Soluble T Cell Receptor Vβ Domains Engineered for High-Affinity Binding to Staphylococcal or Streptococcal Superantigens

Abstract
:1. Overview
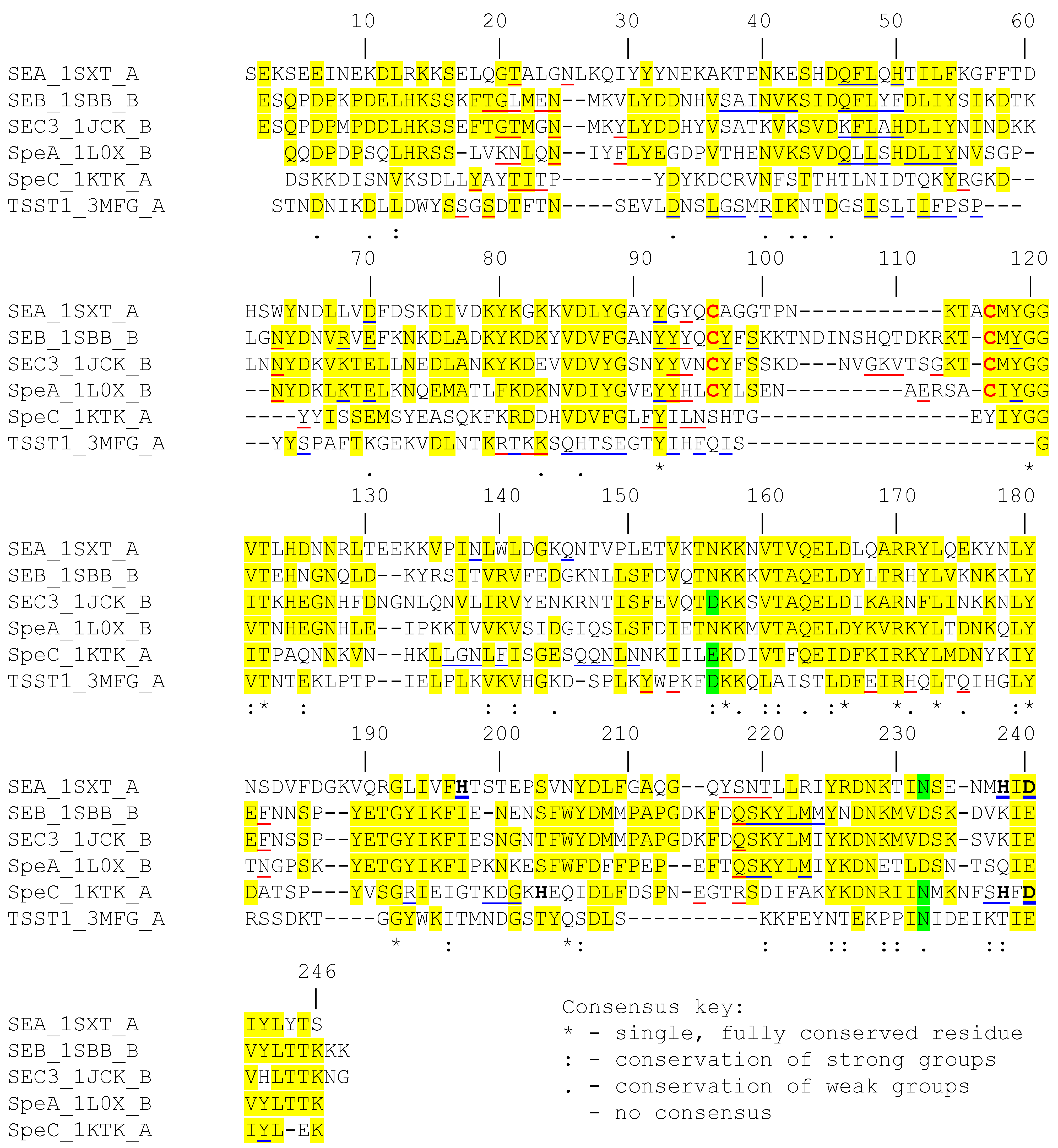
2. Structural Features of the Superantigens



{kind=link}
{kind=link}
{kind=link}
{kind=link}
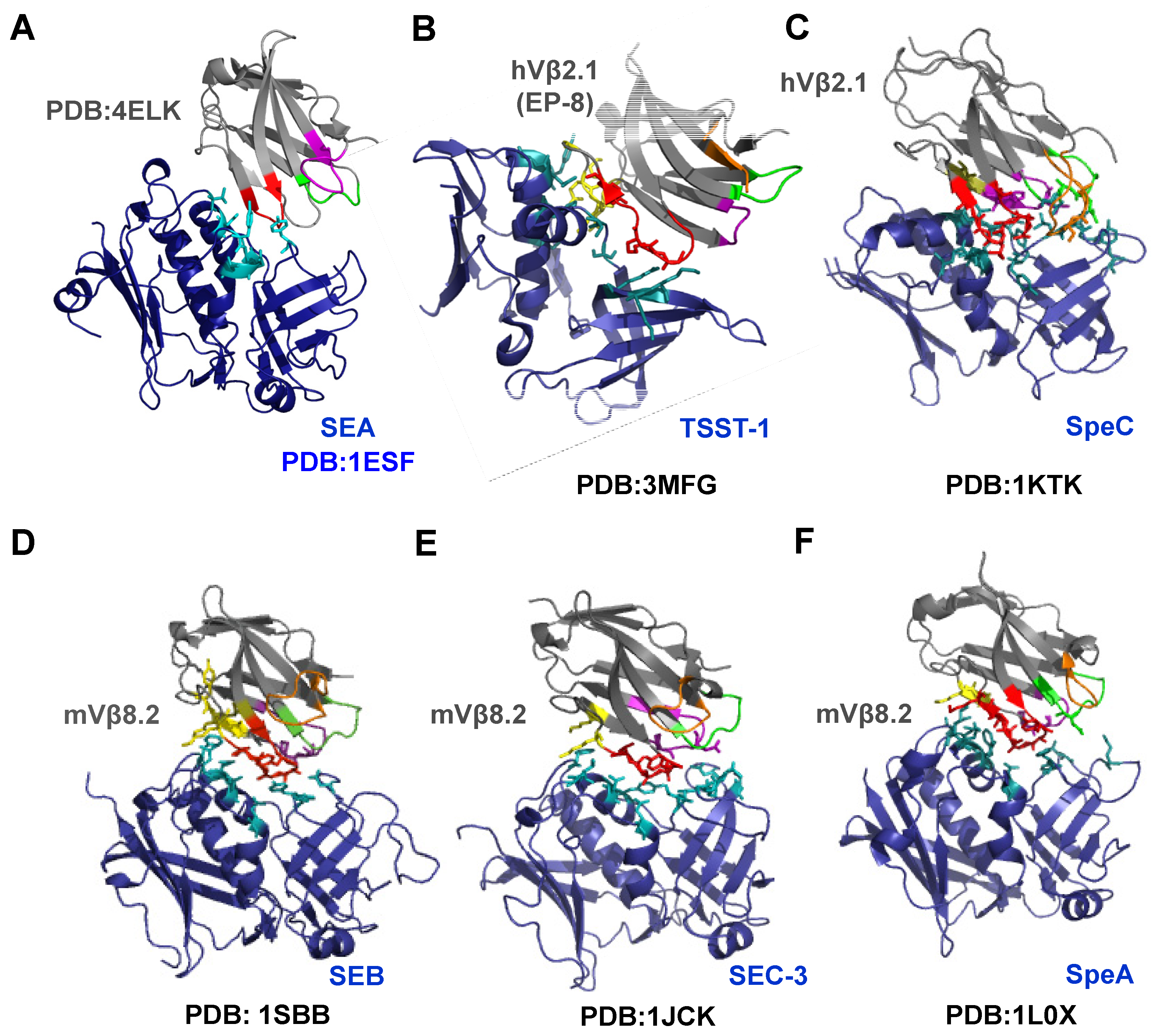{kind=link}
| Organism | SAg | Crystal Structure (PDB code, ligand) | Year | Reference |
|---|---|---|---|---|
| S. aureus | SEA | 1ESF (co-crystallized with Cd2+) | 1995 | [53] |
| S. aureus | 1SXT (co-crystallized with Zn2+) | 1996 | [54] | |
| S. aureus | SEB | 3SEB | 1998 | [55] |
| S. aureus | 1SBB (co-crystallized with mVβ8.2) | 1998 | [39] | |
| S. aureus | 3R8B (co-crystallized with affinity matured mVβ8.2 mutant G5-8) | 2011 | [56] | |
| S. aureus | SEC3 | 1CK1 (co-crystallized with Zn2+) | 2002 | [57] |
| S. aureus | 1JCK (co-crystallized with mVβ8.2) | 1996 | [40] | |
| S. aureus | 2AQ3 (co-crystallized with affinity matured mVβ8.2 mutant L2CM) | 2005 | [58] | |
| S. aureus | TSST1 | 2QIL | 1996 | [59] |
| S. aureus | 2IJ0 (co-crystallized with affinity matured hVβ2.1 mutant D10) | 2007 | [42] | |
| S. aureus | 3MFG (co-crystallized with hVβ2.1 stabilized wild-type EP-8) | 2011 | [56] | |
| S. pyogenes | SpeA | 1FNU (co-crystallized with Cd2+) | 2000 | [60] |
| 1FNV (co-crystallized with Cd2+) | ||||
| 1FNW (co-crystallized with Cd2+) | ||||
| S. pyogenes | 1L0X (co-crystallized with mVβ8.2) | 2002 | [41] | |
| 1L0Y (co-crystallized with mVβ8.2 and Zn2+) | ||||
| S. pyogenes | SpeC | 1AN8 | 1997 | [61] |
| S. pyogenes | 1KTK (co-crystallized with hVβ2.1) | 2002 | [41] |
3. Engineering High-Affinity T Cell Receptor Vβ Domains against Superantigens SEA, SEB, SEC3, TSST-1, SpeA, and SpeC
| Organism | SAg | WT Vβ | High affinity Vβ | Improvement in affinity (fold) | References | ||
|---|---|---|---|---|---|---|---|
| Name | Affinity (µM) | Name | Affinity (pM) | ||||
| S. aureus | SEA | Human Vβ22 | 100 | FL | 4,000 | 25,000 | [37] |
| S. aureus | SEB | Mouse Vβ8.2 | 144 | G5-8 | 50 | 2,880,000 | [33,66] |
| S. aureus | SEC3 | Mouse Vβ8.2 | 3 | L3 | 3,000 | 1,000 | [36,66] |
| S. aureus | TSST1 | Human Vβ2.1 | 2.3 | D10 | 180 | 13,000 | [34] |
| S. pyogenes | SpeA | Mouse Vβ8.2 | 6 | KKR | 270 | 22,000 | [35,66] |
| S. pyogenes | SpeC | Human Vβ2.1 | 20 | HG_FI | 500 | 40,000 | [41,69] |


4. Topology of Vβ:Superantigen Interactions

5. Structural Basis of High-Affinity and Specificity of the Vβ:SAg Interactions
6. High-Affinity Vβ Domains as Neutralizing Agents
Acknowledgements
Conflicts of Interest
References
- Marrack, P.; Kappler, J. The staphylococcal enterotoxins and their relatives. Science 1990, 248, 705–711. [Google Scholar]
- Spaulding, A.R.; Salgado-Pabón, W; Kohler, P.L.; Horswill, A.R.; Leung, D.Y.; Schlievert, P.M. Staphylococcal and streptococcal superantigen exotoxins. Clin. Microbiol. Rev. 2013, 26, 422–447. [Google Scholar] [CrossRef]
- Krakauer, T.; Stiles, B.G. The staphylococcal enterotoxin (SE) family: SEB and siblings. Virulence 2013, 4, 759–773. [Google Scholar] [CrossRef]
- Dinges, M.M.; Orwin, P.M.; Schlievert, P.M. Exotoxins of Staphylococcus aureus. Clin. Microbiol. Rev. 2000, 13, 16–34. [Google Scholar] [CrossRef]
- McCormick, J.K.; Yarwood, J.M.; Schlievert, P.M. Toxic shock syndrome and bacterial superantigens: An update. Ann. Rev. Microbiol. 2001, 55, 77–104. [Google Scholar] [CrossRef]
- Li, H.; Llera, A.; Malchiodi, E.L.; Mariuzza, R.A. The structural basis of T cell activation by superantigens. Ann. Rev. Immunol. 1999, 17, 435–466. [Google Scholar]
- Baker, M.D.; Acharya, K.R. Superantigens: Structure-function relationships. Int. J. Med. Microbiol. 2004, 293, 529–537. [Google Scholar] [CrossRef]
- Papageorgiou, A.C.; Acharya, K.R. Microbial superantigens: From structure to function. Trends Microbiol. 2000, 8, 369–375. [Google Scholar] [CrossRef]
- Fraser, J.D.; Proft, T. The bacterial superantigen and superantigen-like proteins. Immunol. Rev. 2008, 225, 226–243. [Google Scholar] [CrossRef]
- Pless, D.D.; Ruthel, G.; Reinke, E.K.; Ulrich, R.G.; Bavari, S. Persistence of zinc-binding bacterial superantigens at the surface of antigen-presenting cells contributes to the extreme potency of these superantigens as T-cell activators. Infect. Immun. 2005, 73, 5358–5366. [Google Scholar] [CrossRef]
- Bavari, S.; Ulrich, R.G.; LeClaire, R.D. Cross-reactive antibodies prevent the lethal effects of Staphylococcus aureus superantigens. J. Infect. Dis. 1999, 180, 1365–1369. [Google Scholar] [CrossRef]
- Varshney, A.K.; Mediavilla, J.R.; Robiou, N.; Guh, A.; Wang, X.; Gialanella, P.; Levi, M.H.; Kreiswirth, B.N.; Fries, B.C. Diverse enterotoxin gene profiles among clonal complexes of Staphylococcus aureus isolates from the Bronx, New York. Appl. Environ. Microbiol. 2009, 75, 6839–6849. [Google Scholar] [CrossRef]
- Hu, D.L.; Omoe, K.; Inoue, F.; Kasai, T.; Yasujima, M.; Shinagawa, K.; Nakane, A. Comparative prevalence of superantigenic toxin genes in meticillin-resistant and meticillin-susceptible Staphylococcus aureus isolates. J. Med. Microbiol. 2008, 57, 1106–1112. [Google Scholar] [CrossRef]
- Lindsay, J.A.; Ruzin, A.; Ross, H.F.; Kurepina, N.; Novick, R.P. The gene for toxic shock toxin is carried by a family of mobile pathogenicity islands in Staphylococcus aureus. Mol. Microbiol. 1998, 29, 527–543. [Google Scholar]
- Novick, R.P.; Schlievert, P.; Ruzin, A. Pathogenicity and resistance islands of staphylococci. Microbes Infect. 2001, 3, 585–594. [Google Scholar] [CrossRef]
- Fitzgerald, J.R.; Monday, S.R.; Foster, T.J.; Bohach, G.A.; Hartigan, P.J.; Meaney, W.J.; Smyth, C.J. Characterization of a putative pathogenicity island from bovine Staphylococcus aureus encoding multiple superantigens. J. Bacteriol. 2001, 183, 63–70. [Google Scholar] [CrossRef]
- Betley, M.J.; Mekalanos, J.J. Staphylococcal enterotoxin A is encoded by phage. Science 1985, 229, 185–187. [Google Scholar]
- Derzelle, S.; Dilasser, F.; Duquenne, M.; Deperrois, V. Differential temporal expression of the staphylococcal enterotoxins genes during cell growth. Food Microbiol. 2009, 26, 896–904. [Google Scholar] [CrossRef]
- Bachert, C.; Gevaert, P.; Zhang, N.; van Zele, T.; Perez-Novo, C. Role of staphylococcal superantigens in airway disease. Chem. Immunol. Allergy 2007, 93, 214–236. [Google Scholar]
- Strandberg, K.L.; Rotschafer, J.H.; Vetter, S.M.; Buonpane, R.A.; Kranz, D.M.; Schlievert, P.M. Staphylococcal superantigens cause lethal pulmonary disease in rabbits. J. Infect. Dis. 2010, 202, 1690–1697. [Google Scholar] [CrossRef]
- Schlievert, P.M.; Case, L.C.; Strandberg, K.L.; Abrams, B.B.; Leung, D.Y. Superantigen profile of Staphylococcus aureus isolates from patients with steroid-resistant atopic dermatitis. Clin. Infect. Dis. 2008, 46, 1562–1567. [Google Scholar] [CrossRef]
- Macias, E.S.; Pereira, F.A.; Rietkerk, W.; Safai, B. Superantigens in dermatology. J. Am. Acad. Dermatol. 2011, 64, 455–472, quiz 473–454. [Google Scholar] [CrossRef]
- Bohach, G.A.; Fast, D.J.; Nelson, R.D.; Schlievert, P.M. Staphylococcal and streptococcal pyrogenic toxins involved in toxic shock syndrome and related illnesses. Crit. Rev. Microbiol. 1990, 17, 251–272. [Google Scholar] [CrossRef]
- Lappin, E.; Ferguson, A.J. Gram-positive toxic shock syndromes. Lancet Infect. Dis. 2009, 9, 281–290. [Google Scholar] [CrossRef]
- Dhodapkar, K.; Corbacioglu, S.; Chang, M.W.; Karpatkin, M.; DiMichele, D. Purpura fulminans caused by group A beta-hemolytic Streptococcus sepsis. J. Pediatr. 2000, 137, 562–567. [Google Scholar] [CrossRef]
- Tilahun, A.Y.; Holz, M.; Wu, T.T.; David, C.S.; Rajagopalan, G. Interferon gamma-dependent intestinal pathology contributes to the lethality in bacterial superantigen-induced toxic shock syndrome. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Tilahun, A.Y.; Karau, M.J.; Clark, C.R.; Patel, R.; Rajagopalan, G. The impact of tacrolimus on the immunopathogenesis of staphylococcal enterotoxin-induced systemic inflammatory response syndrome and pneumonia. Microbes Infect. 2012, 14, 528–536. [Google Scholar] [CrossRef]
- Kieke, M.C.; Sundberg, E.; Shusta, E.V.; Mariuzza, R.A.; Wittrup, K.D.; Kranz, D.M. High affinity T cell receptors from yeast display libraries block T cell activation by superantigens. J. Mol. Biol. 2001, 307, 1305–1315. [Google Scholar] [CrossRef]
- Garrison, L.; McDonnell, N. Etanercept: therapeutic use in patients with rheumatoid arthritis. Ann. Rheum. Dis. 1999, 58, 165–169. [Google Scholar]
- Combe, B. Update on the use of etanercept across a spectrum of rheumatoid disorders. Biologics 2008, 2, 165–173. [Google Scholar]
- Boder, E.T.; Wittrup, K.D. Yeast surface display for directed evolution of protein expression, affinity, and stability. Meth. Enzymol. 2000, 328, 430–444. [Google Scholar] [CrossRef]
- Boder, E.T.; Wittrup, K.D. Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 1997, 15, 553–557. [Google Scholar] [CrossRef]
- Buonpane, R.A.; Churchill, H.R.; Moza, B.; Sundberg, E.J.; Peterson, M.L.; Schlievert, P.M.; Kranz, D.M. Neutralization of staphylococcal enterotoxin B by soluble, high-affinity receptor antagonists. Nat. Med. 2007, 13, 725–729. [Google Scholar] [CrossRef]
- Buonpane, R.A.; Moza, B.; Sundberg, E.J.; Kranz, D.M. Characterization of T cell receptors engineered for high affinity against toxic shock syndrome toxin-1. J. Mol. Biol. 2005, 353, 308–321. [Google Scholar] [CrossRef]
- Wang, N.; Mattis, D.M.; Sundberg, E.J.; Schlievert, P.M.; Kranz, D.M. A single, engineered protein therapeutic agent neutralizes exotoxins from both Staphylococcus aureus and Streptococcus pyogenes. Clin. Vaccine Immunol. 2010, 17, 1781–1789. [Google Scholar] [CrossRef]
- Mattis, D.M.; Spaulding, A.R.; Chuang-Smith, O.N.; Sundberg, E.J.; Schlievert, P.M.; Kranz, D.M. Engineering a soluble high-affinity receptor domain that neutralizes staphylococcal enterotoxin C in rabbit models of disease. Protein Eng. Des. Sel. 2013, 26, 133–142. [Google Scholar] [CrossRef]
- Sharma, P.; Postel, S.; Sundberg, E.J.; Kranz, D.M. Characterization of the Staphylococcal enterotoxin A: Vβ receptor interaction using human receptor fragments engineered for high affinity. Protein Eng. Des. Sel. 2013, 26, 781–789. [Google Scholar] [CrossRef]
- John, C.C.; Niermann, M.; Sharon, B.; Peterson, M.L.; Kranz, D.M.; Schlievert, P.M. Staphylococcal toxic shock syndrome erythroderma is associated with superantigenicity and hypersensitivity. Clin. Infect. Dis. 2009, 49, 1893–1896. [Google Scholar] [CrossRef]
- Li, H.; Llera, A.; Tsuchiya, D.; Leder, L.; Ysern, X.; Schlievert, P.M.; Karjalainen, K.; Mariuzza, R.A. Three-dimensional structure of the complex between a T cell receptor beta chain and the superantigen staphylococcal enterotoxin B. Immunity 1998, 9, 807–816. [Google Scholar] [CrossRef]
- Fields, B.A.; Malchiodi, E.L.; Li, H.; Ysern, X.; Stauffacher, C.V.; Schlievert, P.M.; Karjalainen, K.; Mariuzza, R.A. Crystal structure of a T-cell receptor b-chain complexed with a superantigen. Nature 1996, 384, 188–192. [Google Scholar] [CrossRef]
- Sundberg, E.J.; Li, H.; Llera, A.S.; McCormick, J.K.; Tormo, J.; Schlievert, P.M.; Karjalainen, K.; Mariuzza, R.A. Structures of two streptococcal superantigens bound to TCR beta chains reveal diversity in the architecture of T cell signaling complexes. Structure 2002, 10, 687–699. [Google Scholar] [CrossRef]
- Moza, B.; Varma, A.K.; Buonpane, R.A.; Zhu, P.; Herfst, C.A.; Nicholson, M.J.; Wilbuer, A.K.; Seth, N.P.; Wucherpfennig, K.W.; McCormick, J.K.; et al. Structural basis of T-cell specificity and activation by the bacterial superantigen TSST-1. EMBO J. 2007, 26, 1187–1197. [Google Scholar] [CrossRef]
- Petersson, K.; Thunnissen, M.; Forsberg, G.; Walse, B. Crystal structure of a sea variant in complex with MHC class II reveals the ability of Sea to crosslink MHC molecules. Structure 2002, 10, 1619–1626. [Google Scholar] [CrossRef]
- Jardetzky, T.S.; Brown, J.H.; Gorga, J.C.; Stern, L.J.; Urban, R.G.; Chi, Y.; Stauffacher, C.; Strominger, J.; Wiley, D.C. Three-dimensional structure of a human class II histocompatibility molecule complexed with superantigen. Nature 1994, 368, 711–718. [Google Scholar] [CrossRef]
- Sundberg, E.J.; Andersen, P.S.; Schlievert, P.M.; Karjalainen, K.; Mariuzza, R.A. Structural, energetic, and functional analysis of a protein-protein interface at distinct stages of affinity maturation. Structure 2003, 11, 1151–1161. [Google Scholar] [CrossRef]
- Papageorgiou, A.C.; Collins, C.M.; Gutman, D.M.; Kline, J.B.; O’Brien, S.M.; Tranter, H.S.; Acharya, K.R. Structural basis for the recognition of superantigen streptococcal pyrogenic exotoxin A (SpeA1) by MHC class II molecules and T-cell receptors. EMBO J. 1999, 18, 9–21. [Google Scholar] [CrossRef]
- Li, Y.; Li, H.; Dimasi, N.; McCormick, J.K.; Martin, R.; Schuck, P.; Schlievert, P.M.; Mariuzza, R.A. Crystal structure of a superantigen bound to the high-affinity, zinc- dependent site on MHC class II. Immunity 2001, 14, 93–104. [Google Scholar] [CrossRef]
- Wang, X.; Xu, M.; Cai, Y.; Yang, H.; Zhang, H.; Zhang, C. Functional analysis of the disulphide loop mutant of staphylococcal enterotoxin C2. Appl. Microbiol. Biotechnol. 2009, 82, 861–871. [Google Scholar] [CrossRef]
- Hovde, C.J.; Marr, J.C.; Hoffmann, M.L.; Hackett, S.P.; Chi, Y.I.; Crum, K.K.; Stevens, D.L.; Stauffacher, C.V.; Bohach, G.A. Investigation of the role of the disulphide bond in the activity and structure of staphylococcal enterotoxin C1. Mol. Microbiol. 1994 13, 897–909.
- Schlievert, P.M.; Jablonski, L.M.; Roggiani, M.; Sadler, I.; Callantine, S.; Mitchell, D.T.; Ohlendorf, D.H.; Bohach, G.A. Pyrogenic toxin superantigen site specificity in toxic shock syndrome and food poisoning in animals. Infect. Immun. 2000, 68, 3630–3634. [Google Scholar] [CrossRef]
- Kim, J.; Urban, R.G.; Strominger, J.L.; Wiley, D.C. Toxic shock syndrome toxin-1 complexed with a class II major histocompatibility molecule HLA-DR1. Science 1994, 266, 1870–1874. [Google Scholar]
- Tiedemann, R.E.; Urban, R.J.; Strominger, J.L.; Fraser, J.D. Isolation of HLA-DR1.(staphylococcal enterotoxin A)2 trimers in solution. Proc. Natl. Acad. Sci. USA 1995, 92, 12156–12159. [Google Scholar] [CrossRef]
- Schad, E.M.; Zaitseva, I.; Zaitsev, V.N.; Dohlsten, M.; Kalland, T.; Schlievert, P.M.; Ohlendorf, D.H.; Svensson, L.A. Crystal structure of the superantigen staphylococcal enterotoxin type A. EMBO J. 1995, 14, 3292–3301. [Google Scholar]
- Sundstrom, M.; Hallen, D.; Svensson, A.; Schad, E.; Dohlsten, M.; Abrahmsen, L. The Co-crystal structure of staphylococcal enterotoxin type A with Zn2+ at 2.7 A resolution. Implications for major histocompatibility complex class II binding. J. Biol. Chem. 1996, 271, 32212–32216. [Google Scholar] [CrossRef]
- Papageorgiou, A.C.; Tranter, H.S.; Acharya, K.R. Crystal structure of microbial superantigen staphylococcal enterotoxin B at 1.5 A resolution: implications for superantigen recognition by MHC class II molecules and T-cell receptors. J. Mol. Biol. 1998, 277, 61–79. [Google Scholar] [CrossRef]
- Bonsor, D.A.; Postel, S.; Pierce, B.G.; Wang, N.; Zhu, P.; Buonpane, R.A.; Weng, Z.; Kranz, D.M.; Sundberg, E.J. Molecular basis of a million-fold affinity maturation process in a protein-protein interaction. J. Mol. Biol. 2011, 411, 321–328. [Google Scholar] [CrossRef]
- Chi, Y.I.; Sadler, I.; Jablonski, L.M.; Callantine, S.D.; Deobald, C.F.; Stauffacher, C.V.; Bohach, G.A. Zinc-mediated dimerization and its effect on activity and conformation of staphylococcal enterotoxin type C. J. Biol. Chem. 2002, 277, 22839–22846. [Google Scholar] [CrossRef]
- Cho, S.; Swaminathan, C.P.; Yang, J.; Kerzic, M.C.; Guan, R.; Kieke, M.C.; Kranz, D.M.; Mariuzza, R.A.; Sundberg, E.J. Structural basis of affinity maturation and intramolecular cooperativity in a protein-protein interaction. Structure 2005, 13, 1775–1787. [Google Scholar] [CrossRef]
- Papageorgiou, A.C.; Brehm, R.D.; Leonidas, D.D.; Tranter, H.S.; Acharya, K.R. The refined crystal structure of toxic shock syndrome toxin-1 at 2.07 A resolution. J. Mol. Biol. 1996, 260, 553–569. [Google Scholar] [CrossRef]
- Earhart, C.A.; Vath, G.M.; Roggiani, M.; Schlievert, P.M.; Ohlendorf, D.H. Structure of streptococcal pyrogenic exotoxin A reveals a novel metal cluster. Protein Sci. 2000, 9, 1847–1851. [Google Scholar] [CrossRef]
- Roussel, A.; Anderson, B.F.; Baker, H.M.; Fraser, J.D.; Baker, E.N. Crystal structure of the streptococcal superantigen SPE-C: Dimerization and zinc binding suggest a novel mode of interaction with MHC class II molecules. Nat. Struct. Biol. 1997, 4, 635–643. [Google Scholar] [CrossRef]
- Saline, M.; Rödström, K.E.; Fischer, G.; Orekhov, V.Y.; Karlsson, B.G.; Lindkvist-Petersson, K. The structure of superantigen complexed with TCR and MHC reveals novel insights into superantigenic T cell activation. Nat. Commun. 2010, 1. [Google Scholar] [CrossRef]
- Petersson, K.; Pettersson, H.; Skartved, N.J.; Walse, B.; Forsberg, G. Staphylococcal enterotoxin H induces V alpha-specific expansion of T cells. J. Immunol. 2003, 170, 4148–4154. [Google Scholar]
- Kappler, J.; Kotzin, B.; Herron, L.; Gelfand, E.W.; Bigler, R.D.; Boylston, A.; Carrel, S.; Posnett, D.N.; Choi, Y.; Marrack, P. V beta-specific stimulation of human T cells by staphylococcal toxins. Science 1989, 244, 811–813. [Google Scholar]
- Thomas, D.; Dauwalder, O.; Brun, V.; Badiou, C.; Ferry, T.; Etienne, J.; Vandenesch, F.; Lina, G. Staphylococcus aureus superantigens elicit redundant and extensive human Vbeta patterns. Infect. Immun. 2009, 77, 2043–2050. [Google Scholar] [CrossRef]
- Malchiodi, E.L.; Eisenstein, E.; Fields, B.A.; Ohlendorf, D.H.; Schlievert, P.M.; Karjalainen, K.; Mariuzza, R.A. Superantigen binding to a T cell receptor b chain of known three-dimensional structure. J. Exp. Med. 1995, 182, 1833–1845. [Google Scholar] [CrossRef]
- Khandekar, S.S.; Bettencourt, B.M.; Wyss, D.F.; Naylor, J.W.; Brauer, P.P.; Huestis, K.; Dwyer, D.S.; Profy, A.T.; Osburne, M.S.; Banerji, J.; et al. Conformational integrity and ligand binding properties of a single chain T-cell receptor expressed in Escherichia coli. J. Biol. Chem. 1997, 272, 32190–32197. [Google Scholar] [CrossRef]
- Andersen, P.S.; Geisler, C.; Buus, S.; Mariuzza, R.A.; Karjalainen, K. Role of TCR-ligand affinity in T cell activation by bacterial superantigens. J. Biol. Chem. 2001, 276, 33452–33457. [Google Scholar]
- Wang, N.; Kranz, D.M. University of Illinois: Urbana, IL, USA, Unpublished work. 2013.
- Kieke, M.C.; Shusta, E.V.; Boder, E.T.; Teyton, L.; Wittrup, K.D.; Kranz, D.M. Selection of functional T cell receptor mutants from a yeast surface- display library. Proc. Natl. Acad. Sci. USA 1999, 96, 5651–5656. [Google Scholar]
- Richman, S.A.; Aggen, D.H.; Dossett, M.L.; Donermeyer, D.L.; Allen, P.M.; Greenberg, P.D.; Kranz, D.M. Structural features of T cell receptor variable regions that enhance domain stability and enable expression as single-chain ValphaVbeta fragments. Mol. Immunol. 2009, 46, 902–916. [Google Scholar] [CrossRef]
- Shusta, E.V.; Kieke, M.C.; Parke, E.; Kranz, D.M.; Wittrup, K.D. Yeast polypeptide fusion surface display levels predict thermal stability and soluble secretion efficiency. J. Mol. Biol. 1999, 292, 949–956. [Google Scholar] [CrossRef]
- Sundberg, E.J.; Deng, L.; Mariuzza, R.A. TCR recognition of peptide/MHC class II complexes and superantigens. Semin. Immunol. 2007, 19, 262–271. [Google Scholar] [CrossRef]
- Nur-ur Rahman, A.K.; Bonsor, D.A.; Herfst, C.A.; Pollard, F.; Peirce, M.; Wyatt, A.W.; Kasper, K.J.; Madrenas, J.; Sundberg, E.J.; McCormick, J.K. The T cell receptor beta-chain second complementarity determining region loop (CDR2beta) governs T cell activation and Vbeta specificity by bacterial superantigens. J. Biol. Chem. 2011, 286, 4871–4881. [Google Scholar]
- Moza, B.; Buonpane, R.A.; Zhu, P.; Herfst, C.A.; Rahman, A.K.; McCormick, J.K.; Kranz, D.M.; Sundberg, E.J. Long-range cooperative binding effects in a T cell receptor variable domain. Proc. Natl. Acad. Sci. USA 2006, 103, 9867–9872. [Google Scholar] [CrossRef]
- Churchill, H.R.; Andersen, P.S.; Parke, E.A.; Mariuzza, R.A.; Kranz, D.M. Mapping the energy of superantigen Staphylococcus enterotoxin C3 recognition of an alpha/beta T cell receptor using alanine scanning mutagenesis. J. Exp. Med. 2000, 191, 835–846. [Google Scholar] [CrossRef]
- Yang, J.; Swaminathan, C.P.; Huang, Y.; Guan, R.; Cho, S.; Kieke, M.C.; Kranz, D.M.; Mariuzza, R.A.; Sundberg, E.J. Dissecting cooperative and additive binding energetics in the affinity maturation pathway of a protein-protein interface. J. Biol. Chem. 2003, 278, 50412–50421. [Google Scholar] [CrossRef]
- Hong-Geller, E.; Mollhoff, M.; Shiflett, P.R.; Gupta, G. Design of chimeric receptor mimics with different TcRVbeta isoforms. Type-specific inhibition of superantigen pathogenesis. J. Biol. Chem. 2004, 279, 5676–5684. [Google Scholar]
- Yang, X.; Buonpane, R.A.; Moza, B.; Rahman, A.K.; Wang, N.; Schlievert, P.M.; McCormick, J.K.; Sundberg, E.J.; Kranz, D.M. Neutralization of multiple staphylococcal superantigens by a single-chainprotein consisting of affinity-matured, variable domain repeats. J. Infect. Dis. 2008, 198, 344–348. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sharma, P.; Wang, N.; Kranz, D.M. Soluble T Cell Receptor Vβ Domains Engineered for High-Affinity Binding to Staphylococcal or Streptococcal Superantigens. Toxins 2014, 6, 556-574. https://doi.org/10.3390/toxins6020556
Sharma P, Wang N, Kranz DM. Soluble T Cell Receptor Vβ Domains Engineered for High-Affinity Binding to Staphylococcal or Streptococcal Superantigens. Toxins. 2014; 6(2):556-574. https://doi.org/10.3390/toxins6020556
Chicago/Turabian StyleSharma, Preeti, Ningyan Wang, and David M. Kranz. 2014. "Soluble T Cell Receptor Vβ Domains Engineered for High-Affinity Binding to Staphylococcal or Streptococcal Superantigens" Toxins 6, no. 2: 556-574. https://doi.org/10.3390/toxins6020556