Immunogenicity of a West Nile Virus DIII-Cholera Toxin A2/B Chimera after Intranasal Delivery

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Expression and Characterization of the DIII-CTA2/B Chimera

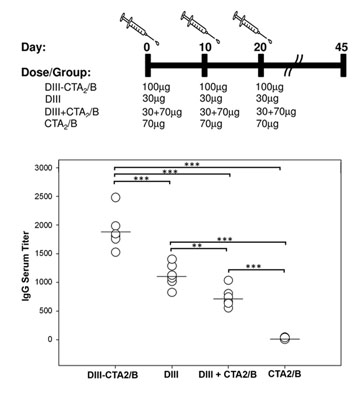
2.2. Immunogenicity after Intranasal Delivery





3. Experimental Section
3.1. Ethics Statement
3.2. Bacterial Strains
3.3. Plasmid Construction
3.4. Expression and Purification of Proteins
3.5. Protein Analysis and Western Blot
3.6. Ganglioside GM1 ELISA Assay
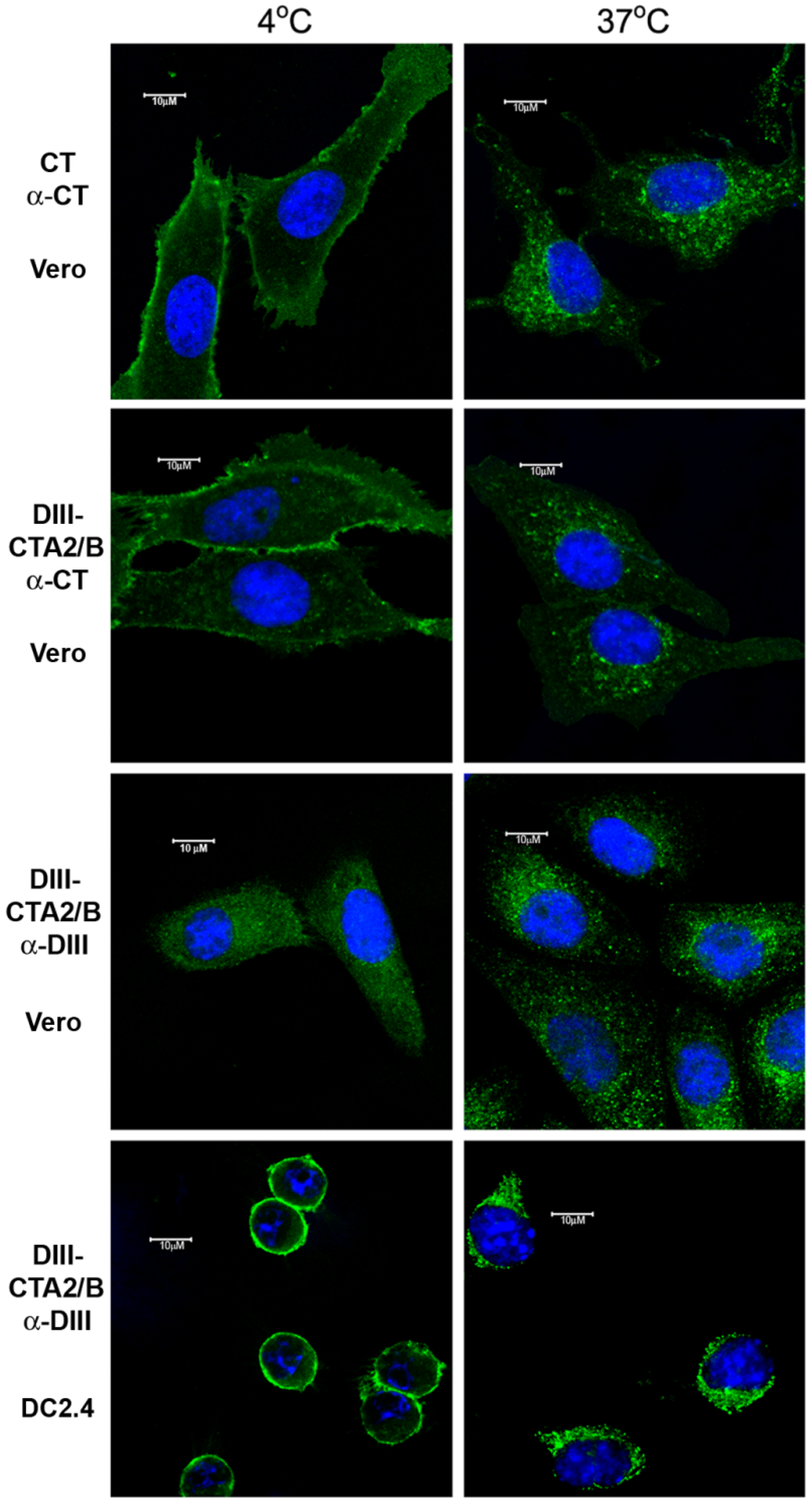
3.7. Cell Culture and Confocal Microscopy
3.8. Mouse Immunizations and Sample Collection
3.9. IgG, IgM and IgA ELISAs
3.10. Serum Bactericidal Assays
3.11. Statistical Analysis
4. Discussion and Conclusions
Acknowledgments
Authors Contributions
Conflicts of Interest
References and Notes
- Smithburn, K.C.; Hughes, T.P.; Burke, A.W.; Paul, K.C. A neuotropic virus isolated from the blood of a native of Uganda. Am. J. Trop. Med. 1940, 20, 471–492. [Google Scholar]
- Lanciotti, R.S.; Roehrig, J.T.; Deubel, V.; Smith, J.; Parker, M.; Steele, K.; Crise, B.; Volpe, K.E.; Crabtree, M.B.; Scherret, J.H.; et al. Origin of the West Nile virus responsible for an outbreak of encephalitis in the northeastern United States. Science 1999, 286, 2333–2337. [Google Scholar] [CrossRef]
- Lindsey, N.P.; Lehman, J.A.; Staples, E.; Fischer, M. West Nile virus and other arboviral diseases—United States, 2012. MMWR Morb Mortal Wkly. Rep. 2013, 62, 513–517. [Google Scholar]
- Beasley, D.W.; Barrett, A.D.; Tesh, R.B. Resurgence of West Nile neurologic disease in the United States in 2012: What happened? What needs to be done? Antiviral Res. 2013, 99, 1–5. [Google Scholar] [CrossRef]
- Sejvar, J.J.; Haddad, M.B.; Tierney, B.C.; Campbell, G.L.; Marfin, A.A.; Van Gerpen, J.A.; Fleischauer, A.; Leis, A.A.; Stokic, D.S.; Petersen, L.R. Neurologic manifestations and outcome of West Nile virus infection. Jama 2003, 290, 511–515. [Google Scholar] [CrossRef]
- Petersen, L.R.; Marfin, A.A. West Nile virus: A primer for the clinician. Ann. Intern. Med. 2002, 137, 173–179. [Google Scholar] [CrossRef]
- Hayes, E.B.; Sejvar, J.J.; Zaki, S.R.; Lanciotti, R.S.; Bode, A.V.; Campbell, G.L. Virology, pathology, and clinical manifestations of West Nile virus disease. Emerg. Infect. Dis. 2005, 11, 1174–1179. [Google Scholar] [CrossRef]
- Lindsey, N.P.; Staples, J.E.; Lehman, J.A.; Fischer, M. Medical risk factors for severe west nile virus disease, United States, 2008–2010. Am. J. Trop. Med. Hyg. 2012, 87, 179–184. [Google Scholar] [CrossRef]
- Lindsey, N.P.; Sejvar, J.J.; Bode, A.V.; Pape, W.J.; Campbell, G.L. Delayed mortality in a cohort of persons hospitalized with West Nile virus disease in Colorado in 2003. Vector Borne Zoonotic Dis. 2012, 12, 230–235. [Google Scholar] [CrossRef]
- Diamond, M.S.; Shrestha, B.; Marri, A.; Mahan, D.; Engle, M. B cells and antibody play critical roles in the immediate defense of disseminated infection by West Nile encephalitis virus. J. Virol. 2003, 77, 2578–2586. [Google Scholar] [CrossRef]
- Byrne, S.N.; Halliday, G.M.; Johnston, L.J.; King, N.J. Interleukin-1beta but not tumor necrosis factor is involved in West Nile virus-induced Langerhans cell migration from the skin in C57BL/6 mice. J. Invest. Dermatol. 2001, 117, 702–709. [Google Scholar] [CrossRef]
- Johnston, L.J.; Halliday, G.M.; King, N.J. Langerhans cells migrate to local lymph nodes following cutaneous infection with an arbovirus. J. Invest. Dermatol. 2000, 114, 560–568. [Google Scholar] [CrossRef]
- Diamond, M.S.; Shrestha, B.; Mehlhop, E.; Sitati, E.; Engle, M. Innate and adaptive immune responses determine protection against disseminated infection by West Nile encephalitis virus. Viral Immunol. 2003, 16, 259–278. [Google Scholar] [CrossRef]
- Diamond, M.S.; Sitati, E.M.; Friend, L.D.; Higgs, S.; Shrestha, B.; Engle, M. A critical role for induced IgM in the protection against West Nile virus infection. J. Exp. Med. 2003, 198, 1853–1862. [Google Scholar] [CrossRef]
- Suthar, M.S.; Diamond, M.S.; Gale, M., Jr. West Nile virus infection and immunity. Nat. Rev. Microbiol. 2013, 11, 115–128. [Google Scholar] [CrossRef]
- Samuel, M.A.; Diamond, M.S. Pathogenesis of West Nile Virus infection: A balance between virulence, innate and adaptive immunity, and viral evasion. J. Virol. 2006, 80, 9349–9360. [Google Scholar] [CrossRef]
- Shrestha, B.; Ng, T.; Chu, H.J.; Noll, M.; Diamond, M.S. The relative contribution of antibody and CD8+ T cells to vaccine immunity against West Nile encephalitis virus. Vaccine 2008, 26, 2020–2033. [Google Scholar] [CrossRef]
- Diamond, M.S.; Pierson, T.C.; Fremont, D.H. The structural immunology of antibody protection against West Nile virus. Immunol. Rev. 2008, 225, 212–225. [Google Scholar] [CrossRef]
- Chu, J.J.; Rajamanonmani, R.; Li, J.; Bhuvanakantham, R.; Lescar, J.; Ng, M.L. Inhibition of West Nile virus entry by using a recombinant domain III from the envelope glycoprotein. J. Gen. Virol. 2005, 86, 405–412. [Google Scholar] [CrossRef]
- Sanchez, M.D.; Pierson, T.C.; McAllister, D.; Hanna, S.L.; Puffer, B.A.; Valentine, L.E.; Murtadha, M.M.; Hoxie, J.A.; Doms, R.W. Characterization of neutralizing antibodies to West Nile virus. Virology 2005, 336, 70–82. [Google Scholar] [CrossRef]
- Nybakken, G.E.; Oliphant, T.; Johnson, S.; Burke, S.; Diamond, M.S.; Fremont, D.H. Structural basis of West Nile virus neutralization by a therapeutic antibody. Nature 2005, 437, 764–769. [Google Scholar] [CrossRef]
- Beasley, D.W.; Barrett, A.D. Identification of neutralizing epitopes within structural domain III of the West Nile virus envelope protein. J. Virol. 2002, 76, 13097–13100. [Google Scholar] [CrossRef]
- Oliphant, T.; Nybakken, G.E.; Austin, S.K.; Xu, Q.; Bramson, J.; Loeb, M.; Throsby, M.; Fremont, D.H.; Pierson, T.C.; Diamond, M.S. Induction of epitope-specific neutralizing antibodies against West Nile virus. J. Virol. 2007, 81, 11828–11839. [Google Scholar] [CrossRef]
- Beasley, D.W. Vaccines and immunotherapeutics for the prevention and treatment of infections with West Nile virus. Immunotherapy 2011, 3, 269–285. [Google Scholar] [CrossRef]
- Iyer, A.V.; Kousoulas, K.G. A review of vaccine approaches for West Nile virus. Int. J. Environ. Res. Public Health 2013, 10, 4200–4223. [Google Scholar] [CrossRef]
- Brandler, S.; Tangy, F. Vaccines in development against West Nile virus. Viruses 2013, 5, 2384–2409. [Google Scholar] [CrossRef]
- Monath, T.P.; Liu, J.; Kanesa-Thasan, N.; Myers, G.A.; Nichols, R.; Deary, A.; McCarthy, K.; Johnson, C.; Ermak, T.; Shin, S.; et al. A live, attenuated recombinant West Nile virus vaccine. Proc. Natl. Acad. Sci. USA 2006, 103, 6694–6699. [Google Scholar] [CrossRef]
- Beasley, D.W. Safety and immunogenicity of a chimeric vaccine for West Nile virus in aged subjects. Expert Rev. Vaccines 2011, 10, 601–604. [Google Scholar] [CrossRef]
- Biedenbender, R.; Bevilacqua, J.; Gregg, A.M.; Watson, M.; Dayan, G. Phase II, randomized, double-blind, placebo-controlled, multicenter study to investigate the immunogenicity and safety of a West Nile virus vaccine in healthy adults. J. Infect. Dis. 2011, 203, 75–84. [Google Scholar] [CrossRef]
- Brandler, S.; Marianneau, P.; Loth, P.; Lacote, S.; Combredet, C.; Frenkiel, M.P.; Despres, P.; Contamin, H.; Tangy, F. Measles vaccine expressing the secreted form of West Nile virus envelope glycoprotein induces protective immunity in squirrel monkeys, a new model of West Nile virus infection. J. Infect. Dis. 2012, 206, 212–219. [Google Scholar] [CrossRef]
- Martina, B.E.; van den Doel, P.; Koraka, P.; van Amerongen, G.; Spohn, G.; Haagmans, B.L.; Provacia, L.B.; Osterhaus, A.D.; Rimmelzwaan, G.F. A recombinant influenza A virus expressing domain III of West Nile virus induces protective immune responses against influenza and West Nile virus. PLoS ONE 2011, 6, e18995. [Google Scholar] [CrossRef]
- Schepp-Berglind, J.; Luo, M.; Wang, D.; Wicker, J.A.; Raja, N.U.; Hoel, B.D.; Holman, D.H.; Barrett, A.D.; Dong, J.Y. Complex adenovirus-mediated expression of West Nile virus C, PreM, E, and NS1 proteins induces both humoral and cellular immune responses. Clin. Vaccine Immunol. 2007, 14, 1117–1126. [Google Scholar] [CrossRef]
- Mason, P.W.; Shustov, A.V.; Frolov, I. Production and characterization of vaccines based on flaviviruses defective in replication. Virology 2006, 351, 432–443. [Google Scholar] [CrossRef]
- Wang, T.; Anderson, J.F.; Magnarelli, L.A.; Wong, S.J.; Koski, R.A.; Fikrig, E. Immunization of mice against West Nile virus with recombinant envelope protein. J. Immunol. 2001, 167, 5273–5277. [Google Scholar]
- Martina, B.E.; Koraka, P.; van den Doel, P.; van Amerongen, G.; Rimmelzwaan, G.F.; Osterhaus, A.D. Immunization with West Nile virus envelope domain III protects mice against lethal infection with homologous and heterologous virus. Vaccine 2008, 26, 153–157. [Google Scholar] [CrossRef]
- Chu, J.H.; Chiang, C.C.; Ng, M.L. Immunization of flavivirus West Nile recombinant envelope domain III protein induced specific immune response and protection against West Nile virus infection. J. Immunol. 2007, 178, 2699–2705. [Google Scholar]
- Zlatkovic, J.; Stiasny, K.; Heinz, F.X. Immunodominance and functional activities of antibody responses to inactivated West Nile virus and recombinant subunit vaccines in mice. J. Virol. 2011, 85, 1994–2003. [Google Scholar] [CrossRef]
- Lieberman, M.M.; Nerurkar, V.R.; Luo, H.; Cropp, B.; Carrion, R., Jr.; de la Garza, M.; Coller, B.A.; Clements, D.; Ogata, S.; Wong, T.; et al. Immunogenicity and protective efficacy of a recombinant subunit West Nile virus vaccine in rhesus monkeys. Clin. Vaccine Immunol. 2009, 16, 1332–1337. [Google Scholar] [CrossRef]
- Watts, D.M.; Tesh, R.B.; Siirin, M.; Rosa, A.T.; Newman, P.C.; Clements, D.E.; Ogata, S.; Coller, B.A.; Weeks-Levy, C.; Lieberman, M.M. Efficacy and durability of a recombinant subunit West Nile vaccine candidate in protecting hamsters from West Nile encephalitis. Vaccine 2007, 25, 2913–2918. [Google Scholar] [CrossRef]
- Pizza, M.; Giuliani, M.M.; Fontana, M.R.; Monaci, E.; Douce, G.; Dougan, G.; Mills, K.H.G.; Rappuoli, R.; Del Giudice, G. Mucosal vaccines: Non toxic derivatives of LT and CT as mucosal adjuvants. Vaccine 2001, 19, 2534–2541. [Google Scholar] [CrossRef]
- Salmond, R.J.; Luross, J.A.; Williams, N.A. Immune modulation by the cholera-like enterotoxins. Expert Rev. Mol. Med. 2002, 1–16. [Google Scholar]
- Holmgren, J.; Adamsson, J.; Anjuere, F.; Clemens, J.; Czerkinsky, C.; Eriksson, K.; Flach, C.F.; George-Chandy, A.; Harandi, A.M.; Lebens, M.; et al. Mucosal adjuvants and anti-infection and anti-immunopathology vaccines based on cholera toxin, cholera toxin B subunit and CpG DNA. Immunol. Lett. 2005, 97, 181–188. [Google Scholar] [CrossRef]
- Apte, S.H.; Redmond, A.M.; Groves, P.L.; Schussek, S.; Pattinson, D.J.; Doolan, D.L. Subcutaneous cholera toxin exposure induces potent CD103(+) dermal dendritic cell activation and migration. Eur. J. Immunol. 2013, 43, 2707–2717. [Google Scholar] [CrossRef]
- Cong, Y.; Weaver, C.T.; Elson, C.O. The mucosal adjuvanticity of cholera toxin involves enhancement of costimulatory activity by selective up-regulation of B7.2 expression. J. Immunol. 1997, 159, 5301–5308. [Google Scholar]
- Hajishengallis, G.; Arce, S.; Gockel, C.M.; Connell, T.D.; Russell, M.W. Immunomodulation with enterotoxins for the generation of secretory immunity or tolerance: Applications for oral infections. J. Dent. Res. 2005, 84, 1104–1116. [Google Scholar] [CrossRef]
- Eriksson, K.; Fredriksson, M.; Nordstrom, I.; Holmgren, J. Cholera toxin and its B subunit promote dendritic cell vaccination with different influences on Th1 and Th2 development. Infect. Immun. 2003, 71, 1740–1747. [Google Scholar] [CrossRef]
- Schnitzler, A.C.; Burke, J.M.; Wetzler, L.M. Induction of cell signaling events by the cholera toxin B subunit in antigen-presenting cells. Infect. Immun. 2007, 75, 3150–3159. [Google Scholar] [CrossRef]
- Holmgren, J.; Czerkinsky, C.; Lycke, N.; Svennerholm, A.M. Strategies for the induction of immune responses at mucosal surfaces making use of cholera toxin B subunit as immunogen, carrier, and adjuvant. Am. J. Trop. Med. Hyg. 1994, 50, 42–54. [Google Scholar]
- Langridge, W.; Denes, B.; Fodor, I. Cholera toxin B subunit modulation of mucosal vaccines for infectious and autoimmune diseases. Curr. Opin. Investig. Drugs 2010, 11, 919–928. [Google Scholar]
- George-Chandy, A.; Eriksson, K.; Lebens, M.; Nordstrom, I.; Schon, E.; Holmgren, J. Cholera toxin B subunit as a carrier molecule promotes antigen presentation and increases CD40 and CD86 expression on antigen-presenting cells. Infect. Immun. 2001, 69, 5716–5725. [Google Scholar] [CrossRef]
- Jobling, M.G.; Holmes, R.K. Fusion proteins containing the A2 domain of cholera toxin assemble with B polypeptides of cholera toxin to form immunoreactive and functional holotoxin-like chimeras. Infect. Immun. 1992, 60, 4915–4924. [Google Scholar]
- Hajishengallis, G.; Hollingshead, S.K.; Koga, T.; Russell, M.W. Mucosal immunization with a bacterial protein antigen genetically coupled to cholera toxin A2/B subunits. J. Immunol. 1995, 154, 4322–4332. [Google Scholar]
- Gockel, C.M.; Russell, M.W. Induction and recall of immune memory by mucosal immunization with a non-toxic recombinant enterotoxin-based chimeric protein. Immunology 2005, 116, 477–486. [Google Scholar]
- Hajishengallis, G.; Michalek, S.M.; Russell, M.W. Persistence of serum and salivary antibody responses after oral immunization with a bacterial protein antigen genetically linked to the A2/B subunits of cholera toxin. Infect. Immun. 1996, 64, 665–667. [Google Scholar]
- Lee, S.F.; Halperin, S.A.; Salloum, D.F.; MacMillan, A.; Morris, A. Mucosal immunization with a genetically engineered pertussis toxin S1 fragment-cholera toxin subunit B chimeric protein. Infect. Immun. 2003, 71, 2272–2275. [Google Scholar] [CrossRef]
- Arlian, B.M.; Tinker, J.K. Mucosal immunization with a Staphylococcus aureus IsdA-cholera toxin A2/B chimera induces antigen-specific Th2-type responses in mice. Clin. Vaccine Immunol. 2011, 18, 1543–1551. [Google Scholar] [CrossRef]
- Tinker, J.K.; Erbe, J.L.; Holmes, R.K. Characterization of fluorescent chimeras of cholera toxin and Escherichia coli heat-labile enterotoxins produced by use of the twin arginine translocation system. Infect. Immun. 2005, 73, 3627–3635. [Google Scholar] [CrossRef]
- Tinker, J.K.; Davis, C.T.; Arlian, B.M. Purification and characterization of Yersinia enterocolitica and Yersinia pestis LcrV-cholera toxin A(2)/B chimeras. Protein Expr. Purif. 2010, 74, 16–23. [Google Scholar] [CrossRef]
- Majoul, I.V.; Bastiaens, P.I.; Soling, H.D. Transport of an external Lys-Asp-Glu-Leu (KDEL) protein from the plasma membrane to the endoplasmic reticulum: studies with cholera toxin in Vero cells. J. Cell. Biol. 1996, 133, 777–789. [Google Scholar] [CrossRef]
- Tinker, J.K. Boise State University: Boise, ID, USA, Unpublished work. 2013.
- Anosova, N.G.; Chabot, S.; Shreedhar, V.; Borawski, J.A.; Dickinson, B.L.; Neutra, M.R.; Neutra, M.R. Cholera toxin, E. coli heat-labile toxin, and non-toxic derivatives induce dendritic cell migration into the follicle-associated epithelium of Peyer’s patches. Mucosal Immunol. 2008, 1, 59–67. [Google Scholar] [CrossRef]
- Kawamura, Y.I.; Kawashima, R.; Shirai, Y.; Kato, R.; Hamabata, T.; Yamamoto, M.; Furukawa, K.; Fujihashi, K.; McGhee, J.R.; Hayashi, H.; et al. Cholera toxin activates dendritic cells through dependence on GM1-ganglioside which is mediated by NF-kappaB translocation. Eur. J. Immunol. 2003, 33, 3205–3212. [Google Scholar] [CrossRef]
- Bastiaens, P.I.; Majoul, I.V.; Verveer, P.J.; Soling, H.D.; Jovin, T.M. Imaging the intracellular trafficking and state of the AB5 quaternary structure of cholera toxin. Embo J. 1996, 15, 4246–4253. [Google Scholar]
- Li, X.; Erbe, J.L.; Lockatell, C.V.; Johnson, D.E.; Jobling, M.G.; Holmes, R.K.; Mobley, H.L. Use of translational fusion of the MrpH fimbrial adhesin-binding domain with the cholera toxin A2 domain, coexpressed with the cholera toxin B subunit, as an intranasal vaccine to prevent experimental urinary tract infection by Proteus mirabilis. Infect. Immun. 2004, 72, 7306–7310. [Google Scholar] [CrossRef]
- Sultan, F.; Jin, L.L.; Jobling, M.G.; Holmes, R.K.; Stanley, S.L., Jr. Mucosal immunogenicity of a holotoxin-like molecule containing the serine-rich Entamoeba histolytica protein (SREHP) fused to the A2 domain of cholera toxin. Infect. Immun. 1998, 66, 462–468. [Google Scholar]
- Marinaro, M.; Staats, H.F.; Hiroi, T.; Jackson, R.J.; Coste, M.; Boyaka, P.N.; Okahashi, N.; Yamamoto, M.; Kiyono, H.; Bluethmann, H.; et al. Mucosal adjuvant effect of cholera toxin in mice results from induction of T helper 2 (Th2) cells and IL-4. J. Immunol. 1995, 155, 4621–4629. [Google Scholar]
- Yamamoto, S.; Kiyono, H.; Yamamoto, M.; Imaoka, K.; Fujihashi, K.; Van Ginkel, F.W.; Noda, M.; Takeda, Y.; McGhee, J.R. A nontoxic mutant of cholera toxin elicits Th2-type responses for enhanced mucosal immunity. Proc. Natl. Acad. Sci. USA 1997, 94, 5267–5272. [Google Scholar] [CrossRef]
- Singh, S.R.; Dennis, V.A.; Carter, C.L.; Pillai, S.R.; Jefferson, A.; Sahi, S.V.; Moore, E.G. Immunogenicity and efficacy of recombinant RSV-F vaccine in a mouse model. Vaccine 2007, 25, 6211–6223. [Google Scholar] [CrossRef]
- Cong, H.; Gu, Q.M.; Yin, H.E.; Wang, J.W.; Zhao, Q.L.; Zhou, H.Y.; Li, Y.; Zhang, J.Q. Multi-epitope DNA vaccine linked to the A2/B subunit of cholera toxin protect mice against Toxoplasma gondii. Vaccine 2008, 26, 3913–3921. [Google Scholar] [CrossRef]
- Ekong, E.E.; Okenu, D.N.; Mania-Pramanik, J.; He, Q.; Igietseme, J.U.; Ananaba, G.A.; Lyn, D.; Black, C.; Eko, F.O. A Vibrio cholerae ghost-based subunit vaccine induces cross-protective chlamydial immunity that is enhanced by CTA2B, the nontoxic derivative of cholera toxin. FEMS Immunol. Med. Microbiol. 2009, 55, 280–291. [Google Scholar] [CrossRef]
- Mehlhop, E.; Whitby, K.; Oliphant, T.; Marri, A.; Engle, M.; Diamond, M.S. Complement activation is required for induction of a protective antibody response against West Nile virus infection. J. Virol. 2005, 79, 7466–7477. [Google Scholar] [CrossRef]
- Lieberman, M.M.; Clements, D.E.; Ogata, S.; Wang, G.; Corpuz, G.; Wong, T.; Martyak, T.; Gilson, L.; Coller, B.A.; Leung, J.; et al. Preparation and immunogenic properties of a recombinant West Nile subunit vaccine. Vaccine 2007, 25, 414–423. [Google Scholar] [CrossRef]
- Ledizet, M.; Kar, K.; Foellmer, H.G.; Wang, T.; Bushmich, S.L.; Anderson, J.F.; Fikrig, E.; Koski, R.A. A recombinant envelope protein vaccine against West Nile virus. Vaccine 2005, 23, 3915–3924. [Google Scholar] [CrossRef]
- Lee, J.W.; Chu, J.J.; Ng, M.L. Quantifying the specific binding between West Nile virus envelope domain III protein and the cellular receptor alphaVbeta3 integrin. J. Biol. Chem. 2006, 281, 1352–1360. [Google Scholar] [CrossRef]
- Chu, J.J.; Ng, M.L. Interaction of West Nile virus with alpha v beta 3 integrin mediates virus entry into cells. J. Biol. Chem. 2004, 279, 54533–54541. [Google Scholar] [CrossRef]
- Schmidt, K.; Keller, M.; Bader, B.L.; Korytar, T.; Finke, S.; Ziegler, U.; Groschup, M.H. Integrins modulate the infection efficiency of West Nile virus into cells. J. Gen. Virol. 2013, 94, 1723–1733. [Google Scholar] [CrossRef]
- Medigeshi, G.R.; Hirsch, A.J.; Streblow, D.N.; Nikolich-Zugich, J.; Nelson, J.A. West Nile virus entry requires cholesterol-rich membrane microdomains and is independent of alphavbeta3 integrin. J. Virol. 2008, 82, 5212–5219. [Google Scholar] [CrossRef]
- Okada, N.; Tsukada, Y.; Nakagawa, S.; Mizuguchi, H.; Mori, K.; Saito, T.; Fujita, T.; Yamamoto, A.; Hayakawa, T.; Mayumi, T. Efficient gene delivery into dendritic cells by fiber-mutant adenovirus vectors. Biochem. Biophys. Res. Commun. 2001, 282, 173–179. [Google Scholar] [CrossRef]
- Harui, A.; Roth, M.D.; Sanghvi, M.; Vira, D.; Mizuguchi, H.; Basak, S.K. Centrifugation enhances integrin-mediated transduction of dendritic cells by conventional and RGD-modified adenoviral vectors. J. Immunol. Methods 2006, 312, 94–104. [Google Scholar]
- Gianni, T.; Campadelli-Fiume, G. AlphaVbeta3-integrin relocalizes nectin1 and routes herpes simplex virus to lipid rafts. J. Virol. 2012, 86, 2850–2855. [Google Scholar] [CrossRef]
- Gianni, T.; Gatta, V.; Campadelli-Fiume, G. AlphaVbeta3-integrin routes herpes simplex virus to an entry pathway dependent on cholesterol-rich lipid rafts and dynamin2. Proc. Natl. Acad. Sci. USA 2010, 107, 22260–22265. [Google Scholar]
- Sanchez, J.; Holmgren, J. Cholera toxin—A foe & a friend. Indian J. Med. Res. 2011, 133, 153–163. [Google Scholar]
- Sun, J.B.; Czerkinsky, C.; Holmgren, J. Mucosally induced immunological tolerance, regulatory T cells and the adjuvant effect by cholera toxin B subunit. Scand. J. Immunol. 2010, 71, 1–11. [Google Scholar] [CrossRef]
- Hagiwara, Y.; Iwasaki, T.; Asanuma, H.; Sato, Y.; Sata, T.; Aizawa, C.; Kurata, T.; Tamura, S. Effects of intranasal administration of cholera toxin (or Escherichia coli heat-labile enterotoxin) B subunits supplemented with a trace amount of the holotoxin on the brain. Vaccine 2001, 19, 1652–1660. [Google Scholar] [CrossRef]
- Van Ginkel, F.W.; Jackson, R.J.; Yuki, Y.; McGhee, J.R. Cutting edge: The mucosal adjuvant cholera toxin redirects vaccine proteins into olfactory tissues. J. Immunol. 2000, 165, 4778–4782. [Google Scholar]
- Van Ginkel, F.W.; Jackson, R.J.; Yoshino, N.; Hagiwara, Y.; Metzger, D.J.; Connell, T.D.; Vu, H.L.; Martin, M.; Fujihashi, K.; McGhee, J.R. Enterotoxin-based mucosal adjuvants alter antigen trafficking and induce inflammatory responses in the nasal tract. Infect. Immun. 2005, 73, 6892–6902. [Google Scholar] [CrossRef]
- Lewis, D.J.; Huo, Z.; Barnett, S.; Kromann, I.; Giemza, R.; Galiza, E.; Woodrow, M.; Thierry-Carstensen, B.; Andersen, P.; Novicki, D.; et al. Transient facial nerve paralysis (Bell’s palsy) following intranasal delivery of a genetically detoxified mutant of Escherichia coli heat labile toxin. PLoS ONE 2009, 4, e6999. [Google Scholar] [CrossRef]
- Mutsch, M.; Zhou, W.; Rhodes, P.; Bopp, M.; Chen, R.T.; Linder, T.; Spyr, C.; Steffen, R. Use of the inactivated intranasal influenza vaccine and the risk of Bell's palsy in Switzerland. N. Engl. J. Med. 2004, 350, 896–903. [Google Scholar] [CrossRef]
- Cuburu, N.; Kweon, M.N.; Song, J.H.; Hervouet, C.; Luci, C.; Sun, J.B.; Hofman, P.; Holmgren, J.; Anjuere, F.; Czerkinsky, C. Sublingual immunization induces broad-based systemic and mucosal immune responses in mice. Vaccine 2007, 25, 8598–8610. [Google Scholar] [CrossRef]
- Tacket, C.O.; Sack, D.A. Cholera Vaccines; Elsievier: Philadelphia, PA, USA, 2008; Volume 5, pp. 127–138. [Google Scholar]
- Czerkinsky, C.; Cuburu, N.; Kweon, M.N.; Anjuere, F.; Holmgren, J. Sublingual vaccination. Hum. Vaccin 2011, 7, 110–114. [Google Scholar] [CrossRef]
- Anjuere, F.; George-Chandy, A.; Audant, F.; Rousseau, D.; Holmgren, J.; Czerkinsky, C. Transcutaneous immunization with cholera toxin B subunit adjuvant suppresses IgE antibody responses via selective induction of Th1 immune responses. J. Immunol. 2003, 170, 1586–1592. [Google Scholar]
- Berry, L.J.; Hickey, D.K.; Skelding, K.A.; Bao, S.; Rendina, A.M.; Hansbro, P.M.; Gockel, C.M.; Beagley, K.W. Transcutaneous immunization with combined cholera toxin and CpG adjuvant protects against Chlamydia muridarum genital tract infection. Infect. Immun. 2004, 72, 1019–1028. [Google Scholar] [CrossRef]
- Czerkinsky, C.; Russell, M.W.; Lycke, N.; Lindblad, M.; Holmgren, J. Oral administration of a streptococcal antigen coupled to cholera toxin B subunit evokes strong antibody responses in salivary glands and extramucosal tissues. Infect. Immun. 1989, 57, 1072–1077. [Google Scholar]
- Hervouet, C.; Luci, C.; Cuburu, N.; Cremel, M.; Bekri, S.; Vimeux, L.; Maranon, C.; Czerkinsky, C.; Hosmalin, A.; Anjuere, F. Sublingual immunization with an HIV subunit vaccine induces antibodies and cytotoxic T cells in the mouse female genital tract. Vaccine 2010, 28, 5582–5590. [Google Scholar] [CrossRef]
- Norton, E.B.; Lawson, L.B.; Freytag, L.C.; Clements, J.D. Characterization of a mutant Escherichia coli heat-labile toxin, LT(R192G/L211A), as a safe and effective oral adjuvant. Clin. Vaccine Immunol. 2011, 18, 546–551. [Google Scholar] [CrossRef]
- Eypper, E.H.; Johnson, P.V.; Purro, E.I.; Hohmann, E.L. Transcutaneous immunization of healthy volunteers with an attenuated Listeria monocytogenes vaccine strain and cholera toxin adjuvant. Vaccine 2013, 31, 3257–3261. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tinker, J.K.; Yan, J.; Knippel, R.J.; Panayiotou, P.; Cornell, K.A. Immunogenicity of a West Nile Virus DIII-Cholera Toxin A2/B Chimera after Intranasal Delivery. Toxins 2014, 6, 1397-1418. https://doi.org/10.3390/toxins6041397
Tinker JK, Yan J, Knippel RJ, Panayiotou P, Cornell KA. Immunogenicity of a West Nile Virus DIII-Cholera Toxin A2/B Chimera after Intranasal Delivery. Toxins. 2014; 6(4):1397-1418. https://doi.org/10.3390/toxins6041397
Chicago/Turabian StyleTinker, Juliette K., Jie Yan, Reece J. Knippel, Panos Panayiotou, and Kenneth A. Cornell. 2014. "Immunogenicity of a West Nile Virus DIII-Cholera Toxin A2/B Chimera after Intranasal Delivery" Toxins 6, no. 4: 1397-1418. https://doi.org/10.3390/toxins6041397