene encoding cytotoxic necrotizing factor 1 of Escherichia coli. Infect. Immun. 1993, 61, 4909–4914. [Google Scholar]
- Pei, S.; Doye, A.; Boquet, P. Mutation of specific acidic residues of the CNF1 T domain into lysine alters cell membrane translocation of the toxin. Mol. Microbiol. 2001, 41, 1237–1247. [Google Scholar] [CrossRef]
- Kim, K.J.; Chung, J.W.; Kim, K.S. 67-kDa laminin receptor promotes internalization of cytotoxic necrotizing factor 1-expressing Escherichia coli K1 into human brain microvascular endothelial cells. J. Biol. Chem. 2005, 280, 1360–1368. [Google Scholar]
- Blumenthal, B.; Hoffmann, C.; Aktories, K.; Backert, S.; Schmidt, G. The cytotoxic necrotizing factors from Yersinia pseudotuberculosis and from Escherichia coli bind to different cellular receptors but take the same route to the cytosol. Infect. Immun. 2007, 75, 3344–3353. [Google Scholar] [CrossRef]
- Contamin, S.; Galmiche, A.; Doye, A.; Flatau, G.; Benmerah, A.; Boquet, P. The p21 Rho-activating toxin cytotoxic necrotizing factor 1 is endocytosed by a clathrin-independent mechanism and enters the cytosol by an acidic-dependent membrane translocation step. Mol Biol Cell 2000, 11, 1775–1787. [Google Scholar] [CrossRef]
- Knust, Z.; Blumenthal, B.; Aktories, K.; Schmidt, G. Cleavage of Escherichia coli cytotoxic necrotizing factor 1 is required for full biologic activity. Infect. Immun. 2009, 77, 1835–1841. [Google Scholar] [CrossRef]
- Etienne-Manneville, S.; Hall, A. Rho GTPases in cell biology. Nature 2002, 420, 629–635. [Google Scholar] [CrossRef]
- Flatau, G.; Lemichez, E.; Gauthier, M.; Chardin, P.; Paris, S.; Fiorentini, C.; Boquet, P. Toxin-induced activation of the G protein p21 Rho by deamidation of glutamine. Nature 1997, 387, 729–33. [Google Scholar] [CrossRef]
- Schmidt, G.; Sher, P.; Wilm, M.; Selzer, J.; Mann, M.; Aktories, K. Gln 63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor-1. Nature 1997, 387, 725–729. [Google Scholar] [CrossRef]
- Lerm, M.; Selzer, J.; Hoffmeyer, A.; Rapp, U.R.; Aktories, K.; Schmidt, G. Deamidation of Cdc42 and Rac by Escherichia coli cytotoxic necrotizing factor 1: Activation of the C-Jun N-terminal kinase in HeLa cells. Infect. Immun. 1999, 67, 496–503. [Google Scholar]
- Rittinger, K.; Walker, P.A.; Eccleston, J.F.; Nurmahomed, K.; Owen, D.; Laue, E.; Gamblin, S.J.; Smerdon, S.J. Crystal structure of a small G protein in complex with the GTPase-activating protein rhoGAP. Nature 1997, 388, 693–697. [Google Scholar] [CrossRef]
- Doye, A.; Mettouchi, A.; Bossis, G.; Clément, R.; Buisson-Touati, C.; Flatau, G.; Gagnoux, L.; Piechaczyk, M.; Boquet, P.; Lemichez, E. CNF1 exploits the ubiquitin-proteasome machinery to restrict Rho GTPase activation for bacterial host cell invasion. Cell 2002, 111, 553–564. [Google Scholar] [CrossRef]
- Weissman, A.M. Themes and variations on ubiquitylation. Nat. Rev. Mol. Cell Biol. 2001, 2, 169–178. [Google Scholar] [CrossRef]
- Marín, I. Animal HECT ubiquitin ligases: Evolution and functional implications. BMC Evol. Biol. 2010, 10, 56. [Google Scholar] [CrossRef]
- Oberoi, T.K.; Dogan, T.; Hocking, J.C.; Scholz, R.P.; Mooz, J.; Anderson, C.L.; Karreman, C.; Meyer zu Heringdorf, D.; Schmidt, G.; Ruonala, M.; et al. IAPs regulate the plasticity of cell migration by directly targeting Rac1 for degradation. EMBO J. 2012, 31, 14–28. [Google Scholar]
- Torrino, S.; Visvikis, O.; Doye, A.; Boyer, L.; Stefani, C.; Munro, P.; Bertoglio, J.; Gacon, G.; Mettouchi, A.; Lemichez, E. The E3 ubiquitin-ligase HACE1 catalyzes the ubiquitylation of active Rac1. Dev. Cell 2011, 21, 959–965. [Google Scholar] [CrossRef]
- Castillo-Lluva, S.; Tan, C.T.; Daugaard, M.; Sorensen, P.H.; Malliri, A. The tumour suppressor HACE1 controls cell migration by regulating Rac1 degradation. Oncogene 2013, 32, 1735–1742. [Google Scholar] [CrossRef]
- Boyer, L.; Turchi, L.; Desnues, B.; Doye, A.; Ponzio, G.; Mege, J.L.; Yamashita, M.; Zhang, Y.E.; Bertoglio, J.; Flatau, G.; et al. CNF1-induced ubiquitylation and proteasome destruction of activated RhoA is impaired in Smurf1−/− cells. Mol. Biol. Cell 2006, 17, 2489–2497. [Google Scholar] [CrossRef]
- Fiorentini, C.; Arancia, G.; Caprioli, A.; Falbo, V.; Ruggeri, F.M.; Donelli, G. Cytoskeletal changes induced in HEp-2 cells by the cytotoxic necrotizing factor of Escherichia coli. Toxicon 1988, 26, 1047–1056. [Google Scholar] [CrossRef]
- Fiorentini, C.; Donelli, G.; Matarrese, P.; Fabbri, A.; Paradisi, S.; Boquet, P. Escherichia coli cytotoxic necrotizing factor 1: Evidence for induction of actin assembly by constitutive activation of the p21 Rho GTPase. Infect. Immun. 1995, 63, 3936–3944. [Google Scholar]
- Fiorentini, C.; Falzano, L.; Fabbri, A.; Stringaro, A.; Logozzi, M.; Travaglione, S.; Contamin, S.; Arancia, G.; Malorni, W.; Fais, S. Activation of rho GTPases by cytotoxic necrotizing factor 1 induces macropinocytosis and scavenging activity in epithelial cells. Mol. Biol. Cell 2001, 12, 2061–2073. [Google Scholar] [CrossRef]
- Falzano, L.; Fiorentini, C.; Donelli, G.; Michel, E.; Kocks, C.; Cossart, P.; Cabanié, L.; Oswald, E.; Boquet, P. Induction of phagocytic behaviour in human epithelial cells by Escherichia coli cytotoxic necrotizing factor type 1. Mol. Microbiol. 1993, 9, 1247–1254. [Google Scholar] [CrossRef]
- Visvikis, O.; Boyer, L.; Torrino, S.; Doye, A.; Lemonnier, M.; Lorès, P.; Rolando, M.; Flatau, G.; Mettouchi, A.; Bouvard, D.; et al. Escherichia coli producing CNF1 toxin hijacks Tollip to trigger Rac1-dependent cell invasion. Traffic 2011, 12, 579–590. [Google Scholar] [CrossRef]
- Fu, Y.; Galán, J.E. A salmonella protein antagonizes Rac-1 and Cdc42 to mediate host-cell recovery after bacterial invasion. Nature 1999, 401, 293–297. [Google Scholar] [CrossRef]
- Caygill, C.P.; Braddick, M.; Hill, M.J.; Knowles, R.L.; Sharp, J.C. The association between typhoid carriage, typhoid infection and subsequent cancer at a number of sites. Eur. J. Cancer Prev. 1995, 4, 187–193. [Google Scholar] [CrossRef]
- Schlumberger, M.C.; Hardt, W.D. Triggered phagocytosis by Salmonella: Bacterial molecular mimicry of RhoGTPase activation/deactivation. Curr. Top. Microbiol. Immunol. 2005, 291, 29–42. [Google Scholar] [CrossRef]
- Ridley, A.J.; Paterson, H.F.; Johnston, C.L.; Diekmann, D.; Hall, A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 1992, 70, 401–410. [Google Scholar] [CrossRef]
- Malorni, W.; Fiorentini, C. Is the Rac GTPase-activating toxin CNF1 a smart hijacker of host cell fate? FASEB J. 2006, 20, 606–609. [Google Scholar] [CrossRef]
- Fiorentini, C.; Matarrese, P.; Straface, E.; Falzano, L.; Donelli, G.; Boquet, P.; Malorni, W. Rho-dependent cell spreading activated by E.coli cytotoxic necrotizing factor 1 hinders apoptosis in epithelial cells. Cell Death Differ. 1998, 5, 921–929. [Google Scholar]
- Fiorentini, C.; Matarrese, P.; Straface, E.; Falzano, L.; Fabbri, A.; Donelli, G.; Cossarizza, A.; Boquet, P.; Malorni, W. Toxin-induced activation of Rho GTP-binding protein increases Bcl-2 expression and influences mitochondrial homeostasis. Exp. Cell Res. 1998, 242, 341–350. [Google Scholar] [CrossRef]
- Duensing, A.; Duensing, S. Centrosomes, polyploidy and cancer. Adv. Exp. Med. Biol. 2010, 676, 93–103. [Google Scholar] [CrossRef]
- Godinho, S.A.; Kwon, M.; Pellman, D. Centrosomes and cancer: How cancer cells divide with too many centrosomes. Cancer Metastasis Rev. 2009, 28, 85–98. [Google Scholar] [CrossRef]
- Falzano, L.; Filippini, P.; Travaglione, S.; Miraglia, A.G.; Fabbri, A.; Fiorentini, C. Escherichia coli cytotoxic necrotizing factor 1 blocks cell cycle G2/M transition in uroepithelial cells. Infect. Immun. 2006, 74, 3765–3772. [Google Scholar] [CrossRef]
- Travaglione, S.; Messina, G.; Fabbri, A.; Falzano, L.; Giammarioli, A.M.; Grossi, M.; Rufini, S.; Fiorentini, C. Cytotoxic necrotizing factor 1 hinders skeletal muscle differentiation in vitro by perturbing the activation/deactivation balance of Rho GTPases. Cell Death Differ. 2005, 12, 78–86. [Google Scholar] [CrossRef]
- Nougayrède, J.P.; Taieb, F.; De Rycke, J.; Oswald, E. Cyclomodulins: Bacterial effectors that modulate the eukaryotic cell cycle. Trends Microbiol. 2005, 13, 103–110. [Google Scholar] [CrossRef]
- Oswald, E.; Nougayrède, J.P.; Taieb, F.; Sugai, M. Bacterial toxins that modulate host cell-cycle progression. Curr. Opin. Microbiol. 2005, 8, 83–91. [Google Scholar]
- Miraglia, A.G.; Travaglione, S.; Meschini, S.; Falzano, L.; Matarrese, P.; Quaranta, M.G.; Viora, M.; Fiorentini, C.; Fabbri, A. Cytotoxic necrotizing factor 1 prevents apoptosis via the Akt/IkappaB kinase pathway: Role of nuclear factor-kappaB and Bcl-2. Mol. Biol. Cell. 2007, 18, 2735–2744. [Google Scholar] [CrossRef]
- Marx, J. Cancer research. Inflammation and cancer: The link grows stronger. Science 2004, 306, 966–968. [Google Scholar] [CrossRef]
- Boyer, L.; Travaglione, S.; Falzano, L.; Gauthier, N.C.; Popoff, M.R.; Lemichez, E.; Fiorentini, C.; Fabbri, A. Rac GTPase instructs nuclear factor-kappaB activation by conveying the SCF complex and IkBalpha to the ruffling membranes. Mol. Biol. Cell 2004, 15, 1124–1133. [Google Scholar]
- Falzano, L.; Quaranta, M.G.; Travaglione, S.; Filippini, P.; Fabbri, A.; Viora, M.; Donelli, G.; Fiorentini, C. Cytotoxic necrotizing factor 1 enhances reactive oxygen species-dependent transcription and secretion of proinflammatory cytokines in human uroepithelial cells. Infect. Immun. 2003, 71, 4178–4181. [Google Scholar]
- Morán, M.; Moreno-Lastres, D.; Marín-Buera, L.; Arenas, J.; Martín, M.A.; Ugalde, C. Mitochondrial respiratory chain dysfunction: Implications in neurodegeneration. Free Radic. Biol. Med. 2012, 53, 595–609. [Google Scholar] [CrossRef]
- Alirol, E.; Martinou, J.C. Mitochondria and cancer: Is there a morphological connection? Oncogene 2006, 25, 4706–4716. [Google Scholar] [CrossRef]
- Abo, A.; Pick, E.; Hall, A.; Totty, N.; Teahan, C.G.; Segal, A.W. Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature 1991, 353, 668–670. [Google Scholar] [CrossRef]
- Sulciner, D.J.; Irani, K.; Yu, Z.X.; Ferrans, V.J.; Goldschmidt-Clermont, P.; Finkel, T. Rac1 regulates a cytokine-stimulated, redox-dependent pathway necessary for NF-kappaB activation. Mol. Cell Biol. 1996, 16, 7115–7121. [Google Scholar]
- Perona, R.; Montaner, S.; Saniger, L.; Sánchez-Pérez, I.; Bravo, R.; Lacal, J.C. Activation of the nuclear factor-kappaB by Rho, CDC42, and Rac-1 proteins. Genes Dev. 1997, 11, 463–475. [Google Scholar] [CrossRef]
- Falzano, L.; Rivabene, R.; Santini, M.T.; Fabbri, A.; Fiorentini, C. An Escherichia coli cytotoxin increases superoxide anion generation via rac in epithelial cells. Biochem. Biophys. Res. Commun. 2001, 283, 1026–1030. [Google Scholar] [CrossRef]
- Munro, P.; Flatau, G.; Doye, A.; Boyer, L.; Oregioni, O.; Mege, J.L.; Landraud, L.; Lemichez, E. Activation and proteasomal degradation of rho GTPases by cytotoxic necrotizing factor-1 elicit a controlled inflammatory response. J. Biol. Chem. 2004, 279, 35849–35857. [Google Scholar]
- Thomas, W.; Ascott, Z.K.; Harmey, D.; Slice, L.W.; Rozengurt, E.; Lax, A.J. Cytotoxic necrotizing factor from Escherichia coli induces RhoA-dependent expression of the cyclooxygenase-2 Gene. Infect. Immun. 2001, 69, 6839–6845. [Google Scholar] [CrossRef]
- Ristimaki, A. Cyclooxygenase 2: From inflammation to carcinogenesis. Novartis Found. Symp. 2004, 256, 215–221. [Google Scholar] [CrossRef]
- Darfeuille-Michaud, A.; Boudeau, J.; Bulois, P.; Neut, C.; Glasser, A.L.; Barnich, N.; Bringer, M.A.; Swidsinski, A.; Beaugerie, L.; Colombel, J.F. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 2004, 127, 412–421. [Google Scholar] [CrossRef]
- Martin, H.M.; Campbell, B.J.; Hart, C.A.; Mpofu, C.; Nayar, M.; Singh, R.; Englyst, H.; Williams, H.F.; Rhodes, J.M. Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology 2004, 127, 80–93. [Google Scholar] [CrossRef]
- Tjalsma, H.; Boleij, A.; Marchesi, J.R.; Dutilh, B.E. A bacterial driver-passenger model for colorectal cancer: Beyond the usual suspects. Nat. Rev. Microbiol. 2012, 10, 575–582. [Google Scholar] [CrossRef]
- Malchiodi-Albedi, F.; Paradisi, S.; Di Nottia, M.; Simone, D.; Travaglione, S.; Falzano, L.; Guidotti, M.; Frank, C.; Cutarelli, A.; Fabbri, A.; et al. CNF1 improves astrocytic ability to support neuronal growth and differentiation in vitro. PLoS One 2012, 7, e34115. [Google Scholar] [CrossRef]
- De Filippis, B.; Fabbri, A.; Simone, D.; Canese, R.; Ricceri, L.; Malchiodi-Albedi, F.; Laviola, G.; Fiorentini, C. Modulation of RhoGTPases improves the behavioral phenotype and reverses astrocytic deficits in a mouse model of Rett syndrome. Neuroopsychopharmacology 2012, 37, 1152–1163. [Google Scholar] [CrossRef]
- Loizzo, S.; Rimondini, R.; Travaglione, S.; Fabbri, A.; Guidotti, M.; Ferri, A.; Campana, G.; Fiorentini, C. CNF1 Increases brain energy level, counteracts neuroinflammatory markers and rescues cognitive deficits in a murine model of Alzheimer’s disease. PLoS One 2013, 8, e65898. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fabbri, A.; Travaglione, S.; Ballan, G.; Loizzo, S.; Fiorentini, C. The Cytotoxic Necrotizing Factor 1 from E. Coli: A Janus Toxin Playing with Cancer Regulators. Toxins 2013, 5, 1462-1474. https://doi.org/10.3390/toxins5081462
Fabbri A, Travaglione S, Ballan G, Loizzo S, Fiorentini C. The Cytotoxic Necrotizing Factor 1 from E. Coli: A Janus Toxin Playing with Cancer Regulators. Toxins. 2013; 5(8):1462-1474. https://doi.org/10.3390/toxins5081462
Chicago/Turabian StyleFabbri, Alessia, Sara Travaglione, Giulia Ballan, Stefano Loizzo, and Carla Fiorentini. 2013. "The Cytotoxic Necrotizing Factor 1 from E. Coli: A Janus Toxin Playing with Cancer Regulators" Toxins 5, no. 8: 1462-1474. https://doi.org/10.3390/toxins5081462